The effect of imine-carbon substituents in bis(imino)pyridine-based ethylene polymerisation catalysts across the transition series†
Theo M.
Smit
,
Atanas K.
Tomov
,
George J. P.
Britovsek
*,
Vernon C.
Gibson
*,
Andrew J. P.
White
and
David J.
Williams
Department of Chemistry, Imperial College London, Exhibition Road, South Kensington, London SW7 2AY, United Kingdom. E-mail: g.britovsek@imperial.ac.uk; Fax: +44-(0)20-75945804; Tel: +44-(0)20-75945863
First published on 19th December 2011
Abstract
The synthesis, characterization and ethylene polymerisation behaviour of a series of first row transition metal complexes of the general formula LMXn (M = Fe, Co, Mn, n = 2, X = Cl; M = V, Cr, Ti, n = 3, X = Cl; M = Ni, n = 2, X = Br) with bis(imino)pyridine ligands L are reported, whereby the ligands contain heteroatom substituents DRm at the imine carbon (D = O, S, m = 1, R = Me, Ph, 2,6-Me2C6H3 or D = N, m = 2, R = Me, Ph). Only the O- and S-substituted complexes show catalytic activity for the polymerisation of ethylene, upon activation with methylaluminoxane (MAO). The Mn- and Ni-based catalysts were found to be inactive under these conditions. The S-substituted complexes are generally more active than the O-substituted complexes and the catalytic activities increase with the size of the substituents. Iron-, vanadium- and chromium-based catalysts give highly active catalyst systems, which in some cases are more active than the well-known ketimine catalysts. All catalysts produce highly linear polyethylene with molecular weights affected by M, D and R. O-substituted catalyst systems are generally less active and produce lower molecular weight polyethylene compared to S-substituted systems.
Introduction
Tremendous advances have been made during the past two decades in the field of olefin polymerisation catalysis, spurred initially by developments in single-site metallocene technology,1,2 and more recently by the discovery and development of highly active non-metallocene systems.3–5 New catalysts are a key driver for the commercial exploitation of new materials as well as for improving the performance of existing polyolefinic materials. Important criteria for new catalysts are not only high propagation rates but also their chain transfer behaviour, which determines the molecular weight and molecular weight distribution as well as chain-end functionalization of the polyolefin product. In addition, environmental and health regulations limit the metals and ligands that can be used for certain polyolefin applications, as catalyst residues normally remain within the polyolefin product.An important class of polymerisation catalysts is based on the bis(imino)pyridine ligand framework (see Fig. 1).6–8 These ligands were initially found to give highly active iron- and cobalt-based ethylene polymerisation and oligomerisation catalysts.9–13 Subsequently, it was shown that other metals such as titanium,14 vanadium,14–16 and chromium17–19 can also be supported by the bis(imino)pyridine ligand and generate active olefin polymerisation systems, although the activities are generally at least an order of magnitude lower compared to iron. All catalysts are generally activated in situ by the addition of an aluminium alkyl reagent such as methylaluminoxane (MAO) or similar co-catalyst systems.20–22 The active species generated upon the addition of the co-catalyst has been investigated by several groups,23–30 but remains a contentious subject of discussion, which is further complicated by the flexible electronic properties of the bis(imino)pyridine ligand.31–34
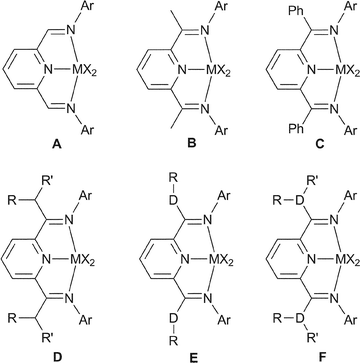 | ||
| Fig. 1 Different imine–carbon substituted bis(imino)pyridine complexes A–F. | ||
There have been many studies on how catalyst behaviour is affected by modifications to the bis(imino)pyridine ligand framework, in particular changes to the substituents at the imine nitrogen donor have been extensively investigated.7,8 Comparatively less effort has been devoted to the effect of changes to the substituents at the imine carbon atom. One likely reason for this is the synthetic difficulties associated with these variations. Catalysts based on aldimine and ketimine ligands of type A and B were amongst the first ligand systems investigated (see Fig. 1).10,12 It was generally found that the aldimine ligands A are an order of magnitude less active and afford lower molecular weight PE products than their ketimine counterparts B.9 A possible reason for this may be that, under the polymerisation conditions, degradation of the catalyst occurs via alkylations by the aluminium alkyl co-catalyst, which are likely to have different rates for A and B. Alkylations of this type have been observed for the free ligands35–37 and for certain metal complexes38 and lead generally to significantly less active catalysts. Further studies have shown that the methyl groups are susceptible to deprotonation, which could also lead to catalyst deactivation.39–41 To avoid these unwanted side-reactions, phenyl groups, as in type C, were introduced to generate iron-,42–44 cobalt-,44 and chromium-based catalysts.17 Improved catalyst stabilities were noted for iron and cobalt,43 but little effect of methyl compared to phenyl was observed in the case of chromium.17 Further modifications such as isopropyl or benzyl substituents at the ketimine position (type D) (R = Me, benzyl and R' = H, Me or benzyl) affect the polymer molecular weight, but had little effect on catalyst activity.45 Interestingly, tertiary-butyl groups at the imine carbon were found to be too sterically hindered to form a stable metal complex.46 A recent report on the application of 2,3,7,8-tetrahydroacridine-4,5(1H,6H) derivatives, whereby the imine carbon is joined with the central pyridine ring at the meta-position to form a six-membered ring, has resulted in a series of iron and cobalt complexes with similar properties to type B.47 The introduction of heteroatom substituents at the imine carbon in ligands of type E was first reported by Bennett for a cobalt catalyst with DR = SMe substituents.48 This was further investigated by us49 for O and S substituents (DR = OMe, SMe, O–2,6-Me2C6H3 and S–2,6-Me2C6H3) and also by others for iron (DR = OMe and OEt)42 and cobalt complexes (DR = OMe).44
The availability of heteroatoms at the imine carbon can have several potential consequences. Firstly, coordination of the heteroatom to the metal centre may occur upon rotation around the pyridine-imine carbon bond. This was indeed observed when small OMe groups were used in combination with very bulky 2,6-diisopropylphenyl groups at the imine nitrogen.44 Secondly, upon activation of the catalyst with a co-catalyst, often aluminium alkyls, binding of the Lewis acidic aluminium centre to the heteroatom could occur and thereby affect the catalyst behaviour. This binding of a Lewis acid may either improve catalyst performance by increasing the electrophilicity at the metal centre or lead to catalyst deactivation. Observations along these lines were previously made by Bazan for nickel complexes with α-diimine-based ligands.50,51 Thirdly, the presence of a heteroatom could affect the rate of alkylation at the imine carbon, which is a known reaction for metal alkyls.37,38 Coordination of the aluminium alkyl to the heteroatom could either provide additional steric protection of the imine carbon centre, or could result in intramolecular alkyl transfer from Al to the imine carbon.52
We have previously reported our initial results on the synthesis and the catalytic ethylene polymerisation behaviour of heteroatom-substituted bis(imino)pyridine iron complexes of type E.49 Extremely high polymerisation activities were observed for the 2,6-dimethylphenylthiolate-substituted iron complex. We have therefore carried out an extensive study on complexes of type E containing ligands with ether and thioether substituents at the imine carbon (D = O, S and R = Me, Ph and 2,6-Me2C6H3) and complexes of type F with amine substituents (D = N, R, R' = Me, Ph) using a range of metals across the 1st row transition series (Fig. 1). The steric requirements of the substituents have been systematically varied from methyl to phenyl to 2,6-dimethylphenyl, in order to establish whether sterics are responsible for the extremely high activities observed in the case of iron. A range of 1st row metals was chosen to investigate whether these structure–activity trends are also observed in related metal complexes.53
Results and discussion
Synthesis of ligands and metal complexes
The ligands 1–11, listed in Fig. 2, were synthesised by the reaction of the imidoyl chloride precursors with two equivalents of NaDR3 (D = O or S, R3 = Me, Ph or 2,6-Me2C6H3) or LiNMePh or LiNPh2 (see Supporting Information). It was not possible to prepare the oxygen analogue of ligand 9via these methods, probably due to the severe steric hindrance between the 2,6-diisopropylphenyl and 2,6-dimethylphenyl groups.
The metal complexes collected in Fig. 3 were synthesized by reaction of the ligand with the appropriate metal salt FeCl2·1.5thf, CoCl2, VCl3·3thf, CrCl3·3thf, TiCl3·3thf, MnCl2·2thf or NiBr2·dme in thf, whereby elevated temperature was required for the reaction with CrCl3·3thf. Metal complexes containing ligands with bulkier substituents were generally obtained in lower yield.
 | ||
| Fig. 3 List of metal complexes of ligands 1–11. | ||
All complexes were characterized by microanalysis, IR spectroscopy, FAB or CI mass spectrometry and magnetic susceptibility measurements (see Supporting Information). In addition, the paramagnetic Fe(II), V(III), Co(II), Ti(III) and Ni(II) complexes were characterized by 1H NMR spectroscopy. The spectra have been assigned on the basis of integrated signal intensities, proximity to the metal centre and by comparison with spectra of previously reported bis(imino)pyridine metal complexes. The paramagnetic 1H NMR spectra of complexes [V(3)Cl3], [Fe(3)Cl2] and [Co(3)Cl2] are shown as representative examples in Fig. 4. The resonances assigned to the protons of the pyridine ring do not only undergo the largest shifts but also show the strongest dependence on the metal centre, with the resonance for the HB protons in meta position shifting from 102 ppm for [Co(3)Cl2] to 79 ppm for [Fe(3)Cl2] and to −2 ppm for [V(3)Cl3]. The resonance of the pyridine para proton HA is located considerably further upfield in [Fe(3)Cl2] (1.4 ppm) compared to the parent bis(imino)pyridine complex [Fe(12)Cl2] (40.1 ppm), which illustrates the different electronic environments in the two complexes.
![Representative 1H NMR spectra of complexes [Fe(3)Cl2], [Co(3)Cl2] and [V(3)Cl3] in CD2Cl2 at 298 K.](/image/article/2012/CY/c2cy00448h/c2cy00448h-f4.gif) | ||
| Fig. 4 Representative 1H NMR spectra of complexes [Fe(3)Cl2], [Co(3)Cl2] and [V(3)Cl3] in CD2Cl2 at 298 K. | ||
Solid state structures
The introduction of heteroatoms (D) at the imine carbon could result in alternative binding modes of the ligand. In addition to the ubiquitous [N,N,N] coordination involving the pyridine and imine donors,7 [N,N,D] or [D,N,D] coordination becomes a distinct possibility, which was already observed on one occasion.44 In order to confirm that the desired [N,N,N] coordination has taken place, the solid state structures of many metal complexes have been determined by X-ray crystallography: [V(4)Cl3], [Cr(4)Cl3], [Mn(4)Cl2], iron(II) dichloride complexes of ligands 1, 2, 3, 4, 5, 6 and 8, cobalt(II) dichloride complexes of ligands 1, 5 and 6, and [Ni(4)Br2]. The molecular structures of [Fe(1)Cl2] and [Fe(8)Cl2] have already been discussed in our previous communication.49 Selected structures for complexes [Co(1)Cl2], [Fe(2)Cl2], [Fe(3)Cl2], [V(4)Cl3] and [Co(5)Cl2] are shown in Fig. 5–10. All other structures are collected in the Supporting Information.![The molecular structure of [Co(1)Cl2]. Selected bond lengths (Å) and angles (°); Co–Cl(1) 2.237(4), Co–Cl(2) 2.236(5), Co–N(1) 2.026(11), Co–N(7) 2.348(11), Co–N(9) 2.261(11), Cl(1)–Co–Cl(2) 121.25(17), Cl(1)–Co–N(1) 114.0(3), Cl(1)–Co–N(7) 99.5(3), Cl(1)–Co–N(9) 95.4(3), Cl(2)–Co–N(1) 124.5(3), Cl (2)–Co–N(7) 91.9(3), Cl(2)–Co–N(9) 101.7(3), N(1)–Co–N(7) 73.9(5), N(1)–Co–N(9) 77.0(5), N(7)–Co–N(9) 150.7(4).](/image/article/2012/CY/c2cy00448h/c2cy00448h-f5.gif) | ||
| Fig. 5 The molecular structure of [Co(1)Cl2]. Selected bond lengths (Å) and angles (°); Co–Cl(1) 2.237(4), Co–Cl(2) 2.236(5), Co–N(1) 2.026(11), Co–N(7) 2.348(11), Co–N(9) 2.261(11), Cl(1)–Co–Cl(2) 121.25(17), Cl(1)–Co–N(1) 114.0(3), Cl(1)–Co–N(7) 99.5(3), Cl(1)–Co–N(9) 95.4(3), Cl(2)–Co–N(1) 124.5(3), Cl (2)–Co–N(7) 91.9(3), Cl(2)–Co–N(9) 101.7(3), N(1)–Co–N(7) 73.9(5), N(1)–Co–N(9) 77.0(5), N(7)–Co–N(9) 150.7(4). | ||
![The molecular structure of [Fe(2)Cl2]. Selected bond lengths (Å) and angles (°); Fe–Cl(1) 2.276(2), Fe–Cl(2) 2.284(2), Fe–N(1) 2.123(4), Fe–N(7) 2.274(4), Fe–N(9) 2.281(4), Cl(1)–Fe–Cl(2) 112.45(7), Cl(1)–Fe–N(1) 119.67(13), Cl(1)–Fe–N(7) 101.46(11), Cl(1)–Fe–N(9) 101.83(12), Cl(2)–Fe–N(1) 127.88(12), Cl(2)–Fe–N(7) 97.01(11), Cl(2)–Fe–N(9) 97.62(12), N(1)–Fe–N(9) 73.0(2), N(1)–Fe–N(7) 73.0(2), N(7)–Fe–N(9) 145.15(14).](/image/article/2012/CY/c2cy00448h/c2cy00448h-f6.gif) | ||
| Fig. 6 The molecular structure of [Fe(2)Cl2]. Selected bond lengths (Å) and angles (°); Fe–Cl(1) 2.276(2), Fe–Cl(2) 2.284(2), Fe–N(1) 2.123(4), Fe–N(7) 2.274(4), Fe–N(9) 2.281(4), Cl(1)–Fe–Cl(2) 112.45(7), Cl(1)–Fe–N(1) 119.67(13), Cl(1)–Fe–N(7) 101.46(11), Cl(1)–Fe–N(9) 101.83(12), Cl(2)–Fe–N(1) 127.88(12), Cl(2)–Fe–N(7) 97.01(11), Cl(2)–Fe–N(9) 97.62(12), N(1)–Fe–N(9) 73.0(2), N(1)–Fe–N(7) 73.0(2), N(7)–Fe–N(9) 145.15(14). | ||
![The molecular structure of [Fe(3)Cl2]. Selected bond lengths (Å) and angles (°); Fe–Cl(1) 2.266(2), Fe–Cl(2) 2.251(2), Fe–N(1) 2.090(4), Fe–N(7) 2.345(4), Fe–N(9) 2.317(4), Cl(1)–Fe–Cl(2) 118.56(9), Cl(1)–Fe–N(1) 112.18(13), Cl(1)–Fe–N(7) 98.93(12), Cl(1)–Fe–N(9) 95.93(12), Cl(2)–Fe–N(1) 128.99(13), Cl(2)–Fe–N(7) 93.27(12), Cl(2)–Fe–N(9) 104.80(12), N(1)–Fe–N(7) 73.62(14), N(1)–Fe–N(9) 73.9(2), N(7)–Fe–N(9) 147.4(2).](/image/article/2012/CY/c2cy00448h/c2cy00448h-f7.gif) | ||
| Fig. 7 The molecular structure of [Fe(3)Cl2]. Selected bond lengths (Å) and angles (°); Fe–Cl(1) 2.266(2), Fe–Cl(2) 2.251(2), Fe–N(1) 2.090(4), Fe–N(7) 2.345(4), Fe–N(9) 2.317(4), Cl(1)–Fe–Cl(2) 118.56(9), Cl(1)–Fe–N(1) 112.18(13), Cl(1)–Fe–N(7) 98.93(12), Cl(1)–Fe–N(9) 95.93(12), Cl(2)–Fe–N(1) 128.99(13), Cl(2)–Fe–N(7) 93.27(12), Cl(2)–Fe–N(9) 104.80(12), N(1)–Fe–N(7) 73.62(14), N(1)–Fe–N(9) 73.9(2), N(7)–Fe–N(9) 147.4(2). | ||
![Overlay of the coordination geometries for the three complexes [Fe(1)Cl2] (solid lines), [Fe(4)Cl2] (dashed lines) and [Fe(8)Cl2] (dotted lines), showing the different inclinations of the pyridine ring.](/image/article/2012/CY/c2cy00448h/c2cy00448h-f8.gif) | ||
| Fig. 8 Overlay of the coordination geometries for the three complexes [Fe(1)Cl2] (solid lines), [Fe(4)Cl2] (dashed lines) and [Fe(8)Cl2] (dotted lines), showing the different inclinations of the pyridine ring. | ||
![The molecular structure of [V(4)Cl3]. Selected bond lengths (Å) and angles (°); V–Cl(1) 2.3418(12), V–Cl(2) 2.2889(12), V–Cl(3) 2.3309(12), V–N(1) 2.074(3), V–N(7) 2.198(3), V–N(9) 2.213(3), Cl(1)–V–Cl(2) 94.68(5), Cl(1)–V–Cl(3) 169.94(5), Cl(1)–V–N(1) 83.28(10), Cl(1)–V–N(7) 89.67(10), Cl(1)–V–N(9) 88.21(9), Cl(2)–V–Cl(3) 95.32(5), Cl(2)–V–N(1) 177.42(10), Cl(2)–V–N(7) 102.96(9), Cl(2)–V–N(9) 106.16(9), Cl(3)–V–N(1) 86.70(10), Cl(3)–V–N(7) 87.05(9), Cl(3)–V–N(9) 90.04(9), N(1)–V–N(7) 75.50(13), N(1)–V–N(9) 75.41(12), N(7)–V–N(9) 150.88(12).](/image/article/2012/CY/c2cy00448h/c2cy00448h-f9.gif) | ||
| Fig. 9 The molecular structure of [V(4)Cl3]. Selected bond lengths (Å) and angles (°); V–Cl(1) 2.3418(12), V–Cl(2) 2.2889(12), V–Cl(3) 2.3309(12), V–N(1) 2.074(3), V–N(7) 2.198(3), V–N(9) 2.213(3), Cl(1)–V–Cl(2) 94.68(5), Cl(1)–V–Cl(3) 169.94(5), Cl(1)–V–N(1) 83.28(10), Cl(1)–V–N(7) 89.67(10), Cl(1)–V–N(9) 88.21(9), Cl(2)–V–Cl(3) 95.32(5), Cl(2)–V–N(1) 177.42(10), Cl(2)–V–N(7) 102.96(9), Cl(2)–V–N(9) 106.16(9), Cl(3)–V–N(1) 86.70(10), Cl(3)–V–N(7) 87.05(9), Cl(3)–V–N(9) 90.04(9), N(1)–V–N(7) 75.50(13), N(1)–V–N(9) 75.41(12), N(7)–V–N(9) 150.88(12). | ||
![The molecular structure of [Co(5)Cl2]. Selected bond lengths (Å) and angles (°); Co–Cl(1) 2.2954(15), Co–Cl(2) 2.2300(14), Co–N(1) 2.048(4), Co–N(7) 2.197(4), Co–N(9) 2.194(4), Cl(1)–Co–Cl(2) 115.82(7), Cl(1)–Co–N(1) 88.19(12), Cl(1)–Co–N(7) 104.78(11), Cl(1)–Co–N(9) 100.43(11), Cl(2)–Co–N(1) 155.81(12), Cl(2)–Co–N(7) 100.43(11), Cl(2)–Co–N(9) 96.72(11), N(1)–Co–N(7) 74.49(15), N(1)–Co–N(9) 74.79(15), N(7)–Co–N(9) 139.18(15).](/image/article/2012/CY/c2cy00448h/c2cy00448h-f10.gif) | ||
| Fig. 10 The molecular structure of [Co(5)Cl2]. Selected bond lengths (Å) and angles (°); Co–Cl(1) 2.2954(15), Co–Cl(2) 2.2300(14), Co–N(1) 2.048(4), Co–N(7) 2.197(4), Co–N(9) 2.194(4), Cl(1)–Co–Cl(2) 115.82(7), Cl(1)–Co–N(1) 88.19(12), Cl(1)–Co–N(7) 104.78(11), Cl(1)–Co–N(9) 100.43(11), Cl(2)–Co–N(1) 155.81(12), Cl(2)–Co–N(7) 100.43(11), Cl(2)–Co–N(9) 96.72(11), N(1)–Co–N(7) 74.49(15), N(1)–Co–N(9) 74.79(15), N(7)–Co–N(9) 139.18(15). | ||
The cobalt methoxy-substituted complex [Co(1)Cl2] is isomorphous with the iron complex [Fe(1)Cl2],49 and with gross structures very similar to that of the iron methyl analogue [Fe(12)Cl2].9 In contrast to the molecular Cs symmetry of the methyl derivative, the structures of [Fe(1)Cl2] and [Co(1)Cl2] are closer to C2 symmetry with both mesityl and methylether substituents being tilted in opposite directions above and below the plane of the ligand. In both complexes, the geometry at the metal is distorted trigonal bipyramidal with the two five-membered chelate rings being essentially planar (the one containing N(7) to within 0.03 Å for [Fe(1)Cl2] and 0.04 Å for [Co(1)Cl2], and the one containing N(9) to within 0.02 Å for both complexes); the metal atom lies only 0.08 Å (Fe), 0.10 Å (Co), out of the N3 plane. The mesityl rings are approximately orthogonal to the ligand plane {ca. 77 and 76° for the N(7)- and N(9)-bound rings in [Fe(1)Cl2] respectively and ca. 76 and 78° respectively in [Co(1)Cl2]}.
The iron complex [Fe(2)Cl2] (Fig. 6) has a structure that is noticeably different from the oxygen analogue [Fe(1)Cl2] (see Fig. S5), having molecular Cs rather than C2 symmetry, the methylthioether substituents being oriented on the same side of the ligand plane. Although the geometry at the iron centre is again distorted trigonal bipyramidal, here the metal sits ca. 0.40 Å out of the plane of the pyridine ring, cf. ca. 0.08 Å in [Fe(1)Cl2]. This distortion is also seen in the five-membered chelate rings where the metal lies ca. 0.12 Å and 0.23 Å out of the planes of the remaining atoms. The mesityl rings are approximately orthogonal to the ligand plane (ca. 87 and 86° for the N(7)– and N(9)-bound rings respectively).
The phenylether complex [Fe(3)Cl2] (Fig. 7) has a core structure virtually identical to that of the methylether complex [Fe(1)Cl2], the only major difference being a change in the torsional twists about the C(7)–OPh and C(9)–OPh bonds, which are ca. 44 and 38° respectively, cf. 20 and 25° for their C(7/9)–OMe counterparts in [Fe(1)Cl2]. The N(7) and N(9) chelate rings are each coplanar to within 0.04 Å and the planes of the associated mesityl rings are inclined by ca. 83 and 79° respectively.
The structure of the iron phenylthioether complex [Fe(4)Cl2] (see Fig. S9) looks superficially quite similar to that of the 2,6-dimethylphenylthioether complex [Fe(8)Cl2] (see Fig. S17). However, the geometry at the metal centre its noticeably different. Whereas [Fe(8)Cl2] has a clear square-based pyramidal geometry, [Fe(4)Cl2] has a geometry intermediate between trigonal bipyramidal (tbp) and square-based pyramidal (sbp). Considered as the latter, the basal plane in [Fe(4)Cl2] is only coplanar to within ca. 0.21 Å, cf. 0.04 Å in [Fe(8)Cl2]. These structural differences are best appreciated in the overlay shown in Fig. 8, which shows a side view of the distortion from tbp to sbp, whereby the FeCl2-moieties have been fixed in one position. Alternatively, if the pyridine moieties had been fixed, the distortion would be a change of the positions of the two chlorine atoms. In [Fe(4)Cl2], the N(1)–Fe–Cl(1) and N(1)–Fe–Cl(2) bond angles are 99.6(2) and 139.46(19)°, while in [Fe(8)Cl2] the corresponding angles are 92.85(11) and 153.64(11)° [the Cl(1)–Fe–Cl(2) angle also changes, from 120.89(12)° in [Fe(4)Cl2] to 113.31(6)° in [Fe(8)Cl2]].
The pyridine ring in [Fe(4)Cl2] is substantially inclined (ca. 23°) with respect to the N3 plane, an unusual feature that was also observed in the structure of [Fe(8)Cl2], where the angle is ca. 24°.49 The two five-membered C2N2Fe chelate rings in [Fe(4)Cl2] are consequently noticeably twisted, with in each case the pyridyl carbon lying substantially out of the plane of the other four atoms; for the N(7) ring the pyridyl carbon lies ca. 0.37 Å out of the plane of the other four atoms which are coplanar to within ca. 0.04 Å, while for the N(9) ring the respective deviations are ca. 0.34 and 0.06 Å. Both mesityl rings are approximately orthogonal with respect to the ligand plane [ca. 85° for the N(7)-bound ring, and ca. 83° for the N(9)-bound ring]. These small twists are such that the complex does not have molecular CS symmetry.
The structure of [Mn(4)Cl2] is isomorphous with that of [Fe(4)Cl2], the r.m.s. deviation of the best fit of the non-hydrogen atoms of the two complexes being ca. 0.05 Å. The pyridine ring is inclined by ca. 24° to the N3 plane, which is noticeably different compared to the bis(imino)pyridine complex [Mn(12)Br2] where the angle is close to zero.54 For the N(7) chelate ring the pyridine carbon lies ca. 0.41 Å out of the plane of the other four atoms which are coplanar to within ca. 0.05 Å, whilst for the N(9) ring the respective deviations are ca. 0.37 and 0.06 Å. The mesityl rings are inclined to ligand plane by ca. 84° for the N(7)-bound ring, and ca. 85° for the N(9)-bound ring.
Although the unit cell determined for a crystal of [Ni(4)Br2] was essentially the same as for [Fe(4)Cl2] and [Mn(4)Cl2], the structure of [Ni(4)Br2] shows some important differences to the other two (see Fig. S11). The most notable difference is a substantial enlargement of the Br(2)–Ni–N(1) angle [151.7(3)°] cf. Cl(2)–Fe–N(1) [139.46(19)°] and Cl(2)–Mn–N(1) [136.54(14)°] in [Fe(4)Cl2] and [Mn(4)Cl2], respectively. Variations are also seen in all the angles involving the X(1) halogen atom and these are a consequence of a geometry that has moved further towards square-based pyramidal, the four basal atoms [N(1),N(7),N(9),Br(2)] being coplanar to within ca. 0.15 Å, cf. 0.21 Å in [Fe(4)Cl2]. The pyridine ring is inclined by ca. 21° to the N3 plane in [Ni(4)Br2], which is different compared to the other bis(imino)pyridine complexes [Ni(12)Cl2], [Ni(13)Cl2] and [Ni(13)Br2].55–57 For the N(7) chelate ring, the pyridine carbon lies ca. 0.34 Å out of the plane of the other four atoms which are coplanar to within ca. 0.02 Å, whilst for the N(9) ring, the respective deviations are ca. 0.31 and 0.05 Å. The mesityl rings are inclined to the ligand plane by ca. 90° for the N(7)-bound ring, and ca. 78° for the N(9)-bound ring.
The structures of the six-coordinate vanadium and chromium complexes [V(4)Cl3] and [Cr(4)Cl3] are isomorphous, (see Fig. 9 and Fig. S11) the complexes having an r.m.s. deviation for the best fit of their non-hydrogen atoms of ca. 0.04 Å. The geometry at the metal centre is distorted octahedral with cis angles in the range 75.41(12)–106.16(9)° for [V(4)Cl3] [76.99(9)–104.05(6)° for Cr], the most acute angles being associated with the five-membered chelate rings. The axial M–Cl bond lengths are, in each case, significantly longer (by ca. 0.04 Å) than those to Cl(2), which is different compared to complexes [V(13)Cl3] and [Cr(13)Cl3], where only one axial M–Cl distance is significantly longer.15,17,41 While the short equatorial M–Cl bond probably reflects the different trans effect of pyridine compared to chloride, the steric influence of the very bulky 2,6-diisopropylphenyl groups is likely responsible for the asymmetry in the axial bond distances. A notable feature of the coordination geometry is that the metal atom lies ca. 0.18 Å [0.16 Å for Cr] out of the plane of the pyridine ring. This distortion causes a twist in the five-membered chelate rings such that the imine nitrogen N(7) lies ca. 0.17 Å [0.17 Å for Cr] “below” the plane of the other four atoms [in the direction of Cl(3)], whilst the N(9) imine nitrogen lies ca. 0.14 Å [0.13 Å for Cr] “above” the plane of the other four atoms [in the direction of Cl(1)]. This results in corresponding “down” and “up” orientations for the associated mesityl rings (which are inclined approximately orthogonally to the chelate planes—ca. 73 and 71° in [V(4)Cl3], and ca. 73 and 72° in [Cr(4)Cl3]). Interestingly, the S–Ph rings adopt a similar “up and down” orientation, but in an opposite sense. The rotations about the C(imine)–S and S–C(phenyl) bonds are, however, very different for the two substituents, and represent the major departure of the conformation of the complex from molecular C2 symmetry.
The phenylether complex [Fe(5)Cl2] has approximate non-crystallographic CS symmetry and a distorted square pyramidal geometry at the iron centre (see Fig. S14). The most striking effect of changing from mesityl in [Fe(3)Cl2] to 2,6-diisopropylphenyl substituents in [Fe(5)Cl2] is a conformational change from a trigonal bipyramidal to a square-based pyramidal coordination geometry, something that was also observed in the case of [Fe(4)Cl2] and [Fe(6)Cl2] (although [Fe(4)Cl2] is quite distorted from ideal tbp) and the parent bis(imino)pyridine ketimine complexes of [Fe(12)Cl2] versus [Fe(13)Cl2].9 The Cl(1)–Fe–Cl(2) angle remains relatively unchanged, being 117.20(5)° in [Fe(5)Cl2] and 118.56(9)° in [Fe(3)Cl2]. In [Fe(5)Cl2], both chelate rings have folded geometries, with C(2) and C(7) lying +0.12 and +0.13 Å respectively out of the [Fe,N(1),N(7)] plane, and C(6) and C(9) lying +0.21 and +0.29 Å respectively out of the [Fe,N(1),N(9)] plane. This in turn leads to the two 2,6-diisopropylphenyl rings (which are approximately orthogonal to the chelate rings—ca. 84 and 82° for N(7)– and N(9)-bound rings respectively) sitting “beneath” the plane of the ligand backbone. The phenylether substituents, by contrast, are both “above” this plane.
The cobalt complex [Co(5)Cl2] (Fig. 10) is isostructural, though not isomorphous, with its iron counterpart [Fe(5)Cl2]. The most noticeable difference in the coordination geometry is an increased pivoting of the MCl2 unit, the N(1)–Co–X angles being 88.19(12) and 155.81(12)° for X = Cl(1) and Cl(2) respectively, cf. 95.73(10) and 147.08(10)° for the corresponding angles in [Fe(5)Cl2]. There is an associated increase in the folding of the five-membered chelate rings with C(2) and C(7) lying +0.28 and +0.36 Å respectively out of the [Co,N(1),N(7)] plane, and C(6) and C(9) lying +0.32 and +0.44 Å respectively out of the [Co,N(1),N(9)] plane, again causing the 2,6-diisopropylphenyl rings to sit “beneath” the plane of the ligand backbone. These rings are, as usual, oriented approximately orthogonally to their associated chelate rings, here by ca. 71 and 83° for the N(7)– and N(9)-bound rings respectively.
The phenylthioether complex [Fe(6)Cl2] has a structure that is similar to that of its oxygen analogue [Fe(5)Cl2] (see Fig. S16). In [Fe(6)Cl2], the FeCl2 moiety exhibits a slightly greater degree of “pivoting” with an associated increase in the folding of the N,N′ chelate rings; C(2) and C(7) lie +0.13 and +0.24 Å respectively out of the [Fe,N(1),N(7)] plane, and C(6) and C(9) lie +0.27 and +0.49 Å respectively out of the [Fe,N(1),N(9)] plane. The N–Ar rings are inclined by ca. 88° [N(7)] and 85° [N(9)] to their parent N3 planes. The most noticeable difference between the conformations of the two structures is a greater inward folding of the phenylthioether substituents cf. O–Ph as a consequence of the greatly reduced angles at sulfur [105.0(2) and 98.2(2)°] compared with those at oxygen [118.3(3) and 120.6(3)°]. The cobalt complex [Co(6)Cl2] is isomorphous with its iron analogue [Fe(6)Cl2].
Ethylene polymerisation studies
The effect of modifications to the ligand framework on the ethylene polymerisation behaviour of the bis(imino)pyridine complexes has been investigated under a range of reaction conditions, using Fisher Porter reaction vessels and a 1L stainless steel reactor. In all experiments, methylaluminoxane (MAO) has been used as the co-catalyst and the polymerisation results have been compared to a benchmark catalyst [M(12)Cln]. In addition, for some catalysts, an initial assessment of the hydrogen response on the polymer molecular weight and the ability to incorporate 1-hexene has been included.Iron-based catalysts
The polymerisation results using the iron(II) complexes containing ligands 1–11 as the catalyst are collected in Table 1, together with the benchmark catalysts [Fe(12)Cl2] and [Fe(13)Cl2]. All catalysts, with the exception of [Fe(1)Cl2], [Fe(10)Cl2] and [Fe(11)Cl2], gave linear polyethylene with relatively broad molecular weight distributions. The thioether-substituted complexes with ligands 2, 4, 6 and 8 showed significantly higher activities than their ether-substituted relatives 3, 5 and 7. The highest value of 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mmol−1 h−1 bar−1 was obtained with [Fe(8)Cl2] (run 7), which is comparable to the bis(imino)pyridine benchmark catalyst [Fe(12)Cl2] (run 9). The inactivity of the methylether derivative [Fe(1)Cl2] and the amine derivatives [Fe(10)Cl2] and [Fe(11)Cl2] is notable, an aspect that we will return to later in this section.
200 g mmol−1 h−1 bar−1 was obtained with [Fe(8)Cl2] (run 7), which is comparable to the bis(imino)pyridine benchmark catalyst [Fe(12)Cl2] (run 9). The inactivity of the methylether derivative [Fe(1)Cl2] and the amine derivatives [Fe(10)Cl2] and [Fe(11)Cl2] is notable, an aspect that we will return to later in this section.
| Run | Complex μmol | T °C | P bar | Activity g mmol−1 h−1 bar−1 | Mne | Mwe | PDIe | sat. endsf | vinyl endsf |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerisation in Fisher Porter reactor; 200 ml toluene, 0.3 mmol MAO scavenger/activator, reaction time 30 min. b Reaction in the presence of 10 ml 1-hexene. c Polymerisation in 1 L stainless steel autoclave; 400 ml toluene, 2.0 mmol MAO scavenger/activator, reaction time 60 min. d Reaction in the presence of 0.3 bar H2. e Determined by GPC at 135 °C. f Results from NMR analysis, given per 1000 carbon atoms. g Not recorded. | |||||||||
| 1 | [Fe(2)Cl2] (1.0) | 25 | 1 | 3800 | 4200 | 55000 |
13.0 | 4.1 | 2.4 |
| 2 | [Fe(3)Cl2] (1.0) | 25 | 1 | 600 | 1800 | 41000 |
22.2 | 3.4 | 2.2 |
| 3 | [Fe(4)Cl2] (1.0) | 25 | 1 | 22600 |
1900 | 4600 | 2.4 | 7.9 | 6.6 |
| 4 | [Fe(5)Cl2] (1.0) | 25 | 1 | 3500 | 6500 | 11000 |
17.0 | 3.7 | 0.5 |
| 5 | [Fe(6)Cl2] (1.0) | 25 | 1 | 10600 |
13000 |
60000 |
4.7 | 3.0 | 0.4 |
| 6 | [Fe(7)Cl2] (1.0) | 25 | 1 | 5400 | 7900 | 164000 |
20.7 | 4.3 | 1.2 |
| 7 | [Fe(8)Cl2] (1.0) | 25 | 1 | 26200 |
6200 | 18000 |
2.8 | 3.4 | 2.2 |
| 8 | [Fe(9)Cl2] (1.0) | 25 | 1 | 8200 | 12000 |
430000 |
36.5 | 3.1 | 0.3 |
| 9 | [Fe(12)Cl2] (1.0) | 25 | 1 | 32000 |
11000 |
45000 |
4.2 | 2.2 | 1.4 |
| 10 | [Fe(13)Cl2] (1.0) | 25 | 1 | 15600 |
23000 |
102000 |
4.4 | 1.3 | 0.3 |
| 11 | [Fe(4)Cl2] (0.5) | 25 | 2 | 17600 |
1400 | 3500 | 2.5 | 7.1 | 5.2 |
| 12 | [Fe(4)Cl2] (0.5)b | 25 | 2 | 18800 |
1600 | 4600 | 2.9 | 7.5 | 4.9 |
| 13 | [Fe(6)Cl2] (0.5) | 25 | 2 | 9000 | 13400 |
36000 |
2.7 | g | g |
| 14 | [Fe(12)Cl2] (0.5) | 25 | 2 | 22100 |
8700 | 26000 |
3.0 | g | g |
| 15 | [Fe(4)Cl2] (0.5)c | 25 | 4 | 29100 |
1500 | 5900 | 3.9 | g | g |
| 16 | [Fe(4)Cl2](0.5)c,d | 25 | 4 | 29500 |
1200 | 2900 | 2.4 | g | g |
An increase in the size of the ortho-aryl substituents from methyl to isopropyl, for example in the benchmark catalysts [Fe(12)Cl2] and [Fe(13)Cl2], results in an increase in the molecular weight of the polymer, but at the expense of a decrease in catalyst activity (see runs 9 and 10).9 This is attributed to congestion at the metal centre, which inhibits monomer coordination and thereby decreases the rate of propagation and the rate of chain transfer. A first order dependence on the rate of propagation is typically observed for these iron-based catalysts.9 A similar decrease in activity is seen here when the related thioether complexes [Fe(4)Cl2] versus [Fe(6)Cl2] and [Fe(8)Cl2] versus [Fe(9)Cl2] are compared. In contrast, runs 2 and 4 show that for the phenylether-substituted catalysts [Fe(3)Cl2] and [Fe(5)Cl2], the ortho-isopropyl substituents improve the activity. The extra steric protection in the case of [Fe(5)Cl2] is likely to suppress alkylations at the imine carbon, which are believed to be responsible for catalyst degradation and the very low activity observed for [Fe(3)Cl2] and the absence of activity for [Fe(1)Cl2].
The molecular weight of the polymer increases with the size of the N-aryl substituents, for example from Mn = 1800 for [Fe(3)Cl2] to 6500 for [Fe(5)Cl2], from 1900 for [Fe(4)Cl2] to 13000 for [Fe(6)Cl2] and from 6200 for [Fe(8)Cl2] to 12000 for [Fe(9)Cl2]. Interestingly, it is shown here that an increase in the size of the backbone substituents has a similar effect, cf. 1800 for [Fe(3)Cl2] versus 7900 for [Fe(7)Cl2] and 1900 for [Fe(4)Cl2] versus 6200 for [Fe(8)Cl2]. The rate of chain transfer is clearly affected by the size of both substituents on the imine moiety. This is in line with our earlier observations regarding the difference between aldimine and ketimine catalysts of type A and B in Fig. 1,9 and our later modifications to bulkier imine–carbon substituents of type D.45 A possible explanation is that the larger imine carbon substituents push the aryl rings forward and thereby increase steric hindrance.
The ratio of saturated versus vinyl end-groups increases with steric hindrance at the metal centre. For example, the saturated/vinyl ratio changes from 1.2–1.7 for mesityl-substituted complexes (runs 2, 3, 6 and 7) to 7.4-10.3 for 2,6-diisopropylphenyl analogues (runs 4, 5 and 8). As the rate of β-H transfer decreases, the rate of chain transfer to aluminium becomes the dominant chain termination process for the more hindered catalysts. This has also been observed for the related ketimine catalysts of type B, and has been explained on the basis of an intimate interaction between AlMe3 and the iron centre in the active species.9,58
The iron catalysts containing thioether substituents generate polyethylene with comparable molecular weight to their ether-analogues, but with much narrower molecular weight distributions. This is believed to be a consequence of the higher affinity of the aluminium co-catalyst for oxygen donors, resulting in binding of the co-catalyst and the formation of several different propagating species. The fact that a broad molecular weight distribution is also obtained with the methylthioether complex [Fe(2)Cl2] seems to support this hypothesis, as the co-catalyst should bind more strongly to the sulfur donor in this case due to a lack of steric hindrance.
None of the catalysts incorporated 1-hexene and the catalyst performance was unaffected by the presence of 1-hexene (cf. runs 11 and 12). Addition of 0.3 bar H2 to the ethylene feed slightly reduced the molecular weight of the polymer produced by [Fe(4)Cl2], but did not significantly affect the activity of the catalyst (cf. runs 15 and 16).
The addition of MAO to a solution of complex [Fe(1)Cl2] in toluene under an ethylene atmosphere resulted in a black precipitate and no polymer was formed, even after prolonged exposure to ethylene. It is likely that AlMe3 or other Lewis acidic sites in MAO, bind to the lone pairs of the relatively exposed oxygen atoms on the ligand. This would make the complex more susceptible to deactivation, for example via alkylation at the imine carbon, as seen in similar related compounds.52 A similar coordination of AlMe3 could also be responsible for the inactivity of the amine complexes [Fe(10)Cl2] and [Fe(11)Cl2]. The slightly more protected phenylether derivative [Fe(3)Cl2] resulted in an activity of 600 g mmol−1 h−1 bar−1 and the activity was further increased to 5400 g mmol−1 h−1 bar−1 by incorporating a bulkier 2,6-dimethylphenylether substituent (cf. complex [Fe(7)Cl2] in run 6). Greater steric protection of the imine carbon centre appears to have a stabilizing effect on the active species.
Vanadium-based catalysts
The ethylene polymerisation results using the bis(imino)pyridine vanadium(III) complexes [V(1)Cl3], [V(3)Cl3], [V(4)Cl3] and [V(6)Cl3] in combination with MAO as the co-catalyst have been determined and compared against the ketimine benchmark catalyst [V(12)Cl3] (see Table 2). All polymerisation experiments were performed at 50 °C as [V(4)Cl3] displayed the highest activity at this temperature (runs 25–27). Complex [V(4)Cl3] shows a remarkably high activity of 11500 g mmol−1 h−1 bar−1, significantly higher than the benchmark catalysts [V(12)Cl3], which shows an activity of 6300 g mmol−1 h−1 bar−1 under the same conditions (runs 20 vs. 23 and also 26 vs. 31). This catalyst is also considerably more active than the related phenylether complex [V(3)Cl3] (runs 19 and 20) or the more sterically demanding 2,6-diisopropylphenyl derivative [V(6)Cl3]. The activity for the vanadium series follows the order [V(4)Cl3] > [V(6)Cl3] > [V(3)Cl3] > [V(1)Cl3], which is the same as the order observed for the analogous iron(II) complexes. All complexes produce linear, relatively low molecular weight polyethylene, in line with previous observations by others.16 The ratio between the saturated and vinyl end groups is close to one, which indicates that β-H transfer is the dominant chain transfer mechanism.
| Run | Complex μmol | T °C | P bar | Activity g mmol−1 h−1 bar−1 | Mnf | Mwf | PDIf | sat. endsg | vinyl endsg |
|---|---|---|---|---|---|---|---|---|---|
| a Toluene solvent. b Polymerisation in Fisher Porter reactor; 200 ml solvent, 4.0 mmol MAO scavenger/activator, reaction time 30 min. c Polymerisation in stainless steel autoclave; 400 ml solvent, 4.0 mmol MAO scavenger/activator, reaction time 60 min. d Reaction in the presence of 10 ml 1-hexene. e Reaction in the presence of 0.3 bar H2. f Determined by GPC at 135 °C. g Results from 13C NMR analysis, given per 1000 carbon atoms. h Not determined. | |||||||||
| 18 | [V(1)Cl3] (10.0)b | 50 | 2 | 35 | 2080 | 5220 | 2.5 | h | h |
| 19 | [V(3)Cl3] (10.0)b | 50 | 2 | 240 | 670 | 2310 | 3.5 | 15.3 | 13.8 |
| 20 | [V(4)Cl3] (1.0)b | 50 | 2 | 11500 |
2060 | 4890 | 2.4 | 5.6 | 5.3 |
| 21 | [V(4)Cl3] (1.0)b,d | 50 | 2 | 15600 |
1980 | 4610 | 2.3 | h | h |
| 22 | [V(6)Cl3] (1.0)b | 50 | 2 | 2000 | 5010 | 17800 |
3.6 | h | h |
| 23 | [V(12)Cl3] (1.0)b | 50 | 2 | 6300 | 1860 | 4610 | 2.5 | 7.8 | 5.1 |
| 24 | [V(4)Cl3] (1.0)b | 50 | 2 | 15600 |
1980 | 4610 | 2.3 | h | h |
| 25 | [V(4)Cl3] (1.0)c | 25 | 4 | 1920 | 1850 | 4330 | 2.3 | h | h |
| 26 | [V(4)Cl3] (1.0)c | 50 | 4 | 12300 |
2210 | 5280 | 2.4 | 7.5 | 4.6 |
| 27 | [V(4)Cl3] (1.0)c | 75 | 4 | 3200 | 2180 | 5030 | 2.3 | h | h |
| 28 | [V(4)Cl3] (1.0)c,e | 50 | 4 | 9400 | 1990 | 4890 | 2.5 | h | h |
| 29 | [V(4)Cl3] (1.0)c | 50 | 2 | 11100 |
1900 | 4750 | 2.5 | h | h |
| 30 | [V(4)Cl3] (1.0)c | 50 | 6 | 12000 |
2050 | 5070 | 2.5 | h | h |
| 31 | [V(12)Cl3](1.0)c | 50 | 4 | 5700 | 1760 | 3970 | 2.3 | h | h |
The catalytic performance of complex [V(4)Cl3] was further studied by polymerisation experiments under different reaction conditions. The polymer yield appears to increase linearly with ethylene pressure up to 6 bar, indicative of a first order dependence on ethylene pressure (runs 29, 26 and 30). Co-polymerisation experiments resulted in an ethylene/1-hexene co-polymer with 8.3 butyl branches per 1000 carbons (run 21). Introducing 0.3 bar H2 in the reaction mixture resulted in a slight reduction in polymer molecular weight and catalyst activity (run 28).
Chromium-based catalysts
During our initial polymerisation experiments using [Cr(4)Cl3], it was found that no catalytic activity was observed at room temperature in heptane (run 32, Table 3), but when the reaction temperature was increased to 70 °C, the activity increased significantly (see run 33). Similar observations were made by Esteruelas and co-workers, who reported that the ethylene polymerisation activity of bis(imino)pyridine chromium(III)-based catalysts is strongly affected by temperature.17 Toluene was found to be a better solvent and this was used for all subsequent reactions (run 34). Previous reports suggested that the catalyst activity was affected by an activation time between mixing of the catalyst and the co-catalyst and the introduction of ethylene.17 In the case of complex [Cr(4)Cl3], an immediate colour change occurred upon addition of MAO from green to brown, indicative of the formation of the active species, and no activation period was required (run 35).| Run | T °C | Solvent | Activation min | Yield gb | Activity g mmol−1 h−1 bar−1b | Mnc | Mwc | PDIc |
|---|---|---|---|---|---|---|---|---|
| a Polymerisation in Schlenk flask, scavenged with 1110 eq. MAO, 2 bar ethylene pressure, 2.0 μmol catalyst preactivated with 140 eq. MAO, 30 min run. b Catalyst produces small amount of oligomers in all cases (15%, 0.95 < K < 1.00), yield is oligomer + polymer fraction. c Determined by GPC at 135 °C. | ||||||||
| 32 | 25 | heptane | 30 | 0 | 0 | — | — | — |
| 33 | 70 | heptane | 30 | 4.1 | 2070 | 650 | 900 | 1.4 |
| 34 | 70 | toluene | 30 | 11.5 | 5740 | 730 | 1200 | 1.7 |
| 35 | 70 | heptane | 0 | 4.4 | 2180 | 720 | 1000 | 1.4 |
The chromium complexes containing ligands 1, 3, 4, 6 and 12 were tested for the polymerisation of ethylene at 70 °C in toluene (Table 4). The ether-substituted catalysts [Cr(1)Cl3] and [Cr(3)Cl3] were both inactive under these conditions. Coordination of the Lewis acidic activator to the oxygen atoms in these complexes is believed to be responsible for catalyst deactivation in these cases. In contrast, complex [Cr(4)Cl3] is highly active when activated with MAO (run 36), similar to the ketimine catalyst [Cr(12)Cl3]. The activity of complex [Cr(6)Cl3] with bulkier isopropyl substituents is approximately 30 times less (run 38). A similar trend has been observed for the ketimine chromium catalysts [Cr(12)Cl3] and [Cr(13)Cl3].17 For comparison, the iron catalyst [Fe(4)Cl2] is twice as active as [Fe(6)Cl2] and the iron ketimine complex [Fe(12)Cl2] is twice as active as the isopropyl substituted analogue [Fe(13)Cl2]. An important trend that is clearly emerging from these studies is that too much steric congestion is detrimental to the catalytic activity of these bis(imino)pyridine-based catalyst systems.
| Run | Catalyst | Pressure bar | Activity g mmol−1 h−1 bar−1 | Mnf | Mwf | PDIf,e | sat. endsg | vinyl endsg | Branchg |
|---|---|---|---|---|---|---|---|---|---|
| a Precatalyst activated prior to injection with 140 equiv. MAO, 1.0 μmol of catalyst used, toluene solvent, 70 °C. b Polymerisation in Fisher Porter reactor; 200 ml solvent, 1.0 mmol MAO scavenger/activator, reaction time 30 min. c Polymerisation in stainless steel autoclave, 400 ml solvent, 4.0 mmol MAO scavenger/activator, reaction time 60 min. d Reaction in the presence of 0.3 bar H2. e Reaction in the presence of 10 ml 1-hexene. f Determined by GPC at 135 °C. g Results from 13C NMR analysis, given per 1000 carbon atoms. h Not recorded. | |||||||||
| 36 | [Cr(4)Cl3]b | 2 | 10300 |
710 | 1210 | 1.7 | h | h | — |
| 37 | [Cr(4)Cl3]b,e | 2 | 7200 | 780 | 1450 | 1.9 | 13.7 | 9.3 | 1.7 |
| 38 | [Cr(6)Cl3]b | 2 | 310 | 1470 | 3240 | 2.2 | h | h | — |
| 39 | [Cr(12)Cl3]b | 2 | 9200 | 700 | 1100 | 1.6 | h | h | — |
| 40 | [Cr(4)Cl3]c | 4 | 12600 |
730 | 1480 | 2.0 | 13.6 | 10.7 | — |
| 41 | [Cr(4)Cl3]c,d | 4 | 7730 | 680 | 1260 | 1.8 | 17.3 | 11.5 | — |
| 42 | [Cr(12)Cl3]c | 4 | 12200 |
670 | 1260 | 1.9 | 12.7 | 10.8 | — |
The chromium catalysts containing ligands 4, 6 and 12 generate linear, low molecular weight polyethylene (Mw = 1000–3000). Increasing the size of the ortho aryl substituents from methyl in [Cr(4)Cl3] to isopropyl in [Cr(6)Cl3] increases the polymer molecular weight (runs 36 and 38). Due to the relatively low molecular weight polymer, all catalysts produce small amounts of a toluene-soluble fraction (<15%).
Catalyst [Cr(4)Cl3] was tested for a hydrogen response on the polymer molecular weight and for 1-hexene incorporation. Addition of 0.3 bar H2 to the ethylene feed leads to a slight reduction in polymer molecular weight (run 41), similar to the iron and vanadium catalysts [Fe(4)Cl2] and [V(4)Cl3]. However, in this case the molecular weight reduction occurs at the expense of a significant drop in catalytic activity. An additional polymerisation experiment with catalyst [Cr(4)Cl3] was performed in the presence of 1-hexene (run 37). Only a very limited amount of 1-hexene was incorporated resulting in 1.7 butyl branches per 1000 carbons. The yield of co-polymer was found to be lower than the polyethylene yield obtained in the absence of 1-hexene (run 36). The saturated/vinyl end group ratio is close to one in all cases and β-hydrogen transfer is therefore the sole chain termination mechanism, resulting in narrow PDIs of approximately 2.
Cobalt-based catalysts
Two sets of experiments were conducted using either Fisher Porter reaction vessels at 2 bar pressure or a 1L steel autoclave at 4 bar pressure with ethylene gas uptake monitoring (Table 5). Complex [Co(1)Cl2] gives the lowest activity in the cobalt series (runs 43 and 52) and results in the formation of oligomers, whereas complex [Co(3)Cl2] gives a mixture of liquid oligomers and solid low molecular weight polyethylene (ratio 1.5/1). At 2 bar ethylene pressure, a familiar activity trend is observed, whereby the thioether catalysts show higher activities compared to their ether analogues, cf. run 45 vs. 44 and run 48 vs. 47, but generally lower activities compared to the ketimine benchmark catalyst [Co(12)Cl2] (run 49). Complex [Co(4)Cl2] shows the best activity, which is similar to the benchmark catalyst. However, at 4 bar ethylene pressure, catalysts [Co(3)Cl2] and [Co(5)Cl2] show higher activities than their thioether relatives [Co(4)Cl2] and [Co(6)Cl2]. In order to explain this observation, a closer look at the avtivity profiles was required.The ethylene uptake over time for runs 52–57 is shown in Fig. 11. The activity profiles for [Co(4)Cl2] and [Co(6)Cl2] illustrate the stability of these thioether catalysts. Only a very gradual loss of activity is observed over time, whereby the profiles of [Co(4)Cl2] and [Co(12)Cl2] are essentially parallel, but [Co(4)Cl2] has a consistently lower ethylene uptake. The kinetic profile of the phenylether complex [Co(3)Cl2] shows a high initial activity, but a rapid decrease, most likely due to catalyst deactivation. The overall activity over one hour is the same for [Co(3)Cl2] and [Co(12)Cl2] (runs 53 and 57). The kinetic profile of [Co(1)Cl2] shows that the low activity of this catalyst is due to a very short catalyst lifetime of approximately 10 min.

The nature of the heteroatom has a significant effect on the molecular weight of the polyethylene. Replacing oxygen with sulfur results in a more than 10-fold increase in molecular weight, for example from 280 for [Co(3)Cl2] to 3090 for [Co(4)Cl2] and from 930 [Co(5)Cl2] to 15300 [Co(6)Cl2]. The molecular weight obtained with ketimine catalyst [Co(12)Cl2] is between those observed for ether catalyst [Co(3)Cl2] and thioether catalyst [Co(4)Cl2].
Catalyst [Co(4)Cl2] was tested for a hydrogen response on the polymer molecular weight. As seen for catalyst [Fe(4)Cl2], the addition of H2 leads to a small reduction in polymer molecular weight (run 59 vs. 54). Co-polymerisation of ethylene/1-hexene does not occur (run 46) and when compared with run 45, it can be seen that the catalyst activity is unaffected by the presence of 1-hexene.
Other metals
Bis(imino)pyridine titanium complexes have been previously investigated as catalysts for the polymerisation of ethylene.14,59 An activity of 168 g mmol−1 h−1 bar−1 was reported for the ketimine complex [Ti(12)Cl3] using MeAlCl2 as co-catalyst and 18 g mmol−1 h−1 bar−1 for [Ti(13)Cl3] in combination with MAO,14 which is similar to the value of 38 g mmol−1 h−1 bar−1 reported for [Ti(13)Cl3]Cl.59 As can be seen in Table 6, complex [Ti(4)Cl3] exhibits low activities with MAO and MeAlCl2 as the co-catalyst and, in the case of MAO, at both 25 and 70 °C.| Run | Complex | T °C | P bar | Activity g mmol−1 h−1 bar−1 | Mnf | Mwf | PDIf | Kg | sat. endsh | vinyl endsh |
|---|---|---|---|---|---|---|---|---|---|---|
| a Toluene solvent, 1.0 μmol of catalyst used. b Polymerisation in Fisher Porter reactor; 200 ml solvent, 0.3 mmol MAO scavenger/activator (300 equiv.), reaction time 30 min. c Polymerisation in stainless steel autoclave; 400 ml solvent, 2.0 mmol MAO scavenger/activator (2000 equiv.), reaction time 60 min. d Reaction in the presence of 0.3 bar H2. e Reaction in the presence of 10 ml 1-hexene. f Determined by GPC at 135 °C. g Determined by GC. h Results from 13C NMR analysis, given per 1000 carbon atoms. i Not recorded. | ||||||||||
| 43 | [Co(1)Cl2]b | 25 | 2 | 230 | — | — | — | 0.84 | i | i |
| 44 | [Co(3)Cl2]b | 25 | 2 | 1320 | 420 | 550 | 1.3 | 0.89 | 19.5 | 19.5 |
| 45 | [Co(4)Cl2]b | 25 | 2 | 4680 | 3000 | 7500 | 2.5 | — | 3.9 | 3.8 |
| 46 | [Co(4)Cl2]b,e | 25 | 2 | 4600 | 2780 | 7300 | 2.6 | — | i | i |
| 47 | [Co(5)Cl2]b | 25 | 2 | 830 | 1500 | 2800 | 1.9 | — | i | i |
| 48 | [Co(6)Cl2]b | 25 | 2 | 990 | 17300 |
38200 |
2.2 | — | i | i |
| 49 | [Co(12)Cl2]b | 25 | 2 | 5490 | 1010 | 2020 | 2.0 | — | i | i |
| 52 | [Co(1)Cl2]c | 25 | 4 | 230 | — | — | — | 0.80 | i | i |
| 53 | [Co(3)Cl2]c | 25 | 4 | 5900 | 280 | 400 | 1.4 | 0.89 | 24.9 | 23.0 |
| 54 | [Co(4)Cl2]c | 25 | 4 | 3900 | 3090 | 8160 | 2.6 | — | 3.6 | 2.9 |
| 55 | [Co(5)Cl2]c | 25 | 4 | 2320 | 930 | 2270 | 2.4 | — | i | i |
| 56 | [Co(6)Cl2]c | 25 | 4 | 1030 | 15300 |
33000 |
2.2 | — | i | i |
| 57 | [Co(12)Cl2]c | 25 | 4 | 5900 | 940 | 2040 | 2.2 | — | 8.1 | 7.0 |
| 58 | [Co(4)Cl2]c | 50 | 4 | 560 | 1670 | 3870 | 2.3 | — | i | i |
| 59 | [Co(4)Cl2]c,d | 25 | 4 | 4800 | 2640 | 6690 | 2.5 | — | i | i |
| 60 | [Co(3)Cl2]c | 25 | 2 | 7700 | 350 | 460 | 1.3 | 0.89 | i | i |
| 61 | [Co(3)Cl2]c | 25 | 6 | 5630 | 310 | 420 | 1.4 | 0.89 | i | i |
| Run | co-catalyst (equiv.) | T/°C | Activity (g mmol−1 h−1 bar−1) | Mnb | Mwb | PDIb |
|---|---|---|---|---|---|---|
| a Tested in Schlenk flask employing 10.0 μmol of catalyst, 100 ml of toluene and 2 bar of ethylene pressure. b Determined by GPC. c Not determined. | ||||||
| 62 | MAO (1000) | 25 | 85 | 2770 | 215000 |
77.7 |
| 63 | MAO (1000) | 70 | 95 | 1230 | 10700 | 8.7 |
| 64 | MeAlCl2 (1000) | 25 | <10 | c | c | c |
The manganese complex [Mn(4)Cl2] was tested under a range of different reaction conditions using various co-catalysts (MAO, triisobutylaluminium/dry-MAO and Me2AlCl/Cl3CCO2Et) and temperatures (25 and 70 °C). None of these conditions resulted in any polymerisation activity. The analogous nickel complex [Ni(4)Cl2] was also found to be inactive for ethylene polymerisation using MAO as the co-catalyst (Al/Ni 500:1).
Conclusions
The synthesis and characterization of an extensive series of first row transition metal complexes of iron, vanadium, chromium and cobalt containing bis(imino)pyridine ligands with heteroatom substituents at the imine carbon atoms has been described. The molecular geometries of the five-coordinate complexes of iron and cobalt show a trigonal bipyramidal geometry, provided the imine substituents are relatively small. Larger aryl substituents, such as 2,6-dimethylphenyl, lead to a significant deviation of the pyridine unit out of the [N, N, N] coordination plane, resulting in coordination geometries that are best describes as square-based pyramidal. None of the complexes studied here show coordination of the O or S heteroatoms to the metal centre and only coordination via the imine nitrogen donors is observed. The ethylene polymerisation activity of the complexes, upon activation with MAO, has been found to be strongly affected by the nature of the heteroatom and the size of the substituents. Whereas the sulfur-based substituents give highly active polymerisation catalysts, in some cases exceeding the ketimine benchmark catalysts, the oxygen-based ligands are generally much less active. Some oxygen-based catalysts with small substituents and the nitrogen-based complexes are inactive. Very large substituents lead to highly active but short-lived catalysts systems. Coordination of the aluminium alkyl co-catalyst to the heteroatom and subsequent alkylation of the bis(imino)pyridine ligand is believed to be responsible for the deactivation. All active catalyst systems (Fe, V, Cr and Co) show a small response to the addition of hydrogen to the polymerisation reaction, resulting in a lowering of the polymer molecular weight. Only the vanadium-based catalysts show significant incorporation of 1-hexene in co-polymerisation experiments.An important outcome of these studies is that for the bis(imino)pyridine-based ethylene polymerisation catalysts, the ligand is the most important aspect of the catalyst and many metals will result in active catalyst systems. However, it remains a mystery why iron-based catalysts are highly active, while others such as Mn and Ni are inactive. We will continue our efforts to further the understanding of these remarkable ethylene polymerization catalysts.
Acknowledgements
We are grateful to INEOS (formerly BP Chemicals Ltd.) for financial support and to Dr J. Boyle and P. Jehoulet for NMR and GPC measurements.References
- H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem. Int. Ed. Engl., 1995, 34, 1143–1170 CrossRef CAS.
- A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98, 2587–2598 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem. Int. Ed. Engl., 1999, 38, 428–447 CrossRef CAS.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1203 CrossRef CAS.
- V. C. Gibson and G. A. Solan, Top. Organomet. Chem., 2009, 26, 107–158 CrossRef CAS.
- V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745–1776 CrossRef CAS.
- C. Bianchini, G. Giambastiani, I. Guerrero Rios, G. Mantovani, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2006, 250, 1391–1418 CrossRef CAS.
- G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728–8740 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849–850 RSC.
- G. J. P. Britovsek, S. Mastroianni, G. A. Solan, S. P. D. Baugh, C. Redshaw, V. C. Gibson, A. J. P. White, D. J. Williams and M. R. J. Elsegood, Chem. Eur. J., 2000, 6, 2221–2231 CrossRef CAS.
- B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049–4050 CrossRef CAS.
- B. L. Small and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 7143–7144 CrossRef CAS.
- B. A. Dorer, (BP Chemicals Ltd.), WO00/47586, 2000.
- D. Reardon, F. Conan, S. Gambarotta, G. Yap and Q. Wang, J. Am. Chem. Soc., 1999, 121, 9318–9325 CrossRef CAS.
- R. Schmidt, M. B. Welch, R. D. Knudsen, S. Gottfried and H. G. Alt, J. Mol. Catal., 2004, 222, 17–25 CAS.
- M. A. Esteruelas, A. M. López, L. Méndez, M. Oliván and E. Onate, Organometallics, 2003, 22, 395–406 CrossRef CAS.
- Y. Nakayama, K. Sogo, H. Yasuda and T. Shiono, J. Polym. Sci. A, 2005, 43, 3368–3375 CrossRef CAS.
- B. L. Small, M. J. Carney, D. M. Holman, C. E. O'Rourke and J. Halfen, Macromolecules, 2004, 37, 4375–4386 CrossRef CAS.
- Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef.
- M. J. Hanton and K. Tenza, Organometallics, 2008, 27, 5712–5716 CrossRef CAS.
- N. V. Semikolenova, V. A. Zakharov, E. P. Talsi, D. E. Babushkin, A. P. Sobolev, L. G. Echevskaya and M. M. Khysniyarov, J. Mol. Catal., 2002, 182–183, 283–294 CrossRef CAS.
- G. J. P. Britovsek, G. K. B. Clentsmith, V. C. Gibson, D. M. L. Goodgame, S. McTavish and Q. A. Pankhurst, Catal. Commun., 2002, 3, 207–211 CrossRef CAS.
- K. P. Bryliakov, N. V. Semikolenova, V. N. Zudin, V. A. Zakharov and E. P. Talsi, Catal. Commun., 2004, 5, 45–48 CrossRef CAS.
- E. P. Talsi, D. E. Babushkin, N. V. Semikolenova, V. N. Zudin, V. N. Panchenko and V. A. Zakharov, Macromol. Chem. Phys., 2001, 202, 2046–2051 CrossRef CAS.
- K. P. Bryliakov, N. V. Semikolenova, V. A. Zakharov and E. P. Talsi, Organometallics, 2004, 23, 5375–5378 CrossRef CAS.
- A. A. Barabanov, G. D. Bukatov, V. A. Zakharov, N. V. Semikolenova, T. B. Mikenas, L. G. Echevskaya and M. A. Matsko, Macromol. Chem. Phys., 2005, 207, 1368 CrossRef.
- A. A. Barabanov, V. A. Zakharov, N. V. Semikolenova, L. G. Echevskaya and M. A. Matsko, Macromol. Chem. Phys., 2005, 206, 2292 CrossRef CAS.
- Y. V. Kissin, C. Qian, G. Xie and Y. Chen, J. Polym. Sci. A, 2006, 44, 6159 CrossRef CAS.
- K. P. Bryliakov, E. P. Talsi, N. V. Semikolenova and V. A. Zakharov, Organometallics, 2009, 28, 3225–3232 CrossRef CAS.
- A. M. Tondreau, C. Milsmann, A. D. Patrick, H. M. Hoyt, E. Lobkovsky, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 15046–15059 CrossRef CAS.
- B. de Bruin, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2000, 39, 2936–2947 CrossRef CAS.
- P. H. M. Budzelaar, B. de Bruin, A. W. Gal, K. Wieghardt and J. H. van Lenthe, Inorg. Chem., 2001, 40, 4649–4655 CrossRef CAS.
- D. Zhu, I. Thapa, I. Korobkov, S. Gambarotta and P. H. M. Budzelaar, Inorg. Chem., 2011, 50, 9879–9887 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson, S. Mastroianni, D. C. H. Oakes, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Eur. J. Inorg. Chem., 2001, 431–437 CrossRef CAS.
- V. C. Gibson, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Organometallics, 2007, 26, 5119–5123 CrossRef CAS.
- M. D. Bruce, V. C. Gibson, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 2523–2524 RSC.
- J. Scott, S. Gambarotta, I. Korobkov and P. H. M. Budzelaar, J. Am. Chem. Soc., 2005, 127, 13019–13029 CrossRef CAS.
- M. W. Bouwkamp, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2006, 45, 2–4 CrossRef CAS.
- D. Reardon, G. Aharonian, S. Gambarotta and G. P. A. Yap, Organometallics, 2002, 21, 786–788 CrossRef CAS.
- H. Sugiyama, G. Aharonian, S. Gambarotta, G. P. A. Yap and P. H. M. Budzelaar, J. Am. Chem. Soc., 2002, 124, 12268–12274 CrossRef CAS.
- A. M. Archer, M. W. Bouwkamp, M.-P. Cortez, E. Lobkovsky and P. J. Chirik, Organometallics, 2006, 25, 4269–4278 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson, O. Hoarau, S. K. Spitzmesser, A. J. P. White and D. J. Williams, Inorg. Chem., 2003, 42, 3454–3465 CrossRef CAS.
- N. Kleigrewe, W. Steffen, T. Blömker, G. Kehr, R. Fröhlich, B. Wibbeling, G. Erker, J.-C. Wasilke, G. Wu and G. C. Bazan, J. Am. Chem. Soc., 2005, 127, 13955–13968 CrossRef CAS.
- S. McTavish, G. J. P. Britovsek, T. M. Smit, V. C. Gibson, A. J. P. White and D. J. Williams, J. Mol. Catal. A: Chem., 2007, 261, 293–300 CrossRef CAS.
- J. E. Steves, M. D. Kennedy, K. P. Chiang, W. S. Kassel, W. G. Dougherty, T. J. Dudley and D. L. Zubris, Dalton Trans., 2009, 1214–1222 RSC.
- V. K. Appukuttan, Y. Liu, B. C. Son, C.-S. Ha, H. Suh and I. Kim, Organometallics, 2011, 30, 2285–2294 CrossRef CAS.
- A. M. A. Bennett, (DuPont), WO98/27124, 1998.
- T. M. Smit, A. K. Tomov, V. C. Gibson, A. J. P. White and D. J. Williams, Inorg. Chem., 2004, 43, 6511–6512 CrossRef CAS.
- Z. J. A. Komon, G. C. Bazan, C. Fang and X. Bu, Inorg. Chim. Acta, 2003, 345, 95–102 CrossRef CAS.
- B. Y. Lee, G. C. Bazan, J. Vela, Z. J. A. Komon and X. Bu, J. Am. Chem. Soc., 2001, 123, 5352–5353 CrossRef CAS.
- Y.-L. Huang, B.-H. Huang, B.-T. Ko and C.-C. Lin, J. Chem. Soc., Dalton Trans., 2001, 1359–1365 RSC.
- N. V. Semikolenova, V. A. Zakharov, L. G. Echevskaya, M. A. Matsko, K. P. Bryliakov and E. P. Talsi, Catal. Today, 2009, 144, 334–340 CrossRef CAS.
- V. C. Gibson, S. McTavish, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 221–226 RSC.
- H. Suzuki, S.-i. Matsumura, Y. Satoh, K. Sogoh and H. Yasuda, React. Funct. Polym., 2004, 58, 77–91 CrossRef CAS.
- Y. Huang, J. Chen, L. Chi, C. Wei, Z. Zhang, Z. Li, A. Li and L. Zhang, J. Appl. Polym. Sci., 2009, 112, 1486–1495 CrossRef CAS.
- R.-Q. Fan, Y.-L. Yang and Z.-W. Lu, Chem. Res. Chin. Univ., 2008, 24, 4–7 CrossRef CAS.
- M. van Meurs, G. J. P. Britovsek, V. C. Gibson and S. A. Cohen, J. Am. Chem. Soc., 2005, 127, 9913–9923 CrossRef CAS.
- F. Calderazzo, U. Englert, G. Pampaloni, R. Santi, A. Sommazzi and M. Zinna, Dalton Trans., 2005, 914–922 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 253597–253608. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cy00448h |
| This journal is © The Royal Society of Chemistry 2012 |