Carbene insertion into transition metal–carbon bonds: a new tool for catalytic C–C bond formation
Nicole M. G.
Franssen†
ab,
Annemarie J. C.
Walters†
a,
Joost N. H.
Reek
a and
Bas
de Bruin
*a
aVan't Hoff Institute for Molecular Sciences (HIMS), Department of Homogeneous and Supramolecular Catalysis, Universiteit van Amsterdam, P. O. Box 94720, 1090 GS Amsterdam, The Netherlands. E-mail: B.deBruin@uva.nl; Fax: +31 20 525 5604; Tel: +31 20 525 6495
bDutch Polymer Institute DPI, P. O. Box 902, 5600 AX Eindhoven, The Netherlands
First published on 10th February 2011
Abstract
In this perspective we highlight the applicability of migratory carbene insertion reactions into TM–C bonds as a new tool for catalytic C–C bond formation. In Section 1 we introduce the reaction, wherein we also discuss the applicability of transition metal carbene formation from reactive carbene precursors. In Section 2 we summarise the available mechanistic information about this elementary step derived from stoichiometric model reactions. In Section 3 we review the available catalytic examples, with a focus on new developments in palladium mediated cross-coupling reactions (thus expanding the substrate scope with carbene precursors) and carbene polymerisation (allowing the synthesis of highly functionalised stereoregular polymers that are difficult to prepare otherwise). Recent developments in these fields in combination with the close analogy of carbene insertion reactions with CO (and alkene) insertions open up new possibilities for the development of interesting new reactions based on carbene insertions.
1. Introduction
Migratory insertion reactions are important bond forming elementary steps, and in particular the reactions involving CO and alkene insertions into transition metal–carbon (TM–C) bonds are ubiquitous in organometallic catalysis as a tool for C–C bond formation. The reaction generally involves the migration of an anionic alkyl- or aryl ligand to a cisoidal π-coordinated neutral ligand, thus forming a new anionic ligand (Scheme 1). Alkene and CO migratory insertions into TM–C bonds are key elementary steps in, among others, the Monsanto process, olefin oligomerisation and (co)polymerisation, olefin/CO copolymerisation, hydroformylation and Heck-type reactions.1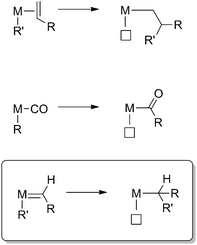
Migratory insertion of an alkene fragment leads to introduction of two additional carbons (C2 unit) to organic fragments, while insertion of CO has been widely exploited as a valuable and powerful method for the introduction of C1 fragments. The analogous insertion of carbenes as a tool for the introduction of functionalised C1 fragments has so far received comparably less attention (Scheme 1). Nonetheless, this elementary step offers ample opportunities in catalysis and has received quite some recent interest in this light. The purpose of this perspective is to give an overview of the field of carbene insertions, focusing on the recent and interesting catalytic developments.
Metal–carbene species can be easily generated from diazo compounds2 and other carbene precursors (e.g. sulfoxonium ylides3 and α,α-dihalocarbons). Diazo compounds are excellent and widely applied carbene precursors due to their ease of preparation and their relative ‘green’ character: the only byproduct upon carbene generation is dinitrogen, while other carbene precursors give rise to formation of byproducts that are not so easily removed from the reaction mixture (e.g. dmso).4,5
The large scale applicability of carbene precursors is somewhat limited in the substrate scope due to some potential safety hazards associated with some diazo compounds. Many diazo compounds are toxic, and some of them are even potentially explosive under certain conditions.6 Especially aliphatic diazoalkanes are very unstable and require handling at low temperatures and low concentrations. Diazocarbonyl compounds (diazoketones or diazoesters), on the other hand, are much more stable and quite safe to work with. Hence they are frequently applied as carbene precursors in several organic transformations, even in large scale industrial reactions. They are excellent substrates for the introduction of highly-functionalised carbon units, since a wide variety of substituted diazocarbonyl compounds is readily available via well-defined synthesis routes developed in the past decades.7 Because the handling of aliphatic carbene precursors is more problematic, much recent effort has been put into the development of safer alternative methods that allow either in situ generation of the inherently unstable aliphaticdiazo compounds (for a review see Fulton et al.8) or avoid the use of diazo compounds entirely (for a review see e.g. Müller9). The availability of these alternative methods to generate (aliphatic) carbenes at transition metals will certainly stimulate further research into the use of carbenes in (catalytic) organic transformations in general. In this light, migratory carbene insertion reactions into TM−C bonds offer valuable new tools to make new C–C bonds, and nowadays we have a large potential substrate scope of several carbene precursors available to develop such new reactivity.
In the present perspective, we give an overview of recent advances in the field of TM catalyzed insertions of carbenes into metal–carbon bonds, giving rise to the formation of new C–C bonds. Catalytic C–C bond formation can also be achieved by carbene insertions into C–H bonds, but this field has already been reviewed by Doyle et al.10 and is beyond the scope of this perspective. We will start with an overview of mechanistic information available from stoichiometric model reactions before we focus on the noteworthy catalytic developments. The migratory carbene insertion elementary step offers interesting new opportunities to enlarge the substrate scope in catalytic Pd-catalyzed cross-coupling reactions. Carbene insertion reactions are also attracting quite some interest over the past years as a tool for the synthesis of highly functionalised polymers which are not accessible via traditional olefin polymerisation routes. The reaction likely also plays an important role in the Fischer–Tropsch reaction.
2. Mechanistic insights from stoichiometric model reactions
Migratory carbene insertions into metal–carbon bonds represent the common theme in this perspective, and play an essential role in several catalytic processes ranging from a variety of Pd-mediated cross-coupling reactions involving diazo compounds (Section 3.1), C1 polymerisation (Sections 3.2.1 and 3.2.2) and Fischer–Tropsch synthesis (Section 3.2.3). This elementary step is at the same time difficult to study. The reaction is often very fast, and hence the cis-carbene–alkyl intermediate is difficult to detect and characterise. However, some model studies focused on slowing-down the reaction in order to detect the carbene intermediate and in the next section we will discuss the mechanistic information available from experimental model studies. The insertion of carbenes into M–H bonds will not be considered here. Migratory insertions into metal–hydride bonds are generally easier than insertions into metal–carbon bonds, and we consider this beyond the scope of the current perspective.11Noteworthy for developing mechanistic insights are the reactions studied by Thorn and Tulip.12,13 They studied the formation of RhIII and IrIIIcis-carbene–allyl species (generated by alcohol elimination from M–CH2–OR species) in the presence of pyridine (2 in Scheme 2).12
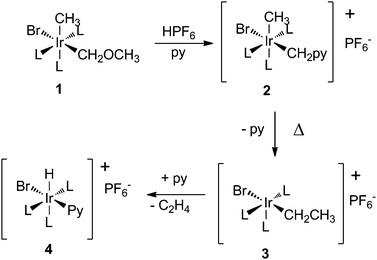 | ||
| Scheme 2 Migratory insertion of a ‘hidden carbene’ into an Ir–C bond. | ||
The intermediate methylene complex is trapped by pyridine. The pyridine adduct is stable at room temperature, but if the temperature is raised to 60 °C an Ir–hydride complex is formed by pyridine loss, methyl migration to the Ir–carbene and subsequent β-hydride elimination with liberation of ethene. A similar reaction occurred when BrCH2OMe was reacted with the Ir-starting material, and the ethyl intermediate formed before β-hydride elimination was isolated in this case. The same end-products were obtained for the analogous RhIII complex, but no intermediates were detected.13
Mechanistic insights into the carbene insertion process were also gained by studying various Pt–alkyl/Pt–aryl complexes14–17 and mixed Pt–(alkyl)X (X = halide) complexes. Exposing various Pt complexes bearing chiral diphosphine ligands [(chiral diphosphine)PtX(Me)] to different diazo compounds in polar solvents leads to mainly the Pt–C inserted products, although for these complexes there is a competition between Pt–C and Pt–X insertion (Scheme 3). The chemoselectivity can be tuned towards Pt–C insertion by increasing the polarity of the solvent and by varying the halides towards better leaving halides (chemoselectivity I < Br < Cl). These observations led to a proposed mechanism involving cationic Pt intermediates, which are formed by dissociation of the halide from the inner sphere of the metal centre. The vacant site is then occupied by the carbene, followed by rapid insertion into the Pt–C bond. Afterwards the halide binds again to the metal, giving rise to Pt(alkyl)X complexes in which the alkyl fragment is extended by one carbon unit. Polar solvents stabilise the cationic intermediates, thereby facilitating carbene insertion into the Pt–C bond. Insertion into the Pt–X bond follows a different pathway that does not involve these cationic species and is therefore not affected by the polarity of the solvent.
 | ||
| Scheme 3 Carbene insertions into Pt–X or Pt–C bonds. | ||
Involvement of cationic Pt complexes in carbene insertions is further emphasized by McCrindle et al., who prepared cationic [(cod)Pt(Me)]+ complexes on purpose by abstraction of the halide in (cod)Pt(Me)(X) complexes.18 When these complexes were subjected to a solution of diazomethane the formation of large amounts of polymethylene was observed, indicating that multiple carbene insertions take place for these species. When phosphine ligands were applied instead of the cod ligand, the intermediate carbene species are stabilised (slowing down carbene insertions) and in that case Pt–(CH2)n–Me (n ≈ 8) species were observed, indicating that indeed the cationic Pt complexes are involved in the catalysis.
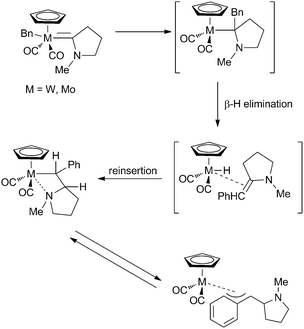
Cooper et al. reported the insertion of carbenes into W+–alkyl bonds, indicating that these reactions also take place for electrophilic (cationic) metal–alkylidene fragments.19,20 The resulting cationic insertion product is rapidly converted to the coordinated olefin complex viaβ-hydride elimination. This strategy was further developed towards W+–aryl insertions, which do not undergo further reactions due to the lack of β-hydrogens.21 Winter and co-workers showed that insertion in neutral W–benzoyl complexes is also possible, although this reaction required harsher reaction conditions (reflux in THF, yield ∼16%) while the analogous Mo complexes readily undergo insertion at room temperature in good yield (53%).22 Later they reported formation of W–benzyl complexes that undergo carbene migrations, although the actual insertion products were never observed.23 Instead, W–alkyl complexes and η3-benzyl complexes were formed, which are proposed to be formed upon rapid β-hydride elimination/reinsertion after carbene migration (Scheme 4). Similar results were obtained with Mo, although here the starting benzyl–Mo–carbene complexes could not be isolated since the insertion takes place more rapidly than is the case for W.
Metallacyclopentene–iron complexes bearing a Cp ligand undergo migratory insertion to form cyclobutyl–iron complexes, which undergo rapid β-hydride elimination to the corresponding olefin complexes (Scheme 5).24 Remarkable about this reaction is that it can also go backwards, i.e. forming a Fe–carbene complex from a Fe–alkyl complex. This is most probably due to a relief of ring strain in the Fe–cyclobutyl complex upon formation of the five-membered metallacycle–carbene complex and has also been predicted (but never observed) for cyclopropyl–iron complexes. This concept has been applied to the synthesis of metal-stabilised bridgehead olefinsin situ, while they are normally very unstable and undergo rapid dimerisation.25
 | ||
| Scheme 5 Carbene insertion/formation in ring-strained iron complexes. | ||
Carbene insertions for other metals have also been studied, but to a much lesser extent. Competitive CO and carbene migratory insertion of alkyl fragments has been studied for osmium complexes and it turned out that these insertions favour the carbene ligand.26 Sharp and Schrock prepared a tantalum carbene complex by alkylidene transfer from phosphoranes. The resulting complexes decompose readily to the corresponding olefin complexes viacarbene insertion and sequential β-hydride elimination (Scheme 6).27
 | ||
| Scheme 6 Carbene insertion/formation for Ta–CH3 complexes followed by β-hydride elimination. | ||
Albéniz and co-workers were able to spectroscopically observe aryl–Pd–carbene species and the subsequent insertion products of the carbenes into the Pd–aryl bonds by applying highly fluorinated aryls.28,29 Solé and co-workers demonstrated insertions of trimethylsilyl diazomethane (TMSDM) and ethyl diazoacetate (EDA) in Pd–aryl bonds, giving rise to the formation of azapalladacycles (Scheme 7).30

3. Catalytic reactions
3.1 Single carbene insertions: cross-coupling reactions
Insertions of carbenes have recently been used as the starting point for various cross-coupling reactions. The interest in cross-coupling reactions as a tool for C–C bond formation has increased rapidly over the last decades, since these reactions contribute to facile synthesis of many biologically active building blocks and functional materials. Pd is often used as catalyst for these reactions, although Ni and Cu catalysed cross-couplings have also been developed. Although a wide variety of products can be synthesised via traditional Heck reactions based on coupling of aryl halides with acrylates, the use of carbenes in this reaction possibly allows for a wider substrate scope. As stated above, many carbene precursors are commercially available or can easily be made according to known procedures, giving rise to easy synthesis of a wide range of compounds extended by one carbon unit.Recently, Van Vranken et al. developed C–C coupling reactions based on insertions of TMSDM into Pd–C bonds generated by oxidative addition of benzyl halides, leading to the synthesis of styrene derivatives in low to moderate yields (Scheme 8).31 The SiMe3 functionalities introduced in the product were easily lost by desilylation while ester functionalities introduced via reaction with EDA remained in the product, giving rise to the synthesis of ethyl cinnamates with various substituents on the phenyl ring in good yields (up to 74%).32 This reaction was further extended to the synthesis of α,β-diarylacrylates by reaction of α-aryldiazoesters with various benzyl halides.33 Both benzyl bromides and chlorides were converted in good yields and stereoselectivities (up to 85% and 81%, respectively, with stereoselectivities > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1) and the reaction is tolerant towards many functional groups on the benzyl halide as well as the aromatic ring of the diazo compound, making this reaction a versatile pathway for the synthesis of many biologically active substrates.
∶1) and the reaction is tolerant towards many functional groups on the benzyl halide as well as the aromatic ring of the diazo compound, making this reaction a versatile pathway for the synthesis of many biologically active substrates.
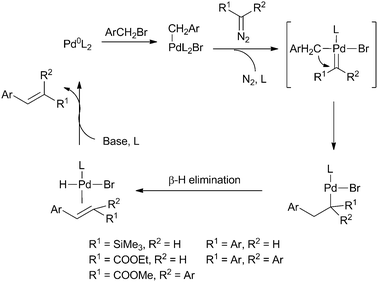 | ||
| Scheme 8 Proposed mechanism for Heck-type C–C bond formation from benzyl halides and carbenes with Pd catalysts. | ||
The proposed mechanism of these reactions is outlined in Scheme 8. The catalytic cycle starts with oxidative addition of the benzyl halide, followed by coordination of the diazo compound. Subsequent nitrogen loss leads to the formation of a benzyl–Pd–carbene species, which undergoes migratory insertion. The final products are formed by β-hydride elimination and the initial catalyst is recovered by treatment with a base.
Insertions into Pd–aryl bonds have also been developed and this concept was exploited as a tool for the synthesis of a variety of products. Kudirka and Van Vranken demonstrated the synthesis of various indenylsilanes viainsertions of TMSDM into Pd–aryl bonds, followed by a carbopalladative cyclisation reaction (Scheme 9).34
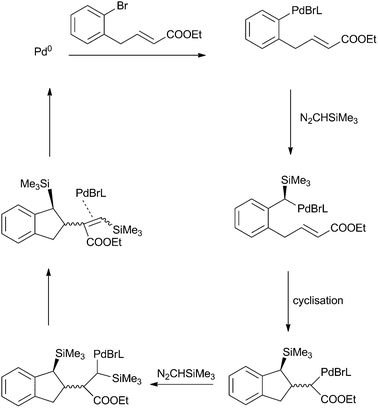 | ||
| Scheme 9 Mechanism for the formation of indenylsilanes. | ||
Due to their instability, the use of diazo compounds without electron-withdrawing substituents in this reaction is rather limited. However, Barluenga and co-workers demonstrated that these substrates can be used in Pd-catalysed cross-coupling reactions by generating them in situ from tosylhydrazones. The conditions for hydrolysis of the tosylhydrazones turned out to be compatible with cross-coupling catalysis. In this way both diazoaryls and diazoalkanes can be generated and coupled to aryl halides in a catalytic fashion, allowing the synthesis of polysubstituted olefins35 and even 4-aryltetrahydropyridines,36 which are valuable building blocks in medicinal chemistry (Scheme 10). Similarly, Wang et al. demonstrated the synthesis of di- and tri- substituted olefins under mild conditions in good yields (>67%) by using these in situ generated diazo compounds directly in Pd-catalysed cross-coupling reactions with benzyl halides as depicted in Scheme 8.37
 | ||
| Scheme 10 Synthesis of polysubstituted olefinsviainsertion of in situ generated carbenes (left) and structure of 4-aryltetrahydropyridines (right). | ||
Pd catalysed cross-coupling of α-diazocarbonyl compounds with arylboronic acids affords 2-phenylacrylates (Scheme 11).38 This concept was extended to stereoselective synthesis of α,β-diarylacrylates by applying bulky bidentate N-donor ligands around the Pd centre.39 In all cases the reaction was selective towards the formation of the E-isomer and mechanistic studies to account for this selectivity are ongoing. Remarkable about this reaction is that it requires solely molecular oxygen to reoxidise the catalyst, while most other processes based on boronic acids require external oxidants (e.g.benzoquinone) to initiate a new catalytic cycle.
 | ||
| Scheme 11 General mechanism for Pd-catalyzed coupling of diazo compounds and arylboronic acids. | ||
Recently, Wang and co-workers have shown that aryl boronic acids can also be coupled to tosylhydrazones, which are assumed to generate aryl diazomethane compounds in situ.40 This reaction further demonstrates the generality of coupling reactions based on carbene insertions. Tosylhydrazones have also been used as carbene precursors in a three-component coupling reaction involving carbenes, aryl halides and terminal alkynes (Scheme 12).41 After insertion of the carbene into the Pd−aryl bond the terminal alkyne is introduced at the Pd centre viatransmetallation from copper. Reductive elimination of both alkyne and alkyl groups yields benzhydryl acetylene derivatives from easily available starting compounds. The yields of the competing Sonogashira reaction between aryl halides and terminal alkynes could be reduced by carefully tuning the reaction conditions. This reaction is an elegant example in which two separate C–C bonds are formed on the same carbene carbon atom and it clearly shows that carbene insertions can be applied in combination with transmetallation from various metal–organic complexes.
 | ||
| Scheme 12 Proposed mechanism for three-component coupling of carbenes, aryl halides and terminal alkynes. | ||
Recently, Devine and Van Vranken prepared vinyl silanes by reacting vinyl halides with TMSDM in presence of a Pd catalyst and a nucleophile (amines42 or stabilised carbon nucleophiles43) (Scheme 13). The resulting vinylsilanes are excellent functional groups for nucleophilic substitution reactions and are therefore useful intermediates for stereospecific organic transformations.44 This concept was extended to the synthesis of α,β-unsaturated γ-amino esters by insertion of EDA into Pd–vinyl bonds, followed by attack of the nucleophile.45 This route allows for a one-step synthesis of various γ-amino esters bearing both natural and non-natural side chains.
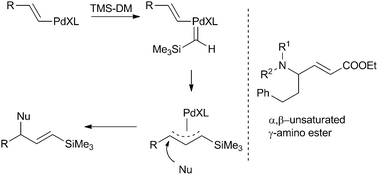 | ||
| Scheme 13 Preparation of vinylsilanes viacarbene insertion followed by nucleophilic attack (left) and structure of α,β-unsaturated γ-amino esters (right). | ||
Carbene insertion has been proposed as a possible pathway in the reaction of allyl halides with α-diazocarbonyl compounds leading to functionalised 1,3-diene compounds (Scheme 14).46 However, different pathways involving non-carbenoid species can lead to the formation of similar products and mechanistic studies for this reaction are ongoing.
 | ||
| Scheme 14 Synthesis of functionalised 1,3-diene compounds viacarbene insertion into Pd–allyl bonds. | ||
Wang et al. have exploited the similarities between CO insertions and carbene insertions to develop a tandem catalytic insertion reaction using both CO and carbenes (Scheme 15).47 The catalytic cycle starts with insertion of CO into a Pd–aryl bond and the key step is believed to be migratory insertion of a carbene unit into the newly formed Pd–acyl bond. In presence of a base, the product formed is an enone, while the saturated counterparts (ketones) are formed in presence of Et3SiH as the external hydrogen source. The reaction is applicable for both carbenes with and without a stabilising carbonyl fragment, thereby giving rise to a large substrate scope. This reaction opens up new possibilities for Pd catalysed carbene transfer reactions.
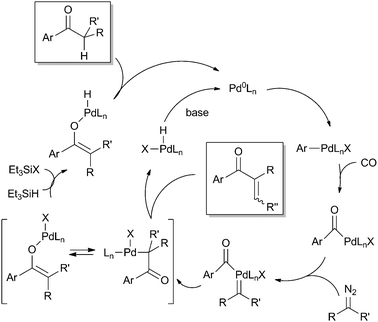 | ||
| Scheme 15 Pd catalysed tandem insertion reactions of CO and carbenes. | ||
3.2 Multiple carbene insertions
Homogeneous and colloidal catalytic systems. If the rate constant for carbene insertion is significantly larger than the rate constant for elimination/termination or other follow-up reactivity, multiple carbene insertions will occur leading to the formation of oligomers or polymers (provided an excess of the carbene precursor is available in the reaction mixture). As such, this process has gained much attention since it provides a valuable alternative to conventional olefin polymerisation based on the coupling of C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond containing monomers, allowing the synthesis of densely functionalised polymers that (so far) cannot be synthesised in any other way. In the next sections we will highlight transition-metal catalysts that allow multiple insertions of both functionalised and non-functionalised carbenes and we will show recent advances in this field. Aside from transition metals several Lewis acids and main group metals are suitable for catalysing this process and the whole field of so-called ‘C1 polymerisation’ has recently been reviewed.48,49
C bond containing monomers, allowing the synthesis of densely functionalised polymers that (so far) cannot be synthesised in any other way. In the next sections we will highlight transition-metal catalysts that allow multiple insertions of both functionalised and non-functionalised carbenes and we will show recent advances in this field. Aside from transition metals several Lewis acids and main group metals are suitable for catalysing this process and the whole field of so-called ‘C1 polymerisation’ has recently been reviewed.48,49
3.2.1.1 Polymers from diazoalkanes. Although many different metals have been attempted as catalysts for the polymerisation of diazoalkanes, most of the earlier reports deal with the activity of copper and gold and have been reviewed (Scheme 16).50,51
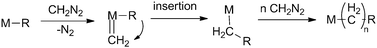 | ||
| Scheme 16 General mechanism for polymerisation of diazomethane by TM catalysts. | ||
In 1909 polymerisation of diazoalkanes was attempted with Cu–bronze. Unfortunately this reaction produced only dimers and short oligomers.52 Later on a broad range of copper reagents (copper metal, CuI and CuII salts) has been applied for the polymerisation of carbenes from diazo compounds. The molecular weights (Mw) of polymethylenes obtained from diazomethane with these copper reagents are up to 20000 Da.51 The active catalyst is a CuI species. If the reaction is started with a CuII catalyst (e.g.copper sulfate, copper stearate) it is thought to be reduced to CuIin situ during the reaction. Several mechanistic proposals for the Cu-mediated polymerisation have been postulated: rapid growth of a cationic chain, step-growthvia repetitive migratory insertion steps of carbene units (generated from the diazo compound) into a CuI–alkyl growing chain, and radical polymerisation by a stabilised CuI radical have all been proposed.50
Colloidal gold particles were later found to be highly active catalysts for the polymerisation of diazomethane.53 The active colloidal gold species were formed upon immediate reduction of the AuCl3 precursor upon reaction with the diazo compound, resulting in almost quantitative formation of polymethylenes with Mw up to 50 kDa. After complete reduction of all AuCl3, new polymer chains with similar lengths were obtained upon addition of fresh diazomethane, indicating that metallic gold is the catalytically active material. Higher diazoalkanes could also be polymerised by this system with some indications for formation of stereoregular (syndiotactic) polymers, albeit with lower reaction rates and lower yields (<35%).54,55
The activity of other metals is less extensively studied, but there are some reports mentioning the activity of nickel and palladium towards polymerisation of diazoalkanes.
Nickel carbene complexes are mainly used as catalysts for cyclopropanation reactions56 and insertion of carbenes into TM–C bonds with these complexes is rare. Nevertheless, Werner and Richards found a highly selective nickelocene complex that is able to polymerise diazomethane to polymethylene in almost quantitative yield.57 The reaction is much faster than reactions carried out by copper powder and ferric chelates and in all cases the catalyst is recovered unchanged after the reaction. The unique feature of this catalyst is that it produces exclusively polymers without the formation of byproducts such as ethene or low-Mw oligomeric material. These results do not change when the reaction is performed in the presence of alkenes or aromatic hydrocarbons, emphasizing the high selectivity of this reaction towards polymerisation over e.g. addition to double bonds. This suggests that no free carbenes are formed during the reaction. The high selectivity is likely the result of extremely fast propagation rates of the polymerisation. The intermediacy of radicals could also be ruled out since addition of radicals does not influence the reaction.
The reactivity of the [Ni(Cp)2] complex towards other diazoalkanes and diazoesters diminishes with increasing steric bulk of the carbon bearing the diazo group, thereby limiting its use. Reaction of phenyldiazomethane yields only a small amount of polymer while diphenyldiazomethane or diazofluorene shows no reaction at all. Ethyl diazoacetate was not polymerised and the main products were the dimers diethyl fumarate and diethyl maleate. In the presence of double bonds, the cyclopropanation product was formed as a third reaction product due to the lack of polymerisation activity.
Polymethylene was also produced by the iridium chloro-carbonyl-bis(triphenylphosphine) complex [IrICl(CO)(PPh3)2] upon reaction with diazomethane.58 By electrophilic attack of the Ir-species on the carbon of the diazomethane a methylene (:CH2) moiety coordinates as a fifth ligand and the resulting complex is stable in air. In contrast with the above-described nickel catalyst, the iridium complex decomposes in solution and the only products obtained after reaction with diazomethane are polymethylene and decomposition products.58
More recently Ihara and co-workers showed that phenyldiazomethane can be converted to small amounts of low-Mw oligomers (Mw ≈ 1500 Da, yield < 20%) in presence of Pd2(dba)3(CHCl3) and pyridine.59 The 1H NMR spectrum of the resulting polymers shows broad resonances for both the aromatic protons and the proton attached to the carbon atom of the polymer backbone, indicating the formation of atactic material. Elemental analysis showed the incorporation of a large amount of azo groups in the polymer (∼one per four monomer insertions), which indicates that the reaction is not fully selective towards carbene insertion (Scheme 17). This, in combination with the low yields and the lack of stereoregularity of the reaction, shows that the reaction needs to be optimised to exploit its full potential.
 | ||
| Scheme 17 Polymerisation of phenyl diazomethane by Pd2(dba)3(CHCl3). | ||
3.2.1.2 Polymers from carbenes bearing polar functionalities. C1 polymerisation of carbenes bearing polar functionalities is much more challenging due to the increased stability of the corresponding carbene precursors and most metal catalysts form only the corresponding dimers upon the decomposition of these precursors.57,60,61
Ihara and co-workers investigated the polymerisation of diazo compounds bearing polar functionalities catalysed by a variety of PdII catalysts. They were able to obtain a variety of highly-functionalised oligomers/polymersvia so-called ‘poly(substituted methylene) synthesis’ by both homo- and copolymerisation of different diazo monomers. Amongst these were mainly diazoesters,59,62,63 diazoketones,59,64 cyclic diazoketones65 and diazoacetamides66 and all could be converted to oligomeric or low-Mw atactic polymeric material. In 2003 they reported that PdCl2 reacts with methyl diazoacetate (MDA) and ethyl diazoacetate in the presence of an amine with formation of low-Mw oligomers (up to 700 Da) that could be isolated as viscous oils in good yields (50–100%).62NMR analysis showed broad resonances indicative of atactic material and the spectra looked similar to those of the product obtained by radical polymerisation of fumarates. However, MALDI-ToF analysis of the resulting materials confirmed that the polymers consisted of repeating carbene units, thereby excluding participation of olefin polymerisation. From the m/z values it could be deducted that polymers bear amine functionalities at both chain ends of the polymer. Since then, several Pd salts have been applied as catalysts for this reaction and it turned out that the results are quite similar in all cases.64 Surprisingly, Pd0 sources such as Pd2(dba)3(CHCl3) show similar activity towards polymerisation. The authors proposed that in this case the active species are formed by in situoxidation of Pd0 to PdII. Later reports deal mainly with the use of PdCl2(MeCN)2 as the catalyst, because this catalyst has a better solubility in organic solvents.
This reaction could be extended to oligomerisation of saturated and unsaturated diazoketones. A clear relation between the structure of the monomer and the ease of polymerisation was observed.59,64 For example, diazo compounds with a double bond next to the carbonyl functionality could be polymerised in good yields (e.g. Mw ≈ 2500 Da, yield ≈ 50%) while the corresponding saturated diazoketones gave only low yields and lower molecular weights under the same conditions with the same catalyst (Mw ≈ 900 Da, yield ≈ 13%).64 This indicates that the presence of a double bond adjacent to the ketone functionality is beneficial for polymerisation, although it is not exactly clear why this is the case.67Polymerisation is hampered by steric hindrance around the metal centre during polymerisation, as is emphasized by very low reactivity of Pd systems towards bulky diazoketones and a variety of cyclic diazoketones (Scheme 18).65 Unsaturated cyclic diazoketones can be polymerised, albeit in low yields (9–24%, Mw = 900–1400 Da), while saturated cyclic diazoketones do not undergo polymerisation reactions.
 | ||
| Scheme 18 Structure of some diazoketones attempted as monomers in the Pd catalysed polymerisation. | ||
Depending on the structure of the diazoketone various amounts of azo functionalities were incorporated in the polymer main chain, sometimes giving rise to rather ill-defined copolymeric structures. Cyclic diazoketones gave rise to higher amounts of azo groups (∼25%) than acyclic diazoketones (<10%), although in the latter case the amounts varied significantly for the different monomers.
Oligomers functionalised with N-substituted carbamoyl groups were available viapolymerisation of certain diazoacetamides, although they were obtained in low yield (∼10%) and with higher azo contents (∼20%) than the oligomers described above.66 These results indicate that the catalyst system is not sensitive to the presence of functional groups in the monomer, emphasizing the wide range of applicability of this technique towards the synthesis of functional materials.
Recently the authors have shown that higher-Mwpolymers of EDA (up to 24000 Da) could be obtained in reasonable yields by applying Pd0(NHC)/BPh4− systems as catalysts.63NMR analysis of the polymeric material showed that the material is atactic, although variations in the structure of the NHC ligand seem to have a slight effect on the tacticity, thereby creating possibilities for stereocontrol. Activation of the Pd0(NHC) catalyst by BPh4− is required to obtain higher-Mwpolymers.
For all catalysts, the authors propose a PdII-based mechanism that does not involve discrete carbene intermediates, thereby formally excluding these systems from the scope of this perspective. According to Ihara and co-workers the initiation takes place via nucleophilic attack on the α-carbon atom of the diazo compound, which leads vianitrogen extrusion to the formation of a PdII–carbon bond. Amines62 are proposed as initiating species but, since the reaction also proceeds in absence of amines, this could well involve the attack of weak nucleophiles such as water, MeCN or the monomer.64 The observation of amines at both chain-ends suggests that two polymer chains might grow simultaneously from one Pd centre and the final polymer could well be formed by reductive elimination of both chains.
Propagation was proposed to involve direct migration of the growing polymer chain onto the coordinated diazo monomer, followed by nitrogen extrusion (Scheme 19). A true carbene polymerisation mechanism involving discrete Pd–carbene intermediates can however not be excluded.
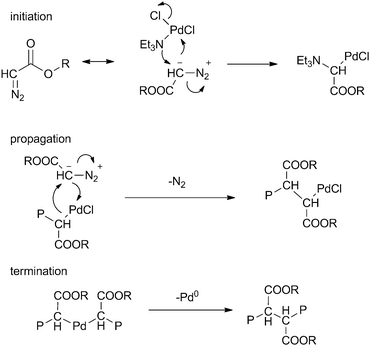 | ||
| Scheme 19 Proposed chain propagation in Pd mediated ‘carbene’ polymerisation from diazo compounds. | ||
The proposed mechanism (Scheme 19) contrasts with other reports suggesting that carbenoid species are intermediates in insertion reactions with diazo compounds catalysed by Pd,31,32 and since there is no direct evidence for direct attack on the diazo compounds, the intermediacy of carbene species in Pd catalysed oligomerisation reactions cannot be ruled out. Since these reports contribute for a large part to the development and understanding of carbene polymerisation (and C1 polymerisation in general) we decided to include them in this perspective, despite the ambiguity about the mechanism.
Recently, de Bruin et al. have shown that most likely low valent Pd species (probably Pd0) are involved as catalytically active species in this reaction.68 Several Pd0 complexes, including heterogeneous Pd0 adsorbed on carbon, catalysed the formation of oligomers from EDA in good yields (up to 80%) and higher Mw (up to 2000 Da) than the previously reported PdII salts. Furthermore, these systems do not incorporate olefins in the growing carbene polymer chain, which would have been expected if PdII–alkyl intermediates were involved. This, in combination with the known reducing power of diazo compounds, makes the involvement of low valent Pd more likely. These results might contribute to a more strategic reaction design, directed to the development of highly active and (stereo)selective Pd-based C1-polymerisation catalysts.
In 2006 polar functionalised carbenes generated from diazo compounds were polymerised in a stereoregular manner by transition metal catalysts, as reported by de Bruin and co-workers.69 By reacting EDA and other diazoesters with RhI(diene) catalysts, syndiotactic (co)polymers are produced with high molecular weights and in high yields (Mw up to 150000 Da, yields up to 85%) (Scheme 20). The remaining products are atactic oligomers (Mw ≈ 1200 Da, Mw/Mn ≈ 3) and diethyl maleate and diethyl fumarate.70–73 The syndiotactic polymers are highly crystalline, and reveal thermotropic and lyotropic liquid crystalline properties.69,73

Rh-mediated stereoselective polymerisation is also applicable to other polar functionalised C1 monomers, such as n-butyl diazoacetate, 3-butenyl diazoacetate,69benzyl diazoacetate and tBu-diazoacetate, and the reaction allows the synthesis of random and [homo-A]–[random-B > A]-type block copolymers (Scheme 21).73
![Formation of random and [homo-A]–[random-B > A] block copolymers by Rh-mediated carbene polymerisation.](/image/article/2011/CY/c0cy00065e/c0cy00065e-s21.gif) | ||
| Scheme 21 Formation of random and [homo-A]–[random-B > A] block copolymers by Rh-mediated carbene polymerisation. | ||
In case iridium analogues of this Rh-catalyst were used for the polymerisation of EDA, only [(N-benzyl-L-prolinate)IrI(cod)] proved active (cod = 1,5-cyclooctadiene).70 In 2007, Buchmeiser et al. studied the reactivity of another IrI-complex, [(N-acetyl-N,N-dipyrid-2-yl)(Cl)IrI(cod)], towards EDA. This did not polymerise EDA at all, while the analogous Rh complex proved quite active in the polymerisation of EDA.74
A DFT study was performed to explain the formation of stereoregular polymer with non-chiral catalysts, suggesting that the propagation steps are chain-end controlled.70 The diene ligand, or a ligand derived from the diene formed under the applied reaction conditions, must be stabilising the active species for polymer formation, as can be derived from the large influence of the applied diene ligand on the obtained polymer lengths and molecular weight distributions.70 No or little chain transfer seems to occur. DFT calculations using the unmodified Rh(cod) catalyst as a simplified model suggest that the reaction proceeds via a migratory insertion mechanism (Scheme 22).70
 | ||
| Scheme 22 Proposed mechanism of propagation steps of the Rh-mediated polymerisation of polar functionalised carbenes (P = growing polymer chain, E = COOMe). | ||
Ligand modification under the applied reaction conditions (or possibly ligand oxidation by H2O/O2 in case of 1,5-dimethylcyclooctadiene)71 cannot be excluded, and in fact the polymerisation reaction with Rh(cod) catalysts is associated with a clear induction period during which the selectivity changes from predominant dimerisation/oligomerisation activity to almost exclusively polymerisation activity.70 Hence, it is clear that not all details of the reaction mechanism are fully understood. Gaining a deeper understanding of the mechanism, especially the processes leading to chain initiation and chain termination/chain transfer, will be important in order to exploit the full potential of this new reaction. Detailed mechanistic studies are therefore necessary.
The clear advantage of Rh-mediated carbene polymerisation over other C1 polymerisation techniques is that it gives stereoregular (syndiotactic) polymers from polar functionalised C1 monomers with high molecular weights.48 Nevertheless, there is room for improvement in terms of yields, initiation efficiencies of the catalysts and in the synthesis of stereoregular polymers with different tacticities.
Catalysis on heterogeneous metal surfaces. Aside from the above-described homogeneous catalysts also some heterogeneous metal surfaces catalyse the polymerisation of carbenes. Encouraged by the findings that colloidal gold particles catalyse polymerisation of diazoalkanes (vide supra) Nasini and co-workers found that polymers can as well be obtained by applying thin films of gold as catalysts in the reaction of various diazoalkanes.53 When diazoethane was used as the carbene precursor, small amounts of stereoregular polyethylidene were formed, although the exact tacticity was not assigned (Scheme 23). Many other metal surfaces have also been explored in their catalytic activity towards diazoethane polymerisation and most of them showed activity (Cu, Ti, Fe, Mg, W, Ni, V, Mn, Ta, Pt, Co, Zn, Cd, Cr, Al, Mo; listed in order of decreasing yield). No activity was obtained for metallic Pd, Rh, Zr and Ag, although a clear reason for this lack of activity could not be found. Remarkably, none of the other active surfaces gave rise to the formation of stereoregular polyethylidene.
 | ||
| Scheme 23 Proposed mechanism of propagation steps for the polymerisation of diazoethane on gold surfaces. | ||
The catalytic activity of gold surfaces in the polymerisation of diazomethane has been used to obtain ultra-thin films of polymethylene on gold surfaces, since these films can be applied in areas such as microelectronics and packaging.75 The films start to grow as nanometre-scale polymethylene assemblies on defect sites at the gold surface but in later stages these assemblies coalesce to uniformly cover the whole surface. Bai and Jennings have exploited this fact and modified the gold surface with either copper or silver, thereby enabling polymer growth uniformly on the whole surface.76 More recently, the same group found that EDA can act as co-catalyst for DM polymerisation on gold surfaces.77 Although homopolymerisation of EDA was not possible, small amounts (1–4%) of esters were incorporated in copolymerisation attempts. The thickness of the films could be tuned by changing the EDA concentration and films up to several hundreds of nanometres could be obtained. The effect of EDA on the polymerisation of DM is thought to be caused by the removal of electron density from the gold surface by adsorbed ethyl ester carbene species and these electron-deficient gold sites are believed to be the active sites for polymerisation. This effect is similar to that of copper-modified surfaces, in which the electropositive copper atoms remove electron density from gold.
Although the mechanism of these reactions is still elusive, most evidence points to the direction of carbene insertions involving MCH2 (or M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) species, which are similar to intermediates in the Fischer–Tropsch synthesis (for an overview of similar C–C bond forming reactions on surfaces see ref. 78 and 79).
CH) species, which are similar to intermediates in the Fischer–Tropsch synthesis (for an overview of similar C–C bond forming reactions on surfaces see ref. 78 and 79).
 | ||
| Scheme 24 Different mechanisms proposed for the Fischer–Tropsch synthesis on heterogeneous catalysts. | ||
More recent papers have indicated that a single reaction pathway cannot be responsible for the observed product distributions and therefore the FTS is presumed to be a combination of at least two parallel reactions, likely involving both carbene insertion and CO insertion steps.83–85 However, there is still much debate on the mechanism, and research will be continued until direct observation of the active species will lead to conclusive evidence for one of the above-described mechanisms.
In principle all group VIII metals show some activity towards C–C bond formation in the FTS process, although the most active catalysts are found to be iron, cobalt, nickel and ruthenium.81 Of these catalysts only (the cheapest) iron and cobalt are used in industrial processes. Ruthenium is the most active metal, but is not economical for FTS due to its high price. The problem with nickel is that it produces mainly methane instead of higher olefins under industrial conditions. Most of the above-described mechanistic studies are performed with industrially relevant iron or cobalt systems, and thereby possible alternative pathways for other metals are mostly being neglected.
Some homogeneous multinuclear complexes proved to be good models for the catalytically active multinuclear metal sites at heterogeneous surfaces in the Fischer–Tropsch process. Curtis et al. reported in 1980 the reaction of diazoalkanes with binuclear compounds containing a metal–metal bond. The reaction led to binuclear μ-alkylidene complexes with a bridging alkylidene-ligand.86,87 The formation of alkylidene species from diazoalkanes at multinuclear complexes is non-trivial, and can follow several pathways and can lead to several alkylidene coordination modes.
Cowie et al. studied binuclear Rh,Os species as synthetic homogeneous models for the active heterogeneous surfaces in the FTS process. They focused on the relevance of the bridging methylene groups and the role of the two different metals (Rh and Os).88Mixing [RhOs(CO)4(dppm)2](BF4) compound 5 with diazomethane led to formation of complex 8, which is likely formed by a series of consecutive carbene insertions (Scheme 25).89 In order to get a better insight into the mechanism of this methylene-coupling reaction, the reaction was performed at different temperatures. This allowed them to obtain more information about the intermediates.
 | ||
| Scheme 25 Products obtained by reacting heterobinuclear complex 5 with diazomethane at varying temperatures. | ||
Mixing
[RhOs(CO)4(dppm)2](BF4) with diazomethane at −78 °C yielded the methylene-bridged [RhOs(CO)3(μ2-CH2)(μ2-CO)(dppm)2](BF4) compound 6. By increasing the temperature from −60 °C to 0 °C compound 6 transformed into the butanediyl species 7. Compound 4 starts to form at higher temperatures between −40 °C and room temperature. Compound 6 can be converted to 7 and 8 by raising the temperature, but 7 and 8 do not interconvert. The mechanism likely proceeds via discrete RhCH2 intermediates at the coordinatively unsaturated Rh site, and after diazomethane activation at this metal, methylene insertion into the Rh–C bond of the bridging methylene follows. The Os–hydrocarbyl bond strength increases by this process, and this is crucial for the chain growth. If otherwise, loss of ethene would stop the methylene coupling sequence. This underlines the importance of the bridging binding modes. The group of Dry described a similar type of methylene coupling sequence to support the carbon–carbon coupling in the Fischer–Tropsch process.90
To gain more information about these insertion reactions, 13C and 2H labelled diazomethane was used. This, however did not provide much more information regarding the formation of 7 and 8.88 Further information was obtained by adding diazomethane to vinylidene-bridged species (Scheme 26). This leads to similar types of carbene insertion reactions with a comparable temperature dependency,91 and shows that carbene insertion into bridging positions is possible. Alkyl ligand migration between the two metals apparently plays an important role. The properties of the metals, the lability and coordinative unsaturation of the Rh atom, and the strong coordination of the hydrocarbyl ligands to Os make these homogeneous hetero-binuclear Rh,Os systems quite suitable to model Fischer–Tropsch-like C–C bond formations.
 | ||
| Scheme 26 Products obtained by reacting diazomethane with vinylidene bridged heterodinuclear Rh,Os compounds. | ||
4. Conclusions and outlook
Migratory carbene insertions into TM–C bonds offer interesting opportunities in catalytic C–C bond formation and this reaction already has some existing applications. Intriguing examples allow the preparation of densely functionalised polymers from C1 monomers, and hence the synthesis of new (stereoregular) polymers that are functionalised with (polar) substituents at every carbon atom of the polymer backbone. Such polymers are not so easily obtained by conventional olefin polymerisation techniques. Carbene insertions most likely also play an important role in the related Fischer–Tropsch synthesis, and therefore renewed mechanistic insights into this reaction based on (stoichiometric) studies of carbene insertion reactions may well contribute to further developments of this catalytic ‘fuel production’ reaction. Several of such model reactions reveal the feasibility of the migratory carbene insertion elementary step, and underline the importance of electronic and steric influences of ligands and (partial) charges on the outcome of the reaction, both in homogeneous systems as on heterogeneous metal-surfaces, thereby offering ample opportunities for catalyst development (catalysis by design). This will be important to gain control over the chain-growth and chain termination/transfer processes in (existing) Fischer–Tropsch and C1 polymerisation reactions, but also holds the promise to develop completely new (catalytic) reactions based on migratory carbene insertions. In this respect, recent developments in Pd catalysed cross-coupling reactions show that transmetallation can be successfully combined with migratory carbene insertions, thus expanding the substrate scope substantially and allowing the efficient synthesis of new molecular compounds.The close analogy of the migratory carbene insertion reaction with migratory CO (and olefin) insertion reactions, combined with the clear and strong steering influence of ligands (homogeneous systems) and other metal-surroundings (heterogeneous systems) give high hopes for the future development of further interesting new reactions. Replacing CO by carbenes in existing catalytic reactions may, for example, allow the development of ‘hydro-carbenylation’ (carbene analog of hydroformylation), ‘methoxy-carbenylation’ (carbene analog of methoxycarbonylation), or olefin/carbene copolymerisation reactions. While all of the individual elementary steps for these (thus far) hypothetical reactions are feasible and existing, the development of such new reactivity will certainly require substantial screening, detailed mechanistic investigations, appropriate ligand design and careful tuning of the catalytic conditions. We are convinced however that these efforts will pay-off, and lead to groundbreaking new discoveries. We hope therefore that this perspective will inspire more researchers to explore the field of migratory carbene insertions as a promising new synthetic tool to speed-up these upcoming and promising developments.
Acknowledgements
We kindly thank the Dutch Polymer Institute DPI, Eindhoven, The Netherlands (projects #646 and #647) and the European Research Council (ERC, EU 7th framework program, grant agreement 202886-CatCIR) for financial support.Notes and references
- (a) Homogeneous Catalysis: Understanding the Art, ed. P. W. N. M. van Leeuwen, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed; (b) Theoretical Aspects of Homogeneous Catalysis, ed. P. W. N. M. van Leeuwen, K. Morokuma and J. H. van Lenthe, Kluwer Academic Publishers, Dordrecht, 1995 Search PubMed.
- Modern Catalytic Methods for Organic Synthesis, ed. M. P. Doyle, M. A. McKervey and T. Ye, John Wiley & Sons, New York, 1998 Search PubMed.
- X.-Z. Zhou and K. J. Shea, J. Am. Chem. Soc., 2000, 122, 11515 CrossRef CAS.
- Green Chemistry: Theory and Practice, ed. P. T. Anastas and J. C. Warner, Oxford University Press, New York, 1998, p. 30 Search PubMed.
- Of course, if we include the energy load and the byproducts generated in their synthesis, diazo compounds are less ‘green’ in terms of atom and energy efficiency.
- Diazo compounds—Properties and Synthesis, ed. M. Regitz and G. Maas, Academic Press, Orlando, 1986 Search PubMed.
- Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS , and references cited therein.
- J. R. Fulton, V. K. Aggarwal and J. de Vicente, Eur. J. Org. Chem., 2005, 1479–1492 CrossRef CAS.
- P. Müller, Acc. Chem. Res., 2004, 37, 243 CrossRef.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS.
- M. T. Whited and R. H. Grubbs, Acc. Chem. Res., 2009, 42, 1607 CrossRef CAS.
- D. L. Thorn and T. H. Tulip, J. Am. Chem. Soc., 1981, 103, 5984 CrossRef CAS.
- D. L. Thorn, Organometallics, 1986, 5, 1897 CrossRef CAS.
- P. Bergamini and E. Costa, Organometallics, 1994, 13, 1058 CrossRef CAS.
- P. Bergamini and E. Costa, Organometallics, 1995, 14, 3178 CrossRef CAS.
- P. Bergamini, E. Costa, C. Ganter, A. G. Orpen and P. G. Pringle, J. Chem. Soc., Dalton Trans., 1999, 861 RSC.
- L. Jánosi and L. Kollár, Transition Met. Chem., 2007, 32, 746 CrossRef CAS.
- R. McCrindle, G. J. Arsenault, R. Farwaha, M. J. Hampden-Smith and A. J. McAlees, J. Chem. Soc., Chem. Commun., 1986, 943 RSC.
- J. C. Hayes, G. D. N. Pearson and N. J. Cooper, J. Am. Chem. Soc., 1981, 103, 4648 CrossRef CAS.
- J. C. Hayes and N. J. Cooper, J. Am. Chem. Soc., 1982, 104, 5570 CrossRef CAS.
- P. Jernakoff and N. J. Cooper, J. Am. Chem. Soc., 1984, 106, 3026 CrossRef CAS.
- H. Adams, N. A. Bailey, C. E. Tattershall and M. J. Winter, J. Chem. Soc., Chem. Commun., 1991, 912 RSC.
- H. Adams, N. A. Bailey, G. W. Bentley, C. E. Tattershall, B. F. Taylor and M. J. Winter, J. Chem. Soc., Chem. Commun., 1992, 533 RSC.
- Y. Senstrøm, A. E. Koziol, G. E. Palenik and W. M. Jones, Organometallics, 1987, 6, 2079 CrossRef.
- R. S. Bly, R. K. Bly, M. M. Hossain, L. Lebioda and M. Raja, J. Am. Chem. Soc., 1988, 110, 7723 CrossRef CAS.
- K. Roder and H. Werner, Angew. Chem., Int. Ed. Engl., 1987, 26, 686 CrossRef.
- P. R. Sharp and R. R. Schrock, J. Organomet. Chem., 1979, 171, 43 CrossRef CAS.
- A. C. Albéniz, P. Espinet, R. Manrique and A. Pérez-Mateo, Angew. Chem., Int. Ed., 2002, 41, 2363 CrossRef CAS.
- A. C. Albéniz, P. Espinet, R. Manrique and A. Pérez-Mateo, Chem.–Eur. J., 2005, 11, 1565 CrossRef CAS.
- D. Solé, L. Vallverdú, X. Solans, M. Font-Bardia and J. Bonjoch, Organometallics, 2004, 23, 1438 CrossRef CAS.
- K. L. Greenman, D. S. Carter and D. L. Van Vranken, Tetrahedron, 2001, 57, 5219 CrossRef CAS.
- K. L. Greenman and D. L. Van Vranken, Tetrahedron, 2005, 61, 6438 CrossRef CAS.
- W.-Y. Yu, Y.-T. Tsoi, Z. Zhou and A. S. C. Chan, Org. Lett., 2009, 11, 469 CrossRef CAS.
- R. Kudirka and D. L. Van Vranken, J. Organomet. Chem., 2008, 73, 3585 CAS.
- J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587 CrossRef CAS.
- J. Barluenga, M. Tomás-Gamasa, P. Moriel, F. Aznar and C. Valdés, Chem.–Eur. J., 2008, 14, 4792 CrossRef CAS.
- Q. Xiao, J. Ma, Y. Yang, Y. Zhang and J. Wang, Org. Lett., 2009, 11, 4732 CrossRef CAS.
- C. Peng, Y. Wang and J. Wang, J. Am. Chem. Soc., 2008, 130, 1566 CrossRef CAS.
- J.-T. Tsoi, Z. Zhou, A. S. C. Chan and W.-Y. Yu, Org. Lett., 2010, 12, 4506 CrossRef.
- X. Zhao, J. Jing, K. Lu, Y. Zhang and J. Wang, Chem. Commun., 2010, 46, 1724 RSC.
- L. Zu, F. Ye, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2010, 132, 13590 CrossRef CAS.
- S. K. J. Devine and D. L. Van Vranken, Org. Lett., 2007, 9, 2047 CrossRef CAS.
- S. K. J. Devine and D. L. Van Vranken, Org. Lett., 2008, 10, 1909 CrossRef CAS.
- H. Inami, T. Ito, H. Urabe and F. Sato, Tetrahedron Lett., 1993, 34, 5919 CrossRef CAS.
- R. Kudirka, S. K. J. Devine, C. S. Adams and D. L. Van Vranken, Angew. Chem., Int. Ed., 2009, 48, 3677 CrossRef CAS.
- S. Chen and J. Wang, Chem. Commun., 2008, 4190 Search PubMed.
- Z. Zhang, Y. Liu, M. Gong, X. Zhao, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1139 CrossRef CAS.
- E. Jellema, A. L. Jongerius, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2010, 39, 1706 RSC.
- E. Ihara, Adv. Polym. Sci., 231, 191 Search PubMed.
- G. W. Cowell and A. Ledwith, Q. Rev. Chem. Soc., 1970, 24, 119 RSC.
- N. Imoto and T. Nakaya, J. Macromol. Sci., Rev. Macromol. Chem., 1972, C7(1), 1 Search PubMed.
- A. Loose, J. Prakt. Chem., 1909, 124, 185.
- A. G. Nasini, L. Trossarelli and G. Saini, Makromol. Chem., 1961, 44, 550 CrossRef.
- A. G. Nasini, G. Saini and L. Trossarelli, Pure Appl. Chem., 1962, 4, 255 CrossRef CAS.
- A. G. Nasini and L. Trossarelli, Chimia, 1962, 127 CAS.
- X. Zhang, Z.-Y. Geng, Y.-C. Wang, W.-Q. Li, Z. Wang and F.-X. Liu, THEOCHEM, 2009, 893, 56 CrossRef CAS , and references therein.
- H. Werner and J. H. Richards, J. Am. Chem. Soc., 1968, 90, 4976 CrossRef CAS.
- F. D. Mango and I. Dvoretzky, J. Am. Chem. Soc., 1966, 88, 1654 CrossRef CAS.
- E. Ihara, M. Kida, M. Fujioka, N. Haida, T. Itoh and K. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1536 CrossRef CAS.
- G. D. Buckley, L. H. Cross and N. H. Ray, J. Chem. Soc., 1950, 2714 RSC.
- K. Lorey, J. Prakt. Chem., 1929, 124, 185 CrossRef.
- E. Ihara, N. Haida, M. Iio and K. Inoue, Macromolecules, 2003, 36, 36 CrossRef CAS.
- E. Ihara, Y. Ishiguro, N. Yoshida, T. Hiraren, T. Itoh and K. Inoue, Macromolecules, 2009, 42, 8608 CrossRef CAS.
- E. Ihara, M. Fujioka, N. Haida, T. Itoh and K. Inoue, Macromolecules, 2005, 38, 2101 CrossRef CAS.
- E. Ihara, T. Hiraren, T. Itoh and K. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1638 CrossRef CAS.
- E. Ihara, T. Hiraren, T. Itoh and K. Inoue, Polym. J. (Tokyo), 2008, 40, 1094 Search PubMed.
- This could well be related to a general higher stability of unsaturated diazo-carbonyl compounds: T. H. Bonge and T. Hansen, Tetrahedron Lett., 2010, 51, 5378 Search PubMed.
- N. M. G. Franssen, J. N. H. Reek and B. de Bruin, Polym. Chem., 2011, 2, 422 RSC.
- D. G. H. Hetterscheid, C. Hendriksen, W. I. Dzik, J. M. M. Smits, E. R. H. van Eck, A. E. Rowan, V. Busico, M. Vacatello, V. Van Axel Castelli, A. Segre, E. Jellema, T. G. Bloemberg and B. de Bruin, J. Am. Chem. Soc., 2006, 128, 9746 CrossRef CAS.
- E. Jellema, P. H. M. Budzelaar, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2007, 129, 11631 CrossRef CAS.
- E. Jellema, A. L. Jongerius, A. J. C. Walters, J. M. M. Smits, J. N. H. Reek and B. de Bruin, Organometallics, 2010, 29, 2823 CrossRef CAS.
- M. Rubio, E. Jellema, M. A. Siegler, A. L. Spek, J. N. H. Reek and B. de Bruin, Dalton Trans., 2009, 8970 RSC.
- E. Jellema, A. L. Jongerius, G. Alberda van Ekenstein, S. D. Mookhoek, T. J. Dingemans, E. M. Reingruber, A. Chojnacka, P. J. Schoenmakers, R. Sprenkels, E. R. H. van Eck, J. N. H. Reek and B. de Bruin, Macromolecules, 2010, 43, 8892 CrossRef CAS.
- B. Bantu, K. Wurst and M. R. Buchmeiser, J. Organomet. Chem., 2007, 692, 5272 CrossRef CAS.
- K. Sheshadri, S. V. Atre, Y.-T. Tao, M.-T. Lee and D. L. Allara, J. Am. Chem. Soc., 1997, 119, 4698 CrossRef CAS.
- W. Guo and K. Jenning, Langmuir, 2002, 18, 3123 CrossRef CAS.
- D. Bai and K. Jennings, J. Am. Chem. Soc., 2005, 127, 3048 CrossRef CAS.
- M. Leconte, J. Mol. Catal., 1994, 86, 205 CrossRef CAS.
- B. Kirste and H. Kurreck, J. Am. Chem. Soc., 1980, 102, 6181 CrossRef.
- J. Lin, C. Chang, C. J. Jenks, M. X. Yank, T. H. Wentzlaff and B. E. Bent, J. Catal., 1994, 147, 250 CrossRef CAS.
- J. P. Hindermann, G. J. Hutchings and A. Kiennemann, Catal. Rev. Sci. Eng., 1993, 35, 1 CrossRef CAS.
- O. R. Inderwildi, S. J. Jenkins and D. A. King, J. Phys. Chem. C, 2008, 112, 1305 CrossRef CAS.
- J. Gaube and H.-F. Klein, J. Mol. Catal. A: Chem., 2008, 283, 60 CrossRef CAS.
- J. Gaube and H.-F. Klein, Appl. Catal., A, 2010, 374, 120 CrossRef CAS.
- X. Lu, D. Hildebrandt, X. Liu and D. Glasser, Ind. Eng. Chem. Res., 2010, 49, 9753 CrossRef CAS.
- L. Messerle and M. D. Curtis, J. Am. Chem. Soc., 1980, 102, 7789 CrossRef CAS.
- M. Dartiguenave, M. J. Menu, E. Deydier, Y. Dartiguenave and H. Siebald, Coord. Chem. Rev., 1998, 178–180, 623 CrossRef.
- S. J. Trepanier, J. N. L. Dennett, B. T. Sterenberg, R. McDonald and M. Cowie, J. Am. Chem. Soc., 2004, 126, 8046 CrossRef CAS.
- S. J. Trepanier, B. T. Sterenberg, R. McDonald and M. Cowie, J. Am. Chem. Soc., 1999, 121, 2613 CrossRef CAS.
- M. E. Dry, Appl. Catal., A, 1996, 138, 319 CrossRef CAS.
- J. R. Wigginton, A. Chokshi, T. W. Graham, R. McDonald, M. J. Ferguson and M. Cowie, Organometallics, 2005, 24, 6398 CrossRef CAS.
Footnote |
| † Equal contributions from AJCW and NMGF. |
| This journal is © The Royal Society of Chemistry 2011 |