
Five coordinated Mn in Ba4Mn2Si2Te9: synthesis, crystal structure, physical properties, and electronic structure†

Abstract
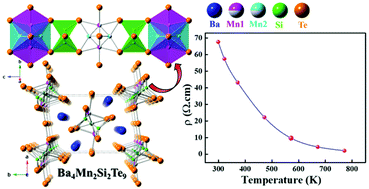
We report the synthesis of single-crystals of a new transition metal-containing quaternary chalcogenide, Ba4Mn2Si2Te9, synthesized by the solid-state method at 1273 K. A single-crystal X-ray diffraction study shows that it crystallizes in the orthorhombic crystal system (space group: Pbam) with cell constants of a = 13.4690(6) Å, b = 8.7223(4) Å, and c = 10.0032(4) Å. The asymmetric unit of the structure consists of eight unique crystallographic sites: one Ba, two Mn, one Si, and four Te sites. In this structure, the two Mn sites, Mn(1) and Mn(2), are disordered, each with fractional occupancy of 50%. The short distance of 2.170(3) Å between Mn(1) and Mn(2) implies that both Mn sites are not occupied simultaneously. The Mn atoms show two types of polyhedra: unique Mn(1)Te5 units along with traditional Mn(2)Te4 tetrahedra. The main motifs of the Ba4Mn2Si2Te9 structure are dimeric Si2Te6 units (with Si–Si single bond), Mn(1)Te5, and Mn(2)Te4 polyhedra. The structure can be described as pseudo-two-dimensional if only Mn(1) atoms are present and one-dimensional when only Mn(2) atoms are filled in the structure. The extended 2∞[Mn(1)Si2Te9]10− layers and 1∞[Mn(2)Si2Te8]8− chains are separated by Ba2+ cations. The direct bandgap for the polycrystalline Ba4Mn2Si2Te9 sample is 0.6(1) eV, as determined from an optical absorption study consistent with the sample's black color. The resistivity study of the polycrystalline Ba4Mn2Si2Te9 also confirms the semiconducting behavior. The thermal conductivity (κ) values are extremely low and decrease with increasing temperature up to 0.46 W m−1 K−1 at 773 K. The DFT studies suggest that the computed bandgap depends on the magnetic ordering of Mn magnetic moments, and the value varies from ∼0.3–1.0 eV. Relative inter-atomic bond strengths of pertinent atom pairs have been analyzed using the crystal orbital Hamilton populations (COHP).
 Please wait while we load your content...
Please wait while we load your content...

