Unique catalytic features of Ag nanoclusters for selective NOx reduction and green chemical reactions
Kenichi
Shimizu†
,
Kyoichi
Sawabe
and
Atsushi
Satsuma
*
Department of Molecular Design and Engineering, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: kshimizu@cat.hokudai.ac.jp; satsuma@apchem.nagoya-u.ac.jp; Fax: +81 52-789-3193; Tel: +81 52-789-4608
First published on 26th February 2011
Abstract
The size control of Ag into nanoclusters generates unique catalytic features of Ag on automotive emission control and environmentally friendly organic reactions. Hydrogen-assisted selective catalytic reduction of NO by hydrocarbons (H2–HC–SCR), which is one of the promising technologies for removal of NO in the diesel engine exhausts, results in the formation of Ag clusters on alumina or zeolites and enhancement of the conversion of NO into N2. A mechanistic study using in situUV-Vis, EXAFS, and FT/IR revealed that the promotion of partial oxidation of hydrocarbons by active oxygen species is the essential role of Ag clusters. Hydrogen is suggested to be indispensable for the formation of the Ag cluster and the active oxygen. The important role of hydride on the formation of H2O2-like active chemical species on Ag clusters was confirmed by density functional theory (DFT) calculation. Knowing the high activity of Ag nanoclusters for the formation of Ag-hydride, supported Ag clusters were applied for various organic reactions. Supported Ag clusters on appropriate metal oxide, especially Al2O3, are recyclable and show very high activity and selectivity for atom-economic reactions, i.e., alcohol dehydrogenation, C–C coupling reaction of alcohols, direct amide synthesis from alcohols and amines, N-benzylation of anilines with alcohols, and selective hydrogenation of nitroaromatics.
1. Introduction
Catalytic technology is an indispensable tool for sustainable chemical technology. There are two important roles of green and environmental catalysis. One is degradation or reduction of pollutants at “the end of pipe”, as exemplified by the catalysis for automotive emission control. Another approach is to establish a safety chemical process with less waste and energy. From both industrial and academic viewpoints, platinum-group-metal (PGM)-based catalysts, including metal particles or complexes of PGM on oxide supports and homogeneous PGM complexes, have been the most important catalysts for environmental pollution control1–4 and green production of chemicals.5–10 Theoretically, partially filled d orbitals account for the high catalytic efficiency of PGM-based catalysts.11 However, knowing that platinum-group-metal (PGM) will soon be in short supply in the near future, the high cost of PGM-based catalysts is a major barrier to worldwide spread of green cars and green chemical processes. In this context, the findings that Au nanoclusters (NCs) show similar or higher catalytic activity for various reactions than PGM catalysts12–19 have attracted much attention, because they will give a new strategy to design PGM-free catalysts. Considering the high price of gold, one may expect that silver as a less expensive group IB metal may be a better candidate for the design of PGM-free catalysts if size control and support selection are successfully undertaken. Unsupported or supported large Ag particles are commercial partial oxidation catalysts for production of ethylene oxide20 and formaldehyde.21 Recently, salts and complexes of Ag(I), as homogeneous soft Lewis acid catalysts, have been used in organic synthesis to promote the reaction involving C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds activation.22–26 A recent theoretical study27 pointed out that metallic Ag clusters may show high catalytic activity comparable to Au clusters. Although silver has been a popular element in a research field of cluster synthesis, surprisingly less attempts have been focused on catalysis of nanoparticles28–36 and NCs of Ag,37–59 compared with the research area of Au catalysis. Claus et al. reported selective hydrogenation by Ag nanoparticle catalysts.28,30 Mitsudome et al.31–35 showed that hydrotalcite- or hydroxyapatite-supported Ag nanoparticles acted as effective catalysts for green organic reactions such as oxidation of silane and deoxygenation of epoxides into alkenes. Among a few reports of Ag NCs catalysts, zeolite-supported Ag3 clusters for coupling of CH4 and C2H4 by Baba et al.,37–39hydrotalcite-supported Ag for dehydrogenation of alcohols,40 and size-selected Ag clusters for propylene epoxidation41 are successful examples. Our group has paid attention to Ag NCs catalysts for green and environmental applications.42–58 Based on the fact that the surface of a metal particle with size below a few nanometres contains a large number of low-coordinated metal atoms having high reactivity, we paid special emphasis on the catalysis of metallic Ag with a size of subnanometre to a few nanometres. This report summarizes our recent work on a size- and support-specific catalysis of Ag clusters for automotive emission control and green organic reactions. First, we show a detailed investigation of a reaction mechanism of H2–HC–SCR over subnanometre-sized Ag clusters using both experimental in situ characterizations and theoretical density functional theory (DFT) calculations. A unique size effect and an important role of Ag clusters with hydride species in oxygen activation are clarified. Next, organic reactions using a few nanometre-sized Ag cluster catalysts are demonstrated, and a design concept of Ag catalysts for green organic synthesis is discussed.
C bonds activation.22–26 A recent theoretical study27 pointed out that metallic Ag clusters may show high catalytic activity comparable to Au clusters. Although silver has been a popular element in a research field of cluster synthesis, surprisingly less attempts have been focused on catalysis of nanoparticles28–36 and NCs of Ag,37–59 compared with the research area of Au catalysis. Claus et al. reported selective hydrogenation by Ag nanoparticle catalysts.28,30 Mitsudome et al.31–35 showed that hydrotalcite- or hydroxyapatite-supported Ag nanoparticles acted as effective catalysts for green organic reactions such as oxidation of silane and deoxygenation of epoxides into alkenes. Among a few reports of Ag NCs catalysts, zeolite-supported Ag3 clusters for coupling of CH4 and C2H4 by Baba et al.,37–39hydrotalcite-supported Ag for dehydrogenation of alcohols,40 and size-selected Ag clusters for propylene epoxidation41 are successful examples. Our group has paid attention to Ag NCs catalysts for green and environmental applications.42–58 Based on the fact that the surface of a metal particle with size below a few nanometres contains a large number of low-coordinated metal atoms having high reactivity, we paid special emphasis on the catalysis of metallic Ag with a size of subnanometre to a few nanometres. This report summarizes our recent work on a size- and support-specific catalysis of Ag clusters for automotive emission control and green organic reactions. First, we show a detailed investigation of a reaction mechanism of H2–HC–SCR over subnanometre-sized Ag clusters using both experimental in situ characterizations and theoretical density functional theory (DFT) calculations. A unique size effect and an important role of Ag clusters with hydride species in oxygen activation are clarified. Next, organic reactions using a few nanometre-sized Ag cluster catalysts are demonstrated, and a design concept of Ag catalysts for green organic synthesis is discussed.
2. Role of Ag clusters in hydrogen-assisted HC–SCR
2.1 Supported Ag catalysts for selective catalytic reduction of NO by hydrocarbons (HC–SCR)
HC–SCR is thought to be a potential method for removal of NO in exhausts from diesel engine cars containing excess oxygen.1–4 After Miyadera and Yoshida reported the high activity of SCR with ethanol over Ag/Al2O3,60,61Ag/Al2O3 has attracted much attention and currently is one of the promising catalysts for HC–SCR. The merit of Ag/Al2O3 is its high HC–SCR activity when higher hydrocarbons such as octane are used as reductants.62–64 Actually, Ag/Al2O3 shows good performances in engine bench tests using secondary fuel injection as an additional reductant.65–69 Stability in hydrothermal conditions and cheaper price than precious metals are also the merit of Ag.The drawback of Ag/Al2O3 catalysts is low activity when lower alkanes are used as reducing agents. For example, the NO conversion of octane–SCR achieves 100% around 623 K, while propane cannot reduce NO below 673 K.64 However, Satokawa et al. found a breakthrough in the SCR by lower hydrocarbons, i.e., the boosting effect on HC–SCR activity by H2 addition.70,71 As shown in Fig. 1, when propane is used as a reductant, Ag/Al2O3 shows only poor activity below 673 K. When H2 is added into the reaction atmosphere, HC–SCR activity is significantly boosted. Interestingly, the hydrogen effect is reversible depending on the addition of H2. This promotion effect of H2 is also observed in Ag-ion exchanged zeolites, such as Ag-MFI, as shown in the figure. It should be noted that the added H2 does not act as a reducing agent of NO, because there is no catalytic activity of H2–SCR below 673 K. The loaded hydrogen acts as a promoter for the reaction between NO and hydrocarbons.
 | ||
Fig. 1 Effect of hydrogen addition on NO conversion to N2 over (circles) Ag/Al2O3 with 2 wt% of Ag loading and (triangles) Ag-MFI with 3.5 wt% of Ag loading (ion-exchange level is 58%). Reaction conditions: 0.1% NO, 0.1% C3H8, 10% O2, 0.5% H2, SV = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. 000 h−1. | ||
This promotion effect has been called the “H2 effect”, and re-examined by other research groups.72–79 The addition of H2 is also effective in HC–SCR by higher hydrocarbons72–74 and NH3–SCR.77–79 Burch reported that the use of octane as a reductant results in more than 90% conversion of NO in the temperature range from 473 to 773 K. The H2-assisted NH3–SCR also successfully reduces NO to N2 with 80–100% conversion in the temperature range of 473 to 773 K. The research project financed by the EU (KNOWNOX) investigated European transient cycle (ETC) tests using a full-scale vehicle equipped with a Ag/Al2O3 converter and a post injection system for Swedish MK1 diesel fuel. The research group clarified that, when hydrogen is added and diesel fuel having a low sulfur content is used, the NOx emissions out of the engine can be reduced by more than 50% in the warm-started ETC cycle and the performance meets the Euro 4 limits.76
2.2 Contribution of Ag clusters to H2-assisted HC–SCR
There are several reports about the mechanism of the “H2 effect”. So far, the following points are claimed as reasons for the promotion effect by H2.(1) Formation of Ag clusters.47–51,80,81
(2) Promotion of partial oxidation of hydrocarbons to oxygenates.44,51,53,82
(3) Decrease in nitrate self-poisoning.43
(4) Formation of H2O2-like species.83
(5) Formation of N-containing species, such as NCO and NH3, as excellent reducing agents for NO.84–86
Among them, our research group demonstrated that the factors (1) and (2) are the main reasons for the “H2 effect”, and the factor (3) supports the boosting effect. The addition of H2 results in the formation of Ag clusters, which enhances partial oxidation of hydrocarbons to surface oxygenates. The experimental results, mainly on Ag-MFI as a model catalyst of Ag/Al2O3, are shown in this part.
Fig. 2 shows diffuse reflectance UV-vis spectra of Ag-MFI quenched after various treatments. After calcination of Ag-MFI in a flow of 10% O2/He (spectrum a), the bands at 210 and 235 nm corresponding to the 4d10 to 4d95s1 transition of Ag+ ions were observed.81,86–88 After exposure to C3H8–SCR conditions without H2 (spectrum b), the bands assignable to Ag+ ions are still predominant. After the exposure to H2-assisted C3H8–SCR conditions (spectrum c), new bands at 260 and 284 nm were observed. Generally, Ag species with a larger number of Ag atoms give UV-Vis bands at a higher wavelength,88 and the bands at 260 and 284 nm are assigned to Agnδ+ (n = 4–8) clusters.47–51,77,80,81,88–90 After the evacuation of H2, the spectra recovered to the original one (isolated Ag+ species, spectrum d). It is clear that the loading of H2 results in the reversible formation of Ag clusters.
 | ||
| Fig. 2 UV–Vis spectra of Ag(58%)-MFI. The spectra were taken after (a) a flow of O2 at 773 K, and quenched after the exposure to (b) C3H8–SCR conditions without H2, (c) C3H8–SCR with 0.5% H2, (d) again C3H8–SCR without H2. Reaction conditions: 0.1% NO, 0.1% C3H8, 10% O2, 0.5% H2, 573 K. Reproduced with permission from Elsevier of ref. 49. | ||
The role of Ag clusters in the H2-assisted HC–SCR was also evaluated by the dynamic response of a UV-Vis band using an in situUV-Vis spectrometer.46 In Fig. 3, the dynamic change in the NO conversion for the C3H8–SCR at 573 K on the loading of H2 is compared with the band intensity at 350 nm assignable to Ag clusters. The NO conversion was very low (1%) in the C3H8–SCR condition (t = 0 min). After the addition of H2, NO conversion immediately increased and finally reached 68% without the formation of N2O. After the removal of H2 from the reaction atmosphere at 16.5 min, NO conversion decreased, and it reached nearly the original conversion level after 40 min. The good agreement between the HC–SCR and the band intensity of Ag clusters can rationalize the fact that the boosting effect is accompanied by the morphology change in Ag species into Ag clusters.
 | ||
| Fig. 3 Effect of hydrogen switching on/off on NO conversion and the UV-vis band height at 350 nm during C3H8–SCR over 2 wt% Ag/Al2O3 at 573 K. Conditions: 0.1% NO, 0.1% C3H8, 0% or 0.5% H2, 10% O2, catalyst weight 50 mg. Reproduced with permission from The American Chemical Society of ref. 46. | ||
It should be noted that the formation of Ag clusters depended on Ag loading. The isolated Ag+ ions are major species in 0.5 wt% Ag/Al2O3, the maximum formation of Ag clusters was observed at 2 wt% of Ag loading, and the further loading of Ag results in the formation of large Ag particles which represents a plasmon band above 500 nm.91 The catalytic activity for H2-assisted C3H8–SCR was also strongly dependent on the Ag loading. It gradually increased with Ag loading, showed the maximum at 2 wt%, and decreased with the further Ag loading. The coincidence of the activity and the Ag cluster formation also suggests that the Ag cluster is the active species for the selective NO reduction.
The structure of Ag clusters in the presence of H2 was identified by Ag K-edge EXAFS.50Table 1 shows the structural parameters derived from curve-fitting analysis of Fourier transforms of k3-weighted EXAFS of Ag-MFI and Ag/Al2O3. As shown in the table, Ag-MFI after 0.5% H2 treatment at 573 K followed by quenching and air-tight shielding shows the Ag–Ag shell with a coordination number of 3.3 and a bond distance of 2.73 Å. The coordination number around 3 with the uniform Ag–Ag distance means the formation of tetrahedral 4-membered Ag species. In the separate experiment, H2-temperature programmed reduction profiles showed two equivalent H2 consumption peaks at 420 K and above 600 K.50,51 Since the H2-treatment was carried out at 573 K, the average valence of Ag in an Ag cluster can be determined as +0.5. Therefore, the species formed in Ag-MFI under the H2-treatment can be determined as a Ag42+ cluster as an average structure. Considering a Ag–Ag distance of 2.73 Å, Ag species in Ag-MFI after H2-assisted C3H8–SCR can also be pictured as Ag42+. In the case of Ag/Al2O3, the Ag–Ag shell with a bond distance of 2.74 Å was also observed under the H2 containing atmosphere. Similar species are formed on Ag/Al2O3, though the contribution of a Ag–O shell suggests that a part of Ag is dispersed as Ag+ ions on the alumina surface.
| Sample | Treatment | Atom | CNa | R b/Å | Rfc |
|---|---|---|---|---|---|
| a Coordination number. b Shell distance. c Residual factor. d Measured after quenching and air-tight shielding.49 e In situ measured at 573 K.46 f Data from X-ray crystallography. | |||||
| Ag(58)-MFI d | 0.5% H2 at 573 K | Ag | 3.3 | 2.73 | 5.3 |
| H2–SCR at 578 K | Ag | 1.0 | 2.73 | 5.0 | |
| 2 wt% Ag/Al2Oe | O2 at 773 K | O | 3.8 | 2.48 | 2.2 |
| Ag | 0.1 | 2.74 | |||
| H2 + O2 at 573 K | O | 4.4 | 2.48 | 4.9 | |
| Ag | 0.6 | 2.74 | |||
| H2 at 573 K for 3 min | O | 4.8 | 2.48 | 4.8 | |
| Ag | 0.6 | 2.74 | |||
| Ag foilf | Ag | (12) | (2.89) | ||
| Ag2SO4f | O | (6) | (2.50) |
The mechanistic investigation by in situ FT/IR revealed that the role of Ag clusters is mainly in the promotion of hydrocarbon activation. It is well known that the HC–SCR on Ag/Al2O3 starts from the oxidation of NO to NO3− (nitrates, NO2 ad-species on a catalyst surface) and the partial oxidation of hydrocarbons to oxygenated species, such as acetate.44 The N2 production proceeds via the reaction between surface nitrates and surface oxygenated species, forming N-containing species, such as cyanide (–CN) and isocyanate (–NCO) as reaction intermediates.80,84,89,92,93 Shibata et al. clearly demonstrated that the addition of H2 significantly promoted the initial parallel oxidations.82 In the absence of H2, the rate of oxidation of NO to nitrates was 66 nmol g−1s−1, while that of C3H8 was below 0.1 nmol g−1s−1 at 473 K over Ag/Al2O3. On the other hand, in the presence of 1000 ppm of H2, the rate of oxidation of NO to nitrates significantly increased to 560 nmol g−1s−1, and that of hydrocarbon to oxygenated species by nitrates also increased to 89 nmol g−1s−1. This rate of partial oxidation to oxygenated hydrocarbons was comparable to that of gaseous NO consumption (68 nmol g−1s−1). Therefore, the role of Ag clusters is to promote the partial oxidation of hydrocarbons. The promotion of oxidation steps measured by in situ FT/IR and the detection of super oxide by ESR46 indicate that the formation of active oxygen species should contribute to the “H2 effect” on Ag clusters. Our research group also presented the similar reaction mechanism of H2-assisted C3H8–SCR by Ag-MFI, in which H2-addition accelerated C3H8 oxidation to acetate, as shown in Scheme 1.53 In this case, the formation of NH4+ plays an important role in the NOx reduction to N2. The rate determining step of the scheme is the partial oxidation of C3H8 to acetate (CH3COO−). Although the contribution of most of the species in Scheme 1 was confirmed by the kinetic study with in situ FT/IR, the chemical species contributing to the promoted oxidation of C3H8 to CH3COO− and NO to NO2 could not be experimentally identified. It can be speculated that the reductive activation of O2 to OOH species is the determining factor for the partial oxidation, which is revealed by the DFT calculation described in Section 3.
 | ||
| Scheme 1 Proposed mechanism for H2-assisted C3H8–SCR by Ag-MFI. | ||
2.3 Ag42+ clusters in MFI as model catalysts
For the investigation of the formation of Ag clusters, we performed a H2 adsorption micro-calorimetric experiment at 573 K to obtain quantitative information on the reduction of Ag+ ions in Ag-MFI.94Fig. 4 shows profiles of the heat of adsorption as a function of loaded H2. The numbers in the parenthesis in the catalyst name indicate the ion-exchange level in %. Ag-MFI showed an initial heat of H2 adsorption above 200 kJ mol−1, then sharply decreased to around 140 kJ mol−1 and very gradually decreased to around 100 kJ mol−1. The changes in the heat may be due to the surface nonuniformity or lateral interactions. Sharp drops of the heat were again observed around 0.06 mmol g−1 for Ag(58)-MFI and 0.08 mmol g−1 for Ag(83)-MFI, respectively. These points correspond to the Ag/H2 ratio of 4. According to the H2/Ag ratio, the formation of Ag clusters can be drawn as in the following equation.| 4Ag+ + H2 → Ag42+ + 2H+. |
 | ||
| Fig. 4 Differential heat of H2 adsorption on Ag(58)-MFI and Ag(83)-MFI at 573 K. The samples are calcined at 823 K before the measurement. | ||
In an oxidative atmosphere, Ag+ ions are fixed on ion-exchange sites of zeolites (Oz−). In a reductive atmosphere, such as in the presence of CH4, Baba et al. clarified the formation of silver hydride (Agn–H).37–39,95 Considering the formation of silver hydride in the presence of H2, silver hydride plays an important role in the aggregation of Ag+ ions into Ag42+ clusters, and the detailed scheme of the Ag42+ cluster formation can be drawn as in the following equation.
| 4Ag+–Oz− + 2H2 → 2Ag+–Oz− + 2Ag–H + 2H+–Oz− → Oz−–Ag42+–Oz− + 2H+–Oz− + H2. |
2.4 Debates on the role of Ag clusters
There are some debates on the role of Ag clusters in H2-assisted HC–SCR. The research group of Sazama et al. and Burch et al. claimed that the formation of Ag clusters is not essential for the enhancement of the H2-assisted HC–SCR activity. Their reasons can be summarized as follows.(1) The slower response of Ag clusters than the NOx conversion during hydrogen switching in the decane–SCR.
(2) The formation of Ag clusters can be observed under the co-feeding CO into HC–SCR, but CO does not enhance the HC–SCR activity.
The dynamic response of Ag clusters was also examined by Sazama et al. using a home-made in situUV-Vis spectrometer.75 They also observed the bands at 323 nm assignable to Agnδ+ clusters under the reaction conditions of H2–decane–SCR. The NOx conversion increased sharply after the addition of H2 into the decane–SCR at 523 K with a sharp increase in the band at 323 nm. After hydrogen was switched off, NOx conversion decreased within 4 min, but the disappearance of the band of Ag clusters took 50 min. Similar results can be seen in our paper as shown in Fig. 3. The slower response of Ag clusters than the NOx conversion is their basis of the counterargument.
As for the latter reason, Breen et al.96 reported the counterevidence using in situEXAFS for the determination of Ag species of Ag/Al2O3 under the H2-assisted HC–SCR. The change of the Ag structure by the presence of H2 was not significant. They found that the Agnδ+ (n is ca. 3) cluster was already present on the alumina support under the HC–SCR conditions without H2. The Agnδ+ cluster was also observed in the presence of CO, although CO did not boost the HC–SCR activity. They concluded that the enhanced activity in H2–HC–SCR is not attributed to structural changes but to a chemical effect.
As for the chemical effect, Sazama and Wichterlova pointed out an important role of H2O2-like species in H2-assisted HC–SCR.83Table 2 shows the effect of hydrogen and hydrogen peroxide on the decane–SCR over Ag/Al2O3 at 473 K. The promotion effect was also observed in decane–SCR with co-feeding of 0.2% of H2O2: the NO conversion significantly increased from nearly zero to 60% with the addition of H2O2. This effect is similar to that when H2 is added into the decane–SCR. They attributed this promotion effect of SCR by hydrogen peroxide to highly reactive hydroxy and hydroperoxy radicals. The higher NO2 yield was attributed to the reaction between hydroxy radicals, HO2 + NO → NO2 + OH.
The effective oxygen activation in the H2-assisted HC–SCR is reasonable, because the main role of H2 is to enhance the reaction rate of the partial oxidation of hydrocarbons to surface oxygenates, as described in Section 2.2. But the contribution of Ag clusters cannot be entirely denied. The idea that “The formation of Ag clusters is not a sufficient condition but a necessary condition” can rationalize the results ever reported as follows. If we consider that the formation of an Ag-hydride cluster results in the formation of H2O2-like active oxygen which results in the promotion of hydrocarbon oxidation, the fact (2) can be rationalized because CO does not have hydrogen. The fact (1) can be rationalized by the idea that Ag clusters having hydride are the true active sites. Although the Ag cluster without hydride is detectable in UV-Vis spectra and causes apparent longer lifetime of the Ag cluster, they are no more active species after the consumption of hydride. The detailed theoretical explanation is described in the next section.
3. Density functional theory (DFT) calculation of the H2 assisted HC–SCR
3.1 Ag4 formation by the H2 addition
In experiments for the study of the H2 promotion effect, it is not clear whether Ag clusters formed by the H2 addition are related with the HC–SCR reactivity directly or indirectly. Therefore, our group carried out the DFT calculation for the H2 promotion effect using the model system of Ag clusters on the MFI zeolite.54 One of the roles of the H2 addition is the promotion of the formation of an Ag4 cluster. Heterolytic dissociation of hydrogen on the Ag-MFI system leads to the formation of neutral AgH species. The diffusion of two AgH molecules to two neighboring Ag–Oz sites (Oz means the adsorption sites of MFI) results in the formation of an HAg4H cluster, followed by the Ag4 cluster and by the desorption of a H2 molecule. The system of H2 and Ag4–Oz is more stable by 33.3 kJ mol−1 than that of HAg4H–Oz. The proposed reaction scheme of the formation of an Ag4 cluster by the addition of hydrogen is as follows:| 4Ag–Oz + 2H2 → 2AgH + 2H–Oz + 2Ag–Oz → HAg4H–Oz + 2H–Oz → Ag4–Oz + H2 + 2H–Oz. |
 | ||
| Fig. 5 Optimized structures of the adsorbed molecules on the MFI cluster model. (a) Ag4–Oz; (b) HAg4H–Oz; (c) O2Ag4–Oz; (d) HOOAg4H–Oz. Reproduced with permission from Elsevier of ref. 54. | ||
3.2 O2 adsorption on Ag4 and HAg4H
Several experiments46,53,75 suggested that the O2activation was required for the HC–SCR and superoxide (O2−1) ions were observed in the ESR experiments.46,53 It was suggested that the H2 addition was necessary for the activation of O2. The optimized geometry of the O2 adsorption on the Ag4–Oz system is shown in Fig. 5(c). The bond distance of the adsorbed oxygen is 1.26 Å which is the same as that of the isolated oxygen. The adsorbed oxygen is neutral and its adsorption energy is 18.2 kJ mol−1. Therefore, the DFT calculation also denies the activation of oxygen by the Ag4 cluster alone.The HAg4H cluster is the intermediate for the formation of the Ag4 cluster by the addition of hydrogen. The additional presence of O2 changes the reaction path of the cluster formation. The reaction of O2 with an HAg4H cluster results in the formation of an HOOAg4H adsorbate. The system of HOOAg4H–Oz is more stable by 101.3 kJ mol−1 than that of the isolated H2 + O2 + Ag4–Oz. The optimized structure of HOOAg4H–Oz is shown in Fig. 5(d). The sum of natural charges of O1, O2 and H2 is −0.71. This means that oxygen is activated by the formation of HOO− species bound to an HAg4 cluster. Sazama and Wichterlova83 reported that H2O2 enhances substantially selective reduction of NOx to nitrogen. Therefore, the HAg4H cluster plays an important role in the SCR reaction by activating oxygen molecules.
3.3 UV absorption of Ag4 and HAg4H clusters
Fig. 6 shows the UV absorption spectra of Ag4–Oz and HAg4H–Oz simulated using the time dependent (TD) DFT calculation. The MFI model cluster alone has weak bands which have the oscillator strength (f) less than 0.01. Therefore, all strong absorption bands observed in this range involve the excitation of the electronic structure of the Ag cluster and/or the charge transfer between the cluster and the zeolite. The HAg4H–Oz system has a weak absorption band at 282 (f = 0.04) nm. On the other hand, the Ag4–Oz system has three strong absorption bands at 286 (f = 0.15), 305 (f = 0.09) and 318 (f = 0.09) nm. The first strong band and the latter two bands correspond to the bands at 260 and 284 nm observed in the UV-Vis spectra (Fig. 2), respectively. When hydrogen atoms adsorb on the Ag4 cluster, partial occupation in the unoccupied orbitals of the Ag4 cluster results in the decrease in the intensity of the absorption bands. Thus, the UV-Vis measurements hardly detect the active species of HAg4H for the HC–SCR reaction. This is consistent with the time dependence of NO conversion for the C3H8–SCR over Ag/Al2O3 as shown in Fig. 3. In this experiment, the addition of H2 immediately increased NO conversion with the presence of O2, whereas the UV bands attributed to the Ag4 cluster appeared a short time after the introduction of H2. This delay is explained by the fact that the UV bands attributed to the Ag4 cluster does not appear until H2O2 is consumed from the HOOAg4H cluster in the SCR reaction. Furthermore, it is also explained that CO unlike H2 did not enhance the SCR reaction in spite of the formation of Ag clusters promoted by the presence of CO.96,97 This is because the HAg4H adsorbate is not produced in the absence of H2. | ||
| Fig. 6 Simulation of UV spectra of (a) HAg4H–Oz and (b) Ag4–Oz. Bars in the figures are the calculated values of TD-DFT. Solid lines are simulated by overlapping Lorentz functions with the FWHM of 10 nm. Reproduced with permission from Elsevier of ref. 54. | ||
3.4 Hydride abstraction by Ag clusters
DFT calculation suggests the potential of the hydride abstraction by the Ag cluster catalysts. The net natural charges of the adsorbates of Ag4 and HAg4H are 1.72 and 1.42, respectively. Both of two hydrogens in the HAg4H are anionic and the net charge of the Ag atoms is 1.9. Therefore, the adsorbates of Ag4 and HAg4H are considered to be Ag42+ and H−Ag42+H−, respectively. This indicates that the abstraction of hydride from the reactants by the Ag42+ cluster forms the cationic species Ag42+H− which is strongly bound to the MFI by the ionic bond. Therefore, cationic Ag clusters on the oxide surfaces are expected to be a candidate for the catalysts using the hydride abstraction. The specific examples are shown in the following sections.4. Green organic reactions by Al2O3-supported Ag NCs
4.1 Catalyst preparation and characterization
Ag NCs supported on Al2O3 (typically 5 wt% Ag) were prepared by impregnating γ-AlOOH (or γ-Al2O3) with an aqueous solution of silver nitrate followed by evaporation to dryness at 120 °C. Before each catalytic or spectroscopic experiment, the precursor was calcined in air at 600 °C, followed by reduction by H2 at 300 °C. An X-ray diffraction (XRD) pattern of Ag/Al2O3 showed the absence of large Ag or Ag2O particles. A high angle annular dark field scanning TEM (HAADF-STEM) image showed Ag particles with size in the range ca. 0.8–4.2 nm.59 However, particle size determination of this sample was unsuccessful, because bright points due to Ag clusters with diameter below 1 nm disappeared during the HAADF-STEM observation possibly due to the reaction of Ag clusters with the carbonaceous residue under a strong electron beam irradiation. Claus and Hofmeister also commented on the difficulty in the electron microscopy observation of extremely small Ag clusters less than about 1 nm.28 Arve et al.98 reported complementary TEM, HAADF, O2-chemisorption experiments to study Ag particle size in Ag/Al2O3 and they emphasized that establishing a structure–reactivity relationship for this type of catalyst requires in situ characterization. On the basis of the systematic TEM and EXAFS studies on particle size determination of Pt/zeolite catalysts, Koningsberger et al.99 concluded that for the size range below 2 nm EXAFS have to be used to determine particle size. From these facts, we used in situEXAFS to determine particle size of Ag clusters on Al2O3.55In situAg K-edge XANES and EXAFS showed that the H2-reduction of pre-oxidised Ag/Al2O3 at 300 °C resulted in the reduction and aggregation of Ag+ ions to metallic Ag clusters. The Ag–Ag coordination number for 5 wt% Ag/Al2O3 (5.9) is lower than that of the bulk silver (12). Using the coordination numbers of Ag–Ag contribution in EXAFS, the average size of metallic silver species was determined to be 0.8–0.9 nm. The Ag–O EXAFS contributions with coordination number of 0.5 suggest that the Ag cluster is not fully reduced and can be present as a cationic nanocluster (Agnδ+) under the interaction with oxygen atoms at the metal surface or the metal–support interface.4.2 Oxidation of alcohols55
Oxidation of alcohols to carbonyl compounds is important topics in catalysis. Recent efforts have been devoted to transition-metal-catalyzed oxidation of alcohols using environmentally friendly oxidants such as oxygen.6–8,10,16,17 From the viewpoint of atom efficiency and safety of the reaction, an oxidant-free catalytic dehydrogenation of alcohols is more ideal, and several examples using PGM catalysts have been reported.100,101 Mitsudome et al. first developed hydrotalcite-supported Ag NCs as extremely active catalysts for the oxidant-free dehydrogenation of alcohols.40 We found that Ag/Al2O3 acts as a recyclable heterogeneous catalyst for dehydrogenation of various alcohols to carbonyl compounds and H2 (eqn (1)). | (1) |
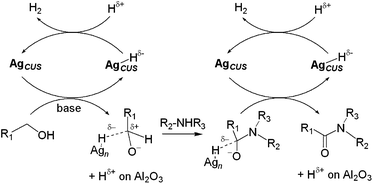
Ag/Al2O3 showed higher activity than commercial PGM catalysts (Ru/C, Ru/Al2O3, Pd/C and Pd/Al2O3). Systematic studies of structure–activity relationships showed that the small size of metallic Ag NCs and the acid–base bifunctional nature of Al2O3 are important factors which affect the catalytic activity. The reaction mechanism is investigated by kinetic studies combined with in situFTIR experiments, and the following mechanism is proposed (Scheme 2): (1) reaction of alcohol with the basic OH group of alumina to yield alkoxide on alumina and adsorbed water molecule, (2) C–H activation of alkoxide species by coordinatively unsaturated Ag atoms (AgCUS) on the silver cluster to form silver hydride species and carbonyl compounds, (3) H2 desorption promoted by an acid site of alumina. The proposed mechanism provides fundamental reasons for the higher activity of Ag NCs on an acid–base bifunctional support (Al2O3) than those on basic (MgO and CeO2) and acidic to neutral (SiO2) supports.
 | ||
| Scheme 2 Proposed mechanism for dehydrogenation of alcohol by Ag/Al2O3. Reproduced with permission from Wiley-VCH of ref. 55. | ||
4.3 Coupling of alcohols with amines to form amides and H257
Direct amidation from alcohols and amines driven by H2 removal, discovered by Milstein et al.,102,103 is of great interest as an ideal method for amide synthesis. To date, only a few homogeneous Ru catalysts with molecularly designed cooperative ligands have been reported.102–105 However, these expensive catalysts have difficulty in catalyst/product separation and the necessity of special handling of metal complexes. We reported the first example of a heterogeneous and PGM-free catalyst for this reaction (eqn (2)). | (2) |
In the presence of Ag/Al2O3 and a co-catalyst (Cs2CO3), various secondary amines were converted to the corresponding amides. Although the turnover number (TON) of the Ag catalyst is lower than Milstein's Ru catalyst, it is noteworthy that this is the first example of direct amide formation from a secondary amine and alcohol as well as the first example of heterogeneous catalysis of this direct amidation. This method tolerates various primary alcohols, including aliphatic primary alcohols (Table 3). The structure–activity relationship suggests that AgCUS sites on Agnδ+ and acid–base sites of Al2O3 are necessary for the reaction. Basic sites at the silver–support interface deprotonate alcohols to give alkoxides. AgCUS sites are required for the C–H cleavage of alkoxide and hemiaminal species. Protonic OH groups adjacent to silver sites facilitate the removal of hydride species from the silver sites to regenerate AgCUS sites (Scheme 3).
| Entry | Alcohol | Amine | Yield (%) |
|---|---|---|---|
| a Alcohol (1 mmol), amine (2 mmol), toluene (2 mL), under reflux, Ag/Al2O3-5 (4 mol%), Cs2CO3 (20 mol%), t = 24 h. b In o-xylene under reflux conditions. c Yield of the corresponding ester. | |||
| 1 | Benzylalcohol | Piperidine | 82 |
| 2 | Benzylalcohol | Pyrrolidine | 92 |
| 3 | Benzylalcohol | N-Methyl benzylamine | 78 (10c) |
| 4 | Benzylalcohol | Morpholin | 93 |
| 5 | 4-Fluorobenzylalcohol | Pyrrolidine | 79 |
| 6 | 4-Methoxybenzyl alcohol | Morpholin | 93 |
| 7 | 4-Methylbenzylalcohol | Morpholin | 84 |
| 8 | 4-Fluorobenzylalcohol | Morpholin | 91 |
| 9b | 1-Hexanol | Morpholin | 83 |
| 10b | 1-Octanol | Morpholin | 71 |
| 11b | Cyclohexanemethanol | Morpholin | 67 (12c) |
 | ||
| Scheme 3 Proposed mechanism for direct amidation of amines by alcohols. Reproduced with permission from Wiley-VCH of ref. 57. | ||
4.4 N-Alkylation of anilines with alcohols58
Amines are intermediates and products of enormous importance for chemical and life science applications. A well-known method for the preparation of N-alkylamines is the reaction of amines with alkyl halides or similar alkylating agents. However, there can be problems with the toxicity of such alkylating agents and control of mono-alkylation can be problematic. We found that, in the presence of a Lewis acidic co-catalyst (polyvalent metal salts such as FeCl3·6H2O), Ag/Al2O3 catalyzed the direct N-alkylation of anilines with benzyl alcohols driven by the oxidation/imination/reduction sequence, the so-called “borrowing hydrogen strategy”106–114 (eqn (3)). | (3) |
In the absence of a Lewis acid, a proton and a hydride reacted to yield H2, and an equimolar amount of imine was produced as a by-product. A Lewis acid (Fe3+ cation) increases the positive charge of immonium cation intermediates, which promotes hydride transfer to an immonium cation to produce the main product. There were some reports on heterogeneous catalysts for direct N-alkylation of amines with alcohols.106–111 For N-alkylation of aniline with benzyl alcohol, TON of Ag/Al2O3 (24) is comparable to that of Ru(OH)x/Al2O3 (19).106
4.5 C–C cross-coupling reaction of alcohols56
The construction of C–C bonds is a fundamental reaction in organic synthesis. Alcohols are not used as starting materials due to the poor leaving group ability of hydroxide. The coupling of enolate derivatives with alkyl halides, as one of the most conventional methods, suffers great disadvantages such as the use of strong bases, the large amount of weight lost with the leaving group of the alkylating agent, thus decreasing the atom efficiency of the process, as well as the problem of generating waste. There are some reports of C–C bonds formation from alcohols based on the PGM-catalyzed borrowing hydrogen strategy.113–118 In the presence of a catalytic amount of Cs2CO3, Ag/Al2O3 acts as a heterogeneous catalyst for the one-pot C–C cross-coupling reaction of various secondary and primary alcohols to give coupled ketones (eqn (4)). | (4) |
This catalyst shows higher activity than Al2O3-supported PGM catalysts. Good yield (74%) was obtained with a small amount of Ag/Al2O3 (0.092 mol%) at 145 °C, and the total TON based on the total Ag content was 800. This value is larger than those for the related reaction (β-alkylation of 1-phenylethanol with benzylalcohol) by homogeneous PGM catalysts (TON = 164 for Cp*Ir,118 TON = 48 for RuCl2(DMSO)4).116
Mechanistic studies indicate that the reaction proceeds via the silver-catalyzed dehydrogenation of alcohols to give aldehyde, ketone and AgCUS–H intermediates, and the electrophilic aldehydes undergo the Cs2CO3-catalyzed aldol reaction with the ketone to give the corresponding α,β-unsaturated ketone, which finally is reduced by AgCUS–H (Scheme 4).
 | ||
| Scheme 4 Proposed mechanism for cross-coupling of secondary and primary alcohols. Reproduced with permission from Wiley-VCH of ref. 56. | ||
4.6 Selective hydrogenation of nitroaromatics59
It is well known that hydrogen interacts only very weakly with extended silver surfaces (single crystals, polycrystalline surfaces), and dissociative chemisorption of H2 does not occur at low temperature.119 This is attributed to the completely filled d-band of silver as well as the position of the d-band center relative to the Fermi level.11 However, Claus et al. first reported excellent selectivity of Ag nanoparticle catalysts for the hydrogenation of a CO group in the presence of a CC bond.28,30 They confirmed experimentally that dissociation of H2 occurs on their catalyst by H2/D2 exchange.30,120 For the selective hydrogenation of crotonaldehyde by TiO2- or SiO2-supported silver particles with a size of 1 to 7 nm, the larger silver particles gave higher selectivity to the desired product, unsaturated alcohol. Bao et al. reported that Ag/SiO2 with a Ag nanoparticle size of 7–9 nm catalyzed the selective hydrogenation of chloronitrobenzenes to their corresponding chloroanilines.29
Selective hydrogenation of a nitro group in the presence of other reducible functional groups (such as olefinic groups) is an important reaction to produce functionalized anilines as industrial intermediates for a variety of specific and fine chemicals.121Catalytic hydrogenation with conventional platinum-group metal catalysts simultaneously hydrogenate both the nitro and olefinic functions. Only a few catalysts, including Au/TiO2,18 succeeded in the selective hydrogenation of nitrostyrene. We found that silver clusters on Al2O3 selectively catalyze the hydrogenation of the nitro group in the presence of CC, CO, or CN groups. For the selective reduction of 4-nitrostyrene to 4-aminostyrene, Ag/Al2O3 showed 96% selectivity of 4-aminostyrene at 100% conversion. The reaction rate and selectivity of Ag/Al2O3–0.9 were close to those of Au/TiO2 supplied from the World Gold Council.
 | (5) |
The Ag particle with smaller size gives higher turnover frequency (TOF), suggesting an important role of AgCUS sites of Agnδ+. The support with basic character (CeO2 and MgO) and that with acidic character (SnO2 and WO3) resulted in lower activity than Al2O3 (result not shown), suggesting that both acidic and basic surface sites are necessary for this reaction. Mechanistic and structural studies showed that the reaction occurs through the consecutive route via a hydroxylamine intermediate, and a cooperation of AgCUS sites and adjacent acid–base pair sites of Al2O3 are responsible for the rate-limiting H2 dissociation step. The basic site also acts as an adsorption site of the NO2-group (Scheme 5).
 | ||
| Scheme 5 Proposed mechanism for selective hydrogenation of nitrostyrene. Reproduced with permission from Elsevier of ref. 59. | ||
We recently found that Au NCs on Al2O3 catalyzed selective reduction of nitrostyrene and found that small Au NCs (2.5 nm) on the acid–base bifunctional support (Al2O3) gave the highest activity.19
4.7 Design concept of co-operative Ag catalysts
It is interesting to note that the structure–activity relationship for alcohol dehydrogenation (eqn (1)) and hydrogenation (eqn (5)) reactions showed similar tendencies, suggesting that an active site with a similar nature is responsible for these C–H and H–H activation reactions. The results summarized in Fig. 7A and B show that the same trends are also observed in one-pot C–C and C–N bond formation reactions (eqn (2)–(4)); Ag NCs with smaller size and acid–base bifunctional nature of the support oxide are preferable. In the research area of organometallic catalysis, attentions have been focused on the cooperation of an acidic or basic ligand in transition metal complex catalyzed organic reactions.100–103,122–125 One of the successful examples is Noyori's Ru catalyst for the hydrogenation of polar bonds, in which cooperation of an acidic hydrogen on an amido ligand with ruthenium hydride plays an important role.123 Milstein's Ru PNP pincer complex, which is effective for various reactions including dehydrogenation of alcohols and amide synthesis from amines and alcohols, possesses a cooperative basic site in a β position to the metal which acts as a co-catalyst.101–103 Taking into account the fact that acidic and basic sites are located close to each other on the surface of γ-Al2O3,126,127 our results suggest that cooperation between AgCUS sites of Ag NCs and acid–base pair sites at the metal–support interface is a key concept for the design of Ag NCs catalysts for dehydrogenation and hydrogenation reactions. This implies that the coordinatively unsaturated PGM center and an adjacent bi-functional ligand in an organometallic catalyst could be replaced by corner and/or edge sites of non-PGM clusters and acid–base sites of inorganic support materials. | ||
| Fig. 7 (A) Turnover frequency (TOF) per surface Ag site versusAg particle size of Ag/Al2O3 (Ag = 1–50 wt%), (B) reaction rates of supported Ag (5 wt%) catalystsversus acidity of supports, and (C) product yields for Al2O3-supported metal (1 wt%) catalystsversus the d-band center relative to the Fermi energy (EF):11 (●) direct amidation from amine and primary alcohol (eqn 2), (△) N-benzylation of aniline (eqn (3)), and (□) C–C cross-coupling of secondary alcohols with primary alcohols (eqn (4)). Catalytic data are re-plotted from ref. 56–58. | ||
To discuss the reason why Ag shows higher activity than other metal species, products yields for various reactions (eqn (2)–(4)) by Al2O3-supported metal catalysts are plotted in Fig. 7C as a function of the d-band center relative to the Fermi energy (EF) for the clean metal surface (Hammer–Nørskov model).11 Taking into account a frequently observed tendency that the further the d-band center is from EF the weaker is the bond of hydrogen to the transition metal,128 the result indicates that the weaker metal–hydrogen bond results in the higher yields for desired products. This suggests the following explanation for higher catalytic efficiency of the Ag catalyst. The less stable metal–hydrogen bond of the Ag catalyst, compared to Pt, Pd, and Au, results in the higher rate of hydride transfer to unsaturated intermediates in reactions (3) and (4), which leads to the high activity and selectivity. For the dehydrogenative reaction (2), the weaker metal–hydrogen bond leads to the higher rate of hydrogen removal from metal sites to regenerate coordinatively unsaturated metal sites.
5. Conclusions
Unique catalytic features of nano- and subnano-sized Ag clusters in selective catalytic reduction of NO by hydrocarbons (HC–SCR) and greener chemical reactions based on oxidant-free selective dehydrogenation were summarized. The HC–SCR activity of Ag/alumina can be significantly boosted by the addition of hydrogen into the reaction atmospheres. By means of in situ measurements of UV-Vis, FT/IR, and EXAFS with the aid of DFT calculation, the important role and reaction mechanism of Ag clusters in the hydrogen-assisted HC–SCR are clarified. The formation and the higher catalytic activity of Ag clusters were related to formation of Ag-hydride and that of hydrogen peroxide-like species on Ag clusters, respectively. The proposed mechanism can be rationalized by various proposals, a part of which has been under debate, about the active species of H2-assisted HC–SCR. Knowing this unique catalytic behaviour of Ag clusters, a Ag cluster supported on alumina was applied as a heterogeneous recyclable catalyst for green chemical reactions, such as oxidant-free dehydrogenation of alcohols, direct C–C cross-coupling of alcohols, direct dehydrogenative amide synthesis, and selective hydrogenation. It was proposed that cooperation between AgCUS sites and acid–base pair sites at a metal–support interface is a key concept for the design of Ag cluster catalysts for these organic reactions.Acknowledgements
A part of this study was supported by the Grant-in-Aid from the Ministry of Education, Science, Sports and Culture in Japan.Notes and references
- S. Matsumoto, Catal. Today, 2004, 90, 183–190 CrossRef CAS.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal., B, 2002, 39, 283–303 CrossRef CAS.
- H. Shinjoh, Catal. Surv. Asia, 2009, 13, 184–190 CrossRef CAS.
- M. Bowker, Chem. Soc. Rev., 2008, 37, 2204–2211 RSC.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- K. Kaneda, K. Ebitani, T. Mizugaki and K. Mori, Bull. Chem. Soc. Jpn., 2006, 79, 981–1016 CrossRef CAS.
- T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem.–Asian J., 2008, 3, 196–214 CrossRef CAS.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC.
- K. Yamaguchi and N. Mizuno, Synlett, 2010, 2365–2382 CAS.
- B. Hammer and J. K. Nørskov, Adv. Catal., 45, 71–129 Search PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–406 CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef.
- G. J. Hutchings, Chem. Commun., 2008, 1148–1164 RSC.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- C. D. Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS.
- K. Shimizu, Y. Miyamoto, T. Kawasaki, T. Tanji, Y. Tai and A. Satsuma, J. Phys. Chem. C, 2009, 113, 17803–17810 CrossRef CAS.
- R. A. Van Santen and H. P. C. E. Kuipers, Adv. Catal., 35, 265–321 Search PubMed.
- I. E. Wachs, Surf. Sci., 2003, 544, 1–4 CrossRef CAS.
- M. Naodovic and H. Yamamoto, Chem. Rev., 2008, 108, 3132–3181 CrossRef CAS.
- J. M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149–3173 CrossRef CAS.
- M. Álvarez-Corral, M. Munoz-Dorado and I. Rodriguez-Garcia, Chem. Rev., 2008, 108, 3174–3198 CrossRef CAS.
- Y. Yamamoto, Chem. Rev., 2008, 108, 3199–3222 CrossRef CAS.
- C. Wei, Z. Li and C.-J. Li, Org. Lett., 2003, 5, 4473–4475 CrossRef CAS.
- T. Jiang, D. J. Mowbray, S. Dobrin, H. Falsig, B. Hvolbæk, T. Bligaard and J. K. Nørskov, J. Phys. Chem. C, 2009, 113, 10548–10553 CrossRef CAS.
- P. Claus and H. Hofmeister, J. Phys. Chem. B, 1999, 103, 2766–2775 CrossRef CAS.
- Y. Chen, C. Wang, H. Liu, J. Qiu and X. Bao, Chem. Commun., 2005, 5298–5300 RSC.
- M. Bron, D. Teschner, A. Knop-Gericke, F. C. Jentoft, J. Kröhnert, J. Hohmeyer, C. Volckmar, B. Steinhauer, R. Schlögl and P. Claus, Phys. Chem. Chem. Phys., 2007, 9, 3559–3569 RSC.
- T. Mitsudome, S. Arita, H. Mori, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 7938–7940 CrossRef CAS.
- T. Mitsudome, Y. Mikami, H. Mori, S. Arita, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 3258–3260 RSC.
- Y. Mikami, A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Lett., 2010, 39, 223–225 CrossRef CAS.
- T. Mitsudome, A. Noujima, Y. Mikami, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2010, 49, 5545–5548 CrossRef CAS.
- Y. Mikami, A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Tetrahedron Lett., 2010, 51, 5466–5468 CrossRef CAS.
- H. Cong, C. F. Becker, S. J. Elliott, M. W. Grinstaff and J. A. Porco, Jr., J. Am. Chem. Soc., 2010, 132, 7514–7518 CrossRef CAS.
- T. Baba, Y. Tohjo, T. Takahashi and H. Sawada, Catal. Today, 2001, 66, 81–89 CrossRef CAS.
- T. Baba, H. Sawada, T. Takahashi and M. Abe, Appl. Catal., A, 2002, 231, 55–63 CrossRef CAS.
- T. Baba, Y. Iwase, K. Inazu, D. Masih and A. Matsumoto, Microporous Mesoporous Mater., 2007, 101, 142–147 CrossRef CAS.
- T. Mitsudome, S. Arita, H. Mori, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 138–141 CrossRef CAS.
- Y. Lei, F. Mehmood, S. Lee, J. P. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlögl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef CAS.
- K. Shimizu, T. Higashimata, M. Tsuzuki and A. Satsuma, J. Catal., 2006, 239, 117–124 CrossRef CAS.
- K. Shimizu, J. Shibata and A. Satsuma, J. Catal., 2006, 239, 402–409 CrossRef CAS.
- K. Shimizu and A. Satsuma, Phys. Chem. Chem. Phys., 2006, 8, 2677–2695 RSC.
- K. Shimizu, M. Tsuzuki and A. Satsuma, Appl. Catal., B, 2007, 71, 80–84 CrossRef CAS.
- K. Shimizu, M. Tsuzuki, K. Kato, S. Yokota, K. Okumura and A. Satsuma, J. Phys. Chem. C, 2007, 111, 950–959 CrossRef CAS.
- K. Shimizu and A. Satsuma, J. Phys. Chem. C, 2007, 111, 2259–2264 CrossRef CAS.
- K. Shimizu, M. Hashimoto, J. Shibata, T. Hattori and A. Satsuma, Catal. Today, 2007, 126, 266–271 CrossRef CAS.
- J. Shibata, Y. Takada, A. Shichi, S. Satokawa, A. Satsuma and T. Hattori, J. Catal., 2004, 222, 368–376 CrossRef CAS.
- J. Shibata, K. Shimizu, Y. Takada, A. Shichi, H. Yoshida, S. Satokawa, A. Satsuma and T. Hattori, J. Catal., 2004, 227, 367–374 CrossRef CAS.
- A. Satsuma, J. Shibata, K. Shimizu and T. Hattori, Catal. Surv. Asia, 2005, 9, 75–85 CrossRef CAS.
- K. Shimizu, K. Sugino, K. Kato, S. Yokota, K. Okumura and A. Satsuma, J. Phys. Chem. C, 2007, 111, 1683–1688 CrossRef CAS.
- K. Shimizu, K. Sugino, K. Kato, S. Yokota, K. Okumura and A. Satsuma, J. Phys. Chem. C, 2007, 111, 6481–6487 CrossRef CAS.
- K. Sawabe, T. Hiro, K. Shimizu and A. Satsuma, Catal. Today, 2010, 153, 90–94 CrossRef CAS.
- K. Shimizu, K. Sugino and A. Satsuma, Chem.–Eur. J., 2009, 15, 2341–2351 CrossRef CAS.
- K. Shimizu, R. Sato and A. Satsuma, Angew. Chem., Int. Ed., 2009, 48, 3982–3986 CrossRef CAS.
- K. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977–9980 CrossRef CAS.
- K. Shimizu, M. Nishimura and A. Satsuma, ChemCatChem, 2009, 1, 497–503 Search PubMed.
- K. Shimizu, Y. Miyamoto and A. Satsuma, J. Catal., 2010, 270, 86–94 CrossRef CAS.
- T. Miyadera and K. Yoshida, Chem. Lett., 1993, 1483–1484 CAS.
- T. Miyadera, Appl. Catal., B, 1993, 2, 199–205 CrossRef CAS.
- K. Shimizu, A. Satsuma and T. Hattori, Appl. Catal., B, 2000, 25, 239–247 CrossRef CAS.
- K. Shimizu, J. Shibata, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2001, 3, 880–896 RSC.
- K. Shimizu, J. Shibata, H. Yoshida, A. Satsuma and T. Hattori, Appl. Catal., B, 2001, 30, 151–162 CrossRef CAS.
- T. Kikuchi and M. Kumagai, Sekiyu Gakkaishi, 1998, 41, 173–174 Search PubMed.
- T. Nakatsuji, R. Yasukawa, K. Tabata, K. Ueda and M. Niwa, Appl. Catal., B, 1998, 17, 333–345 CrossRef CAS.
- K. Eränen, L.-E. Lindfors, A. Niemi, P. Elfving, L. Cider, SAE paper, 2000-01-2813.
- L.-E. Lindfors, K. Eranen, F. Klingstedt and D. Yu. Murzin, Top. Catal., 2004, 28, 185–189 CrossRef CAS.
- J. F. Thomas, S. A. Lewis, G. B. Bunting, J. M. Storey, R. L. Graves, P. W. Park, SAE paper, 2005-01-1082.
- S. Satokawa, Chem. Lett., 2000, 294–295 CAS.
- S. Satokawa, J. Shibata, K. Shimizu, A. Satsuma and T. Hattori, Appl. Catal., B, 2003, 42, 179–186 CrossRef CAS.
- R. Burch, J. P. Breen, C. J. Hill, B. Krutzsch, B. Konrad, E. Jobson, L. Cider, K. Eranen, F. Klingstedt and L.-E. Lindfors, Top. Catal., 2004, 30/31, 19–25 CrossRef.
- R. Burch, Catal. Rev. Sci. Eng., 2004, 46, 271–333 CrossRef CAS.
- J. P. Breen and R. Burch, Top. Catal., 2006, 39, 53–58 CrossRef CAS.
- P. Sazama, L. Čapek, H. Drobná, Z. Sobalík, J. Ddeček, K. Arve and B. Wichterlová, J. Catal., 2005, 232, 302–317 CrossRef CAS.
- F. Klingstedt, K. Arve, K. Eränen and D. Y. Murzin, Acc. Chem. Res., 2006, 39, 273–282 CrossRef CAS.
- M. Richter, U. Bentrup, R. Eckelt, M. Schneider, M.-M. Pohl and R. Fricke, Appl. Catal., B, 2004, 51, 261–274 CrossRef CAS.
- M. Richter, R. Fricke and R. Eckelt, Catal. Lett., 2004, 94, 115–118 CrossRef CAS.
- E. V. Kondratenko, V. A. Kondratenko, M. Richter and R. Fricke, J. Catal., 2006, 239, 23–33 CrossRef CAS.
- K. Sato, T. Yoshinari, K. Kintaichi, M. Haneda and H. Hamada, Appl. Catal., B, 2003, 44, 67–78 CrossRef CAS.
- S. T. Korhonen, A. M. Beale, M. A. Newton and B. M. Weckhuysen, J. Phys. Chem. C, 2011, 115, 885–896 CrossRef CAS.
- J. Shibata, K. Shimizu, S. Satokawa, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2003, 5, 2154–2160 RSC.
- P. Sazama and B. Wichterlova, Chem. Commun., 2005, 4810 RSC.
- S. Chansai, R. Burch, C. Hardacre, J. Breen and F. Meunier, J. Catal., 2010, 276, 49–55 CrossRef CAS.
- F. Thibault-Starzyk, E. Seguin, S. Thomas, M. Daturi, H. Arnolds and D. A. King, Science, 2009, 324, 1048–1051 CrossRef CAS.
- (a) S. Tamm, H. H. Ingelsten and A. E. C. Palmqvist, J. Catal., 2008, 255, 304–312 CrossRef CAS.
- Z. M. Wang, M. Yamaguchi, I. Goto and M. Kumagai, Phys. Chem. Chem. Phys., 2000, 2, 3007–3015 RSC.
- C. Chi, M. Cheng, Z. Qu and X. Bao, Appl. Catal., B, 2004, 51, 171–181 CrossRef CAS.
- H. He, J. Wang, Q. Feng, Y. Yu and K. Yoshida, Appl. Catal., B, 2003, 46, 365–370 CrossRef CAS.
- B. Wicherlova, Top. Catal., 2004, 28, 131–140 CrossRef.
- S. Arsalane, M. Ziyad, G. Coudurier and J. C. Védrine, J. Catal., 1996, 159, 162–169 CrossRef CAS.
- S. Sumiya, M. Saito, H. He, Q. C. Feng, N. Takezawa and K. Yoshida, Catal. Lett., 1998, 50, 87–91 CrossRef CAS.
- A. Abe, N. Aoyama, S. Sumiya, N. Kakuta and K. Yoshida, Catal. Lett., 1998, 51, 5–9 CrossRef CAS.
- K. Shimizu, T. Hiro, K. Sawabe, A. Satsuma, unpublished data.
- A. Ausavasukhi, A. Suwannaran, J. Limtrakul and T. Sooknoi, Appl. Catal., A, 2008, 345, 89–96 CrossRef CAS.
- J. P. Breen, R. Burch, C. Hardacre and C. J. Hill, J. Phys. Chem. B, 2005, 109, 4805–4807 CrossRef CAS.
- B. Wichterlová, P. Sazama, J. Breen, R. Burch, C. Hill, L. Čapek and Z. Sobalík, J. Catal., 2005, 235, 195–200 CrossRef CAS.
- K. Arve, K. Svennerberg, F. Klingstedt, K. Eraänen, L. R. Wallenberg, J.-O. Bovin, L. Čapek and D. Yu. Murzin, J. Phys. Chem. B, 2006, 110, 420–427 CrossRef CAS.
- J. de Graaf, A. J. van Dillen, K. P. de Jong and D. C. Koningsberger, J. Catal., 2001, 203, 307–321 CrossRef CAS.
- S. Arita, T. Koike, Y. Kayaki and T. Ikariya, Chem.–Asian J., 2008, 3, 1479–1485 CrossRef CAS.
- J. Zhang, M. Gandelman, L. J. W. Shimon, H. Rozenberg and D. Milstein, Organometallics, 2004, 23, 4026–4033 CrossRef CAS.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS.
- D. Milstein, Top. Catal., 2010, 53, 915–923 CrossRef CAS.
- L. U. Nordstrom, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef CAS.
- J. Zhang, M. Senthilkumar, S. C. Ghosh and S. H. Hong, Angew. Chem., Int. Ed., 2010, 49, 6391–6395 CrossRef CAS.
- J. W. Kim, K. Yamaguchi and N. Mizuno, J. Catal., 2009, 263, 205–208 CrossRef CAS.
- J. W. Kim, K. Yamaguchi and N. Mizuno, Chem. Lett., 2010, 39, 1182–1183 CrossRef.
- T. Ishida, N. Kawakita, T. Akita and M. Haruta, Gold Bull., 2009, 42, 267–274 CAS.
- A. Corma, T. Ródenas and M. J. Sabater, Chem.–Eur. J., 2010, 16, 254–260 CrossRef CAS.
- L. He, X. Lou, J. Ni, Y. Liu, Y. Cao, H. He and K. Fan, Chem.–Eur. J., 2010, 16, 13965–13969 CrossRef CAS.
- R. Martínez, D. J. Ramón and M. Yus, Org. Biomol. Chem., 2009, 7, 2176–2181 RSC.
- G. Guillena, D. J. Ramn and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS.
- T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC.
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS.
- G. Guillena, D. J. Ramn and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 CrossRef CAS.
- R. Martinez, D. J. Ramon and M. Yus, Tetrahedron, 2006, 62, 8982–8987 CrossRef CAS.
- K. Taguchi, H. Nakagawa, T. Hirabayashi, S. Sakaguchi and Y. Ishii, J. Am. Chem. Soc., 2004, 126, 72–73 CrossRef CAS; K. Motokura, D. Nishimura, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 5662–5663 CrossRef CAS.
- K.-i. Fujita, C. Asai, T. Yamaguchi, F. Hanasaka and R. Yamaguchi, Org. Lett., 2005, 7, 4017–4019 CrossRef CAS.
- A. Montoya, A. Schlunke and B. S. Haynes, J. Phys. Chem. B, 2006, 110, 17145–17158 CrossRef CAS.
- J. Hohmeyer, E. V. Kondratenko, M. Bron, J. Kroehnert, F. C. Jentoft, R. Schloegl and P. Claus, J. Catal., 2010, 269, 5–14 CrossRef CAS.
- H. U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 Search PubMed.
- Y. Shvo, D. Czarkie and Y. Rahamim, J. Am. Chem. Soc., 1986, 108, 7400–7402 CrossRef CAS.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS.
- M. Ito and T. Ikariya, Chem. Commun., 2007, 5134–5142 RSC.
- T. Zweifel, J. Naubron and H. Grutzmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54–68 CrossRef CAS.
- C. Morterra and G. Magnacca, Catal. Today, 1996, 27, 497–532 CrossRef CAS.
- V. Pallassana, M. Neurock, L. B. Hansen, B. Hammer and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 6146–6154 CrossRef CAS.
Footnote |
| † Present address: Hokkaido University Catalysis Research Center Kita21, Nishi10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan. Tel: +81 11 716 9104; fax: +81 11 706 9110. |
| This journal is © The Royal Society of Chemistry 2011 |