Selectivity viacatalyst or substrate control in catalytic asymmetric transformations of bifunctional symmetrical substrates
David S.
Glueck
*
6128 Burke Laboratory, Department of Chemistry, Dartmouth College, Hanover, New Hampshire, USA. E-mail: glueck@dartmouth.edu; Fax: +1 603-646-3946; Tel: +1 603-646-1568
First published on 4th April 2011
Abstract
Catalytic asymmetric transformations of bifunctional symmetrical substrates pose interesting problems in selectivity. In one extreme, catalyst control leads to identical selectivity at both sites, and amplification of chirality in the product. This improved enantiopurity comes at the expense of reduced yield; the undesired meso byproduct must be separated. Alternatively, substrate control may modify the inherent selectivity of the catalyst. The first transformation may change the substrate to make the second transformation less selective (negative cooperativity), while positive cooperativity can result in selectivity greater than that afforded by simple catalyst control. Similar behavior may occur in polyfunctional substrates with three or more identical reactive sites. Substrate vs. catalyst control and positive vs. negative cooperativity plausibly depends upon the structure of the substrate and the linker between the active sites, but has rarely been investigated systematically. This perspective presents several examples of structure–selectivity relationships with the eventual goal of designing substrates for positive cooperativity and enhanced selectivity in asymmetric catalysis.
 David S. Glueck | David Glueck received A.B. and A.M. degrees in 1986 from Harvard University, where he did research with John Cooper. After a Ph.D. at U.C. Berkeley in 1990 (with Robert Bergman), he did postdoctoral research at Oxford with Malcolm Green. He joined the faculty at Dartmouth College in 1992, and served as Chemistry Department Chair from 2007–2010. His research interests include asymmetric catalysis, reaction mechanisms in homogeneous catalysis, and metal-phosphine chemistry. |
Introduction. Symmetrical bifunctional molecules and their role in stoichiometric and catalytic Horeau duplication
Symmetrical bifunctional molecules are frequent synthetic targets. Common examples include the large class of C2-symmetric natural products1 and the most common group of bidentate ligands used in metal catalysts for asymmetric synthesis, also bearing C2 symmetry.2 These compounds may be prepared either by combination of two subunits, or by two identical reactions on a symmetrical bifunctional substrate. Both approaches share the goal of enantiomerically pure products and result in the same stereochemical consequences, which are the topic of this perspective.Reaction of an enantiomerically enriched substrate (R/S mixture) with an achiral bifunctional linker (Y∼Y), assumed to be unselective (statistical), may be used to increase the enantiopurity of the substrate (Scheme 1; this process is often called the Horeau duplication).3 The major product (R∼R) arises from attachment of the major substrate enantiomer to both ends of the linker. The meso (R∼S) isomer forms when both the major (R) and the minor (S) enantiomers bind to the linker. Finally, on statistical grounds, little of the S∼S product is expected. After separation of the rac (R∼R/S∼S) products from the meso one, removal of the linker yields an enantiomerically enriched R substrate. If its original enantiomeric ratio (er) was x/(1 − x), where x is the mole fraction of the R-enantiomer, the upgraded er is (x/(1 − x))2. Thus, material of 80% enantiomeric excess (ee; 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 er) yields product of 97.6% ee (81∶1 er), at the cost of 2x(1 − x) (here 18%) of the material, as some of the R isomer is sacrificed to consume the unwanted S isomer in formation of the meso compound.†
∶1 er) yields product of 97.6% ee (81∶1 er), at the cost of 2x(1 − x) (here 18%) of the material, as some of the R isomer is sacrificed to consume the unwanted S isomer in formation of the meso compound.†
 | ||
| Scheme 1 The Horeau duplication. | ||
Scheme 2 shows how this process was used to increase the enantiomeric purity of an alcohol. 1-Naphthylethanol (96∶4 er) was attached to oxalyl chloride; after separation of the minor meso isomer by recrystallization, hydrolysis gave the pure alcohol in 78% recovery, with 99.8∶0.2 er.4
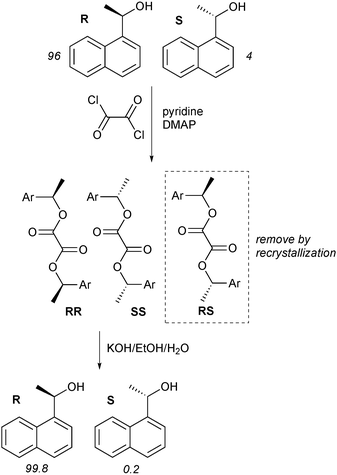 | ||
| Scheme 2 Increasing the enantiomeric purity of an alcoholvia Horeau duplication. | ||
The same advantageous process operates in catalytic transformations of bifunctional symmetrical substrates Z∼Z (Scheme 3).5‡ Identical selectivity at both sites, typically controlled by a chiral catalyst, leads to the intermediates R∼Z and S∼Z, and then to enantiomerically and diastereomerically enriched products (R∼R/S∼S/R∼S) without the added step of adding and removing a linker.6
 | ||
| Scheme 3 “Duplication” via asymmetric catalysis: conversion of Z moieties in bifunctional symmetrical Z∼Z into chiral groups (R or S). | ||
Such processes are most synthetically useful with highly selective catalysts, when little or no unwanted meso product is formed. For example, iridium-catalyzed asymmetric hydrogenation of triene 1 gave essentially only one enantiomer of 2, which is potentially useful in tocopherol synthesis (Scheme 4).7§
 | ||
| Scheme 4 Enantioselective hydrogenation of a triene. | ||
If the meso products can be readily separated, catalytic reactions are also useful even with reduced selectivity, as seen in two remarkable transformations used in total syntheses (Schemes 5 and 6). After a 17-step preparation of substrate 3, a selective Pd(Tol-Binap)-catalyzed Heck reaction yielded 4 in 62% yield and 90% ee, along with 21% meso isomers (Scheme 5). Enriched 4 was subsequently converted to the natural products quadrigemine C and psycholeine.8 In another palladium-catalyzed reaction, a mixture of rac and meso isomers of 5 yielded highly enriched 6, a precursor to cyanthiwigin F, along with separable meso-7 (Scheme 6).9
 | ||
| Scheme 5 Asymmetric Heck reaction of a diene. | ||
 | ||
| Scheme 6 Pd-catalyzed double enantioselective alkylation. | ||
These ideas may be extended (with a selective catalyst) to polyfunctional substrates. For example, Shi epoxidation of tetra-ene 8, a key step in asymmetric synthesis of glabescrol, gave enriched 9 along with its diastereomers; epoxidation of each double bond was estimated to proceed with ca. 20∶1 selectivity (Scheme 7).10 Similarly, osmium-catalyzed Sharpless asymmetric dihydroxylation (AD) of squalene yielded poly-alcohol 10 in high yield as one of 36 possible diastereomers (Scheme 8);11 the er of 10 was estimated to be about 1010!5
 | ||
| Scheme 7 Selective epoxidation of a tetra-ene. | ||
 | ||
| Scheme 8 Osmium-catalyzed asymmetric dihydroxylation of squalene. | ||
Selectivity viacatalyst or substrate control
Definitions and the general problem
These processes exploit catalyst control, where changes in the substrate structure do not affect catalytic selectivity. However, this is frequently not the case, and selectivity often depends on substrate control. To analyze this more generally than in Scheme 3, consider again a bifunctional symmetrical substrate with two reactive sites, Z∼Z (Scheme 9). The first asymmetric transformation will convert a Z group (which may be achiral or chiral but racemic)12 into a chiral group, R or S. | ||
| Scheme 9 Product ratios for catalystvs. substrate control in reactions of a bifunctional symmetrical substrate Z∼Z. | ||
The mole fractions of the intermediates R∼Z and S∼Z are then defined as x and 1 − x. In general, the second asymmetric transformation may have different selectivity than the first (substrate control), which may be described by the mole fractions a and b. The resulting product ratios are listed in Scheme 9 (substrate control); the rac/meso ratio (diastereomeric ratio or dr) and the er of the rac product are given by eqn (1) and (2).
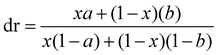 | (1) |
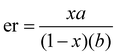 | (2) |
In the special case of catalyst control, both steps have the same selectivity (x = a = 1 − b), and the product ratios simplify to the catalyst control values in Scheme 9, as in Scheme 3. This process is an attractive way to get highly enantiomerically enriched materials, because catalyst mistakes are converted into the meso product R∼S. After rac/meso separation, the highly enriched R∼R product can be isolated.
Under substrate control, the catalyst recognizes and processes the intermediates R∼Z and S∼Z differently; matched and mis-matched pairs frequently occur, so that a and 1 − b are often different.13 This may result in positive cooperativity, where the formation of the first chiral group promotes formation of the second with higher selectivity, or in negative cooperativity, leading to meso products.
For practical applications, a highly selective catalyst is ideal; the resulting catalyst control will lead to a bifunctional chiral product in high dr and er. Substrate control has not been investigated systematically, but, in principle, it might lead to selectivity even higher than that available from catalyst control, in cases of positive cooperativity. This perspective describes several examples of catalytic reactions of this type, with the eventual goal of identifying structure–reactivity–selectivity relationships to enable the design of highly selective catalytic asymmetric processes.¶
To establish quantitative structure–selectivity relationships it is useful to measure the parameters x, a and b. This may be accomplished in several ways, all of which require measuring the final product ratios:
1. Direct measurement is possible if the intermediates R∼Z and S∼Z may be isolated, which may occur when the second step is slower than the first and/or when stoichiometry is controlled, or when these intermediates may be prepared independently as a mixture of known composition.
2. Alternatively, the ratio x/1 – x may be measured at low conversion.
3. The selectivity of the first step may be modeled with a single-site substrate U∼Z, where U is an unreactive group.
4. When catalyst control is suspected, one may check to see if the product ratios fit a given x-value. Especially in cases where substrate control is unlikely, for example when the two Z groups are separated by a long, flexible linker, finding an x value which is consistent with the observed product ratios likely indicates catalyst control (but does not rule out substrate control).
As shown in the following examples, the possibility of catalystvs. substrate control is strongly substrate-dependent. Intuitively, substrate control is expected for molecules in which the two Z groups are connected by a short, rigid linker, or in cases where the substrate has a distinct conformational preference, such as in cyclic systems.
Structure–selectivity relationships
Ru(Binap)-catalyzed asymmetric hydrogenations of the dienes in Scheme 10 demonstrate how substrate control and its degree may vary dramatically even with small changes in the substrate structure (Table 1).14 | ||
| Scheme 10 Ru-catalyzed asymmetric hydrogenations of dienes 11 and 12. | ||
| x | 1 − x | a | 1 − a | 1 − b | b | |
|---|---|---|---|---|---|---|
| R = H (11) | 0.95 | 0.05 | 0.98 | 0.02 | 0.62 | 0.38 |
| R = Ph (12) | 0.24 | 0.76 | 0.53 | 0.47 | 0.03 | 0.97 |
These data indicate that, for substrate 11, the upper channel is governed by catalyst control (x ≈ a). In the lower channel, mistake correction is less efficient (a > 1 − b). Phenyl-substituted 12 behaves quite differently. Its first hydrogenation is far less selective and favors the opposite configuration, while the second hydrogenation operates with positive cooperativity (b > 1 − x).
Such observations are not limited to homogeneous catalysts. For example, asymmetric hydrogenation of acetylacetone was carried out with a tartaric acid-modified RANEY® Ni catalyst. While catalyst control prevailed in the major channel, when one mistake occurred, the catalyst was not very good at correcting it (Scheme 11).15
 | ||
| Scheme 11 Selectivity of asymmetric hydrogenation of acetylacetone with a heterogeneous RANEY® Ni catalyst. | ||
With the same diketone substrate and a Ru(Binap) catalyst, a striking linker effect was observed. While acetylacetone was hydrogenated with very high selectivity under catalyst control, hydrogenation of biacetyl was meso-selective. However, the minor C2-symmetric product was obtained in very high ee (Scheme 12).16
 | ||
| Scheme 12 Ru(Binap)-catalyzed asymmetric hydrogenation of diketones. | ||
Attempts to prepare the pseudo-C2 symmetric core of the zaragozic acids using Os-catalyzed AD of meso-tartrate-derived 13 were thwarted by selective formation of the meso product 14 (Scheme 13). The structural basis for this extreme substrate control is not clear, but it could be overcome by modifying the group connecting the two alkenes (Scheme 13). With alkyne-linked 15, sequential catalyst-controlled AD gave the desired pseudo-C2-symmetric product. The alkyne was then converted to the desired pseudo-mesodiol by hydrogenation to an alkene followed by OsO4-catalyzed dihydroxylation.17
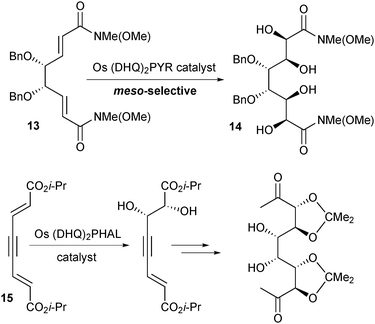 | ||
| Scheme 13 Os-catalyzed asymmetric dihydroxylation of dienes 13 and 15. | ||
As in this case, meso-selectivity with a chiral catalyst is rarely desired or synthetically useful, which may account for the infrequent reports of such processes. However, quantitative analyses of these transformations may provide structure–selectivity relationships of potential value for understanding the structural basis of substrate control. The Pt-catalyzed asymmetric phosphination of bis(secondary phosphine) 16b in Scheme 14 is a rare example of this sort of negative cooperativity. Although the platinum catalyst alkylates the first phosphorus center with reasonable selectivity (∼2∶1), this stereocenter, three bonds away from the second reactive site, is somehow able to reverse the selectivity in the second alkylation. Thus, intermediate RP-17 was alkylated with ∼1∶3 selectivity, favoring the S-configuration, while the diastereomeric SP-17, which differs only in the absolute configuration of the pendant tertiary phosphine group, was alkylated with ca. 7∶1 selectivity for the R-configuration, resulting in selective formation of the meso-bis(phosphine).18
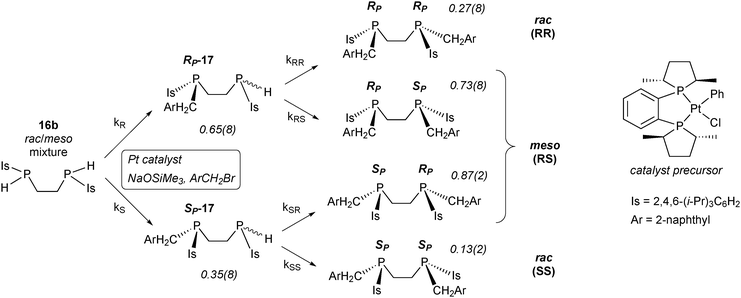 | ||
| Scheme 14 Meso-selective Pt-catalyzed asymmetric alkylation of bis(isitylphosphino)ethane (16b) with 2-bromomethylnaphthalene. | ||
In an unusual systematic study of linker effects on catalystvs. substrate control, related alkylations of substrate 16b (Scheme 14) and its analogous bis(phosphino)alkanes 16a and 16c–e were investigated (Scheme 15).19Catalyst control (x ≈ 0.68) was observed for the substrates 16d–e with longer linkers (n = 4–5). In contrast, alkylation of ethane-linked 16b was meso-selective, presumably because of negative cooperativity, as seen in alkylation of the same phosphine with a related substrate, 2-bromomethylnaphthalene, in Scheme 14. The selectivity of the slower catalytic alkylation of C1-bridged 16a could not be determined quantitatively, because the background (catalyst-free) reaction was competitive, but it was also consistent with substrate control. Finally, propane-bridged 16c showed intermediate behavior, with weak substrate control. The selectivity of the first alkylation was similar to that of 16d–e (x = 0.69); it resulted in small but distinct changes in the selectivity of the second phosphine alkylation (x ≠ a ≠ 1−b, Scheme 16).
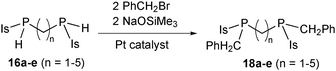 | ||
| Scheme 15 Pt-catalyzed asymmetric alkylation of a series of bis(phosphino)alkanes. | ||
 | ||
| Scheme 16 Selectivity of Pt-catalyzed asymmetric alkylation of bis(isitylphosphino)propane (16c) with benzyl bromide. | ||
In a similar study of Os-catalyzed AD of dienes, the length and nature of the linker were also found to affect the selectivity of dihydroxylation (Scheme 17). The x-values listed were found for the first step, conversion of 19 to 20. Although insufficient product ratio data were reported to enable calculation of a and b values, comparison of the observed enantiopurity of 21 with that predicted from the x-values, assuming catalyst control, showed that catalystvs. substrate control depended on the linker. As in Schemes 15 and 16, reducing the linker length by a single carbon (19avs.19b) resulted in a change from catalyst control to substrate control, while the reduced selectivity for 19c, approximately isosteric with 19b, is striking.20
 | ||
| Scheme 17 Os-catalyzed AD of dienes 19a–c (ligand = (DHQ)2PYR). | ||
Origin of substrate control?
Although systematic studies like those discussed above are rare, understanding the relationship between the substrate structure and catalyst selectivity should, in principle, enable rational design of substrates and catalysts to improve selectivity in asymmetric catalysis. This is a challenging problem, since detailed understanding of the origins of enantioselection or diastereoselection is difficult to obtain even for a single substrate, without considering structural variations.21However, some systems have been reported in which, after the fact, substrate-controlled selectivity has been rationalized, often on the basis of preferred substrate conformations. For example, when a Sharpless-style titanium catalyst was used for asymmetric oxidation of ketene dithioacetal 22, both high diastereoselectivity and high enantioselectivity were observed in formation of bis(sulfoxide) 24 (Scheme 18). Sulfoxide 23 was also isolated as a minor product. If its 85% ee results from the selectivity of a single oxidation (x = 0.925), rac-24 should be formed, under catalyst control, in 152∶1 er (99% ee), consistent with the experimental results. This analysis predicts that 14% of meso-bis(sulfoxide) 25 would be formed, but it was not observed.

Further studies revealed substantial substrate control in the second oxidation: treatment of 22 with achiral MCPBA led only to rac-24. Oxidation of enantioenriched 23 (86% ee), which had been formed from Ti-(+)-DET-catalyzed oxidation of 22, with a Ti-(−)-DET catalyst did not yield primarily meso-25, as would be expected from catalyst control. Instead, rac-24 was formed, with substantial enriched 23 recovered (Scheme 18). Although the missing meso product and the incomplete mass balance prevented detailed analysis of these results, it was clear that substrate control could occur with positive cooperativity, favoring formation of the desired rac-24 over meso-25.22
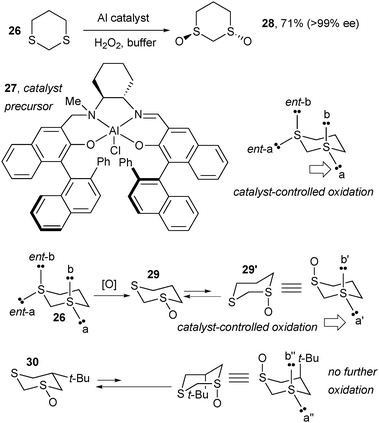
In a related system (Scheme 19), conformational preferences of the cyclic substrate were used to rationalize similar substrate-controlled selectivity. Oxidation of disulfide 26 mediated by a catalyst formed from Al-salalen complex 27 and hydrogen peroxide gave dioxide 28 in high enantiopurity. Although details of the catalyst structure and its interactions with the substrate are not known, this catalyst is evidently able to differentiate between the four sulfur lone pairs in 26, preferring to oxidize equatorial lone pair a (Scheme 19) to yield monoxide 29. In the preferred conformation of 29, the oxygen is equatorial, but oxidizing the remaining equatorial S lone pair to make the cis-dioxide is very slow. However, interconversion of 29 to the higher-energy conformer 29′, with an axial oxygen, can occur readily, enabling oxidation of the equatorial S lone pair (a′, with a geometry similar to a in the initial substrate 26) to selectively give trans-dioxide 28. This is consistent with observations on the t-Bu-substituted substrate 30, which cannot undergo ring flip to make available the preferred equatorial a″ S lone pair to the catalyst, and therefore cannot be oxidized a second time (Scheme 19).23
In a related example, again featuring conformational dynamics of six-membered rings, Cr-catalyzed asymmetric ring opening of the C2h-symmetric epoxide 31, under catalyst control, selectively gave mono-epoxide 32 (x = 0.9, Scheme 20). The second ring-opening step showed strong match–mismatch effects and a competition between catalyst and substrate control. In the upper channel, substrate control (the preferred conformer of 32 contains equatorial OSiMe3 and N3groups), the required axial attack of the nucleophile to open the epoxide, and catalyst control, with selective attack at the R-epoxide carbon, are matched. This leads almost exclusively to product 33 (a ≈ 1). After the catalyst makes one “mistake”, yielding intermediate ent-32, correcting it is made difficult by substrate control. To enable the catalyst-preferred axial attack at the R-epoxide carbon, both the azide and OSiMe3 substituents must be axial, leading to regio-isomer 34. This process is presumably disfavored by a 1,3-diaxial interaction, but nonetheless gives about half the product in the lower channel (b ≈ 0.5). In contrast, a second mistake by the catalyst, with formation of the minor enantiomer ent-33, is favored by the substrate's preference (equatorial azide and OSiMe3 substituents during axial attack, Scheme 20).24
 | ||
| Scheme 20 Selectivity of Cr-catalyzed asymmetric ring opening of diepoxide 31. | ||
Substrate control may arise not only from conformational preferences in six-membered rings, as in these examples, but from similar considerations in the formation of cyclic products. Scheme 21 shows an example observed in cyclizations of diastereomeric tetraols 35 and 36 using Pd π-allyl chemistry.25
 | ||
| Scheme 21 Selectivity of Pd-catalyzed asymmetric alkylation of tetraols 35 and 36. | ||
The increased diastereoselectivity for cyclization of all-syntetraol 35vs. anti–syn–antitetraol 36 was rationalized on the basis of conformational preferences. In both cases, catalyst-controlled cyclization, matched with substrate steric preference to form an equatorial vinyl group, was proposed to yield the C15 pyrans 37 and 38 selectively. The second step for 37, however, is mismatched, since catalyst control would lead to the RR product 39, with one equatorial and one axial vinyl group, while substrate control would lead to the thermodynamically preferred meso-40, with two equatorial vinyl groups. In contrast, catalyst control in the second cyclization of 38 should be more favorable, since it leads to only one axial substituent in the product 41; the absence of a 1,3-diaxial interaction in 41, as compared to 39, was proposed to explain the increased diastereoselectivity.||
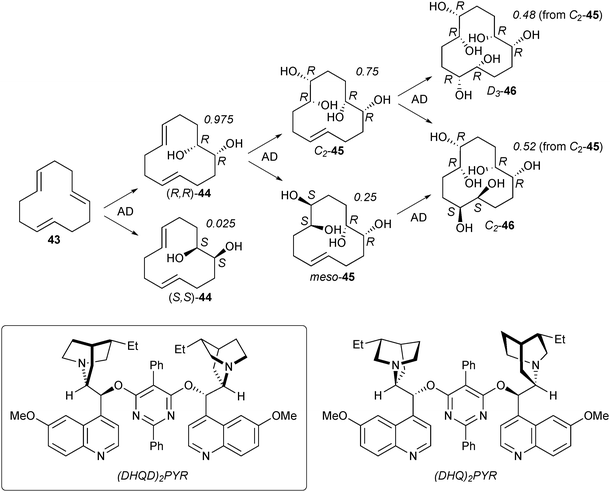
Such conformational effects are not limited to six-membered rings, as shown by studies of the selectivity of Os-catalyzed AD of all-transcyclododecatriene 43 (Scheme 22).26 Dihydroxylation of 43 with an Os-(DHQD)2PYR catalyst gave, at low conversion, diene–diol (R,R)-44 in 95% ee (x = 0.975). If this reaction was carried out for a longer time, (R,R)-44 was obtained in higher enantiopurity as a result of substrate control; the minor enantiomer (S,S)-44 reacted in a second AD process with the catalyst faster than did (R,R)-44, so that mistakes in the first AD were destroyed by further reaction.**
 | ||
| Scheme 22 Selectivity of Os-catalyzed AD of cyclic triene 44. | ||
Such substrate control was investigated in more detail using enantiomerically pure diene–diol (R,R)-44. With an achiral ligand, quinuclidine, dihydroxylation was highly meso-selective, yielding meso-45 in 97∶3 dr. Molecular mechanics calculations showed that the si,si and re,re faces of the two double bonds in (R,R)-44 were presented to the outside (pro S,S) and the inside (pro R,R) of the ring, respectively, explaining the preference for reaction with the more accessible alkene face. However, this substrate control could be partially overcome by the chiral Os-(DHQD)2PYR catalyst, which gave a 75∶25 mixture of C2- and meso-45 (Scheme 22). This selectivity apparently results from a mismatch between the catalyst preference, leading to C2-45, and substrate control, which favors meso-45. Consistent with this idea, use of the pseudo-enantiomeric (DHQ)2PYR ligand led to highly selective formation of meso-45 (99.7∶0.3 dr, matched case).
Finally, double AD on (R,R)-44 with the Os-(DHQD)2PYR catalyst gave a 36∶64 mixture of all-R D3-symmetric hexaol 46 and C2-symmetric 46 (Scheme 22).†† The selectivity of this catalyst in the third AD is lower than in the first two, with an R∶S ratio of only 48∶52, as compared to 75∶25 for the second AD and 97.5∶2.5 for the first. Presumably the reduced selectivity in the third step results from substrate conformational preferences, as in step 2.
Conclusions
How can highly selective catalytic asymmetric reactions of bi- or polyfunctional substrates be designed? Ideally, a universal catalyst with equally high selectivity for every substrate would be developed. Since this has historically been difficult, another approach seeks to harness substrate control to obtain positive cooperativity, exploiting matched selectivity favored by both substrate and catalyst control. In the few cases where this has been proposed (after the fact), such processes often involve strong conformational preferences of the substrate, as seen in cyclohexanes, or in reactions involving ring formation. Systematic investigation of structure–selectivity relationships for such compounds may enable rational improvement of selectivity in catalytic asymmetric processes.Acknowledgements
I thank the US National Science Foundation for support.Notes and references
- M. Vrettou, A. A. Gray, A. R. E. Brewer and A. G. M. Barrett, Tetrahedron, 2007, 63, 1487–1536 CrossRef CAS.
- J. K. Whitesell, Chem. Rev., 1989, 89, 1581–1590 CrossRef CAS; Phosphorus Ligands in Asymmetric Catalysis. Synthesis and Applications, ed. A. Börner, Wiley-VCH, Weinheim, 2008 Search PubMed.
- J. P. Vigneron, M. Dhaenens and A. Horeau, Tetrahedron, 1973, 29, 1055–1059 CrossRef CAS.
- I. Fleming and S. K. Ghosh, J. Chem. Soc., Chem. Commun., 1994, 99–100 RSC; T. R. Hoye, M. J. Mayer, T. J. Vos and Z. Ye, J. Org. Chem., 1998, 63, 8554–8557 Search PubMed.
- V. Rautenstrauch, Bull. Soc. Chim. Fr., 1994, 131, 515–524 CAS; S. El Baba, K. Sartor, J.-C. Poulin and H. B. Kagan, Bull. Soc. Chim. Fr., 1994, 131, 525–533.
- F. Lagasse, M. Tsukamoto, C. J. Welch and H. B. Kagan, J. Am. Chem. Soc., 2003, 125, 7490–7491 Search PubMed.
- S. Bell, B. Wüstenberg, S. Kaiser, F. Menges, T. Netscher and A. Pfaltz, Science, 2006, 311, 642–644 CrossRef CAS.
- A. D. Lebsack, J. T. Link, L. E. Overman and B. A. Stearns, J. Am. Chem. Soc., 2002, 124, 9008–9009 CrossRef CAS.
- J. A. Enquist Jr and B. M. Stoltz, Nature, 2008, 453, 1228–1231 CrossRef.
- Z. Xiong and E. J. Corey, J. Am. Chem. Soc., 2000, 122, 9328–9329 CrossRef CAS.
- G. A. Crispino, P. T. Ho and K. B. Sharpless, Science, 1993, 259, 64–66 CAS.
- J. T. Mohr, D. C. Ebner and B. M. Stoltz, Org. Biomol. Chem., 2007, 5, 3571–3576 RSC.
- S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Angew. Chem., Int. Ed. Engl., 1985, 24, 1–30 CrossRef.
- H. Muramatsu, H. Kawano, Y. Ishii, M. Saburi and Y. Uchida, J. Chem. Soc., Chem. Commun., 1989, 769–770 RSC.
- A. Tai, K. Ito and T. Harada, Bull. Chem. Soc. Jpn., 1981, 54, 223–227 CAS.
- M. Kitamura, T. Ohkuma, S. Inoue, N. Sayo, H. Kumobayashi, S. Akutagawa, T. Ohta, H. Takaya and R. Noyori, J. Am. Chem. Soc., 1988, 110, 629–631 CrossRef CAS.
- Y. Wang, S. Gang, A. Bierstedt, M. Gruner, R. Fröhlich and P. Metz, Adv. Synth. Catal., 2007, 349, 2361–2367 CrossRef CAS.
- T. W. Chapp, D. S. Glueck, J. A. Golen, C. E. Moore and A. L. Rheingold, Organometallics, 2010, 29, 378–388 Search PubMed.
- T. W. Chapp, A. J. Schoenfeld and D. S. Glueck, Organometallics, 2010, 29, 2465–2473 Search PubMed.
- H. Takahata, S. Takahashi, S. Kouno and T. Momose, J. Org. Chem., 1998, 63, 2224–2231 CrossRef CAS.
- P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis, University Science Books, Sausalito, 2009 Search PubMed.
- V. K. Aggarwal, R. M. Steele, Ritmaleni, J. K. Barrell and I. Grayson, J. Org. Chem., 2003, 68, 4087–4090 Search PubMed; V. K. Aggarwal, B. N. Esquivel-Zamora, G. R. Evans and E. Jones, J. Org. Chem., 1998, 63, 7306–7310 CrossRef CAS.
- J. Fujisaki, K. Matsumoto, K. Matsumoto and T. Katsuki, J. Am. Chem. Soc., 2011, 133, 56–61 Search PubMed.
- A. Ironmonger, P. Stockley and A. Nelson, Org. Biomol. Chem., 2005, 3, 2350–2353 RSC.
- B. S. Lucas and S. D. Burke, Org. Lett., 2003, 5, 3915–3918 CrossRef CAS.
- H. Becker, M. A. Soler and K. B. Sharpless, Tetrahedron, 1995, 51, 1345–1376 CrossRef CAS.
Footnotes |
| † The increased enantiopurity and the required separation of the meso isomer may be depicted graphically; see P. Wyatt and S. Warren, Organic Synthesis. Strategy and Control, John Wiley and Sons Ltd., Chichester, England, 2007, pp. 391–393. |
| ‡ For additional discussion of the historical development of such reactions, see ref. 5. For more information on double asymmetric induction, see refs. 13, 21 and O. I. Kolodiazhnyi, Tetrahedron, 2003, 59, 5953–6018. |
| § The other enantiomer of triene 1 underwent hydrogenation with equally high selectivity, demonstrating the catalyst-controlled nature of this process, and showing that the formally inequivalent prochiral alkenes in 1 are treated identically by the catalyst. |
| ¶ Many examples of Horeau duplication in catalysis are known; because of the present focus on understanding the structural origins of substrate vs. catalyst control, comprehensive coverage is not attempted here. Moreover, the related desymmetrization of bifunctional symmetrical meso substrates, which presents similar issues of selectivity, is not included (see M. C. Willis, J. Chem. Soc., Perkin Trans. 1, 1999, 1765–1784 for a review). |
| || Higher selectivity was observed for 36 on addition of NEt3, but the data in Scheme 21 were selected for comparison of selectivity under the same reaction conditions. |
| ** For related editing processes in meso-desymmetrization, see S. L. Schreiber, T. S. Schreiber, and D. B. Smith, J. Am. Chem. Soc. 1987, 109, 1525–1529 and C. S. Poss and S. L. Schreiber, Acc. Chem. Res. 1994, 27, 9–17. |
| †† The reported 36∶64 ratio of D3-46 to C2-46 is consistent with conversion of all of meso-45 to the illustrated enantiomer of C2-46. |
| This journal is © The Royal Society of Chemistry 2011 |