Liquid electrolytes for dye-sensitized solar cells
Ze
Yu
a,
Nick
Vlachopoulos
c,
Mikhail
Gorlov
b and
Lars
Kloo
*a
aInorganic Chemistry, Royal Institute of Technology (KTH), S-100 44, Stockholm, Sweden
bOrganic Chemistry, Royal Institute of Technology (KTH), S-100 44, Stockholm, Sweden
cPhysical Chemistry, Uppsala University, Box 259, S-751 05, Uppsala, Sweden
First published on 22nd September 2011
Abstract
The present review offers a survey of liquid electrolytes used in dye-sensitized solar cells from the beginning of photoelectrochemical cell research. It handles both the solvents employed, and the prerequisites identified for an ideal liquid solvent, as well as the various effects of electrolyte solutes in terms of redox systems and additives. The conclusions of the present review call for more detailed molecular insight into the electrolyte-electrode interface reactions and structures.
 Ze Yu | Ze Yu received his M.E. in Applied Chemistry at Dalian University of Technology (DUT), China, in 2007. He then joined the group of Prof. Lars Kloo as a PhD candidate in Inorganic Chemistry at the Royal Institute of Technology (KTH), Stockholm, Sweden. His research interest has mainly been focused on the optimization of liquid electrolytes and the development of alternative redox mediators for dye-sensitized solar cells. |
 Nick Vlachopoulos | Nick Vlachopoulos obtained a chemical engineering degree from the National Technical University of Athens, Greece (1979) and two doctorates from University of Pennsylvania, USA (1984) and Swiss Federal University of Technology (EPFL)- Lausanne (1990) in chemical engineering and physical chemistry, respectively. He worked as a researcher in Prof. Michael Grätzel's group in Lausanne for several years (1986–99) in the area of photochemical solar energy conversion, for shorter periods in universities of France, Denmark and Germany in various areas of analytical and physical electrochemistry, and as research and development scientist in NTERA Inc. in electrochromic display technology from 2004–2007. Since 2008 he has been employed as a researcher in Uppsala University in the team of Prof. Anders Hagfeldt in the physical chemistry of dye-sensitised solar cells and electrochemical supercapacitors. He has 30 publications in scientific journals and 4 patents. |
 Mikhail Gorlov | Mikhail Gorlov was born in Saint Petersburg, Russia and received his PhD in Chemistry from the Saint Petersburg State Institute of Technology in 2002. He was a postdoctoral fellow (2003–2004) at the Royal Institute of Technology (KTH), Stockholm, Sweden, and he is currently a researcher at Organic Chemistry Department, KTH. His research interest focuses on water splitting processes and dye-sensitized solar cells. |
 Lars Kloo | Lars Kloo received his PhD in Inorganic Chemistry at Lund University, Sweden, in 1990, and moved to his current position as Prof. in Inorganic Chemistry at the Royal Institute of Technology (KTH) in Stockholm in 1998. During 1991–95 he was in shorter or longer periods visiting scientist in other laboratories in the UK, USA and New Zealand. He has been working with liquid electrolytes and electrode interfaces in dye-sensitized solar cells since about 2000, and with engineering of liquid reaction and electrolyte media since the PhD. |
1. Introduction
The dye-sensitized solar cell (DSC) can be regarded as a simple electrochemical device, consisting of two electrodes and an electrolyte. At least one of the electrodes is photosensitive generating energy-rich photoelectrons upon irradiation. The third component, the electrolyte, takes care of the charge transport between the electrodes. As can be noted from the historical survey below, the typical DSC electrolyte is liquid. Also, in the paper regarded as the birth of modern DSC research, a liquid electrolyte based on organic carbonates was chosen.1Today, there is a vivid research with the aim to find and explore new electrolyte media avoiding the main disadvantage of the typical liquid DSC electrolytes; volatility.2 The efforts involve both gelation/solidification of liquid electrolytes and solid, polymeric or molecular hole-conductors. Another direction of research exploits the main advantage of ionic liquid-based electrolytes3,4 in terms of an almost negligible vapour pressure. The main disadvantage of ionic liquids is the rather high viscosity linked to low ion mobility. This typically gives mass-transport problems at higher levels of irradiation. Strategies to overcome this problem have recently been presented involving solvent modifications, such as eutectic mixtures,5 deep eutectic solvents6 or incompletely solvated ionic liquids.7
In 1991 O'Regan and Grätzel obtained a remarkable efficiency of almost 8% using a very rudimentary electrolyte essentially only consisting of an organic solvent and a dissolved redox couple; the iodide/triiodide system.1 This type of electrolyte has over the years been optimized by the inclusion of various additives, having an effect on both electrode surface electronics and structure, as well as on the electrolyte chemistry itself. Still today, this type of electrolyte provides DSCs with the highest conversion efficiencies, up to 12%.8 All alternatives, so far, lag behind in terms of conversion efficiencies, although they may offer advantages in terms of long-term endurance. The present review covers the field of liquid electrolytes, partitioned into solvents, additives, redox couples and dye-electrolyte interactions.
2. Molecular solvents for dye-sensitized solar cell electrolytes
The origin of research on photochemical solar conversion systems producing electricity or energy-rich chemicals ties to studies of the natural photosynthesis processes. At a first glance, dyes, electrolytes and mediators very similar to those of nature would be of primary interest, i.e. involving neutral aqueous electrolytes. However, the energy output and stability requirements of artificial systems would be more restrictive in the sense that synthetic dyes require either aqueous electrolytes with non-neutral pH, usually acidic, or non-aqueous electrolytes all together. In this respect, the field benefits from substantial progress achieved in the last half-century in the understanding of the mechanisms of chemical, including electrochemical, reactions in non-aqueous solvents. In particular, research related to the development of non-aqueous, high-energy-density batteries and supercapacitors provides important guidelines regarding solvents suitable for DSCs. The availability of high-purity organic solvents from the chemical industry has been a positive factor in such developments.The following general requirements should be fulfilled for the solvents of interest to DSC research and development:
(i) Melting point below −20 °C and boiling point above 100 °C, so that the electrolyte prepared with these solvents will not evaporate under cell operating conditions, especially in an outdoor environment. For precise cell design, the effect of added electrolytes and other additives on melting and boiling points should be taken into account.
(ii) Low light absorption; this condition is usually fulfilled for the solvents mostly used in their pure form, the light absorption normally originates from redox mediators or other additives.
(iii) Sufficient chemical stability in the dark and under illumination; in particular, the solvents should be stable toward oxidation either by the photooxidized dye or by photoproduced holes in the oxide substrate, generated by direct excitation by the ultraviolet (UV) and near-visible component of sunlight, and should not contain photooxidizable impurities. Usually, a filter is added in the cell in order to remove UV and near-visible light (up to ∼420 nm), but filtering is never 100% efficient. The redox potential of photoproduced holes can be as high as 3V vs. the standard hydrogen electrode (NHE) depending on the solution composition. In particular, these affect the hydrogen ion activity and any strongly chemisorbed species. A result of parasitic oxidation of the solvent or solvent impurities is the progressive decrease of the amount of the oxidized form of the redox mediator; for example, in solar cells containing I−/I3− in addition to the main reaction, with the oxidation of I− to I3−, the solvent or its impurity is oxidised. Therefore, the amount of I3− reduced at the counter electrode is larger than the amount of I3− generated at the photoelectrode, and the concentration of the I3− is progressively decreased. An indication of stability towards oxidation and reduction is provided by the electrochemical ‘window of stability’, determined by the potentials at which the electrolyte undergoes oxidative and reductive decomposition at an inert electrode, usually Pt. For water, the theoretical window of stability is 1.23V, the practical one is in the range of 1.5 to 2V depending on the electrode used. Such stability data are of interest to battery and supercapacitor development, as well as to the less widespread field of electrochromical windows and displays, and they are of intrinsic importance to the DSC field. However, the requirements could be more stringent for DSC systems in several aspects, since batteries are hermetically sealed, so that the contents are protected from light and not directly exposed to the ambient environment.
(iv) High dielectric constant, so that electrolyte salts are sufficiently soluble and exist in a fully dissociated state. Good solubility for the redox mediators of interest.
(v) Low viscosity, so that redox mediators of interest possess high diffusion coefficients.
(vi) Good conductivity of dissolved electrolytes; a requirement usually fulfilled for thin-layer electrochemical photovoltaic cells (thickness below 100 μm). This is related to the previous two requirements.
(vii) Poor solubility with respect to the organic sealants used to seal the DSC.
viii) Low toxicity and environmental hazard, especially for application-oriented electrolytes.
(ix) Inertness with respect to the surface-attached dye, including the dye-metal-oxide (or other semiconductor) bond.
The electrolyte conductivity, the diffusion coefficient of dissolved species and the solvent viscosity [relating to the requirements (v) and (vi)] are related by the following equations derived from irreversible thermodynamics.9
(A) Conductivity vs. diffusion coefficients of electrolyte ions.
 | (1) |
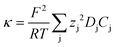 | (2) |
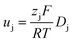 | (3) |
(B) Diffusion coefficient and conductivity vs. viscosity.
The following relations are obtained by applying a classical mechanics model for viscous flow around a sphere to solvated molecules in the electrolyte, charged or not.
(B1) Diffusion coefficient vs. viscosity:
 | (4) |
(B2) Conductivity vs. viscosity:
Inserting the Djvs. η expression (eqn (4)) into the κ vs. Dj expression (eqn (2)), the following relation for the product of conductivity and viscosity is obtained:
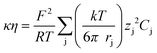
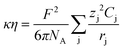 | (5) |
 | (6) |
The above equations have better applicability in the case of dilute electrolytes, when the migration of each ion (movement in electrical field) can be regarded as independent of those of the other ions. Moreover, the correlation of diffusion coefficient with viscosity is more accurate when comparing data in structurally related solvents (e.g. straight-chain alcohols or nitriles).
At present, no single solvent or solvent mixture fulfills all these aforementioned requirements. Therefore, the choice of electrolyte will depend on the particular use of the cell under consideration. In the following section, the utilization of particular classes of solvents will be discussed in detail. Useful information about solvents and their uses in electrochemistry is provided by the books by Sawyer et al.10 and Conway.11
2.1 Water
In the initial phase of research, from the early 1970s to 1985,12 concerning sensitization of semiconductor electrodes, water was the solvent of choice. Such investigations were focused on single semiconductor-electrolyte interfaces with the photoelectrode connected to a reference electrode in a potentiostatic circuit. The addition of a supporting electrolyte was essential in the absence of a redox mediator in an ionic form. Usually, the photocurrent efficiencies based on incident light (IPCE) were of the order of 1%, due to the low real-to-geometric surface area (roughness factor). The investigations by the Tsubomura group13,14 focused on Rose Bengal adsorbed on polycrystalline ZnO prepared from sintered powder were a notable exception. Those experiments showed a maximal IPCE of 22% at 562 nm and a monochromatic (562 nm) light-to-electricity conversion efficiency of 2.5% for electricity generation. The first systematic investigations of photoelectrochemical dye sensitization of porous TiO2 electrodes by the Grätzel group in Lausanne, Switzerland,15–18 also involved aqueous electrolytes of a pH in the range 2–7. For TiO2 substrates acidification of the electrolyte is necessary for two reasons. Firstly, concerning efficient chemisorptions of the sensitizing dye molecules. The presence of a negatively charged carboxylate anchoring group of the dye molecules necessitates a positive charge of the semiconductor surface. This is attained when the electrolyte pH is below that corresponding to the isoelectric point of TiO2, ca. pH = 5. Secondly, the position of the TiO2 oxide conduction band edge at the TiO2 surface shifts in the negative direction (with respect to the unbound vacuum state) in the energy scale with decreasing pH. This means that the driving force for electron transfer from the dye excited state to TiO2 increases, and excited state deactivation by emission of heat or fluorescent light is suppressed. This has been exemplified in a TiO2 sensitization study by Zn porphyrins involving both electrodes and colloidal particles.16 In this early phase of dye sensitization studies, the maximum IPCE exceeded 70% and a monochromatic light-to-electricity conversion efficiency of 12% was obtained in a NaI-based electrolyte (pH = 3) with a regenerative (agitated beaker) cell in a LiBr-based electrolyte (pH = 3), with I− or Br− as redox mediator components, respectively.17Following the above studies, the focus in DSC research shifted to non-aqueous electrolytes. In general, higher efficiencies have been obtained in water-free electrolytes than in purely aqueous or mixed electrolytes containing water. Very few studies have been devoted to a comparison of aqueous and non-aqueous systems. However, in industrial applications of DSCs the rigorous exclusion of water is not always feasible. Moreover, imperfect sealing results in continuous water uptake from the environment. In a systematic study by Lindquist et al. with TiO2 electrodes coated with one of the commonly used Ru-based dyes (N3)19 the deliberate addition of small amounts of water in a non-aqueous liquid electrolyte caused a small decrease in photocurrent and an increase in photovoltage.19 It has been argued that water causes desorption of dye molecules due to a hydrolysis of the surface dye-oxide bond or a weakening of the interaction between the dye and the electrode oxide material. In fact, special dyes have to be considered for either pure or mixed aqueous electrolytes. With respect to the increase of cell potential with water content, useful insights are provided in the study of Enright et al.,20 who studied the shift of the conduction band edge of a pure non-aqueous electrolyte with the progressive addition of water. A recent publication by O'Regan et al.21 indicates that for the Ru-based dyes an addition of up to 20% of water into a non-aqueous electrolyte slightly enhances the energy conversion efficiency from 5.5% to 5.7% (AM1.5, 1000 Wm−2) without deterioration of the long-term stability. The efficiency decreases upon further water addition, so that for pure water the efficiency is 2.4%. To date, this is the best conversion efficiency obtained for a pure aqueous solvent. An alternative explanation for the long-term instability of dye-sensitised solar cells in the presence of water has been attributed to the oxidation of iodide to iodate (IO3−) instead of triiodide in the presence of water,22 which cannot be reduced at the counterelectrode, resulting in a progressive I3− depletion and decrease in cell performance.
2.2 Alcohols
An early example of an ethanol-based (EtOH) electrolyte (LiI/I2) is a study of a Ru-trimer dye by the Grätzel group,23 in which for the first time energy conversion efficiency data for a thin-layer DSC cell were presented (11.3% at 520 nm). However, in the following studies by the same group aprotic polar electrolytes were used. The disadvantages of EtOH and other lower alcohols is the hydrophilicity, which may affect the dye-oxide bond in the same way as water, as described in the previous section, and the lower chemical stability towards both oxidation and reduction, as compared to several aprotic solvents.2.3 Nitriles
Acetonitrile (methyl cyanide, AN) is considered as the best electrolyte for fundamental electrochemical studies due to its excellent chemical stability, as indicated by the electrochemical stability window24 (more than 4V), the low viscosity and the capability to dissolve a large number of components; both salts and organic molecules. Therefore, its use in solar cells as the sole or principal solvent is advantageous, since the maximum mass transport-limiting current will be substantially higher than that obtained under AM1.5 illumination. However, its low boiling point (78 °C) in combination with the relatively high toxicity will prevent utilization in industrial solar cells. Nevertheless, AN is the solvent of choice for laboratory investigations involving new sensitizers, when the effects of photochemical processes are to be investigated without influence from mass-transport limitations. Glutaronitrile (pentanedinitrile, GN) has been considered for some time for DSC research and development due to the high boiling point (285 °C), but its use has been discontinued because of toxicity considerations.25 Higher nitriles, mainly propionitrile (ethyl cyanide, PN) and valeronitrile (butyl cyanide, VN) have been used, often mixed with AN, but cost and toxicity have to be considered. An energy-conversion efficiency for AM1.5-1000 W m−2 with illumination exceeding 11%8,26 has been repeatedly obtained with an electrolyte based on AN-VN (85%–15% vol).Another class of nitriles containing methoxy groups are less toxic and have a higher boiling point than acetonitrile. Methoxyacetonitrile (MAN) and 3-methoxypropionitrile (MPN) have been extensively used as DSC electrolytes; at present, MPN is the primary solvent of choice for solar cells to be used in applications, with a boiling point of 164 °C, melting point of −63 °C and a viscosity of 2.5 cP, as compared to 82 °C, −44 °C and 0.33 cP for acetonitrile; the water viscosity is 0.89 cP. MPN has sufficiently good chemical stability to be used in long-term DSC stability tests27 (7.6% efficiency, aging 100 h, continuous light, simulated AM1.5-1000 W m−2). However, the stability of the CH3–O–C2H4– ether bond in tests of longer duration and higher temperature has to be thoroughly investigated.
2.4 Esters, lactones and related compounds
Compounds in this category have been extensively used in battery and supercapacitor technology.11 Several members of this group have the advantage of a high boiling point, often exceeding 200 °C, and a high dielectric constant, sometime higher than that of water. Two cyclic esters of carbonic acid, ethylene carbonate (EC) and propylene carbonate (PC), and a related compound, N-methyloxazolidinone (3-methyl-2-oxazolidinone, NMO) have been widely used in battery technology and in the initial phase of DSC development. Usually, those compounds have an elevated boiling point. On the other hand, EC and NMO have rather elevated melting points, above that of water (36 °C and 15 °C, respectively), so the addition of a solvent with a lower melting point may be necessary, e.g. PC or AN (melting points −49 °C and −44 °C, respectively).1,16,28,29 The electrolyte for the first efficient DSC device based on a Ru-trimer dye (energy conversion efficiency 7%, AM1.5-1000 W m−2) contained an EC-AN mixture as solvent (80%–20% vol).1 In the first device with a standard Ru-based red dye (N3) with good spectral overlap to the red part of the visible spectrum and reaching 10% conversion efficiency for the first time (AM1.5-1000 W m−2), the solvent employed was an AN-NMO mixture (90%–10% vol).28 At that time, the sealing technique for DSC cells was not well developed and the presence of a low-volatility electrolyte was necessary. However, in several long-term experiments DSCs containing this type of solvents displayed solvent decomposition, and consequently their use has been discontinued;29 this with one exception, which is studies of novel redox mediators soluble only in these media. In electrochemical research, PC is known to have a high electrochemical window of stability (above 4 V), but in in situ spectroelectrochemical measurements using a reflectance spectroscopy technique (SNIFTIRS) degradation products have been detected at potentials within the window of stability, where no solvent-based electrochemical reactions would normally be expected.9 Another frequently used solvent in DSC research and development work, in particular for long-term outdoor tests, is γ-butyrolactone,30,31 with favourable melting and boiling points (−44 °C and 204 °C, respectively) and viscosity (1.7 cP). However, the long-term stability is inferior to that of the ionic-liquid electrolytes described below. Moreover, for medical reasons it is not readily available for purchase.2.5 Electron donor–acceptor (generalized acid–base) properties of molecular solvents.
All the solvents discussed above can be characterised as electron acceptors. Usually, an increase in electron-donor (or basic) character will lead to higher open-circuit potentials and lower photocurrents32–34 when used in electrolytes in DSCs. Usually, the Gutmann donor number (DN)35 is used for solvent classification with respect to DSC studies. It is defined as the enthalpy of reaction, expressed in kcal units, of the solvent with a strong electron acceptor, SbCl5, at infinite dilution in 1,2-dichloroethane; a poor electron-donor solvent. Examples of electron-acceptor solvents are N,N′-dimethylformamide (DN = 26.6), N-dimethylsulfoxide (DN = 29.8) and pyridine (DN = 33.1). These solvents are often encountered in electrochemical research but never used as major components in efficient dye-sensitised solar cells. In comparison, water, AN and EC display DN = 18.0, 14.1 and 16.4, respectively. The effect of solvent basicity can be rationalised by considering the state of the electrode metal-oxide surfaces,36e.g. for TiO2 the amphoteric character has to be considered according to the equilibria:| Ti–OH2+ ⇄ Ti–OH + H+(solution) | (7) |
| Ti–OH ⇄ Ti–O−+ H+(solution) | (8) |
In this respect, it has been argued that an electron-donating (basic) solvent will decrease the amount of surface-bound protons, resulting in a more negatively charged metal-oxide surface. Therefore, a shift of the conduction band edge for the semiconductor surface in the positive direction (unbound vacuum scale) and a decrease of the driving force for electron transfer from the dye excited state to the semiconductor, resulting in a lower short-circuit photocurrent, will be observed. Moreover, the onset of dark currents will occur at more negative electrode potentials corresponding to the aforementioned shift of the conduction band, resulting in higher open-circuit potentials. The above effects of basicity on photocurrents and photopotentials could be also rationalized by considering a proposition by Grätzel,28 according to which an electron-donating solvent will be bound more strongly to electron-accepting TiO2 sites and will in this way passivate them. This will cause hindering of both electron injection from the dye to the semiconductor and, on the other side, electron recombination from the semiconductor surface to the oxidized component of the redox mediator. Such effects are expected to result in lower photocurrents and higher open-circuit potentials, as argued above. Apart from the effects discussed above, the interactions of electrolytes with particular redox mediators should be considered. For example, in the case of the I−/I3−, or I−/I2, the bond between the electron-donating solvent molecules and I3− or I233,37 results in a decrease of their effective (available) concentration and an increase in cell potential.
The above donor–acceptor discussions are of importance for the formulation of electrolyte mixtures aiming to obtain the highest possible potential without compromising the photocurrent. At first approximation, the donor number of a solvent mixture can be estimated as the mole-fraction-weighted average of the component donor numbers (eqn 9).
 | (9) |
However, the particular properties of the dye and the electrode material should be taken into account; no single electrolyte will fulfill all requirements posed by any dye or electrode material. An electrolyte optimization is consequently required for every new system to be studied, in order to exploit its maximum potential for light-to-electricity conversion. Apart from the acid–base properties of the solvent, the corresponding properties of the added electrolyte components must be taken into account as well. For example, higher currents and lower potentials should be obtained using an iodine-based redox system involving LiI/I2 than using [n-Bu4N]I/I2 at the same concentration, due to the electron-acceptor (Lewis acid) character of Li+. More details will be given in the following section on electrolyte additives.
Similar considerations are also valid for solid polymer and gel electrolytes, a subject that will not be covered in the present review. For example, if a particular solid matrix has acidic properties, i.e. contains acidic pendant groups, the addition of a base at higher molar fraction would be necessary in comparison with a solid matrix that is relatively neutral.
2.6 Ionic liquids
Ionic liquids have emerged as interesting alternative solvents for liquid DSC electrolytes. The main advantages are related to chemical and electrochemical stability, and, most importantly, ionic liquids have extremely low vapour pressure making evaporation less of a problem.38 The vapour pressure of molecular solvents constitutes a practical problem upon DSC device assembly and a significant challenge regarding DSC sealing for long-term endurance. The use of ionic liquids in DSCs has recently been reviewed and will therefore only be mentioned briefly in the present overview of liquid electrolytes.3,4It is clear that ionic liquids can not easily be characterized in terms of donor numbers etc. In spite of exhibiting more complex aggregation in the liquid close to the melting point, ionic liquids can be regarded as molten salts and share many characteristics with such media. This also means that they should not be classified as a single type of solvent; like nitriles, alcoholsetc. Of course, interactions in ionic liquids are dominated by electrostatics, strongly influencing both local structure and macroscopic properties. In terms of Lewis acid–base behaviour, both the character of the (most often) organic cation and the (most often) inorganic anion play significant roles. The cations are typically weak Lewis acids, thus potentially coordinating electron-rich species in the ionic liquid solutions. Of course, the inherent chemical character of the cations affect the ionic-liquid properties significantly, and one clear distinction can be made between ionic liquids based on aliphatic and aromatic cations. One example of a clear cation effect is the observed lowering of available triiodide for dye regeneration in ionic-liquid media, as compared to organic solvent-based electrolytes.39 With respect to anions, you can crudely divide the ionic liquids into two rather different types; those with halide/pseudohalide anions and those with complex, and typically weakly coordinating, anions,40 such as the various borates, triflate derivatives etc. The halides/pseudohalides are well-known Lewis bases, for instance interacting with organometallic sensitizers causing ligand exchange, whereas the other types most commonly are quite weak Lewis bases. As a consequence, it is difficult to treat ionic liquids as a single type of liquid; the choice of cation and anion, and the combination of both, will have a significant impact on DSC performance. Ionic liquids remain a promising alternative to molecular solvents as the solvents for DSC electrolytes, and their full potential remains to be explored. So far, their main drawback has been high viscosity and low ion mobility, and in spite of several good strategies to overcome this problem these fundamental restrictions have limited conversion efficiencies for ionic liquid-based DSCs.
2.7. Conclusions regarding DSC solvents
The choice of solvents for DSCs has been rather restricted, because of the complex requirements for such devices with respect both high efficiency at full sunlight and long-term stability. These requirements are in several respects contradictory. The studies focused on new highly efficient sensitizing dyes involve volatile acetonitrile-based electrolytes, while for long-term stability studies involve the high-boiling 3-methoxypropionitrile or butyrolactone. In both cases, the donor–acceptor (acid–base) properties are important. If both high current and potential have to be achieved, at least one additive, either as co-solvent or solute, is required. Up to the present time, such solvents had to be compatible with the iodide/triiodide redox couple; the ubiquitous redox system. However, recently alternative mediators have been demonstrated to provide high-efficiency devices, so that the compatibility of solvents with such mediators, if ultimately to replace the iodide/triiodide system, should be considered; in particular, regarding solubility and fast diffusion. However, molecular solvents are not the only choice. As an alternative media, ionic liquids possess substantial advantages, and in several cases mixed solvents involving both ionic liquids and molecular solvents might offer the best solution. A survey of common molecular solvents used in DSCs is given in Table 1.| Name | Formula | Melting Point/°C | Boiling Point/°C | Viscosity/cp | Dielectric constant | Donor number |
|---|---|---|---|---|---|---|
| a All data at 25 °C and 760 mmHg unless otherwise indicated. b Values not available in the literature. | ||||||
| Water | H2O | 0 | 100 | 0.89 | 78 | 33 (liq.) |
| Ethanol | CH3CH2OH | −114 | 78 | 1.08 | 25 | 29 |
| Acetonitrile | CH3CN | −44 | 82 | 0.33 (30 °C) | 36 | 14.1 |
| Propionitrile | CH3CH2CN | −93 | 97 | 0.39 (30 °C) | 27 (20 °C) | 16.1 |
| Valeronitrile | CH3(CH2)3CN | −96 | 139 | 0.78 (19 °C) | 21 | b |
| Glutaronitrile |

|
−29 | 287 | 5.3 | 37 | b |
| Ethylene Carbonate |

|
36 | 238 | 90 | 90 | 16.4 |
| 3-Methoxy-propionitrile | CH3OCH2CH2CN | −63 | 164 | 1.1 | 36 | 16.1 |
| Propylene carbonate |

|
−49 | 241 | 2.5 | 64 | 15.1 |
| N-Methyl-oxazolidinone |

|
15 | 87–90 (1mmHg) | 1.9 | 78 | a |
| γ-Butyrolactone |
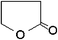
|
−44 | 204 | 1.7 | 39 (20 °C) | 18.0 |
3. Redox couples
Redox-couples (RCs), or redox mediators, play an extremely important role in DSCs. The open-circuit voltage (Voc) is determined by the difference between the Fermi energy of the illuminated, transparent, conducting substrate with a nanocrystalline semiconductor film (working electrode; WE) and the electrochemical potential of the RC. In DSCs, the RCs are responsible for charge transport between the photoanode and the counter electrode. In general, the working mechanism of the RC may be represented as follows.The reduced form of the RC is supposed to regenerate oxidized dye (D+) molecules:
| RC + D+ → RC+ + D | (10) |
After that, the oxidized form of the RC (RC+) diffuses to the surface of the counter electrode to be reduced back to its initial and reduced form.
| RC+ + e−/CE → RC | (11) |
Diffusion processes play a crucial role; the rate of the diffusion strongly depends on several factors: (1) the size of the RC molecule or ion, (2) the viscosity of the solvent, and (3) the concentration of the RC.
3.1 The iodide/triiodide system
The best working RC known so far is the iodide/triiodide system. The most efficient DSCs have been constructed with the I−/I3−redox couple dissolved in volatile organic solvents. The unique performance of I−/I3− based liquid electrolytes is mainly attributed to the favourable penetration into the nanoporous semiconductor film, very fast dye regeneration, and relatively slow recombination losses through reaction with injected photoelectrons. However, there are several negative features limiting industrial application of the I−/I3− RC: (1) iodine is extremely corrosive toward metals such as copper or silver, which are used as current collectors in some DSCs; (2) iodine has a relatively high vapour pressure, which makes proper encapsulation of the cells challenging; (3) the I3− ion absorbs a significant part of visible light, stealing photons from the sensitizing dye; (4) the redox potential of the I−/I3− limits the photovoltage.During the last two decades, a number of articles and reviews dedicated to the properties and behaviour of the iodide/triiodide RC have been published;2,41–45 in this review we will mainly focus on alternative RCs.
3.2 Bromide/tribromide, inter- and pseudohalogen systems
The bromide/tribromide RC has attracted increasing attention during the last years; first of all due to its more positive redox potential (about 1.1 V vs.NHE) in comparison to the I−/I3− RC (about 0.35 V vs.NHE).41 Thus, Wang et al. investigated four different dyes (three organic and N719) in combination with either I−/I3− or Br−/Br3− as redox systems.46 The authors found that the Br−/Br3− RC is much more suitable than I−/I3− in terms of photovoltage when using eosin Y as the sensitizing dye (see Fig. 1).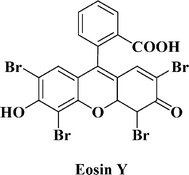 | ||
| Fig. 1 Chemical structure of the eosine Y dye. | ||
According to current/potential (I/V) data obtained at full sunlight illumination, the Br−/Br3− system shows a much higher photovoltage and overall efficiency (0.4 M LiBr + 0.04 M Br2 electrolyte: Isc = 4.63 mA cm−2, Voc = 813 mV, FF = 0.693, η = 2.61%; 0.4 M LiI + 0.04 M I2 electrolyte: Isc = 5.15 mA cm−2, Voc = 451 mV, FF = 0.721, η = 1.67%).
In 2009, Teng and co-authors showed that a photovoltage above 1 V may be obtained by using a Br−/Br3− RC in combination with organic dyes (Fig. 2).47
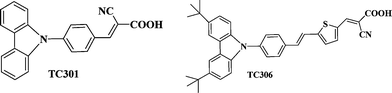 | ||
| Fig. 2 Chemical structures of the TC301 and TC306 dyes. | ||
The authors compared the photovoltaic properties of the cells containing either Br−/Br3− or I−/I3− RCs in an acetonitrile-based electrolyte. The I/V data show a very high photovoltage (TC301 dye, full sunlight) for the Br−/Br3− system (Isc = 4.00 mA cm−2, Voc = 1156 mV, FF = 0.796, η = 3.68%), whereas for the I−/I3− system I/V data showed a slightly higher photocurrent but much lower photovoltage (Isc = 4.42 mA cm−2, Voc = 696 mV, FF = 0.767, η = 2.36%). Moreover, for the TC306 dye, a conversion efficiency of 5.22% was achieved by using the bromide/tribromide-based electrolyte.47
Ning et al. reported the syntheses of three mononuclear polypyridine iridium dyes. The authors noted that Voc was significantly enhanced when I−/I3− was replaced by Br−/Br3− as the RC in the electrolyte (ca. 300 mV enhancement).48
The mechanism of charge recombination in the ZnO–EosynY-based DSCs containing I−/I3− or Br−/Br3− as the RC has also been investigated.49 It was found that the charge recombination mechanism between photoinduced electrons and bromine is of first-order, whereas in the case of I−/I3− it was reported as second-order. The authors suggest, on the basis of IPCE measurements, that the dye regeneration by Br− is less efficient than by I−.
A new RC, SeCN−/(SeCN)3−, was described by the Grätzel group.50 Relatively high power conversion efficiencies of 7.5–8.3% were recorded under different light intensities (between 997 and 95 W m−2). It should be noted that a low-viscous ionic liquid 1-ethyl-3-methylimidazolium thiocyanate (EMISCN) was used as a non-volatile solvent in combination with the Z-907 sensitizing dye. The diffusion coefficient of (SeCN)3− is 1.28 × 10−6 cm2s−1, which is about 7 times higher than the corresponding value for I3− in 1-propyl-3-methylimidazolium iodide (PMII).
Oskam et al. compared two pseudohalogen RCs, SeCN−/(SeCN)3− and SCN−/(SCN)3−.51 Their equilibrium potentials are more positive than for the I−/I3− RC, 0.19 and 0.43 V, respectively. It was found that the regeneration rates with respect to the ruthenium-based N3 dye adsorbed to the nanostructured TiO2 electrodes decrease in the order I− > SeCN− > SCN−. The IPCE values (400–520 nm) were found to be 80, 20 and 4% for the I−-, SeCN−- and SCN−-based electrolytes, respectively.
The pseudohalogen RCs SeCN−/(SeCN)3− and SCN−/(SCN)3− were also studied in SnO2-based DSCs.52 Three different bipyridine, pyrazine and bipyrazine ruthenium-based dyes were investigated. The authors found that cell efficiencies and photovoltages were higher using the alternative RC SeCN−/(SeCN)3− as compared to the I−/I3− one.
Interhalogen salts and ionic liquids containing IBr2− and I2Br− anions have been studied as alternative RCs in DSCs with the N719 sensitizing dye.53 It was shown by means of mass and Raman spectroscopy that there are complex equilibria between the different electrochemically active species in organic solvents. Overall, light-to-electricity conversion efficiencies up to 6.4%, 5.0%, and 2.4% at 1000 W m−2 were achieved using electrolytes based on interhalogen ionic salts with γ-butyrolactone, glutaronitrile and native ionic liquids as solvents, respectively.
3.3 Sulphur-based systems
The challenge of finding non-corrosive RCs, alternative to the halogen-based ones, has encouraged researchers to test halogen-free systems. The first successful result was recently published, although this system was known before.54 A disulfide/thiolate RC, Fig. 3, was introduced; thus mimicking the homoatomic bond-making/breaking, two-electron process of the halogen-based RCs.
T− represents the 5-mercapto-1-methyltetrazole anion, and T2 is its dimer. In this work, the T−/T2 RC was studied in combination with the ruthenium-based dye Z907–Na. It was found that the composition of the electrolytes plays a crucial role in these systems. The best result was obtained for an electrolyte with the composition T−/T2, 4-tert-butylpyridine (TBP), LiClO4 (0.4 M/0.4 M, 0.5 M, 0.05 M), a WE film thickness of 11 (nano-TiO2) + 5 (scattering layer) μm. The photovoltaic characteristics of these cells at full sunlight are: Isc = 16.18 mA cm−2, Voc = 681 mV, FF = 0.58 and η = 6.44%.54
A similar system was developed by Tian and co-workers.55 This sulphur-containing RC is based on 2-mercapto-5-methyl-1,3,4-thiadiazole (McMT), Fig. 4.
 | ||
| Fig. 4 Chemical structure of the McMT−>/BMT RC used in Ref. 55. | ||
The optimized electrolyte contains McMT−, BMT, LiClO4 and TBP (0.2 M, 0.2 M, 0.05 M, 0.5 M) in an acetonitrile/ethylene carbonate mixture (50%–50% vol). The standard redox potential of the McMT−/BMT RC was recorded to be 0.155 V vs.NHE. The I/V data of this system (organic sensitizer TH305 was used56) are: Isc = 12.2 mA cm−2, Voc = 762 mV, FF = 0.42, η = 4.0%. For comparison, the authors investigated an identical electrolyte but with the I−/I3− RC instead of the McMT−/BMT one. Surprisingly, the recorded Voc was almost the same, 774 mV, in spite of the substantial difference in redox potentials between the two RCs. The authors explain this phenomenon by a negative shift of the quasi-Fermi level in the case of the McMT−/BMT RC.56
Another promising sulphur-based RC was described by Liet al.57 In this work, tetramethylthiourea (TMTU) and its oxidized dimer, consisting of the tetramethylformaminium disulfide dication ([TMFDS]2+), were prepared and investigated in DSCs (Fig. 5).
 | ||
| Fig. 5 Chemical structure of the tetramethylthiourea-based RC used in Ref. 57. | ||
The most important advantage of this RC, apart from its low price and non-corrosive nature, is the possibility to explore it in combination with carbon electrodes on different substrates. Moreover, the glassy carbon counter electrode used shows higher reversibility of the TMTU/[TMFDS]2+ reaction than Pt-based counter electrodes under the same measurement conditions. The photovoltaic characteristics of the best cells at full sunlight are: Isc = 9.45 mA cm−2, Voc = 660 mV, FF = 0.49, η = 3.1%.57
It is necessary to highlight that in spite of promising results obtained for new sulphur-based RCs, no information regarding their long-term stability appears to be available so far. Obviously, the stability issue is crucial for potential industrial application of the DSCs.
3.4 Metal-based systems
One of the first metal-based RCs successfully used in DSCs was based on mononuclear Co(II)/Co(III) complexes. The advantage of this class of RCs is that they are non-volatile, non-corrosive and only lightly coloured. In 2004, Cameron et al. reviewed the electrochemical properties of the redox mediator Co(II)/Co(III)(dbbip)2 [dbbip = 2,6-bis(1-butylbenzimidazol-2-yl)pyridine] and its application as RCs in DSCs.48 The rate constants for electron transfer were determined by different techniques. In addition, different materials (platinum, gold, FTO and compact TiO2) were compared as electrode materials. The authors concluded that it is essential to use a compact TiO2 blocking layer on the WE substrate surface in order to prevent charge losses.58A number of cobalt complexes of substituted polypyridine ligands have been investigated by Sapp et al.59 These authors found that the optimal Co(II)/Co(III) ratio is 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The effects of lithium salts, as well as nitrogen-containing heterocyclic additives, in the electrolytes were also studied. In combination with the N3 dye, a maximal conversion efficiency of 1.58% at full sun-light illumination was obtained.
:1. The effects of lithium salts, as well as nitrogen-containing heterocyclic additives, in the electrolytes were also studied. In combination with the N3 dye, a maximal conversion efficiency of 1.58% at full sun-light illumination was obtained.
A light-to-electricity conversion efficiency of 5.2% was recorded by using the mononuclear, one-electron redox mediator Co(II)-bis[2,6-bis(1′-butylbenzimidazol-2′-yl)pyridine] in combination with the Ru-based dyes N719 and Z316 [cis-RuII(2,2-bipyridyl-4,4-dicarboxylic acid)(4-methyl-4-hexadecyl-2,2-bipyridyl)(NCS)2].60 The oxidized form of the cobalt RC was obtained by adding NO[BF4]in situ to the solution of the Co(II) precursor. The authors noted that diffusion problems limiting the performance of the cells may be solved by modification of the organic RC ligands.
Yanagida et al. reported two different Co-based RCs based on propylene-1,2-bis(o-iminobenzylideneaminato)cobalt(II) and tris(4,4-di-tert-butyl-2,2-bipyridine)cobalt(II) diperchlorate.61 It was observed that the presence of Li salts in the electrolyte plays an important role; the electron lifetime was observed to increase with increasing Li+ concentration. This phenomenon was attributed to the decrease in local concentration of Co(III) at the TiO2 electrode surface.
A series of new Co complexes of the general formula [Co(LLL)2X2], where L are aromatic N-donor ligands and X = ClO4, PF6 or OTf, have been synthesized and explored as RCs in DSCs.62 It was discovered that not only the nature of the ligands influences the performance of the cells, but also the nature of the counterion. Among the compounds investigated, the complex [Co(dbbip)2](ClO4)2 [dbbip = 2, 6-bis(1′-butylbenzimidazol-2′-yl)pyridine], showed the best conversion efficiency. The overall power conversion efficiency reached 8% at 100 W m−2 light intensity and more than 4% at 1000 W m−2. It was also found that the nature of the Ru-based sensitizing dye is essential in these systems. Thus, three dyes were screened: cis-[RuII(4,4′-dicarboxylate-H-2,2′-bipyridine)2(NCS)2](TBA)2 (1), cis-[RuII(2,2′-bipyridine-4,4′-dicarboxylic acid)(4-methyl-4′-hexadecyl-2,2′-bipyridine)](NCS)2 (2), and cis-[RuII(2,2′-bipyridine-4,4′-dicarboxylic acid)(4,4′-dinonyl-2,2-bipyridine)](NCS)2 (3); (TBA = tetrabutylammonium). Complexes 2 and 3 showed much higher photocurrent than 1, which may be explained by two factors: (1) 2 and 3 bear long alkyl chains, and (2) ruthenium-containing fragments in 2 and 3 are positively charged, which leads to the repulsion of the Co RC from the TiO2 surface.
Three one-electron bipyridyl (bpy) cobalt complexes, [Co(t-Bu2bpy)3]3+/2+, [Co(Me2bpy)3]3+/2+, and [Co(bpy)3]3+/2+, presumed to regenerate the dye through an outer-sphere mechanism, were investigated by Klahr et al. in combination with the N3 and [Ru(bpy)2(4,4′-dicarboxy-bpy)](PF6)2 dyes.63 In order to improve the quantum yields, the atomic layer deposition (ALD) method was used to deposit alumina on the surface of the nano-TiO2 material. It is important to note that minor variations of the ligand substituents significantly affect the rates of recombination losses, which limit the IPCE obtained for different electrolytes. However, coating the TiO2 nanoparticles with an ultrathin alumina layer generally improves the IPCE. The maximal conversion efficiency obtained was 1.3% at 1000 Wm−2 in combination with the N3 and [Ru(bpy)2(4,4′-dicarboxy-bpy)](PF6)2 sensitizing dyes.63
Just as for alternative main-group RCs, the nature of the counter electrode is essential for the use of Co-based electrolytes. Thus, Ghamouss et al. used screen-printed carbon electrodes associated with tris(4,4′-di-tert-butyl-2,2′-bpy)cobalt(II)/cobalt(III) as the redox mediator together with two ruthenium-based dyes, Ru535-bisTBA and Z907.64 However, the cells containing platinized counter electrodes still showed higher conversion efficiencies, 1.8% in contrast to 1.2% in the case of carbon electrodes. A similar approach used an ultrathin alumina coating, in combination with [Co(1,10-phenanthroline)3](ClO4)2 and its derivatives as RCs (the N3 dye was used).65 The cells containing the redox couple Co(5-nitro-phen)33+/2+, displaying a more positive redox potential and with an alumina coating, showed the highest Voc, amounting to 0.844 V.
A thorough investigation of the transport and interfacial transfer of electrons in DSCs based on the Co(dbbip)2/3redox couple and the N719 dye was performed by Wang et al.66 Intensity-modulated photocurrent and photovoltage spectroscopy (IMPS/IMVS) together with in situ near-IR absorption spectroscopy were used to determine the diffusion length. It was found that the electron diffusion length is significantly reduced when the classical iodide/triiodide electrolyte is replaced by the Co(II)/Co(III) redox system.66
Recently, redox mediators based on cobalt polypyridines were tested in combination with organic dyes.67 Two organic triphenylamine dyes, D29 and D35, were investigated.68 Several cobalt-based RCs with different redox potentials were prepared: cobalt(II/III) tris(2,2′-bpy), [Co(bpy)3]3+/2+, cobalt(II/III) tris(4,4′-dimethyl-2,2′-bipyridine), [Co(dmb)3]3+/2+, cobalt(II/III) tris(4,4′-ditert-butyl-2,2′-bipyridine), [Co(dtb)3]3+/2+, and cobalt(II/III) tris(1,10-phenanthroline), [Co(phen)3]3+/2+. The best result was obtained for the D35-[Co(bpy)3]3+/2+ combination: at full sun-light irradiation a Voc of 0.92 V and an overall conversion efficiency of 6.7% were recorded. Such a high Voc may be explained by the presence of four bulky butoxy chains in the triphenylamine unit of the D35 dye, in contrast to the D29 dye. Interestingly, the corresponding iodide/triiodide system is less efficient: Voc = 0.91, overall efficiency = 5.5%.
Apart from cobalt-based RCs, complexes and clusters of other metals have also been investigated. Thus, the Ni(III)/Ni(IV) bis(dicarbollide) complexes have recently been tested in combination with the N719 sensitizing dye.69 The authors compared the photovoltaic properties of this RC with the Fc/Fc+ system in acetonitrile solution: The Ni(III)/Ni(IV) bis(dicarbollide) RC shows open-circuit voltage almost three times higher than Fc/Fc+ (580 mV vs. 200 mV). In addition, the ALD of alumina allows an increase of the Voc up to 640 mV, of the Isc to 3.76 mA cm−2 and of the overall efficiency to 1.5%.
Hattori and co-workers studied mononuclear, blue copper complexes as alternative RCs for DSCs.70 Three types of complexes were studied: [(−)-sparteine-N,N′](maleonitriledithiolato-S,S′)copper(I/II), [Cu(SP)(mmt)]−/0, bis(2,9-dimethy-1,10-phenanthroline)copper(I/II), [Cu(dmp)2]+/2+, and bis(1,10-phenanthroline)copper(I/II), [Cu(phen)2]+/2+. Maximal IPCEs of about 40% were obtained for the [Cu(SP)(mmt)]−/0 system. The highest overall conversion efficiency recorded for [Cu(dmp)2]+/2+ in combination with the N719 dye under a low light intensity of 200 W m−2 was 2.2%.70
Recently, Bai and co-workers introduced a promising electron mediator, bis(2,9-dimethyl-1,10-phenanthroline)copper(I/II).71 In this work, the authors explored the organic dye C218, Fig. 6.

The electrolyte used contained 0.2 M Cu(dmp)2TFSI (dmp = 2,9-dimethyl-1,10-phenantroline,TFSI = bis(trifluoromethanesulfonyl)imide), 0.05 M Cu(dmp)2TFSI chloride, 0.5 M 4-tert-butylpyridine (additive) and 0.1 M LiTFSI in acetonitrile was used; a control cell with the I−/I3− RC showed 6.5% conversion efficiency. The I/V data for the copper-based electrolyte studied under full sun-light irradiation are quite impressive: Isc = 11.29 mA cm−2, Voc = 0.932 mV, FF = 0.66, η = 7%. Again, the high photovoltage recorded may probably be explained by the presence of the bulky, branched alkyl and alkoxy chains in the structure of the sensitizing dye helping to retard electron recombination loss processes.71
The classical ferrocene/ferrocenium redox couple has also attracted attention in DSC application. The Fc/Fc+ redox potential (0.62 V) is more positive than that of I−/I3− (0.35 V vs.NHE), which has the ‘potential’ to substantially increase the Voc of the DSCs. The major challenge, however, is the very rapid interfacial recombination losses of injected photoelectrons. Different approaches have been introduced to overcome this problem: passivation of the TiO2 films by silicon-based polymers72 and ALD of alumina.73 It was proven, however, that in spite of decreasing the rate of recombination, the regeneration of the dye molecules also becomes slower.74
In 2011 Daeneke and co-authors reported DSCs based on the Fc/Fc+ redox shuttle showing a conversion efficiency of 7.5% at full sun-light illumination.75 In this work, again a bulky organic dye (Fig. 7) was used, in contrast to the more ‘standard’ ruthenium-based dyes studied before.72–74
 | ||
| Fig. 7 Chemical structure of the Carbz-PAHTDTT sensitizer used in Ref. 75. | ||
The non-corrosive nature of the ferrocenium-based RCs and the possibility to easily tune the redox potential by ligand modifications are very attractive intrinsic features of these systems. For the best results, the authors used a dye co-absorbent, chenodeoxycholic acid: I/V data showed Isc = 12.2 mA cm−2, Voc = 842 mV, FF = 73% and efficiency of 7.5%.75
Recent publications clearly show that classical iodide/triiodide and other halogen-based redox couples may be successfully replaced by alternative systems. Cheap, non-corrosive and almost transparent cobalt and iron complexes are probably the most promising candidates. On the other hand, pure organic, sulphur containing redox shuttles also have large potential. However, the thorough investigation of long-term stability of the new RCs in combination with different dyes is absolutely necessary from a practical application point of view.
4. Additives
A great deal of effort has been devoted to the optimization of photovoltaic performance for DSCs. In addition to the modification of electrode materials and sensitizers, the additives in liquid electrolytes play a key role to enhance photovoltaic performance. Specific cations or compounds in the electrolytes are expected to adsorb onto the semiconductor surface (typically TiO2). The adsorption of additives have been found to exert dramatic influences on many parameters, such as modification of the semiconductor surface charge,76,77 shift of the conduction band edge (CB), recombination kinetics etc., thus affecting the fundamental photovoltaic parameters and consequently the overall conversion efficiency.The Voc is a very important parameter in determining the overall photovoltaic performance of DSCs. The Voc of DSCs is defined as the difference between the quasi-Fermi level of the electrons in the TiO2 film and the redox potential of the electrolyte. One class of the most frequently used additives, which has been mainly dedicated to the improvement of the Voc, is nitrogen-containing heterocyclic compounds, such as TBP. TBP was first employed as an additive by Grätzel and co-workers in 1993, resulting in a significant improvement of the Voc.28 The effect of TBP in liquid electrolytes has been intensely studied and several mechanisms have been proposed. Huang et al. found that the addition of TBP and pyridine derivatives may reduce the electron recombination rate by 1–2 orders of magnitude.78 Intensity modulated photovoltage spectroscopy (IMVS) results later indicated that the dramatic increase of Voc could be mainly ascribed to the negative shift of the CB in the TiO2 film of the WE.79 The addition of TBP into the electrolyte was further studied in detail by Boschloo et al.80 It was found that the increase of the Voc could be attributed to a combined effect of the shift of the CB towards a negative direction (with respect to the unbound vacuum state) as well as longer electron lifetimes in the CB. A great number of alkylpyridine and alkylaminopyridine derivatives have also been tested as additives in liquid electrolytes, exhibiting very similar effects to TBP.81,82
Pyridine analogues, and derivatives of N-alkylbenzimidazole, have also been frequently used in liquid electrolytes, particularly in ionic liquid (IL) based electrolytes.3,5,83 Kusama and Arakawa have screened a large number of benzimidazole derivatives as additives in liquid electrolytes.84Benzimidazole derivatives were found to behave in a similar way to TBP. A large series of other nitrogen-containing heterocyclic compounds, such as aminotriazole, pyrimidine, aminothiazole, pyrzaole and quinolineetc., have also been studied by Kusama, Arakawa and co-workers.85–90
In contrast to the nitrogen-containing heterocyclic compounds, another class of most frequently used additives contains specific cations, such as lithium ions (Li+) or guanidinium [C(NH2)3+, abbreviated G+] ions. The adsorption of specific cations in the electrolytes onto the TiO2 surface could shift the CB towards the more positive direction (with respect to the unbound vacuum state). Several studies have been carried out in order to investigate the effect of Li+ in liquid electrolytes.19,91–95 These results indicate that the positive shift of the CB of TiO2, arising from the adsorption of Li+ onto the TiO2 surface and possibly through surface layer intercalation, leads to an increase of the electron injection yield from the excited state of the sensitizing dye to the CB of TiO2. The higher electron injection yield results in an increase of the short-circuit current (Isc). However, Kopidakis et al. discovered that lithium ions could irreversibly intercalate into the dye-sensitized TiO2 films under normal light intensities, which strongly affects both the electron recombination and electron transport properties in the WEs of the DSCs.96
Guanidinium thiocyanate (GSCN) is another commonly used additive in liquid electrolytes for DSCs. The guanidinium cation is not expected to intercalate into the WE TiO2 film, and has been employed to enhance the photovoltaic performance in DSCs. It was reported that guanidinium cations facilitate the self-assembly of a compact dye layer, thus reducing the dark current, which results in a significant improvement in the Voc.97 Here, a low dark current must be considered as strongly correlated to low recombination losses under irradiation. Kopidakis et al. investigated the influence of the guanidinium cation on the recombination kinetics and the conduction band edge movement, and found that a collective effect of the slower electron recombination rate and a positive shift of the CB in TiO2 gives rise to an overall improvement of the Voc.98 Zhang et al. discovered that a significant enhancement of Isc is observed when guanidinium cations are added into the electrolyte due to an increase of the electron injection yield.99
The state-of-the-art DSCs based on liquid electrolytes normally employ these two kinds of additives simultaneously in the electrolytes,5,83,100,101 in spite of the fact that they function in opposite ways with respect to the CB of the TiO2 surface. The expected added benefit is thus the combination of CB shift and simultaneous surface blocking with respect to recombination losses. Durrant and co-workers investigated the influence of electrolyte compositions on the DSC device performance.93 It was revealed that the electrolyte containing both lithium ions and TBP gives the highest device photovoltaic performance, which is better than two other electrolytes containing only one additive. By varying the concentrations of lithium ions and TBP, it was further shown that optimum overall device efficiency is strongly correlated to additive concentrations in the electrolyte.94 The effects of the additives GSCN and N-methylbenzimidazole (MBI) on the photovoltaic performance of DSCs in IL-based electrolytes have also been elucidated. A synergistic effect is observed when GSCN and MBI are used together in an IL-based electrolyte, resulting in an optimal photovoltaic performance.102
Essentially, electrolytes incorporating only one kind of additive mainly maximizes one photovoltaic parameter (photovoltage or photocurrent). The reason for the “optimum” photovoltaic performance, benefiting from the cooperative effect of these two kinds of additives, is still not unambiguously understood at a molecular level. The modulation and control of the balance between electron injection dynamics and electron recombination kinetics is considered to play a crucial role for the enhancement of DSC device performance. In this sense, to get a deeper understanding of the mechanism of additive components in the electrolytes at the semiconductor/electrolyte interface is imperative to obtain even more efficient DSCs.
5. Interactions with the dye
In the following section, the direct interaction between the sensitizing dye molecules and various components of a liquid electrolyte is reviewed. One problem of selection going through this literature is that there are many publications that report photophysical results and as a conclusion indicate an interaction with the dye; thus, the conclusions tend in fact to be hypotheses that offer a plausible explanation for the effects observed. However, direct investigation at a molecular level is typically missing. A broad search using a selection of keywords relating to the dye-electrolyte interaction generates more than 1200 hits. However, out of these less than 50 describe the results from direct investigations. One area where this phenomenon occurs often is the proposed ‘blocking’ effect of bulky substituents on organic and organometallic dyes. This type of indirect study is not included in the review below, and as a consequence the number of relevant papers is low.5.1 Dye-cation interactions.
Furube et al. showed in a combined FT-IR and calculational study, using a semi-empirical method, that photovoltaic effects could be linked to an interaction between a coumarin dye (NKX-2311) and Li+ ions. Also the injection efficiency was improved by the presence of Li+ ions.103 Grätzel and co-workers reported the use of a designed cation-coordinating dye, containing ethylenoxide entities, together with hydrophobic alkyl groups (K68). The interaction of Li+ with the dye was verified using FT-IR spectroscopy. The main effect was an enhanced stability.104 This work is based on the non-hydrophobic predecessor K51, which was described as ion-trapping (with respect to cations, such as Li+) and thus prevents a positive shift of the semiconductor conduction band with good photovoltages as the main result,105 see Fig. 8. | ||
| Fig. 8 Molecular structures of sensitizers K51 and K68. | ||
5.2 Dye–iodine interactions.
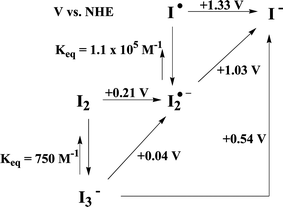 | ||
| Fig. 9 Diagram showing relevant standard thermodynamic potentials and equilibrium constants for the iodine redox species in water solution from Ref. 111. | ||
In an early RRS study, Hagfeldt and co-workers showed that the degradation of the dye N719 could be linked to a loss of thiocyanate ligands proposed to form a pseudo-trihalide of the type I2SCN−.119 A later study by the same group showed that the presence of Li+ accelerated the dye degradation through ligand exchange of the thiocyanate ligands.120 A similar conclusion was drawn on the basis of an HPLC-coupled mass-spectroscopic investigation of the Ru-based N719 and Z907 dyes, showing ligand exchange of thiocyanate for various electrolyte components, also including 4-tert-butylpyridine and the solvent molecules.121,122 However, a microscopic RRS study by Falaras and others showed comparably small, inconclusive spectral changes even after more than 6000 h exposure to light and an operating temperature of over 50 °C.123
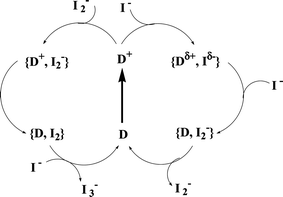
The outer-sphere reaction mechanism was later shown to be compatible also with organic dyes.126 An extension to also other halides later verified the general model of regeneration identified by Privalov et al.127 In a later study, Kusama et al. showed that N-containing heterocycles, such as the common electrolyte additive 4-tert-butylpyridine, actually weaken the dye-iodide interaction and thus suppress regeneration.128 The extension to periodic DFT calculations, in order to include the effects of cations and electrolyte additives, showed that additive molecules interact weakly with iodine-containing anions.129
As a final curiosity in this field, it can be noted that Mayer and his groups used X-ray photoelectron spectroscopy (XPS) to verify the rather obvious expectation that the solvent acetonitrile stabilizes the dye energy levels. The effect is reversible upon evaporation of the solvent.130
It is remarkable that so few direct studies of the direct interaction between the sensitizing dye and the electrolyte components exist. In particular considering that this interaction represents one of the fundamental processes in the DSC system. Clearly, there is an open field for improving our understanding.
6. Concluding remarks
The DSC is a seemingly simple but in reality very complex device. Many different processes must be tuned and integrated in order to provide a cell with high conversion efficiency. In spite of this, the DSC also tends to be very tolerant to the exchange of a single component; still providing reasonable conversion efficiencies even in the absence of any optimization and in the absence of any attempt to understand the mechanisms affected by the new cell component.In this review, the liquid electrolytes have been scrutinized in order to extract information useful to further analysis and improvement of the DSC. Although many steps has been taken to both provide better conversion efficiencies and better understanding at a molecular level, there is still a long walk to sufficient insight. In particular, there is an obvious need for studies at a molecular level in operating DSC devices to be linked to the mass of macroscopic photoelectrochemical data in the literature.
Acknowledgements
The authors gratefully acknowledge support from the Swedish National Research Council, Swedish Energy Agency, as well as the Knut and Alice Wallenberg Foundation.References
- B. O'regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- M. Gorlov and L. Kloo, Dalton Trans., 2008, 2655–2666 RSC.
- S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2009, 19, 2187–2202 CrossRef CAS.
- Y. Bai, Y. M. Cao, J. Zhang, M. Wang, R. Z. Li, P. Wang, S. M. Zakeeruddin and M. Grätzel, Nat. Mater., 2008, 7, 626–630 CrossRef CAS.
- H. R. Jhong, D. S. H. Wong, C. C. Wan, Y. Y. Wang and T. C. Wei, Electrochem. Commun., 2009, 11, 209–211 CrossRef CAS.
- Z. Yu, N. Vlachopoulos, A. Hagfeldt and L. Kloo, unpublished results.
- Q. Yu, Y. Wang, Z. Yi, N. Zu, J. Zhang, M. Zhang and P. Wang, ACS Nano, 2010, 4, 6032–6038 CrossRef CAS.
- J. Koryta, J. Dvorak and L. Kavan, Wiley, Chicester, Editon edn, 1993, pp. 330–335.
- D. T. Sawyer, A. Sobkowiak and J. J. L. Roberts, Wiley, New York, Editon edn, 1995, pp. 299–341.
- B. E. Conway, in Electrochemical Supercapacitors, ed. Kluwer, Plenum, New York, Editon edn, 1999, pp. 335–375 Search PubMed.
- R. Memming, Prog. Surf. Sci., 1984, 17, 7–73 CrossRef CAS.
- M. Matsumura, S. Matsudaira, H. Tsubomura, M. Takata and H. Yanagida, Ind. Eng. Chem. Prod. Res. Dev., 1980, 19, 415–421 CrossRef CAS.
- H. Tsubomura, M. Matsumura, Y. Nomura and T. Amamiya, Nature, 1976, 261, 402–403 CrossRef CAS.
- J. Desilvestro, M. Grätzel, L. Kavan, J. Moser and J. Augustynski, J. Am. Chem. Soc., 1985, 107, 2988–2990 CrossRef CAS.
- K. Kalyanasundaram, N. Vlachopoulos, V. Krishnan, A. Monnier and M. Grätzel, J. Phys. Chem., 1987, 91, 2342–2347 CrossRef CAS.
- N. Vlachopoulos, P. Liska, J. Augustynski and M. Grätzel, J. Am. Chem. Soc., 1988, 110, 1216–1220 CrossRef CAS.
- P. Liska, N. Vlachopoulos, M. K. Nazeeruddin, P. Comte and M. Grätzel, J. Am. Chem. Soc., 1988, 110, 3686–3687 CrossRef CAS.
- Y. Liu, A. Hagfeldt, X. R. Xiao and S. E. Lindquist, Sol. Energy Mater. Sol. Cells, 1998, 55, 267–281 CrossRef CAS.
- B. Enright, G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1994, 98, 6195–6200 CrossRef CAS.
- C. H. Law, S. C. Pathirana, X. O. Li, A. Y. Anderson, P. R. F. Barnes, A. Listorti, T. H. Ghaddar and B. C. O'Regan, Adv. Mater., 2010, 22, 4505–4509 CrossRef CAS.
- H. Tributsch, Coord. Chem. Rev., 2004, 248, 1511–1530 CrossRef CAS.
- M. K. Nazeeruddin, P. Liska, J. Moser, N. Vlachopoulos and M. Grätzel, Helv. Chim. Acta, 1990, 73, 1788–1803 CrossRef CAS.
- M. C. Buzzeo, C. Hardacre and R. G. Compton, ChemPhysChem, 2006, 7, 176–180 CrossRef CAS.
- G. E. Tulloch, J. Photochem. Photobiol., A, 2004, 164, 209–219 CrossRef CAS.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, T. Bessho and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS.
- L. Andrade, S. M. Zakeeruddin, M. K. Nazeeruddin, H. A. Ribeiro, A. Mendes and M. Grätzel, ChemPhysChem, 2009, 10, 1117–1124 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- N. Kato, Y. Takeda, K. Higuchi, A. Takeichi, E. Sudo, H. Tanaka, T. Motohiro, T. Sano and T. Toyoda, Sol. Energy Mater. Sol. Cells, 2009, 93, 893–897 CrossRef CAS.
- N. Kato, K. Higuchi, H. Tanaka, J. Nakajima, T. Sano and T. Toyoda, Sol. Energy Mater. Sol. Cells, 2011, 95, 301–305 CrossRef CAS.
- A. Fukui, R. Komiya, R. Yamanaka, A. Islam and L. Y. Han, Sol. Energy Mater. Sol. Cells, 2006, 90, 649–658 CrossRef CAS.
- J. H. Wu, Z. Lan, J. M. Lin, M. L. Huang and P. J. Li, J. Power Sources, 2007, 173, 585–591 CrossRef CAS.
- J. H. Wu, Z. Lan, S. C. Hao, P. J. Li, J. M. Lin, M. L. Huang, L. Q. Fang and Y. F. Huang, Pure Appl. Chem., 2008, 80, 2241–2258 CrossRef CAS.
- V. Gutmann, Electrochim. Acta, 1976, 21, 661–670 CrossRef CAS.
- M. Grätzel, in Heterogeneous Photochemical Electron Transfer, CRC Press, Boca Raton, Florida, Editon edn, 1989 Search PubMed.
- Z. Kebede and S. E. Lindquist, Sol. Energy Mater. Sol. Cells, 1999, 57, 259–275 CrossRef CAS.
- H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef.
- Z. Yu, M. Gorlov, J. Nissfolk, G. Boschloo and L. Kloo, J. Phys. Chem. C, 2010, 114, 10612–10620 CAS.
- I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721–2744 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS.
- J. G. Rowley, B. H. Farnum, S. Ardo and G. J. Meyer, J. Phys. Chem. Lett., 2010, 1, 3132–3140 CrossRef CAS.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- J. M. Gardner, M. Abrahamsson, B. H. Farnum and G. J. Meyer, J. Am. Chem. Soc., 2009, 131, 16206–16214 CrossRef CAS.
- S. Yanagida, Y. Yu and K. Manseki, Acc. Chem. Res., 2009, 42, 1827–1838 CrossRef CAS.
- Z. S. Wang, K. Sayama and H. Sugihara, J. Phys. Chem. B, 2005, 109, 22449–22455 CrossRef CAS.
- C. Teng, X. Yang, C. Yuan, C. Li, R. Chen, H. Tian, S. Li, A. Hagfeldt and L. Sun, Org. Lett., 2009, 11, 5542–5545 CrossRef CAS.
- Z. J. Ning, Q. Zhang, W. J. Wu and H. Tian, J. Organomet. Chem., 2009, 694, 2705–2711 CrossRef CAS.
- S. Rani, P. Suri and R. M. Mehra, Prog. Photovoltaics, 2011, 19, 180–186 CAS.
- P. Wang, S. M. Zakeeruddin, J.-E. Moser, R. Humphry-Baker and M. Grätzel, J. Am. Chem. Soc., 2004, 126, 7164–7165 CrossRef CAS.
- G. Oskam, B. V. Bergeron, G. J. Meyer and P. C. Searson, J. Phys. Chem. B, 2001, 105, 6867–6873 CrossRef CAS.
- B. V. Bergeron, A. Marton, G. Oskam and G. J. Meyer, J. Phys. Chem. B, 2004, 109, 937–943 CrossRef.
- M. Gorlov, H. Pettersson, A. Hagfeldt and L. Kloo, Inorg. Chem., 2007, 46, 3566–3575 CrossRef CAS.
- M. Wang, N. Chamberland, L. Breau, J.-E. Moser, R. Humphry-Baker, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Nat. Chem., 2010, 2, 385–389 CrossRef CAS.
- H. Tian, X. Jiang, Z. Yu, L. Kloo, A. Hagfeldt and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 7328–7331 CrossRef CAS.
- H. Tian, X. Yang, J. Cong, R. Chen, J. Liu, Y. Hao, A. Hagfeldt and L. Sun, Chem. Commun., 2009, 6288–6290 RSC.
- D. Li, H. Li, Y. Luo, K. Li, Q. Meng, M. Armand and L. Chen, Adv. Funct. Mater., 2010, 20, 3358–3365 CrossRef CAS.
- P. J. Cameron, L. M. Peter, S. M. Zakeeruddin and M. Grätzel, Coord. Chem. Rev., 2004, 248, 1447–1453 CrossRef CAS.
- S. A. Sapp, C. M. Elliott, C. Contado, S. Caramori and C. A. Bignozzi, J. Am. Chem. Soc., 2002, 124, 11215–11222 CrossRef CAS.
- H. Nusbaumer, J.-E. Moser, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2001, 105, 10461–10464 CrossRef CAS.
- S. Nakade, Y. Makimoto, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2005, 109, 3488–3493 CrossRef CAS.
- H. Nusbaumer, S. M. Zakeeruddin, J.-E. Moser and M. Grätzel, Chem.–Eur. J., 2003, 9, 3756–3763 CrossRef CAS.
- B. M. Klahr and T. W. Hamann, J. Phys. Chem. C, 2009, 113, 14040–14045 CAS.
- F. Ghamouss, R. Pitson, F. Odobel, M. Boujtita, S. Caramori and C. A. Bignozzi, Electrochim. Acta, 2010, 55, 6517–6522 CrossRef CAS.
- M. J. DeVries, M. J. Pellin and J. T. Hupp, Langmuir, 2010, 26, 9082–9087 CrossRef CAS.
- H. Wang, P. G. Nicholson, L. Peter, S. M. Zakeeruddin and M. Grätzel, J. Phys. Chem. C, 2010, 114, 14300–14306 CAS.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS.
- D. P. Hagberg, X. Jiang, E. Gabrielsson, M. Linder, T. Marinado, T. Brinck, A. Hagfeldt and L. Sun, J. Mater. Chem., 2009, 19, 7232–7238 RSC.
- T. C. Li, A. M. Spokoyny, C. She, O. K. Farha, C. A. Mirkin, T. J. Marks and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 4580–4582 CrossRef CAS.
- S. Hattori, Y. Wada, S. Yanagida and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 9648–9654 CrossRef CAS.
- Y. Bai, Q. Yu, N. Cai, Y. Wang, M. Zhang and P. Wang, Chem. Commun., 2011, 47, 4376–4378 RSC.
- B. A. Gregg, F. Pichot, S. Ferrere and C. L. Fields, J. Phys. Chem. B, 2001, 105, 1422–1429 CrossRef CAS.
- T. W. Hamann, O. K. Farha and J. T. Hupp, J. Phys. Chem. C, 2008, 112, 19756–19764 CAS.
- S. M. Feldt, U. B. Cappel, E. M. J. Johansson, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2010, 114, 10551–10558 CAS.
- T. Daeneke, T. H. Kwon, A. B. Holmes, N. W. Duffy, U. Bach and L. Spiccia, Nat. Chem., 2011, 3, 211–215 CrossRef CAS.
- S. Kambe, S. Nakade, T. Kitamura, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2002, 106, 2967–2972 CrossRef CAS.
- S. Nakade, Y. Saito, W. Kubo, T. Kanzaki, T. Kitamura, Y. Wada and S. Yanagida, Electrochem. Commun., 2003, 5, 804–808 CrossRef CAS.
- S. Y. Huang, G. Schlichthorl, A. J. Nozik, M. Grätzel and A. J. Frank, J. Phys. Chem. B, 1997, 101, 2576–2582 CrossRef CAS.
- G. Schlichthorl, S. Y. Huang, J. Sprague and A. J. Frank, J. Phys. Chem. B, 1997, 101, 8141–8155 CrossRef.
- G. Boschloo, L. Haggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144–13150 CrossRef CAS.
- H. Kusama, Y. Konishi, H. Sugihara and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2003, 80, 167–179 CrossRef CAS.
- H. Kusama and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2004, 81, 87–99 CrossRef CAS.
- D. Shi, N. Pootrakulchote, R. Li, J. Guo, Y. Wang, S. M. Zakeeruddin, M. Grätzel and P. Wang, J. Phys. Chem. C, 2008, 112, 17046–17050 CAS.
- H. Kusama and H. Arakawa, J. Photochem. Photobiol., A, 2004, 162, 441–448 CrossRef CAS.
- H. Kusama and H. Arakawa, J. Photochem. Photobiol., A, 2004, 164, 103–110 CrossRef CAS.
- H. Kusama and H. Arakawa, J. Photochem. Photobiol., A, 2003, 160, 171–179 CrossRef CAS.
- H. Kusama and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2004, 82, 457–465 CrossRef CAS.
- H. Kusama and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2005, 85, 333–344 CrossRef CAS.
- H. Kusama and H. Arakawa, J. Photochem. Photobiol., A, 2004, 165, 157–163 CrossRef CAS.
- H. Kusama, M. Kurashige and H. Arakawa, J. Photochem. Photobiol., A, 2005, 169, 169–176 CrossRef CAS.
- C. A. Kelly, F. Farzad, D. W. Thompson, J. M. Stipkala and G. J. Meyer, Langmuir, 1999, 15, 7047–7054 CrossRef CAS.
- D. F. Watson and G. J. Meyer, Coord. Chem. Rev., 2004, 248, 1391–1406 CrossRef CAS.
- S. A. Haque, E. Palomares, B. M. Cho, A. N. M. Green, N. Hirata, D. R. Klug and J. R. Durrant, J. Am. Chem. Soc., 2005, 127, 3456–3462 CrossRef CAS.
- S. E. Koops, B. C. O'Regan, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS.
- J. R. Jennings and Q. Wang, J. Phys. Chem. C, 2010, 114, 1715–1724 CAS.
- N. Kopidakis, K. D. Benkstein, J. van de Lagemaat and A. J. Frank, J. Phys. Chem. B, 2003, 107, 11307–11315 CrossRef CAS.
- M. Grätzel, J. Photochem. Photobiol., A, 2004, 164, 3–14 CrossRef.
- N. Kopidakis, N. R. Neale and A. J. Frank, J. Phys. Chem. B, 2006, 110, 12485–12489 CrossRef CAS.
- C. N. Zhang, Y. Huang, Z. P. Huo, S. H. Chen and S. Y. Dai, J. Phys. Chem. C, 2009, 113, 21779–21783 CAS.
- Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Y. Han, Jpn. J. Appl. Phys., 2006, 45, L638–L640 CrossRef CAS.
- F. Gao, Y. Wang, D. Shi, J. Zhang, M. K. Wang, X. Y. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 10720–10728 CrossRef CAS.
- Z. Yu, M. Gorlov, G. Boschloo and L. Kloo, J. Phys. Chem. C, 2010, 114, 22330–22337 CAS.
- A. Furube, R. Katoh, K. Hara, T. Sato, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2005, 109, 16406–16414 CrossRef CAS.
- D. Kuang, C. Klein, H. J. Snaith, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, Inorg. Chim. Acta, 2008, 361, 699–706 CrossRef CAS.
- D. B. Kuang, C. Klein, H. J. Snaith, J. E. Moser, R. Humphry-Baker, P. Comte, S. M. Zakeeruddin and M. Grätzel, Nano Lett., 2006, 6, 769–773 CrossRef CAS.
- C. Bauer, G. Boschloo, E. Mukhtar and A. Hagfeldt, J. Phys. Chem. B, 2002, 106, 12693–12704 CrossRef CAS.
- I. Montanari, J. Nelson and J. R. Durrant, J. Phys. Chem. B, 2002, 106, 12203–12210 CrossRef CAS.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Grätzel and J. R. Durrant, J. Phys. Chem. C, 2007, 111, 6561–6567 CAS.
- A. Y. Anderson, P. R. F. Barnes, J. R. Durrant and B. C. O'Regan, J. Phys. Chem. C, 2010, 114, 1953–1958 CAS.
- J. M. Gardner, J. M. Giaimuccio and G. J. Meyer, J. Am. Chem. Soc., 2008, 130, 17252–17253 CrossRef CAS.
- J. G. Rowley, B. H. Farnum, S. Ardo and G. J. Meyer, J. Phys. Chem. Lett., 2010, 1, 3132–3140 CrossRef CAS.
- M. C. Bernard, H. Cachet, P. Falaras, A. Hugot-Le Goff, M. Kalbac, I. Lukes, N. T. Oanh, T. Stergiopoulos and I. Arabatzis, J. Electrochem. Soc., 2003, 150, E155–E164 CrossRef CAS.
- M. C. Bernard, H. Cachet, P. Falaras, A. Hugot-Le Goff, N. T. T. Oanh and T. Stergiopoulos, in Organic Photovoltaics III, ed. Z. H. Kafafi, Spie-Int. Soc. Optical Engineering, Bellingham, Editon edn, 2003, 4801, pp. 87-98 Search PubMed.
- T. Stergiopoulos, M. C. Bernard, A. H. L. Goff and P. Falaras, Coord. Chem. Rev., 2004, 248, 1407–1420 CrossRef CAS.
- A. G. Kontos, T. Stergiopoulos, G. Tsiminis, Y. S. Raptis and P. Falaras, Inorg. Chim. Acta, 2008, 361, 761–768 CrossRef CAS.
- P. H. Svensson and L. Kloo, J. Chem. Soc., Dalton Trans., 2000, 2449–2455 RSC.
- C. W. Shi, S. Y. Dai, K. J. Wang, X. Pan, F. T. Kong and L. H. Hu, Vib. Spectrosc., 2005, 39, 99–105 CrossRef CAS.
- T. Marinado, PhD Thesis, KTH 2009, 2009.
- H. Greijer, J. Lindgren and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 6314–6320 CrossRef CAS.
- H. G. Agrell, J. Lindgren and A. Hagfeldt, J. Photochem. Photobiol., A, 2004, 164, 23–27 CrossRef.
- F. Nour-Mohammadi, H. T. Nguyen, G. Boschloo and T. Lund, J. Photochem. Photobiol., A, 2007, 187, 348–355 CrossRef CAS.
- P. T. Nguyen, R. Degn, H. T. Nguyen and T. Lund, Sol. Energy Mater. Sol. Cells, 2009, 93, 1939–1945 CrossRef CAS.
- V. Likodimos, T. Stergiopoulos, P. Falaras, R. Harikisun, J. Desilvestro and G. Tulloch, J. Phys. Chem. C, 2009, 113, 9412–9422 CAS.
- J. Preat, D. Jacquemin and E. A. Perpete, Environ. Sci. Technol., 2010, 44, 5666–5671 CrossRef CAS.
- G. B. Timofei Privalov, A. Hagfeldt, Per H. Svensson and L. Kloo, J. Phys. Chem. C, 2009, 113, 783–790 Search PubMed.
- J. Nyhlen, G. Boschloo, A. Hagfeldt, L. Kloo and T. Privalov, ChemPhysChem, 2010, 11, 1858–1862 CAS.
- C. H. Hu, A. M. Asaduzzaman and G. Schreckenbach, J. Phys. Chem. C, 2010, 114, 15165–15173 CAS.
- H. Kusama, H. Sugihara and K. Sayama, J. Phys. Chem. C, 2010, 114, 11335–11341 CAS.
- A. M. Asaduzzaman and G. Schreckenbach, Phys. Chem. Chem. Phys., 2010, 12, 14609–14618 RSC.
- K. Schwanitz, U. Weiler, R. Hunger, T. Mayer and W. Jaegermann, J. Phys. Chem. C, 2007, 111, 849–854 CAS.
| This journal is © The Royal Society of Chemistry 2011 |