Application of chiral ligands: carbohydrates, alkaloids–lanthanides and other Lewis acid complexes to control regio- and stereoselectivity of the dipolar cycloaddition reactions of nitrile oxides and crotonamides and cinnamides
Mirosław
Gucma
and
W. Marek
Gołębiewski
*
Institute of Industrial Organic Chemistry, Annopol 6, Warsaw, 03-236, Poland. E-mail: golebiewski@ipo.waw.pl
First published on 23rd August 2011
Abstract
Chiral Lewis acid mediated 1,3-dipolar cycloaddition reactions of aryl nitrile oxides and secondary α- or β-substituted acrylamides and cinnamides were examined. Excellent enantioselectivities with moderate to good regioselectivities were achieved for crotonamides with complexes of carbohydrates with Yb(OTf)3, TiCl4, Mg(OTf)2, and CsF as well as with the (−)-sparteine–Yb(OTf)3 system. High enantiomeric excess and high regioselectivity were observed for cinnamides in reactions mediated by Yb(OTf)3 complexes with carbohydrate, R-BINOL, and (−)-sparteine.
Introduction
We have reported application of chiral lanthanide–(−)-sparteine and lanthanide–1,1′-bi-2-naphthol (BINOL) complexes as catalysts in 1,3-dipolar cycloadditions of nitrile oxides to unsaturated alcohols, where enantioselectivities up to 89% were obtained.11,3-Dipolar cycloaddition of nitrile oxides to alkenes is the most convenient method of preparation for 2-isoxazolines2 which can be easily reduced to several synthetically important compounds such as β-hydroxy ketones, β-hydroxy esters, α,β-unsaturated carbonyl compounds or imino ketones.3 Reactions of monosubstituted and 1,1-disubstituted alkenes furnish regioselectively 5-substituted 2-isoxazolines while 1,2-disubstituted olefins usually afford mixtures of regio- and stereoisomers.
To achieve a better control of the reaction in one approach optically active reagents were applied, more often dipolarophiles than dipoles.4 Chiral auxiliaries linked by an ester bond such as camphor5 and menthol derivatives6 or an amide bond such as the Rebeck benzoxazole derivative,7 oxazoline derivatives and oxazolidin-2-ones,8 imidazolidin-2-ones,9 proline derivatives,10 Oppolzer's camphorsultam11 and its borane derivative were used.12
In the second more promising approach chiral metal catalysts were applied. Ukaji and Inomata performed the first asymmetric metal-catalyzed 1,3-dipolar cycloaddition reaction of nitrile oxides to γ-substituted allylic alcohols with diethylzinc and (R,R)-diisopropyl tartrate13 and with an N-sulfonylated (S,S)-2,3-diaminosuccinate derivative.14 Sibi achieved excellent regio- and enantioselectivity for cycloadditions of pivalo- and aryl nitrile oxides to crotonamide and acrylimides by attaching an achiral template of pyrazolidinone type and using a bulky chiral Lewis acid obtained from magnesium iodide and a bisoxazoline derivative.15 Faita and Quadrelli applied a polymer supported chiral oxazolidin-2-one–magnesium catalyst.16 Yamamoto et al. reported on asymmetric 1,3-dipolar cycloadditions of benzonitrile oxides to acrylamides bearing chiral auxiliaries of oxazolidinone and imidazolidinone types, and a chiral complex comprising a magnesium salt or ytterbium triflate and 2,6-bis[(4R)-4-phenyl-2-oxazolinyl]pyridine (PyBOX) ligand in equimolar amounts.17 Kanemasa achieved highly efficient enantioselective cycloaddition of nitrile oxides generated with molecular sieves to pyrazole acrylamide in the presence of R,R-DBFOX/Ph-nickel(II) catalyst.18
Carbohydrates have been used for some time as chiral synthons in organic synthesis, as auxiliaries in asymmetric reactions, organocatalysts, and as chiral ligands in metal complexes, which was possible after modifications of the reactive functionalities.19 Titanium enolates containing carbohydrate moieties were used in highly enantioselective aldol addditions.20 Derivatives of 6-deoxy glucose with bulky 2-O and 3-O substituents (O-pivaloyl, O-tert-butyldimethylsilyl) were efficient auxiliaries in Diels–Alder reactions of acrylates with cyclopentadiene as well as 1,3-dipolar cycloadditions with nitrile oxides.21
(+)-(4,6-Benzylidene)methyl-α-D-glucopyranoside (A) and 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose (B) were amply used in the first two roles. Carbohydrate (A) derived ketones were used as organocatalysts in the enantioselective epoxidation of arylalkenes. Sugar B-modified chiral salen–Mn(III) complexes were used as catalysts in the epoxidation of unfunctionalized alkenes. 1,2:3,4-Di-O-isopropylidene-α-D-galactopyranose (C) was used as a chiral component of titanium(IV) alkoxo compounds in the asymmetric epoxidation of cinnamyl alcohol.22 Carbohydrate A was used to prepare a chiral catalyst of the azacrown ether type for enantioselective Michael reactions of 2-nitropropane with substituted chalcones.23A was also used to prepare chiral chromium complexes applied as ligands in the synthesis of rhodium catalysts for the enantioselective hydroboration of styrene derivatives.24 Reaction of racemic biaryldicarboxylic dichloride with A was an example of a new method of optical resolution of axially- and planar chiral dicarboxylates.25 Carbohydrate B was used as an auxiliary or a chiral ligand in Cu(I) catalyzed 1,4-adddition of Grignard reagents to α,β-unsaturated carbonyl compounds.26 It was also applied as a chiral catalyst in sodium borohydride asymmetric reduction of prochiral ketones.
In the quest for controlling stereochemistry and regiochemistry of 1,3-dipolar cycloaddition reactions of nitrile oxides to amides we would like to present new catalytic systems: ytterbium triflate–carbohydrates A–C, ytterbium perchlorate–carbohydrates A,C, ytterbium trichloride–carbohydrate C, ytterbium triflate–(+)-cinchonine F, ytterbium triflate–(−)-cinchonidine G, titanium tetrachloride–carbohydrates A,C, titanium tetrachloride–(−)-sparteine E, magnesium triflate–carbohydrate A, magnesium bromide–carbohydrate C, caesium fluoride–carbohydrates A,C, caesium fluoride–R-BINOL D (Fig. 1).
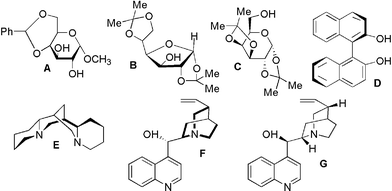 | ||
| Fig. 1 Chiral ligands used in nitrile oxide 1,3-dipolar cycloaddition reactions. | ||
Results and discussion
We have examined 1,3-dipolar cycloaddition reaction of 4-trifluoromethylbenzonitrile oxide to secondary α or β-substituted acrylamides (seven dipolarophiles) and cinnamides (two dipolarophiles) mediated by chiral Lewis acid systems (Schemes 1 and 2). Yields of the reaction were acceptable for dipolarophiles with electron-donating substituents. As ligands, carbohydrates A–C, alkaloids (−)-sparteine, (−)-cinchonidine and (+)-cinchonine, and R-BINOL were used (Fig. 1). The complex of carbohydrate B with ytterbium triflate was not an effective chiral catalyst. We have applied as Lewis acids, apart from known effective salts of ytterbium and magnesium,17 also titanium tetrachloride and caesium fluoride. | ||
| Scheme 1 Nitrile oxide cycloaddition reactions to crotonamides 2a–e. | ||
 | ||
| Scheme 2 Nitrile oxide cycloaddition reactions to α,β-unsaturated carboxamides. | ||
Eighteen chiral Lewis acids were used and the results are presented in Tables 1 and 2.
| Entry | 3, 4 | Rp | R | Chiral catalyst | Yielda (%) | Rs 3/4b | % ee reg. 3 | % ee reg. 4 |
|---|---|---|---|---|---|---|---|---|
| a Isolated yield of amides 3 and 4. b Regioisomer 3/regioisomer 4. c Reaction conditions: 1 mmol amounts of both substrates, 1 mmol of catalyst (for Yb(OTf)3–D system 0.1 mmol was used), reactions in CH2Cl2 at room temp. for 20 h, yield 86–25%, regioselectivity 100/0–60/40, enantioselectivity 99.2–0%. | ||||||||
| 1 | a | CF3 | C6H4-2-OMe | Yb(OTf)3–A | 74 | 40/60 | 0.4 | 95 |
| 2 | a | CF3 | C6H4-2-OMe | Yb(OTf)3–C | 84 | 70/30 | 0 | 95 |
| 3 | a | CF3 | C6H4-2-OMe | Yb(ClO4)3–C | 76 | 79/21 | 0.4 | 76 |
| 4 | a | CF3 | C6H4-2-OMe | Yb(ClO4)3–A | 86 | 24/76 | 0 | 71 |
| 5 | a | CF3 | C6H4-2-OMe | YbCl3–H2O–A | 63 | 42/58 | 0.6 | 1 |
| 6 | a | CF3 | C6H4-2-OMe | TiCl4–A | 43 | 24/76 | 45 | 92 |
| 7 | a | CF3 | C6H4-2-OMe | TiCl4–C | 66 | 74/26 | 1.4 | 86 |
| 8 | a | CF3 | C6H4-2-OMe | Mg(OTf)2–A | 69 | 38/62 | 0 | 99.2 |
| 9 | a | CF3 | C6H4-2-OMe | Mg(OTf)2–C | 94 | 67/33 | 0.2 | 96 |
| 10 | a | CF3 | C6H4-2-OMe | MgBr2–C | 69 | 40/60 | 1 | 9.2 |
| 11 | a | CF3 | C6H4-2-OMe | CsF–A | 74 | 28/72 | 28 | 92 |
| 12 | a | CF3 | C6H4-2-OMe | CsF–C | 84 | 32/68 | 2.2 | 99 |
| 13 | a | CF3 | C6H4-2-OMe | Yb(OTf)3–D | 54 | 43/57 | 0.2 | 60.2 |
| 14 | a | CF3 | C6H4-2-OMe | CsF–D | 76 | 36/64 | 4.4 | 72.2 |
| 15 | a | CF3 | C6H4-2-OMe | Yb(OTf)3–E | 51 | 60/40 | 1.2 | 95 |
| 16 | a | CF3 | C6H4-2-OMe | TiCl4–(−)-E | 31 | 77/27 | 0 | 93 |
| 17 | a | CF3 | C6H4-2-OMe | Yb(OTf)3–G | 85 | 29/71 | 0.6 | 4.6 |
| 18 | a | CF3 | C6H4-2-OMe | Yb(OTf)3–F | 55 | 63/37 | 1.4 | 2.2 |
| 19 | b | i-Pr | C6H4-4-OMe | Yb(OTf)3–A | 40 | 79/21 | 20 | 98 |
| 20 | c | CF3 | C6H4-4-OMe | Yb(OTf)3–D | 58 | 5/95 | 99 | 24 |
| 21 | d | CF3 | CH2-2-furyl | Yb(OTf)3–F | 55 | 0/100 | — | 0.2 |
| 22 | d | CF3 | CH2-2-furyl | Yb(OTf)3–A | 53 | 36/64 | 3 | 94 |
| 23 | e | CF3 | C6H4-4-sec-Bu | Yb(OTf)3–D | 53 | 40/60 | 0 | 2 |
| 24 | a | CF3 | C6H4-2-OMe | — | 25 | 67/33 | — | — |
| Entry | 6, 7 | R | R1 | Chiral catalyst | Yielda (%) | Rs 6/7b | % ee reg. 6 | % ee reg. 7 |
|---|---|---|---|---|---|---|---|---|
| a Isolated yield of amides 6 and 7. b Regioisomer 6/regioisomer 7. c Reaction conditions: 1 mmol amounts of both substrates, 1 mmol of catalyst (for Yb(OTf)3–D system 0.1 mmol was used), reactions in CH2Cl2 at room temp. for 20 h, yield 86–23%, regioselectivity 100/0–84/16, enantioselectivity 99–0%. | ||||||||
| 1 | a | Et | H | Yb(OTf)3–A | 34 | 0/100 | — | 1 |
| 2 | a | Et | H | Yb(OTf)3–C | 36 | 0/100 | — | 0 |
| 3 | b | H | Me | Yb(OTf)3–A | 86 | 0/100 | — | 3 |
| 4 | b | H | Me | Yb(OTf)3–F | 76 | 0/100 | — | 3 |
| 5 | c | H | H | Yb(OTf)3–A | 66 | 0/100 | — | 68.2 |
| 6 | d | C6H4-4-CF3 | H | Yb(OTf)3–C | 48 | 18/82 | 59 | 93 |
| 7 | e | C6H4-4-i-Pr | H | Yb(OTf)3–A | 50 | 100/0 | 1 | — |
| 8 | d | C6H4-4-CF3 | H | Yb(OTf)3–D | 38 | 16/84 | 45 | 99 |
| 9 | d | C6H4-4-CF3 | H | Yb(OTf)3–E | 35 | 16/84 | 0 | 89 |
| 10 | e | C6H4-4-i-Pr | H | Yb(OTf)3–G | 23 | 100/0 | 5 | — |
In most cases mainly 5-carbamoylisoxazolines were obtained as opposed to the uncatalyzed reaction where the expected regioselectivity was observed (Table 124). Similarly 4-carbamoylisoxazolines were the major regioisomers for crotonates Table 12, 7, and the only isomer for cinnamides Table 27, 10. Good, but opposite, regiochemistry was found for cinnamides Table 26, 8, 9. Excellent unexpected 5-regioselectivity was observed for crotonamides Table 120, 21 and for 2-pentenecarboxamides Table 21, 2, as well as for acrylamide Table 25 and methacrylamides Table 23, 4, as was expected.
The regiochemistry of nitrile oxide 1,3-dipolar cycloaddition to α,β-unsaturated amides depends on orbital interactions and steric factors.27 While primary and secondary amides afford mainly 4-carbamoyl substituted isoxazolidines in uncatalyzed reactions due to the preferred HOMO dipolarophile–LUMO dipole binding, tertiary amides give in preponderance 5-carbamoyl derivatives as a result of steric interactions of the β-substituent in the transition state.
Reversal of the regioselectivity for several of the examined secondary amides, where 5-carbamoyl isoxazolidines were the major products, against the orbital control, clearly indicates subtle effects of the catalysts.
Our results show also higher regioselectivity for 2-pentenecarboxamides and cinnamides than for the corresponding crotonamides with the same catalytic systems. It can be explained by the steric factors.
Characteristic was also the opposite regioselectivity (as well as enantioselectivity) of cycloadditions mediated by Yb(OTf)3–(−)-cinchonidine (Table 210) and Yb(OTf)3–(+)-cinchonine, the pseudo-enantiomer of (−)-cinchonidine (Table 121).
Enantioselectivities of Lewis acid mediated aryl nitrile oxide 1,3-dipolar cycloaddition reactions to α,β-unsaturated secondary amides are shown in Tables 1 and 2. High enantioselectivity (ee of 99.2–92%) (Table 11, 2, 19, 22) for crotonamides was achieved with complexes of carbohydrates A and C with ytterbium triflate for the 5-regioisomer, complexes with TiCl4 (Table 16), magnesium triflate (Table 18, 9), caesium fluoride (Table 111, 12), and for complexes of (−)-sparteine with ytterbium triflate and TiCl4 (Table 115, 16). The corresponding 4-regioisomers exhibited only very small enantiomeric excess. The exception was the high asymmetric induction observed for the 4-regioisomer in the reaction mediated by the (R)-BINOL–ytterbium triflate complex (Table 120).
Lower enantioselectivity was observed with the catalytic systems carbohydrate C–ytterbium perchlorate (71% ee, Table 14), carbohydrate A–ytterbium perchlorate (76% ee, Table 13), and (R)-BINOL–caesium fluoride (72.2% ee, Table 114). Similar enantiomeric excess was found also for acrylamide with the carbohydrate A–ytterbium triflate complex (68.2% ee, Table 25). On the other hand very weak chiral induction was found in reactions mediated by carbohydrate A–ytterbium chloride and with (R)-BINOL–ytterbium triflate (Table 15, 23) for crotonamide, with the carbohydrate A–ytterbium triflate complex (Table 23) for methacrylamide, with Cinchona alkaloids (+)-cinchonine– and (−)-cinchonidine–ytterbium triflate complexes for crotonamides (Table 117, 18, 21) and for methacrylamide (Table 24). Similarly very low asymmetric induction was found for 2-pentenecarboxamides in the presence of both carbohydrates and ytterbium triflate (Table 21, 2).
High enantioselectivity was found in the case of cinnamides in the presence of carbohydrate A–ytterbium triflate (Table 26), (R)-BINOL–ytterbium triflate (Table 28), and (−)-sparteine–ytterbium triflate systems (Table 29).
Analysis of the data of Table 11, 3 and 5 indicates that the character of the Lewis acid anion is important, since ee dropped in the order Yb(OTf)3, Yb(ClO4)3, YbCl3, corresponding to diminishing size and softness of the salts.
Studying the effect of the catalyst on regio- and stereoselectivity of the 1,3-dipolar cycloaddition reaction we examined ESI-mass spectra. This method is gaining popularity as a tool to obtain insight into transient intermediates present in solution in metal-catalyzed transformations.28 First, ytterbium triflate was analyzed and then mixtures of two, three, and four components prepared in dichloromethane were recorded in acetonitrile and methanol solution. An equimolar mixture of ytterbium triflate and carbohydrate C showed peaks of the carbohydrate at m/z 283 [C+Na]+, the catalytic complex at m/z 839 [Yb(OTf)2+C+CH2Cl2+Na], and m/z 988 [Yb(OTf)3+C+Na]. When an equivalent amount of amide 2a was added to the above mixture new signals appeared at m/z 813 [Yb(OTf)3+2a+H], 923 [Yb(OTf)2+C+2a], and 1072 corresponding to the ion of m/z [Yb(OTf)3+C+2a]. When finally the fourth component, nitrile oxide 1, was added new signals were detected consistent with a free adduct of m/z 379 [3/4a+H], the adduct bound to ytterbium 733 [3/4a+Yb(OTf)+MeOH] and a nitrile oxide dimer peak at m/z 374. These results confirm formation of an ytterbium–carbohydrate C complex binding the amide dipolarophile.
The observed enantioselectivity of the reaction leading to 5-carbamoyl derivatives could be tentatively explained by binding of the amides to the chiral catalytic complex of ytterbium triflate, interacting also with ytterbium, followed by a preferential attack of nitrile oxide from the upper re-face of the alkene opposite to the 1,2-isopropylidene group (Scheme 3). Cisoid conformation of the α,β-unsaturated amides was assumed as is generally established.27b Absolute configuration was proposed as 4R,5R via Li-Selectride reduction to the known isoxazoline methanol derivative 8.13a This direction of enantioselectivity was found also for the other catalytic systems: Yb(OTf)3–carbohydrates A,C, Yb(OTf)3–sparteine, Mg(OTf)2–carbohydrates A and C, TiCl4–carbohydrates A and C, TiCl4–sparteine.
 | ||
| Scheme 3 Proposed mechanism of chiral induction with the Yb(OTf)3–C system. | ||
The much lower enantioselectivity observed for the 4-regioisomers could be rationalized by the lack of nitrile oxide binding to ytterbium and loss of facial selectivity. Such a difference in enantioselectivities between 4- and 5-regioisomers was recorded before.15a
The opposite chirality was observed for the catalytic systems CsF–carbohydrates A and C, CsF–R-BINOL, Yb(OTf)3–R-BINOL, Yb(ClO4)3–carbohydrate A. The participation of the Yb(OTf)3–R-BINOL complex in 1,3-dipolar cycloaddition of nitrile oxide 1 with amide dipolarophile benzyl-3-E-pent-2-enoyloxazolidin-2-one (9)1b was examined by us with the help of ESI-MS spectra. An equimolar mixture of ytterbium triflate and R-BINOL in ethyl ether and acetonitrile showed inter alia a peak at m/z 1044 [Yb(OTf)3+BINOL+Et2O+Na]. After addition of amide 9 signals at m/z 1249 [Yb(OTf)3+BINOL+9+2xCH3CN+H] and at 1360 [Yb(OTf)3+BINOL+9+2xEt2O+H] were detected. Finally after addition of nitrile oxide 1 new peaks at m/z 447 corresponding to [adduct+H] and at 723 of [Yb(OTf)2+1+CH3CN+Na] as well as at m/z 1249 were recorded. This result showed exchange of amide and nitrile oxide ligands bound to the metal center. In this case the observed enantioselectivity could be explained by preferred attack of the nitrile oxide from the lower si-face of the dipolarophile opposite to the chirally twisted R-BINOL–Yb(OTf)3 complex affording isoxazolines of 4S,5S configuration (Scheme 4).
 | ||
| Scheme 4 Proposed mechanism of chiral induction with the Yb(OTf)3–BINOL system. | ||
Conclusion
We have applied new chiral Lewis acids to study the 1,3-dipolar cycloaddition reaction of aryl nitrile oxides and secondary α,β-unsaturated amides: complexes of carbohydrates A and C with Yb(OTf)3, Yb(ClO4)3, YbCl3, TiCl4, Mg(OTf)2, MgBr2, CsF, complexes of (+)-cinchonine and (−)-cinchonidine with Yb(OTf)3, (−)-sparteine with TiCl4, and the R-BINOL–CsF system achieved high enantioselectivity and regioselectivity for some systems without any auxiliaries. In most cases 4R,5R-trisubstituted isoxazolidines were obtained; only in carbohydrates A,C–Yb(ClO4)3, –CsF, and R-BINOL–Yb(OTf)3 mediated or catalyzed reactions were 4S,5S-isoxazolidines synthesized. We are continuing research to diminish the amount of chiral Lewis acid from equimolar to catalytic quantities which so far were effective only for R-BINOL–Yb(OTf)3 and carbohydrate A–Yb(OTf)3 systems.Experimental
Hydroximinoyl acid chlorides were prepared from the corresponding aryl aldehyde oximes and NCS in DMF.29,30 The corresponding nitrile oxides were generated in situ by dehydrohalogenation with triethylamine or on an Amberlyst A-21 column.15Reagent grade chemicals were used without further purification unless otherwise noted.
Spectra were obtained as follows: IR spectra on a JASCO FTIR-420 spectrometer, 1H NMR spectra on Varian 500 UNITY plus-500 and Varian 200 UNITY plus 200 spectrometers in deuterated chloroform using TMS as an internal standard, ESI and HR ESI mass spectra on a Micromass LCT spectrometer. Flash-chromatography was carried out using silica gel S 230–400 mesh (Merck). Elemental analyses were performed at Microanalysis Laboratory of Institute of Organic Chemistry, Polish Academy of Sciences, Warsaw.
General procedure for dipolarophile amide synthesis
A solution of an aniline derivative (1.2 mmol) in anhydrous toluene (or anhydrous dichloromethane) (10 ml) was added with stirring to an acid chloride (1.0 mmol) followed by dry triethylamine (30 mmol). The obtained solution was stirred under reflux for 1 h and overnight at rt. Water (10 ml) was added, the organic layer was separated, washed with 3% hydrochloric acid solution and water, and was dried over magnesium sulfate. Product was extracted with dichloromethane and purified by flash chromatography.N-(Furan-2-yl-methyl)crotonamide (2c)
It was obtained as a colorless powder in 50% yield, mp = 52–54 °C (from CH2Cl2–hexane). IR (KBr, cm−1): 3420, 3285, 3080, 3015, 2980, 2920, 1665, 1622, 1548, 1501, 1450, 1320, 1227, 1198, 1149, 1105, 1082, 1025, 1005, 975, 905, 820, 745, 729, 695. 1H NMR (200 MHz, CDCl3): δ 7.31 (dd, J = 1.8; 0.8 Hz, 1H, H-5′), 6.82 (dt, J = 15.4; 6.8 Hz, 1H, H3C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 6.68 (s, 1H, NH), 6.29 (dd, J = 3.2; 2.0 Hz, 1H, H-4′), 6.19 (dd, J = 3.2; 0.6 Hz, 1H, H-3′), 5.88 (dq, J = 15.4; 1.7 Hz, 1H, OCC), 4.44 (d, J = 5.4 Hz, 2H,
C), 6.68 (s, 1H, NH), 6.29 (dd, J = 3.2; 2.0 Hz, 1H, H-4′), 6.19 (dd, J = 3.2; 0.6 Hz, 1H, H-3′), 5.88 (dq, J = 15.4; 1.7 Hz, 1H, OCC), 4.44 (d, J = 5.4 Hz, 2H, ![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) NH), 1.81 (dd, J = 6.8; 1.7 Hz, 3 H, H3C) ppm. HR ESI-MS m/z calcd for C9H11NO2Na: 188.0687. Found: 188.0686.
NH), 1.81 (dd, J = 6.8; 1.7 Hz, 3 H, H3C) ppm. HR ESI-MS m/z calcd for C9H11NO2Na: 188.0687. Found: 188.0686.
N-(4-sec-Butylphenyl)crotonamide (2d)
It was obtained as a brownish semisolid in 45% yield. IR (KBr, cm−1): 3420, 3301, 3295, 3200, 3140, 3050, 2958, 2920, 2860, 1672, 1646, 1604, 1539, 1515, 1450, 1430, 1413, 1348, 1300, 1254, 1200, 1180, 1105, 1018, 1000, 956, 938, 831, 730, 669. 1H NMR (CDCl3, 200 MHz): δ 7.62 (s, 1H, NH), 7.49 (d, J = 8.2 Hz, 2H, H-2′, H-6′), 7.11 (d, J = 8.2 Hz, 2H, H-3′, H-5′), 6.96 (dq, J = 15.1; 6.8 Hz, 1H, H3CC), 5.98 (dd, J = 15.1; 1.7 Hz, 1H, OCC), 2.55 (sept, J = 7.1 Hz, 1H, CH3), 1.86 (dd, J = 6.8; 1.7 Hz, 3H, ![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) CHC), 1.56 (quint, J = 7.1 Hz, 2H, CHCH3), 1.20 (d, J = 7.1 Hz, 3H, CHAr), 0.80 (t, J = 7.1 Hz, 3H, CH2). HR ESI-MS m/z calcd for C14H19NONa: 240.1364. Found: 240.1367.
CHC), 1.56 (quint, J = 7.1 Hz, 2H, CHCH3), 1.20 (d, J = 7.1 Hz, 3H, CHAr), 0.80 (t, J = 7.1 Hz, 3H, CH2). HR ESI-MS m/z calcd for C14H19NONa: 240.1364. Found: 240.1367.
(2E)-N-(4-Methoxyphenyl)pent-2-enamide (5a)
It was obtained as a brownish semisolid in 25% yield. IR (KBr, cm−1): 3308, 3130, 340, 2966, 2940, 2840, 2600, 2500, 1665, 1631, 1600, 1512, 1470, 1411, 1355, 1338, 1300, 1241, 1174, 1110, 1090, 1031, 980, 970, 850, 824, 790, 730, 700. 1H NMR (200 MHz, CDCl3): δ 8.30 (s, 1H, NH), 7.56 (d, J = 8.2 Hz, 2H, H-2′, H-6′), 6.98 (dt, J = 15.2; 6.9 Hz, 2H, H2CCH), 6.82 (d, J = 8.2 Hz, 2H, H-3′, H-5′), 6.07 (d, J = 15.2 Hz, 1H, OCC), 3.77 (s, 3H, H3CO), 2.21 (quint, J = 7.0 Hz, 2H, H3CCHC), 1.05 (t, J = 7.0 Hz, 3H, H3C). HR ESI-MS m/z, calcd for C12H15NO2Na: 228.1001. Found: 228.1006.
N-(4-Methoxyphenyl)-4-trifluoromethylcinnamide (5d)
It was obtained as a greenish semisolid in 83% yield. IR (KBr, cm−1): 3440, 3295, 3000, 2940, 2920, 2840, 1659, 1624, 1600, 1540, 1513, 1467, 1411, 1328, 1300, 1236, 1180, 1164, 1126, 1150, 1070, 1033, 1018, 982, 834, 818, 785, 725, 703. 1H NMR (200 MHz, CDCl3): δ 10.34 (s, 1H, NH), 7.83 (d, J = 8.5 Hz, 2H, H-3′, H-5′), 7.65 (d, J = 9.0 Hz, 2H, H-2′′, H-6′′), 7.69–7.52 (d, 1H, ArC), 7.52 (d, J = 8.5 Hz, 2H, H-2′, H-6′), 7.35 (d, J = 15.2 Hz, 1H, OCC), 6.82 (d, J = 9.0 Hz, 2H, H-3′′, H-5′′), 3.76 (s, 3H, H3C). ESI-MS m/z calcd for C17H14NO2F3Na:=344.0874. Found: 344.0881.
N-(4-Methoxyphenyl)-4-isopropylcinnamide (5e)
It was obtained as a brownish semisolid in 70% yield. 1H NMR (200 MHz, CDCl3): δ 7.71 (d, J = 15.6 Hz, 1H, ArC), 7.71 (s, 1H, NH), 7.53 (d, J = 8.5 Hz, 2H, H-2′′, H-6′′), 7.41 (d, J = 8.4 Hz, 2H, H-2′, H-6′), 7.19 (d, J = 8.5 Hz, 2H, H-3′′, H-5′′), 6.85 (d, J = 8.4 Hz, 2H, H-3′, H-5′), 6.53 (d, J = 15.6 Hz, 1H, OCC), 3.77 (s, 3H, OAr), 2.90 (sept, J = 6.9 Hz, 1H, (CH3)2, 1.24 (d, J = 6.9 Hz, 6H, CH()2. HR ESI-MS m/z calcd for C19H21NO2Na: 318.1470. Found: 318.1468.
General procedure for the cycloaddition reactions
A mixture of carbohydrate A (1.0 mmol) and Yb(OTf)3 (1.0 mmol) in dry dichloromethane was stirred at rt for 30 min. Dipolarophile (1 mmol) was added dropwise followed by a solution of dipole in the same solvent generated by passing a hydroximinoyl chloride solution through a column of Amberlyst 21 over 20–30 min. The solution was stirred at rt for ca. 20 h, and water was added to quench the reaction followed by the usual work-up. The crude product was purified by flash column chromatography on silica gel and the enantiomeric excess of separated regioisomers was determined by HPLC analysis (AD-H column).Regioisomers 6a, 6b, 6c, and 7e were not formed.
N-(2-Methoxyphenyl)-5-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-4-carboxamide (3a)
It was obtained as a yellowish semisolid. IR (KBr, cm−1): 3440, 3263, 3200, 3150, 3080, 2970, 2950, 2840, 1658, 1598, 1546, 1494, 1462, 1440, 1420, 1380, 1327, 1290, 1261, 1230, 1162, 1119, 1071, 1050, 1030, 1015, 944, 900, 853, 810, 751, 723, 680. 1H NMR (500 MHz, CDCl3): δ 8.22 (dd, J = 8.0; 1.8 Hz, 1H, H-6′′), 7.95 (s, 1H, NH), 7.92 (d, J = 8.3 Hz, 2H, H-5′, H-3′), 7.67 (d, J = 8.3 Hz, H-2′, H-6′), 7.05 (td, J = 8.0; 1.8, 1H, H-5′′), 6.93 (td, J = 8.0; 1.0 Hz, 1H, H-4′′), 6.82 (dd, J = 8.0; 1.0 Hz, H-3′′), 5.22 (qd, J = 6.5; 5.0 Hz, 1H, H-5), 4.09 (d, J = 5.0 Hz, 1H, H-4), 3.75 (s, 3H, H3CO), 1.52 (d, J = 6.5 Hz, 3H, H3CC). 13C NMR (CDCl3, 50.3 MHz): δ 166.08, 154.56, 148.23, 132.17, 127.49, 126.79, 126.17 (q, J13C–19F = 4.4 Hz), 124.98, 121.22, 120.22, 110.21, 83.86, 63.34, 55.91. HR ESI-MS m/z calcd for C27H25N2O3F3Na): 401.1089. Found: 401.1085.N-(4-Methoxyphenyl)-3-(4-isopropylphenyl)-5-methyl-4,5-dihydroisoxazole-4-carboxamide (3b)
It was obtained as a yellowish semisolid. [α]25D = +70.1 (c 1.07 in acetone) [24.0% ee, (S,S) rich].1H NMR (CDCl3, 200 MHz): δ 7.73 (d, J = 8.0 Hz, 2H, H-5′, H-3′), 7.31 (d, J = 9.2 Hz, 2H, H-2′′, H-6′′), 7.28 (d, J = 9.2 Hz, 2H, H-2′, H-6′), 6.76 (d, J = 9.1 Hz, 2H, H-3′′, H-5′′), 5.15 (qd, J = 6.4; 3.6 Hz, 1H, H-5), 4.03 (d, J = 3.6 Hz, 1H, H-4), 3.76 (s, 3H, H3CO), 2.90 (sept, J = 6.8 Hz, 1H,(CH3)2), 1.44 (d, J = 6.4 Hz, 3H, CH3), 1.24 (d, J = 6.8 Hz, 6H, C()). Anal. calcd for C21H24N2O3: C 71.57, H 6.86, N 7.95. Found: C 71.28, H 6.73, N 7.85.
N-(4-Methoxyphenyl)-5-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-4-carboxamide (3c)
It was obtained as a yellowish semisolid. [α]25D = +12.5 (c 1.7 in acetone) [24.0% ee, (S,S) rich]. 1H NMR (CDCl3, 200 MHz): δ 7.90 (d, J = 8.3 Hz, 2H, H-5′, H-3′), 7.69 (s, 1H, NH), 7.64 (d, J = 8.3 Hz, 2H, H-2′, H-6′), 7.33 (d, J = 9.2 Hz, 2H, H-2′′, H-6′′), 6.81 (d, J = 9.2 Hz, 2H, H-5′′, H-3′′), 5.17 (qd, J = 6.4; 5.2 Hz, 1H, H-5), 4.08 (d, J = 5.2 Hz, 1H, H-4), 3.77 (s, 3 H, CH3O), 1.48 (d, J = 6.4 Hz, 3H, CH3). Anal. calcd for C19H17F3N2O3: C 60.31, H 4.53, N 7.41. Found: C 60.14, H 4.62, N 7.49.N-(2-Furylmethyl)-5-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-4-carboxamide (3d)
It was obtained as a brownish semisolid. 1H NMR (200 MHz, CDCl3): δ 7.79 (d, J = 8.1 Hz, 2H, H-6′, H-2′), 7.54 (d, J = 8.1 Hz, 2H, H-5′, H-3′), 7.26 (d, J = 0.9 Hz, 1H, H-5′′), 6.30 (dd, J = 3.2; 2.0 Hz, 1H, H-4′′), 6.26 (d, J = 3.2 Hz, 1H, H-3′′), 5.07 (m, 1H, H-5), 4.45 (d, J = 5.6 Hz, 2H, CH2NH), 4.26 (m, 1H, H-4), 1.81 (dd, J = 6.8; 1.4 Hz, 3H, H3C). Anal. calcd for C17H15F3N2O3: C 57.97, H 4.29, N 7.95. Found: C 58.14, H 4.13, N 7.82.N-(4-sec-Butylphenyl)-5-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-4-carboxamide (3e)
It was obtained as a yellowish semisolid. 1H NMR (CDCl3, 200 MHz): δ 8.54 (s, 1H, NH), 7.83 (d, J = 8.2 Hz, 2H, H-6′, H-2′), 7.55 (d, J = 8.2 Hz, 2H, H-5′, H-3′), 7.45–7.25 (m, 4H, H-6′′, H-2′′, H-5′′, H-3′′), 5.14 (dq, J = 6.5; 5.8 Hz, 1H, H-5), 4.20 (d, J = 5.8 Hz, 1H, H-4), 2.53 (sept, J = 6.5 Hz, 1H, H3CCH2), 1.53 (sept. J = 6.8 Hz, 2H, CHCH3), 1.42 (d, J = 6.8 Hz, 3H, CH), 1.17 (d, J = 7.2 Hz, 3H, CHAr), 0.78 (t, J = 7.2 Hz, 3H, CH2). HR ESI-MS m/z calcd for C22H23N2O2F3Na: 427.1609. Found: 427.1592.
N-(2-Methoxyphenyl)-4-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-5-carboxamide (4a)
It was obtained as a colorless glass. mp 93–96 °C (from CH2Cl2–hexane); [α]25D = −149.8 (c 0.47 in acetone) [99.0% ee, (R,R) rich]; [α]25D = +56.2 (c 1.5 in acetone) [60.2% ee, (S,S) rich]. IR (KBr, cm−1): 3398, 2980, 2820, 2840, 1687, 1603, 1537, 1485, 1462, 1420, 1324, 1290, 1250, 1225, 1170, 1137, 1120, 1070, 1040, 950, 920, 875, 850, 741, 660, 620 cm−1. 1H NMR (200 MHz, CDCl3): δ 9.12 (s, 1H, NH), 8.35 (dd, J = 7.9; 1.5 Hz, 1H, H-6′′), 7.83 (d, J = 8.2 Hz, 2H, H-5′, H-3′), 7.68 (d, J = 8.2 Hz, 2H, H-6′, H-2′), 7.08 (td, J = 7.9; 1.5 Hz, 1H, H-5′′), 6.96 (td, J = 7.9; 1.5 Hz, 1H, H-4′′), 6.88 (td, J = 7.9; 1.5 Hz, 1H, H-3′′), 4.88 (d, J = 3.7 Hz, 1H, H-5), 4.18 (dq, J = 7.3; 3.8 Hz, 1H, H-4), 3.89 (s, 3H, H3CO), 1.48 (d, J = 6.5 Hz, 3H, H3CCH). 13C NMR (CDCl3, 50.3 MHz): δ 168.36, 160.96, 148.54, 132.76, 131.15, 127.86, 126.55, 126.06 (q, J13C–19F = 4.0 Hz), 124.75, 121.02, 119.84, 110.22, 86.67, 55.59, 47.12, 29.82, 18.65. HR ESI-MS m/z calcd for C19H17O3N2F3Na: 401.1089. Found: 401.1086.N-(4-Methoxyphenyl)-3-(4-isopropylphenyl)-4-methyl-4,5-dihydroisoxazole-5-carboxamide (4b)
It was obtained as a white-brownish semisolid. IR (KBr, cm−1): 3339, 3300, 2961, 1669, 1600, 1518, 1458, 1414, 1380, 1300, 1261, 1231, 1178, 1100, 1038, 950, 880, 860, 827, 800, 695. 1H NMR (CDCl3, 200 MHz): δ 8.44 (s, 1H, NH), 7.63 (d, J = 8.4 Hz, 2H, H-6′, H-2′), 7.46 (d, J = 9.1 Hz, 2H, H-2′′, H-6′′), 7.28 (d, J = 8.4 Hz, 2H, H-5′, H-3′), 6.85 (d, J = 9.1 Hz, 2H, H-3′′, H-5′′), 4.80 (d, J = 3.0 Hz, 1H, H-5), 4.16 (qd, J = 7.2; 3.0 Hz, 1H, H-4), 3.78 (s, 3H, CH3O), 2.93 (sept, J = 7.0 Hz, 1H,(CH3)2), 1.47 (d, J = 7.0 Hz, 3H, CH3), 1.26 (d, J = 7.0 Hz, 6H, C()). 13C NMR (CDCl3, 50.3 MHz): δ 168.65, 162.08 , 156.72 , 152.11, 129.92, 127.55, 127.15, 124.78, 121.56, 114.18, 85.83, 55.47, 47.53, 34.13, 23.74, 18.78. Anal. calcd for C21H24N2O3: C 71.57, H 6.86, N 7.95. Found: C 71.36, H 6.69, N 7.79.
N-(4-Methoxyphenyl)-4-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-5-carboxamide (4c)
It was obtained as a colorless glass. [α]25D = +53.8 (c 1.4 in acetone) [14% ee, (S,S) rich]. IR (KBr, cm−1): 3430, 3340, 3290, 2970, 2920, 2840, 1668, 1620, 1599, 1525, 1460, 1405, 1325, 1254, 1176, 1135, 1118, 1070, 1020, 950, 930, 880, 842, 810, 800, 740, 699. 1H NMR (CDCl3, 200 MHz): δ 8.37 (s, b. 1H, NH), 7.82 (d, J = 8.3 Hz, 2H, H-5′, H-3′), 7.69 (d, J = 8.3 Hz, 2H, H-6′, H-2′), 7.47 (d, J = 9.0 Hz, 2H, H-2′′, H-6′′), 6.86 (d, J = 9.0 Hz, 2H, H-3′′, H-5′′), 4.87 (d, J = 3.5 Hz, 1H, H-5), 4.18 (qd, J = 7.3; 3.5 Hz, 1H, H-4), 3.79 (s, 3H, H3CO), 1.48 (d, J = 7.3 Hz, 3H, CH3). 13C NMR (CDCl3, 50.3 MHz): δ 168.06, 162.00, 156.92, 129.79, 127.78, 127.00, 126.05, 125.98, 121.60, 114.28, 86.37, 31.95, 29.72, 29.68, 22.71,18.55, 14.12. HR ESI-MS m/z calcd for C19H17O3N2F3Na: 401.1089. Found: 401.1077.N-(2-Furylmethyl)-4-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-5-carboxamide (4d)
It was obtained as a white-brownish semisolid. 1H NMR (200 MHz, CDCl3): δ 7.80 (d, J = 8.4 Hz, 2H, H-6′, H-2′), 7.69 (d, J = 8.4 Hz, 2H, H-5′, H-3′), 7.37 (d, J = 1.0 Hz, 1H, H-5′′), 7.08 (m, 1H, –NH–), 6.32 (dd, J = 3.2; 1.6 Hz, 1H, H-4′′), 6.27 (d, J = 3.2 Hz, 1H, H-3′′), 4.78 (d, J = 3.4 Hz, 1H, H-5), 4.49 (d, J = 5.8 Hz, 2H, CH2NH), 4.10 (dd, J = 7.2; 3.4 Hz, 1H, H-4), 1.43 (d, J = 7.2 Hz, 3H). Anal. calcd for C17H15F3N2O3: C 57.97, H 4.29, N 7.95. Found: C 58.11, H 4.40, N 8.09.N-(4-sec-Butylphenyl)-4-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-5-carboxamide (4e)
It was obtained as a white-brownish semisolid. [α]25D = −6.8 (c 1.2 in acetone) [2.0% ee, (R,R) rich]. IR (KBr, cm−1): 3425, 3342, 2960, 2920, 2850, 1668, 1620, 1594, 1529, 1450, 1414, 1330, 1250, 1166, 1131, 1110, 1072, 1020, 945, 880, 846, 775, 705, 650, 600, 550 1H NMR (CDCl3, 200 MHz): δ 8.45 (s, 1H, NH), 7.81 (d, J = 8.4 Hz, 2H, H-6′, H-2′), 7.68 (d, J = 8.4 Hz, 2H, H-5′, H-3′), 7.48 (d, J = 8.5 Hz, 2H, H-6′′, H-2′′), 7.13 (d, J = 8.5 Hz, 2H, H-5′′, H-3′′), 4.87 (d, J = 3.3 Hz, 1H, H-5), 4.18 (dq, J = 7.2; 3.3 Hz, 1H, H-4), 2.56 (sept, J = 7.2 Hz, 1H, H3CCH2), 1.56 (sept. J = 7.2 Hz, 2H, CHCH3), 1.47 (d, J = 7.2 Hz, 3H, CH), 1.19 (d, J = 7.2 Hz, 3H, CHAr), 0.78 (t, J = 7.2 Hz, 3H, CH2). 13C NMR (CDCl3, 50.3 MHz): δ 168.21, 161.13, 144.60, 134.32, 130.85, 127.74, 127.64, 126.00 (q, J13C–19F = 4.0 Hz), 119.88, 86.30, 47.06, 41.19, 31.12, 21.87, 18.51, 12.17. 19F NMR (CDCl3, 471 MHz): δ −63.45 (s, 3F). HR ESI-MS m/z calcd for C22H23N2O2F3Na: 427.1609. Found: 427.1608.
N-(4-Methoxyphenyl)-3-(4-trifluoromethylphenyl)-5-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-4-carboxamide (6d)
It was obtained as a glass.1H NMR (200 MHz, CDCl3): δ (s, 1H, NH), 7.99–7.10 (m, 18H), 6.15 (d, 1H, H-5), 4.40 (d, 1H, H-4), 3.83 (s, 3H, H3CO). Anal. calcd for C25H18F6N2O3: C 59.06, H 3.57, N 5.51. Found: C 59.27, H 3.70, N 5.39.N-(4-Methoxyphenyl)-5-(4-isopropylphenyl)-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-4-carboxamide (6e)
It was obtained as a yellowish glass. IR (KBr, cm−1): 3433, 3300, 3150, 3080, 2959, 2920, 2860, 1652, 1620, 1603, 1540, 1512, 1459, 1440, 1413, 1355, 1325, 1245, 1237, 1169, 1126, 1070, 1035, 1008, 951, 920, 850, 830, 805, 690. 1H NMR (200 MHz, CDCl3): δ 7.91 (d, J = 8.4 Hz, 2H, H-5′, H-3′), 7.66 (d, J = 8.4 Hz, 2H, H-6′, H-2′), 7.35 (dd, J = 8.9; 2.1 Hz, 2H, H-6′′′, H-2′′′), 7.29 (d, J = 8.6 Hz, 2H, H-2′′, H-6′′), 7.25 (d, J = 8.6 Hz, 2H, H-5′′, H-3′′), 6.85 (dd, J = 8.9; 2.1 Hz, 2H, H-3′′′, H-5′′′), 6.01 (d, J = 5.4 Hz, 1H, H-5), 4.44 (d, J = 5.4 Hz, 1H, H-4), 3.82 (s, 3H, H3CO), 2.92 (sept, J = 7.2 Hz, 1H, HC(CH3)2), 1.35 (d, J = 7.2 Hz, 6H, (CH3)2CH). 13C NMR (125.9 MHz, CDCl3): δ 166.03, 136.15, 127.32, 127.19, 126.02, 125.42, 122.09, 88.24, 64.53, 55.51, 33.90, 31.60, 23.90, 22.66, 14.13.19F NMR (471 MHz, CDCl3): δ −63.46 (s, 3F). HR ESI-MS m/z calcd for C27H25N2O3F3Na: 505.1715. Found: 505.1722.N-(4-Methoxyphenyl)-4-ethyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-5-carboxamide (7a)
It was obtained as a greenish glass. IR (KBr, cm−1): 3400, 3349, 2960, 2920, 2830, 1671, 1620, 1595, 1528, 1415, 1328, 1250, 1160, 1135, 1105, 1069, 1040, 1020, 845, 830, 770, 695. 1H NMR (500 MHz, CDCl3): δ 8.39 (s, 1H, NH), 7.81 (d, J = 8.0 Hz, 2H, H-5′, H-3′), 7.69 (d, J = 8.0 Hz, H-6′, H-2′), 7.47 (dd, J = 7.0; 2.5 Hz, 2H, H-6′′, H-2′′), 6.86 (d, J = 7.0, 2.5 Hz, 2H, H-5′′, H-3′′), 4.94 (d, J = 3.5 Hz, 1H, H-5), 4.10 (td, J = 8.0; 3.5 Hz, 1H, H-4), 3.78 (s, 3H, H3CO), 1.89 (m, 1H,![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) CH3), 1.70 (m, 1H,
CH3), 1.70 (m, 1H, ![[b with combining low line]](https://www.rsc.org/images/entities/char_0062_0332.gif) CH3), 1.04 (t, J = 7.3 Hz, 3H, CH2). 13C NMR (50.3 MHz, CDCl3): δ 168.41, 156.81, 129.82, 127.73, 126.04, 125.96, 121.47, 114.21, 83.90, 55.47, 53.52, 31.59, 24.65, 22.61, 10.38. 19F NMR (471.04 MHz, CDCl3): δ −63.44 (s, 3F). HR ESI-MS m/z calcd for C20H19N2O3F3Na: 415.1245. Found: 415.1253.
CH3), 1.04 (t, J = 7.3 Hz, 3H, CH2). 13C NMR (50.3 MHz, CDCl3): δ 168.41, 156.81, 129.82, 127.73, 126.04, 125.96, 121.47, 114.21, 83.90, 55.47, 53.52, 31.59, 24.65, 22.61, 10.38. 19F NMR (471.04 MHz, CDCl3): δ −63.44 (s, 3F). HR ESI-MS m/z calcd for C20H19N2O3F3Na: 415.1245. Found: 415.1253.
N-(4-Methoxyphenyl)-5-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-5-carboxamide (7b)
It was obtained as a white-yellowish glass. [α]25D = −14.6 (c 1.8 in acetone) [3% ee, (R,R) rich]. IR (KBr, cm−1): 3410, 3340, 3300, 2960, 2940, 2840, 1665, 1623, 1600, 1521, 1460, 1412, 1328, 1252, 1167, 1132, 1110, 1072, 1032, 913, 827, 700. 1H NMR (200 MHz, CDCl3): δ 8.54 (s, 1H, NH), 7.73 (d, J = 8.5 Hz, 2H, H-6′, H-2′), 7.66 (d, J = 8.5 Hz, 2H, H-5′, H-3′), 7.50 (ddd, J = 6.8; 3.5; 2.0 Hz, 2H, H-6′′, H-2′′), 6.86 (ddd, J = 6.8; 3.5; 2.0 Hz, 2H, H-5′′, H-3′′), 3.96 (d, J = 17.5 Hz, 1H, Ha-4), 3.78 (s, 3H, H3CO), 3.31 (d, J = 17.5 Hz, 1H, Hb-4), 1.83 (s, 3H, H3CC). 13C NMR (50.3 MHz, CDCl3): δ 171.23, 157.32, 156.95, 132.85, 132.27, 130.18, 127.33, 126.04, 125.96, 121.68, 114.38, 88.40, 55.64, 45.16, 24.29. 19F NMR (471.04 MHz, CDCl3): δ −63.39 (s, 3F). HR ESI-MS m/z calcd for C19H17N2O3F3Na: 401.1089. Found: 401.1072.N-(4-Methoxyphenyl)-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-5-carboxamide (7c)
It was obtained as a white-yellowish glass. [α]25D = −114.0 (c 1.1 in acetone) [68.2% ee, (R,R) rich]. IR (KBr, cm−1): 3432, 3340, 2930, 2840, 1667, 1623, 1601, 1533, 1525, 1418, 1328, 1252, 1160, 1132, 1115, 1070, 1040, 901, 842, 830, 707. 1H NMR (200 MHz, CDCl3): δ 8.38 (s, 1H, NH), 7.81 (d, J = 8.8 Hz, 2H, H-5′, H-3′), 7.69 (d, J = 8.8 Hz, 2H, H-6′, H-2′), 7.48 (ddd, J = 8.7; 3.7; 2.0 Hz, 2H, H-6′′, H-2′′), 6.87 (ddd, J = 8.7; 3.7; 2.0 Hz, 2H, H-5′′, H-3′′), 5.31 (dd, J = 11.0; 6.7 Hz, 1H, H-5), 3.79 (s, 3H, H3CO), 3.78 (d, J = 11.0 Hz, 1H, H-4a), 3.78 (d, J = 6.7 Hz, 1H, H-4b). 13C NMR (50.3 MHz, CDCl3): δ 168.42, 157.06, 156.78, 131.78, 129.87, 127.55, 126.11, 126.03, 121.80, 114.43, 79.52, 55.64, 39.56. 19F NMR (471 MHz, CDCl3): δ −63.42 (s, 3F). HR ESI-MS m/z calcd for C18H15N2O3F3Na: 387.0932. Found: 387.0916.N-(4-Methoxyphenyl)-3-(4-trifluoromethylphenyl)-4-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-5-carboxamide (7d)
It was obtained as a white-yellowish glass. IR (KBr, cm−1): 3420, 3370, 3350, 3282, 3200, 3150, 3080, 3020, 2920, 2840, 1678, 1653, 1620, 1550, 1530, 1513, 1470, 1414, 1327, 1247, 1168, 1247, 1168, 1125, 1070, 1030, 1020, 870, 850, 829. 1H NMR (500 MHz, CDCl3): δ 8.41 (s, 1H, NH), 7.73 (d, J = 8.0 Hz, 2H, H-5′, H-3′), 7.64 (d, J = 8.0 Hz, 2H, H-5′′, H-3′′), 7.59 (d, J = 8.0 Hz, 2H, H-6′, H-2′), 7.50 (d, J = 9.0 Hz, 2H, H-2′′′, H-6′′′), 7.45 (d, J = 8.0 Hz, 2H, H-6′′, H-2′′), 6.88 (dd, J = 9.0; 2.1 Hz, 2H, H-3′′′, H-5′′′), 5.32 (d, J = 3.3 Hz, 1H, H-5), 5.04 (d, J = 3.3 Hz, 1H, H-4), 3.80 (s, 3H, H3CO). 13C NMR (125.9 MHz, CDCl3): δ 167.14, 158.30, 157.04, 141.20, 132.77, 132.50, 131.00, 130.74, 130.56, 129.61, 127.94, 127.92, 126.67 (q, J = 3.8 Hz), 125.98 (q, J = 3.8 Hz), 124.83, 124.60, 122.67, 122.44, 121.61, 114.44, 114.31, 87.40, 57.60, 55.51. 19F NMR (471 MHz, CDCl3): δ −63.17 (s, 3F), −63.58 (s, 3F). HR ESI-MS m/z calcd for C25H18N2O3F6Na: 531.1119. Found: 531.1091.3-(4-Trifluoromethylphenyl)-4,5-dihydroisoxazole-4-methyl-5-methylol (8)
LiBEt3H (0.4 mL of 1.0 M solution in THF) was added dropwise at 0 °C to a solution of N-(2-methoxyphenyl)-4-methyl-3-(4-trifluoromethylphenyl)-4,5-dihydroisoxazole-5-carboxamide (4a) (4R,5R, ee 96%, 0.077 g) in THF (4 mL). The reaction mixture was stirred for 15 min at 0 °C, then 50 min at rt, cooled in an ice bath and quenched with water. Product was extracted with dichloromethane and purified by column chromatography on silica gel using hexane–ethyl acetate as eluent. It was obtained as a brownish oil in 25% yield. [α]25D = −147.0 (c 0.8 in acetone) [95% ee, (R,R) rich]. 1H NMR (200 MHz, CDCl3): δ 7.66 (d, J = 8.0 Hz, 2H, H-6′, H-2′), 7.30 (d, J = 8.0 Hz, 2H, H-5′, H-3′), 4.38 (m, 1H, H-5), 3.90 (m, 2H, CH2OH), 3.78 (m, 1H, H-4), 1,36 (d, J = 7,6 Hz, 3H). ESI-MS m/z 260 (M+H)+, 283 (M+H+Na)+. HR ESI-MS m/z calcd for C12H12NO2F3Na: 282.5766. Found: 282.5761.Acknowledgements
This work was supported in part by the Polish Ministry of Science and Higher Education Research (Grant N N209 003 638), which is gratefully acknowledged.References
- (a) M. Gucma and W. M. Gołębiewski, J. Heterocycl. Chem., 2008, 45, 241–245 CrossRef CAS; (b) W. M. Gołębiewski and M. Gucma, J. Heterocycl. Chem., 2008, 45, 1687–1693 CrossRef.
- M. Christl and R. Huisgen, Chem. Ber., 1973, 106, 3345–3367 CrossRef.
- P. Caramella and P. Grunanger, in 1,3-Dipolar Cycloaddition Chemistry, John Wiley & Sons, New York, NY, 1984, vol. 1 and 2 Search PubMed.
- (a) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863–909 CrossRef CAS; (b) H. Pellisier, Tetrahedron, 2007, 63, 3235–3285 CrossRef.
- T. Olsson, K. Stern, G. Westman and S. Sundell, Tetrahedron, 1990, 46, 2473–2482 CrossRef CAS.
- D. P. Curran, B. P. Kim, H. P. Piyasena, R. J. Loncharich and K. N. Houk, J. Org. Chem., 1987, 52, 2137–2141 CrossRef CAS.
- D. P. Curran and M.-H. Yoon, Tetrahedron, 1997, 53, 1971–1982 CrossRef CAS.
- D. J. Ager, J. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–875 CrossRef CAS.
- S. Minakata, T. Ezoe, K. Nakamura, I. Ryu and R. Komatsu, Tetrahedron Lett., 1998, 39, 5205–5208 CrossRef CAS.
- H. Waldman, Liebigs Ann. Chem., 1990, 1013–1017 CrossRef.
- D. P. Curran and T. A. Heffner, J. Org. Chem., 1990, 55, 4585–4595 CrossRef CAS.
- J. Liu, A. Eddings and R. H. Wallace, Tetrahedron Lett., 1997, 38, 6795–6798 CrossRef CAS.
- (a) Y. Yoshida, Y. Ukaji, S. Fujinami and K. Inomata, Chem. Lett., 1998, 1023–1024 CrossRef CAS; (b) M. Tsuji, Y. Ukaji and K. Inomata, Chem. Lett., 2002, 1112–1113 CrossRef CAS.
- M. Serizawa, Y. Ukaji and K. Inomata, Tetrahedron: Asymmetry, 2006, 17, 3075–3083 CrossRef CAS.
- (a) M. P. Sibi, K. Itoh and C. P. Jasperse, J. Am. Chem. Soc., 2004, 126, 5366 CrossRef CAS; (b) M. P. Sibi, Z. Ma, N. Prabagaran, K. Itoh and C. P. Jasperse, Org. Lett., 2005, 7, 2349–2352 CrossRef CAS.
- G. Faita, A. Paio, P. Quadrelli, F. Rancati and P. Seneci, Tetrahedron, 2001, 57, 8313–8322 CrossRef CAS.
- H. Yamamoto, S. Hayashi, M. Kubo, M. Harada, M. Hasegava, M. Noguchi, M. Sumumoto and K. Hori, Eur. J. Org. Chem., 2007, 2859–2864 CrossRef CAS.
- F. Ono, Y. Ohta, M. Hasagawa and S. Kanemasa, Tetrahedron Lett., 1997, 50, 2111–2114 CrossRef.
- M. K. Boysen, Chem.–Eur. J., 2007, 13, 8648–8659 CrossRef CAS.
- R. O. Duthaler and A. Hafner, Chem. Rev., 1992, 92, 807–832 CrossRef CAS.
- K. Totani, K. Takao and K. Tadano, Synlett, 2004, 2066–2080 CAS.
- (a) J. M. Vega-Perez, M. Vega-Holm, I. Perinan, C. Palo-Nieto and F. Iglesias-Guerra, Tetrahedron, 2011, 67, 364–372 CrossRef CAS; (b) J. Zhao, Y. Zhang, F. Han and S. Zhao, Carbohydr. Res., 2009, 344, 61–66 CrossRef CAS; (c) Y. Pérez, S. Morante-Zarcero, I. Del Hierro, I. Sierra, M. Fajardo and A. Otero, Chirality, 2006, 18, 44–48 CrossRef.
- T. Bako, P. Bako, A. Szollosy, M. Czugler, G. Keglevicha and L. Toke, Tetrahedron: Asymmetry, 2002, 13, 203–209 CrossRef CAS.
- S. U. Son, H.-Y. Jang, J. W. Han, I. S. Lee and Y. K. Chung, Tetrahedron: Asymmetry, 1999, 10, 347–354 CrossRef CAS.
- T. Itoh and S. Shirakami, J. Synth. Org. Chem. Jpn., 2002, 60, 232–239 CrossRef CAS.
- (a) M. Spescha and G. Rihs, Helv. Chim. Acta, 1993, 76, 1219–1230 CrossRef CAS; (b) K. Totani, K. Takao and K. Tadano, Synlett, 2004, 2066–2080 CAS.
- (a) M. A. Weidner-Wells, S. A. Fraga-Spano and I. J. Turchi, J. Org. Chem., 1998, 63, 6319–6328 CrossRef CAS; (b) P. Caramella, D. Reami, M. Falzoni and P. Quadrelli, Tetrahedron, 1999, 55, 7027–7044 CrossRef CAS.
- L. S. Santos, L. Knaack and J. O. Metzger, Int. J. Mass Spectrom., 2005, 246, 84–104 CrossRef CAS.
- K.-C. Liu, B. R. Howe and R. K. Shelton, J. Org. Chem., 1980, 45, 3916–3918 CrossRef CAS.
- W. M. Gołębiewski and M. Gucma, Synthesis, 2007, 3599–3619 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |