Purine 5′,8-cyclonucleoside lesions: chemistry and biology†
Chryssostomos
Chatgilialoglu
*,
Carla
Ferreri
and
Michael A.
Terzidis
ISOF, Consiglio Nazionale delle Ricerche, Via P. Gobetti 101, 40129 Bologna, Italy. E-mail: chrys@isof.cnr.it; Fax: +39 051 639 8349; Tel: +39 051 639 8309
First published on 11th January 2011
Abstract
5′,8-Cyclo-2′-deoxyadenosine and 5′,8-cyclo-2′-deoxyguanosine in their 5′R and 5′S diastereomeric forms are tandem-type lesions observed among the DNA modifications and identified in mammalian cellular DNAin vivo. These lesions result from the chemistry of the C5′ radicals generated by the attack of HO˙ radicals to 2-deoxyribose units. Quantitative determination of these lesions in biological samples as biomarkers of free radical damage is a challenge. Results reported for irradiated samples of calf thymus DNA have been critically reviewed, underlining the need of further research for the potential involvement of these lesions in human health (76 references).
 Chryssostomos Chatgilialoglu | Chryssostomos Chatgilialoglu (born 1952) is director of research at the Consiglio Nazionale delle Ricerche (Bologna) since 1991. He was the winner of the Reagent of the Year 1990. His research interests lie in free radical reactions and in the last decade have been increasingly addressed to applications in life sciences, such as biomimetic chemistry of radical stress and related biomarkers. Author and editor of 4 books, author and co-author of over 240 papers, and invited speaker over 160 times at congresses and institutions, he is the Chairman of the COST Action on Free Radicals in Chemical Biology. He is President and co-founder of the spin-off company Lipinutragen. |
 Carla Ferreri | Carla Ferreri started her scientific career in synthetic organic chemistry, being appointed as research fellow at the University of Napoli “Federico II” in 1984. In 2001 she moved to the Consiglio Nazionale delle Ricerche in Bologna, where she is currently Senior Researcher. Her present research interests are in the field of biomimetic chemistry, investigating free radical transformations of biomolecules related to biomarker discovery. She is also co-founder of a spin-off company on lipidomics and nutraceuticals. |
 Michael A. Terzidis | Michael A. Terzidis received his Bachelor in Chemistry (2005) and gained his PhD (2010) in Organic Chemistry on “Synthesis and structural studies of chromone derivatives with possible biological interest” (Prof. J. Stephanidou-Stephanatou) from the Department of Chemistry of University of Thessaloniki (Greece). In April 2010 he moved to Bologna joining Dr Chatgilialoglu's research group under the Short-Term Scientific Mission of COST Action CM0603 “Free Radicals in Chemical Biology”. His research interests focus on heterocyclic chemistry and particularly mechanistic studies of free radical reactions, nucleosides chemistry, organocatalysis and multicomponent reactions. |
1. Introduction
1.1 Free radical stress and DNA damage
Reactive oxygen species (ROS), including the most harmful hydroxyl radical, are chemically-reactive molecules produced in a wide range of physiological processes (oxygen metabolism).1 The hydroxyl radicals (HO˙) are known for their reactivity and ability to cause chemical modifications to DNA through the formation of strand breaks and nucleobase modifications. The antioxidant network, which is able to cope with the HO˙ radical production by enzymatic and molecular mechanisms, represents the biological counterpart of this damage biasing the possible negative effects at a very sustainable threshold.2 In addition to the induction of DNA damage through the metabolism of oxygen, DNA damage may also be induced through other environmental insults such as ionising radiation, UV light and chemical mutagens. Although the majority of DNA damage induced through oxidative metabolism are single lesions, there are also types of multiple lesions, such as tandem or clustered lesions and DNA/DNA or DNA/protein cross-link, that may challenge the repair machinery of the cell. Indeed, enzymatic systems like base excision repair (BER) and nucleotide excision repair (NER) are known to remove the majority of DNA lesions and look after the integrity of the genome.3 However, the lesions may accumulate in tissues, mainly due to the progressive loss of protective systems and consequent poor repair, as it occurs in the aging process.4 Also enzymatic deficiencies can create accumulation damages that are linked to certain pathologies.As far as the site of DNA attack is concerned, diffusible HO˙ radicals are known to either abstract a hydrogen atom from the 2-deoxyribose units or add to the base moieties. The majority of HO˙ attacks occur at the base moieties and it is estimated to account for as much as 90%. However, experimental evidence on direct strand scission suggest the transfer of the radical center from the base moieties to the sugar backbone.5 The order of reactivity of HO˙ radical towards the various hydrogen atoms of the 2-deoxyribose moiety is currently under debate.6 A proposed order that also parallels the exposure to solvent of the 2-deoxyribose hydrogen atoms (i.e., H5′ > H4′ > H3′ ≈ H2′ ≈ H1′) is generally accepted.7–9 Indeed, the positioning of the C4′- and C5′-hydrogen atoms on the edge of the minor groove renders them accessible to diffusible species. The attack at H5′ of DNA by HO˙ radicals is estimated to be 55% of all possible sugar positions.10 Abstraction of a hydrogen atom from 2-deoxyribose produces a carbon-centered radical whose fate depends upon the environment. Many of the recent studies focus on the selective generation of these species that allowed quantitative data to be obtained.11,12 This subject is attracting a growing interest for the genetic toxicology of oxidative stress and inflammation.6 The chemistry of the C5′ radical is very peculiar with respect to the other positions of 2-deoxyribose in the sense that it does not generate an abasic site and unique cyclic base-sugar adducts are formed (purine 5′,8-cyclo-2′-deoxynucleosides). These tandem-type lesions are observed among the DNA modifications and have also been identified in mammalian cellular DNAin vivo.
1.2 Initial studies of the C5′–C8 bond formation in purine nucleosides
The chemistry of purine 5′,8-cyclonucleosides has its origin in the literature of ionizing radiation since these compounds have been first observed as products of the reaction of HO˙ radicals with adenosine and related derivatives.13Radiolysis of neutral water leads to eaq−, HO˙ and H˙ together with H+ and H2O2 as shown in eqn (1).14 The values in parentheses represent the radiation chemical yields (G) in units of μmol J−1. The reactions of these transient species dominate when DNA is irradiated in dilute aqueous solution. However, in “high” DNA concentration or in cells a direct ionization of DNA also plays a role, causing the formation of clustered lesions.5H2O ![[radiolysis arrow - arrow with voltage kink]](https://www.rsc.org/images/entities/char_e116.gif) eaq−(0.27), HO˙(0.28), H˙(0.062), H+(0.27), H2O2(0.07) eaq−(0.27), HO˙(0.28), H˙(0.062), H+(0.27), H2O2(0.07) | (1) |
In 1968 Keck discovered that the attack of HO˙ radicals to adenosine-5′-monophosphate leads, among other products, to the 5′S and 5′R diastereomers of 5′,8-cycloadenosine-5′-monophosphate (5′,8-cAMP).15 The effect of pH on the formation of the two diastereomers has been studied in detail.16,17 The X-ray structure of (5′S)-5′,8-cAMP was reported and turned out that only the 5′R diastereomer is enzymatically active.18 In the late seventies, 5′,8-cyclo-2′-deoxyadenosine was identified as one of the principal products of γ-radiolysis of 2′-deoxyadenosine in aqueous solutions in absence of oxygen,19 and the 5′S and 5′R diastereomers of the protected 5′,8-cycloadenosine were prepared in a one-pot procedure upon photolysis of 2′,3′-O-isopropylidene-8-phenylthioadenosine derivatives.20
2. Purine 5′,8-cyclo-2′-deoxynucleosides
2.1 Structural features and stabilities of diastereomers
Apart from the usual glycosidic bond, in purine 5′,8-cyclo-2′-deoxynucleosides there is an additional base-sugar linkage between the C8 position of purine and the C5′ position of the 2′-deoxyribose (Fig. 1). Both 5′,8-cyclo-2′-deoxyadenosine (5′,8-cdAdo) and 5′,8-cyclo-2′-deoxyguanosine (5′,8-cdGuo) exist in two diastereomeric forms, which differ from each other in the configuration of the C5′ position. Both sets of diastereomeric forms have been synthesized and fully characterized.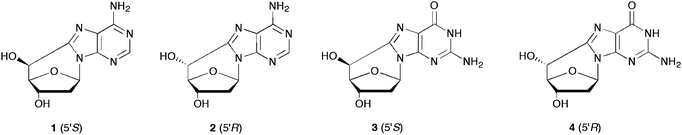 | ||
| Fig. 1 The following abbreviations refer to the four compounds: (5′S)-5′,8-cdAdo (1), (5′R)-5′,8-cdAdo (2), (5′S)-5′,8-cdGuo (3) and (5′R)-5′,8-cdGuo (4). | ||
A detailed NMR characterization is reported for all four compounds by 1H NMR (400 MHz) in DMSO or D2O.21,22 In D2O the chemical shifts are 0.2–0.4 ppm up-field compared to DMSO. It is worth mentioning, that the 5′S and 5′R configurations were assigned by 1H-NMR analysis on the basis of the value of the H4′, H5′ coupling constant. The J4′,5′ value >6 Hz is diagnostic of the (5′S) configuration, since the 4′,5′ dihedral angle is 30°, while a J4′,5′ value <1 Hz is diagnostic for the (5′R) configuration, where the dihedral angle is ∼90°.
In all reported HPLC analyses (reversed phase silica gel column) the 5′R diastereomer of either 5′,8-cdAdo or 5′,8-cdGuo eluted before the related 5′S diastereomer. Structural information of all four compounds was also obtained from multiple fragmentation experiments by mass spectrometry techniques. In the positive ionization mode, the protonated molecular ion (MH+) at m/z 250 for 5′,8-cdAdo and at m/z 266 for 5′,8-cdGuo exhibits a characteristic intense fragmentation at m/z 164 and 180, respectively. In both cases the fragmentation corresponds to the splitting of the 2-deoxyribose moiety, as the result of concomitant C1′–N1 and C4′–C5′ bond cleavages (Fig. 2).23
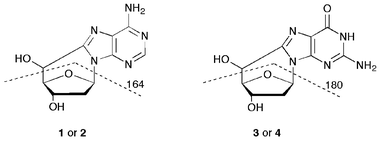 | ||
| Fig. 2 Characteristic fragmentations of the two sets of cyclonucleosides in MS. | ||
Two interesting properties of 5′,8-cdAdo were recently evidenced in aqueous medium. The first pertains the stability of the two diastereomers under acidic conditions24,25 and the second the influence of sun radiation (UVB-radiation) on both 5′R and 5′S diastereomers.26 The glycosidic bond was found to be <40-fold more resistant to acidic hydrolysis than that in 2′-deoxyadenosine (exposure of diastereomer 1 to 66% formic acid at 37 °C for 72 h) and no adenine was released after the glycosidic bond break.24 Similar results are also obtained for 5′R diastereomer 2.25 Under sunlight irradiation, the 5′S diastereomer 1 photoisomerises to the 5′R diastereomer 2, whereas the latter compound does not isomerise under the same conditions.26 This one-way photoisomerization of 1 to 2 has been explained by invoking a heterolytic cleavage of the C–O bond leading to benzyl-type cations, which subsequently undergo nucleophilic trapping by water with concomitant isomerization (Scheme 1). So far no biological implication can be argued, however it is worth noting that isomer 2 is more easily enzymatically repaired when this type of lesion is formed in DNA (vide infra). Density functional theory (DFT) has been used to calculate the geometry, charge distribution and dipole moment of the four compounds in Fig. 1 in gaseous and aqueous phases.27
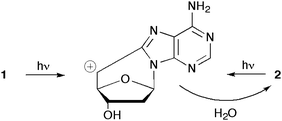 | ||
| Scheme 1 One-way isomerisation of 5′,8-cdAdo under sunlight irradiation. | ||
2.2 The reaction of HO˙ radicals with purine 2′-deoxynucleosides
It is well known that the reaction of HO˙ radicals with 2′-deoxyguanosine (5) occurs mainly at the base moiety by hydrogen abstraction from the exocyclic NH2 group (60–70%) and by addition at the C8 position (ca. 17%).28,29 To a minor extent, hydrogen abstraction at the deoxyribose unit is also reported.22,30 The two diastereomeric forms 5′S and 5′R of 5′,8-cdGuo (3+4) were identified in the γ-irradiation of a N2O-saturated aqueous solution of 2′-deoxyguanosine in a (5′R)/(5′S) ratio of 8.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and in an overall yield of 8–10% (Scheme 2).22 Therefore, the hydrogen abstraction from the C5′ position is the most important path regarding the sugar moiety of the nucleoside.
:1, and in an overall yield of 8–10% (Scheme 2).22 Therefore, the hydrogen abstraction from the C5′ position is the most important path regarding the sugar moiety of the nucleoside.
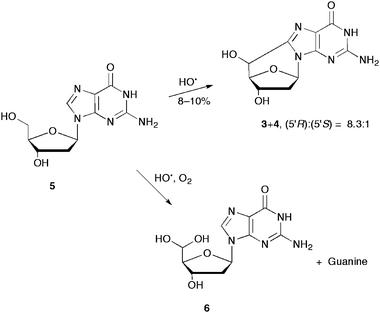 | ||
| Scheme 2 The reaction of hydroxyl radicals with 2′-deoxyguanosine (5) in the presence or absence of molecular oxygen. | ||
The scenario of the reaction of HO˙ radicals with the sugar moiety of 5 in the presence of 2.66 × 10−4 M of O2 changes dramatically (Scheme 2). Indeed, the consumption of 5 was accompanied by the formation of two main products, identified as guanine and the hydrated 5′-aldehyde 6.31 The majority of cyclonucleosides 3+4 were replaced by hydrated 5′-aldehyde 6 (∼8%), which suggests that the oxygen concentration would be high enough to trap most of the C5′ radicals prior to cyclization. In summary, HO˙ radicals are partitioned between the base and sugar moieties of 5 with an overall rate constant of 5.7 × 109 M−1 s−1.29Alkyl radicals derived from the sugar moiety are trapped by oxygen to give hydrated 5′-aldehyde 6via the C5′ radicals, and guaninevia the other sugar radicals (C1′, C3′ and C4′).11
The reaction of HO˙ radicals with 2′-deoxyadenosine (7) occurs with a rate constant of 4.6 × 109 M−1 s−1.14 The most important path is the addition to the base,5 although hydrogen abstraction at the sugar moiety is also documented. The two diastereomeric forms 5′S and 5′R of 5′,8-cdAdo (1+2) were identified in the γ-irradiation of a N2O-saturated aqueous solution of 7 in a (5′R)/(5′S) ratio of 6:1 (Scheme 3).19,31 In the presence of 2.66 × 10−4 M of O2 the consumption of 7 was accompanied by the formation of adenine and the hydrated 5′-aldehyde 8. The cyclonucleosides 1+2 were replaced almost in the same amounts (ca. 11%) by the hydrated 5′-aldehyde 8. The C5′ radicals are trapped by oxygen to give 8 in 11% yield, whereas the remaining C1′, C3′ and C4′ radicals react with oxygen to give a similar amount of adenine (ca. 10%).31
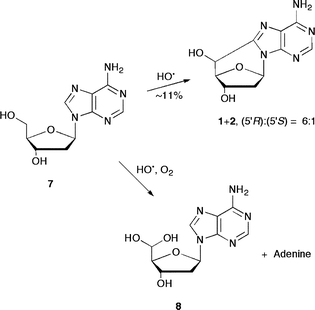 | ||
| Scheme 3 The reaction of hydroxyl radicals with 2′-deoxyadenosine (7) in the presence or absence of molecular oxygen. | ||
2.3 One-pot synthesis by radical cascade
A synthetically useful radical cascade process was developed that allows in a one-pot procedure the conversion of 8-bromo-2′-deoxyadenosine (9) to 5′,8-cdAdo (1+2) as shown in Scheme 4.21,32 In particular, γ-radiolysis of aqueous solutions of 9 and in the presence of K4Fe(CN)6 afforded 5′,8-cdAdo in a 67% yield (based on 38% converted material) and in a diastereomeric ratio (5′R):(5′S) = 6:1.21 Time-resolved spectroscopic studies allowed the reaction mechanism to be defined in some detail and to measure the rate constant for cyclization (see the next section for details). By analogy, the UV photolysis of 9 has been investigated in different solvents and in the presence of additives like halide anions. Photolytic cleavage of the C–Br bond leads to formation of the C8 radical. In methanol, subsequent hydrogen abstraction from the solvent is the main radical reaction; however, in water or acetonitrile, the cyclonucleosides 1 and 2 were obtained. Product studies from steady-state photolysis in acetonitrile have shown the total conversion of 9 to 1+2 in 65% yield and in a diastereomeric ratio (5′R):(5′S) = 1.7:1.32
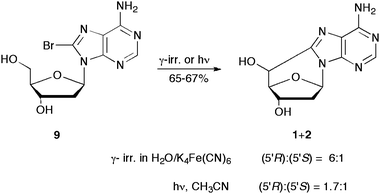 | ||
| Scheme 4 One-pot synthesis of 5′,8-cdAdo from 8-bromo-2′-deoxyadenosine (9). | ||
The analogous reactions with 8-bromo-2′-deoxyguanosine (10) does not work equally well. However, conditions were found that the UV photolysis of 10 in aqueous solution and in the presence of NaI (2 mM) afforded the 5′,8-cdGuo in 26% yield and in a diastereomeric ratio (5′R):(5′S) = 8:1 (Scheme 5).22 For comparison, the analogous reaction with 8-bromo-2′-deoxyinosine (11) is also reported in Scheme 5 affording 5′,8-cdIno in 48% yield and in a diastereomeric ratio (5′R):(5′S) = 4:1.33
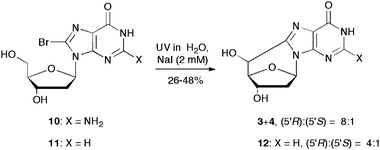 | ||
| Scheme 5 One-pot syntheses of 5′,8-cdGuo (3+4) from 8-bromo-2′-deoxyguanosine (10) and 5′,8-cdIno (12) from 8-bromo-2′-deoxyinosine (11). | ||
The one-pot syntheses described above were also tailored for the reaction of 5′-carbaldehyde 13 with (TMS)3SiH as the mediator for radical reactions (Scheme 6).34,35 The cyclization reaction was performed with 5 equiv. of (TMS)3SiH and 1 equiv. AIBN at 85 °C in 70–75% degassed or aerated conditions. The product 14 consists of a single diastereomer with the 5′S configuration. Photolysis with 254 nm light of 20 mM solution of 14 in a 8:3 CH2Cl2–CH3OH mixture afforded the O5′-desylilated compound 15 and the 2′,5′-dideoxy cyclic derivative 16 in 56% and 14% yields, respectively.
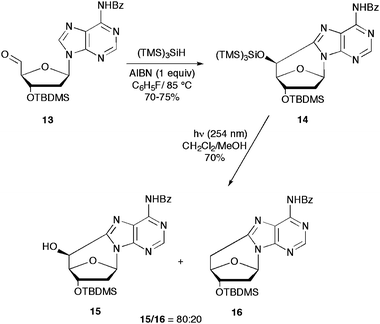 | ||
| Scheme 6 Photolysis-induced deprotection of the cyclopurine derivative 14 derived from radical-based reaction of aldehyde 13 with (TMS)3SiH. | ||
2.4 Selective generation of C5′ radicals and the cyclization step
Specific generation of the C5′ radicals in both pyrimidine- and purine-substituted nucleosides has recently achieved by synthesis of the corresponding 5′-tert-butyl ketones 17 and 20 (Scheme 7 and Scheme 8).36 Photolysis of derivatives 17 and 20 leads to the formation of thymidin-5′-yl (18) and 2′-deoxyguanosin-5′-yl (21) radicals, respectively. In the thymidine system, the C5′ radical 18 is fully quenched to give 19 in the presence of physiological concentrations of thiols. In the 2′-deoxyguanosine system, under similar conditions, the (5′S)-5′,8-cyclo-2′-deoxyguanosine derivative 24 is the obtained product. However, conditions were found in which the C5′ radical 21 is partitioned between two reaction channels, i.e., the reaction with thiol to give 22 and the intramolecular attack onto the C8–N7 double bond of guanine. This approach, known as free-radical clock methodology,37 under pseudo-first-order conditions, allowed the rate constant of this highly stereoselective cyclization 21 → 23 to be estimated and found to be ca. 1 × 106 s−1.36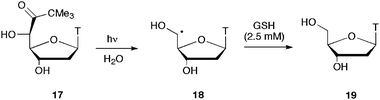 | ||
| Scheme 7 Photolysis of 5′-tert-butyl ketone 17 and quenching of the C5′ radical 18 by glutathione. | ||
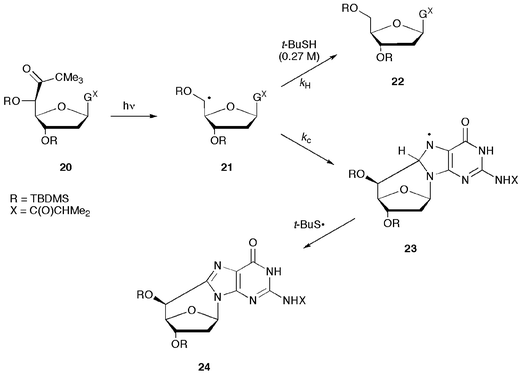 | ||
| Scheme 8 Photolysis of 5′-tert-butyl ketone 20 and the partition of the C5′ radical 21 between hydrogen abstraction from thiol and cyclization to give radical 23. | ||
As reported in the previous section, a one-pot synthesis was developed starting from 8-bromopurine derivatives under continuous radiolysis or photolysis. These procedures involve a radical cascade reaction that mimics the DNA damage causing the formation of 5′,8-cdAdo and 5′,8-cdGuo lesions. The mechanism of these reactions was investigated by time-resolved spectroscopy to provide important information on the fate of C5′ radicals.21,32,38 Some of these findings, summarized in Scheme 9, are as follows: 8-bromo-2′-deoxyadenosine 9 captures electrons and rapidly loses a bromide ion to give the corresponding C8 radical 25.21,38 Radical 25 can also be obtained by homolytic cleavage of a C–Br bond under photolysis.32 This intermediate intramolecularly abstracts a hydrogen atom from the C5′ position, selectively affording the 2′-deoxyadenosin-5′-yl radical 26. Radical 26 undergoes cyclization with a rate constant of kc = (1.6 ± 0.2) × 105 s−1 to give the heteroaromatic radical 27. Similar experiments and results were reported for 2′-deoxyinosin-5′-yl radical and a related rate constant of (1.4 ± 0.2) × 105 s−1 was found.33 It is worth also mentioning that the cyclizations of the ribo-analogues adenosin-5′-yl, inosin-5′-yl, and xanthosin-5′-yl radicals occur with rate constants of one-order of magnitude slower, suggesting conformational changes in going from ribo to 2′-deoxyribo derivatives.39,40
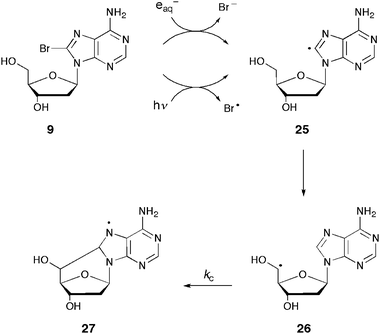 | ||
| Scheme 9 Selective generation of C5′ radical 26via radical translocation and the cyclization step. | ||
The UV-visible spectrum of radical 23 contains two characteristic bands centred at 360 nm with ε = 9600 M−1 cm−1 and at 470 nm with ε = 1500 M−1 cm−1.21 This optical spectrum was computed with the time-dependent DFT method and fitted fairly well with the experimental one. The higher intensity transition was computed at 350 nm and it is due to an excitation from the SOMO localized at the pπ orbital of N7 to the antibonding π* of the N1–C2 double bond (α spin transition). Transition from the out of phase mixing of the pπ N1 and N3 orbitals to the SOMO is computed to occur at higher wavelengths (440 nm) and should be responsible for the weak absorption observed at 470 nm.21
DFT calculations (UB3LYP/6-31G*) on the chair transition states in the pro-(5′S) and pro-(5′R) conformers of the cyclization of 2′-deoxyadenosin-5′-yl radical were carried out in some detail (Fig. 3).21 The reaction barriers (and reaction enthalpies) were calculated to be 8.4 (−9.3) and 13.5 (−11.0) kcal mol−1 for the formation 5′R and 5′S diastereomers, respectively. It is worth mentioning, that the barrier of transition state in the pro-(5′R) arrangement is 4.9 kcal mol−1 lower than the pro-(5′S), due to hydrogen bonding with the oxygen in the sugar ring.
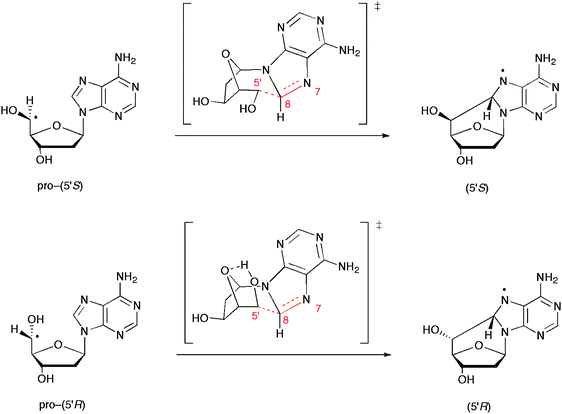 | ||
| Fig. 3 DFT calculations on the chair transition states in the pro-(5′S) and pro-(5′R) conformers of the cyclization. The angle of attack in the transition states are 110.4° and 108.6°, respectively.21 | ||
The cyclization of pro-(5′S) conformer of both 2′-deoxyadenosin-5′-yl and 2′-deoxyguanosin-5′-yl radicals were also studied theoretically using the DFT B3LYP formalism with 6-311++G(d,p) basis set and including CPCM bulk solvation effects.41 The reaction barriers (and reaction enthalpies) were calculated to be 9.4 (−8.8) and 9.7 (−9.7) kcal mol−1 for the Ado and Guo systems, respectively. It was found that the solvation effects on reaction and activation energies of the 5′,8-cdAdo radical formation are significant, but have little influence on the 5′,8-cdGuo radical formation. It is worth mentioning that more than half of the spin density of the unpaired electron in the cyclised radicals is localized on N7, the remaining being delocalized on the six-membered ring.41
A detailed experimental investigation on the stereochemical outcome of the cyclization is also reported.42 In Fig. 4 some examples are selected of 8-Br-dAdo derivatives that produce the corresponding 5′,8-cdAdo upon photolysis. Depending on the substrate and the experimental conditions, the isomeric ratio (5′S)/(5′R) substantially changes in 5′,8-cdAdo derivatives. In CH3CN and for R1 = H the formation of 5′R and 5′S diastereomers is approximately the same. In aqueous solution only the pro-(5′R) conformer can be stabilized by hydrogen bonding, involving either the N3 of the base (29) or the oxygen of the sugar ring (30), as shown in Fig. 5 for 2′-deoxyadenosin-5′-yl radical. The (5′R)/(5′S) isomer ratio of 8:1 were obtained in water for 5′,8-cdGuo. On other hand, bulky substituents like 5′-O-TBDMS prefer to stay in the equatorial position affording a diastereomeric ratio in favour of the (5′S) isomer. The (5′S) stereoselectivity of the cyclization reaction in Scheme 6 and 8 can be explained in the same way.
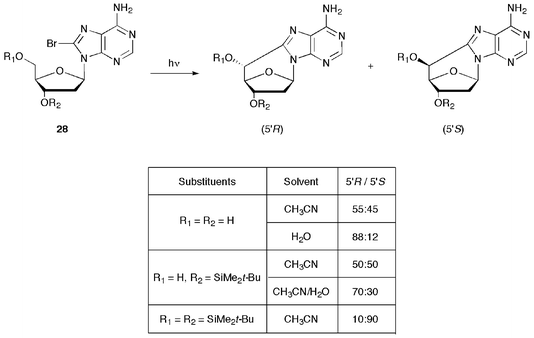 | ||
| Fig. 4 The effect of substituents and solvent in the stereochemistry of cyclization (taken from ref. 42). | ||
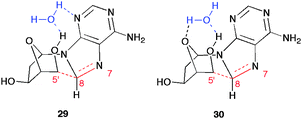 | ||
| Fig. 5 Examples of pro-(5′R) conformer stabilization in aqueous solution by hydrogen bonding. | ||
2.5 Redox reactions of the radicals intermediates involved in the cyclization
The reactivity of 2′-deoxyadenosin-5′-yl radical (26) towards a variety of substrates was studied using pulse radiolysis techniques in competition with the cyclization process, as shown in Scheme 9. In Table 1 are collected the rate constants of the hydrogen abstraction from cysteine (CySH) or glutathione (GSH), the addition to molecular oxygen and one-electron oxidation from K3Fe(CN)6 or MV2+, whereas in Scheme 10 the related reactions are reported.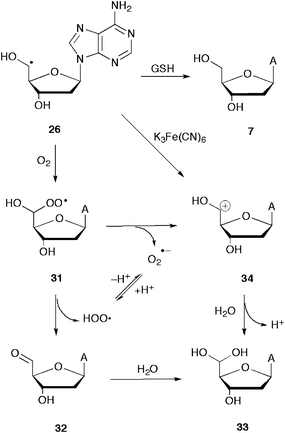 | ||
| Scheme 10 Products derived from reduction or oxidation of the C5′ radical 26 by various trapping agents. The two-oxidation reactions give the same hydrated 5′-aldehyde as the final product. | ||
| Trapping agent | k, M−1 s−1 |
|---|---|
| CySH | (2.1 ± 0.5) × 107 |
| GSH | (4.9 ± 0.6) × 107 |
| MV2+ | (2.2 ± 0.4) × 108 |
| O2 | (1.8 ± 1) × 109 |
| Fe(CN)63− | (4.2 ± 0.4) × 109 |
The reactivity of radical 26 under aerobic conditions has also been elucidated (Scheme 10).31 Using an oxygen concentration in the range of 13–266 μM (typical for oxygenated tissues), the hydrated 5′-aldehyde 33 is accompanied by the 5′,8-cdAdo. The formation of 5′,8-cdAdo was relevant in all experiments, and the yields increased by decreasing O2 concentration. The proposed mechanism involves the formation of peroxyl radical 31 that decays either via a cyclic transition state leading to HOO˙ radical and aldehyde 32, or via heterolytic cleavage to generate the carbocation 34 and superoxide radical anion. The two pathways are formally identical. Trapping C5′ radicals 26 with Fe(CN)63− gave also the corresponding hydrated 5′-aldehydes in good yields (Scheme 10).31 Similar experiments and results were reported for the 2′-deoxyinosin-5′-yl radical.33
The intermediate radical 27 derived from the cyclization of the C5′ radical should be further oxidized in order to afford the final product 5′,8-cdAdo (1+2) (cf.Scheme 11). Information on this elementary step is limited and only very recently the oxidation induced by molecular oxygen was addressed theoretically.23 Hybrid meta DFT calculations were carried out to determine the energy profiles (also in water solution) for this aromatization process in the case of (5′R)-cdAdo. The path shown in Scheme 11 is computed to be highly exothermic (ΔEr = −24.9 kcal mol−1). Hydrogen abstraction (ΔE≠ = 11.6 kcal mol−1) is favoured versus addition. Hydrogen-bonding interaction with one-water molecule lowers sizably the energy barrier for the hydrogen abstraction by molecular oxygen (ΔE≠ = 7.5 kcal mol−1). The rate constant for this process is evaluated to be about 103 M−1 s−1 at room temperature, assuming a “normal” frequency factor (logA/M−1 s−1 = 8.5) for the bimolecular process. The energy barrier and the reaction energy are also computed for the reaction of superoxide radical anions O2˙− with radical 27 and found to be −33.6 and 20.4 kcal mol−1, respectively.23 Radical 27 was found to react promptly with the oxidant Fe(CN)63−, with a rate constant of (8.3 ± 0.3) ×108 M−1 s−1 to give the cation 34 that deprotonates rapidly affording the 5′,8-cdAdo.23
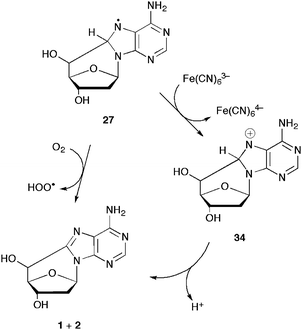 | ||
| Scheme 11 Oxidation steps for the aromatization of radical 27. | ||
3. Multistep synthesis approach to oligonucleosides
The first attempt for the construction of modified oligonucleotides (3′ → 5′ DNA synthesis) concerned the synthesis of (5′S)- and (5′R)-5′,8-cdAdo phosphoramidite synthones following a methodology developed by Matsuda et al.43 for the synthesis of ribo-analogues (5′,8-cAdo).44,45 It involved a multistep synthesis (Scheme 12), starting from N6-benzoyl-2′-deoxyadenosine 31, transformed to N6-benzoyl-5′-tosyl-2′-deoxyadenosine and, consecutively, to N6-benzoyl-5′-thiophenyl-2′-deoxyadenosine (36). After UV-irradiation of 36 (λ = 254 nm) under argon atmosphere and protection of 3′-hydroxyl group with TBDMS-Cl, N6-benzoyl-2′,5′-dideoxyadenosine (37) was obtained. Oxidation with selenium dioxide and selective reduction of C5′ carbonyl group by NaBH4 afforded 38. Protection of the 5′-hydroxyl group with DMTr-Cl, selective removal of the 3′-O-TBDMS protective group and coupling of the free secondary 3′-hydroxyl group with 2-cyanoethyl N,N-diisopropylphosphoramidite chloride in presence of N,N-diisopropylammonium tetrazolate, finally generated the phosphoramidite 39. The overall yield from 35 to 39 was 3.5%. The synthesis of (5′R) diastereomer 40 was obtained by inversion of the C5′ configuration of compound 38. Mesylation and subsequent treatment with potassium dioxide (Corey method) followed by tritylation of the obtained compound 40, selective removal of 3′-O-TBDMS protective group by TBAF and phosphitylation, finally gave the phosphoramidite 41. The overall yield from 35 to 41 was 1.6%. A similar approach was followed by the same research group for the synthesis of the (5′S)-5′,8-cdGuo (42) and (5′R)-5′,8-cdGuo (43) phosphoramidite synthones in overall yields of 4.0% and 2.7%, respectively (Fig. 6).45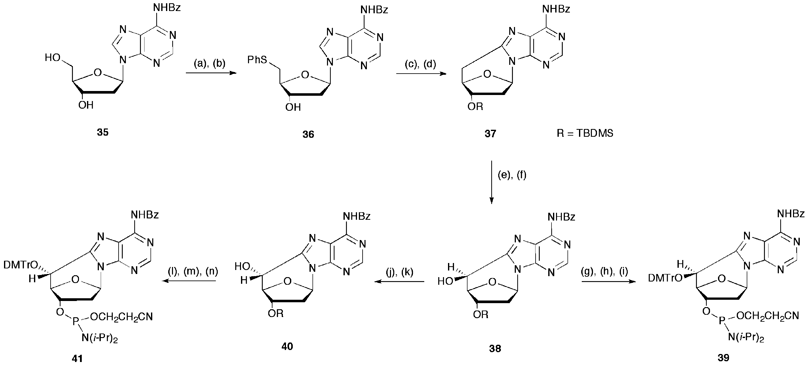 | ||
| Scheme 12 Synthesis of (5′S)-5′,8-cdAdo (39) and (5′R)-5′,8-cdAdo (41) phosphoramidite synthones (refs. 44 and 45). Steps: (a) TsCl, py, −20 °C, 24 h, 82%; (b) PhSH, CH3ONa, CH3OH, reflux, 2 h, 51%; (c) hν (254 nm), (EtO)3P, CH3CN, Ar, 10 h, 51%; (d) TBDMSCl, imid, DMF, 20 h, 79%; (e) SeO2, 1,4-dioxane, reflux, 30 min, 87%; (f) NaBH4, CH3OH, 1 h, 90%; (g) DMTrCl, py, 70 °C, 4 h, 60%; (h) TBAF, THF, 4 h, 72%; (i) iPr2NP(Cl)O(CH2)2CN, N,N′-diisopropylammonium tetrazolate, CH2Cl2, Ar, 4 h, 62%; (j) MsCl, Et3N, CH2Cl2, −20 °C, 1 h, 94%; (k) KO2, 18-crown-6, DMSO/DME 1:1, 0 °C, 20 min, 58%; (l) DMTrCl, py, 80 °C, 4 h, 47%; (m) TBAF, THF, 4 h, 59%; (n) iPr2NP(Cl)O(CH2)2CN, DIEA, CH2Cl2, Ar, 4 h, 80%. | ||
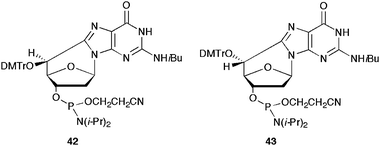 | ||
| Fig. 6 (5′S)-5′,8-cdGuo (42) and (5′R)-5′,8-cdGuo (43) phosphoramidite synthones. | ||
For the reverse oligonucleotide synthesis (5′ → 3′), Brooks et al.46 prepared the 5′-O-phosphoramidite 48 by photochemical conversion of a derivatized 5′-phenylthio-2′,5′-dideoxyadenοsine (45). Molecular model studies indicated that the achievement of the insertion of the bulky 4,4′-DMT group onto the (5′S)-hydroxyl group could have been difficult. Therefore, the strategy involved protection by the TBDMS group of the 3′-hydroxyl group of 5′-O-dimethoxytrityl-2′-deoxyadenosine (44), the removal of the 5′-O-dimethoxytrityl group, and conversion of the 5′-OH to 5′-SPh, affording 5′-phenylthio-3′-O-TBDMS-2′-,5′-dideoxyadenosine (45). This thio-derivative was submitted to photolysis at 254 nm under an argon atmosphere in the presence of trimethyl phosphite. The oxidation by treatment with selenium dioxide in refluxing pyridine gave compound 46. Selective reduction of the carbonyl group of 46 by KBH4 led to the (5′S)-8,5′-cyclo-2′-deoxyadenosine 3′-O-TBDMS ether (47). Treatment with dimethylformamide dimethyl acetal for the protection of the amino group and phosphitylation of the 5′-hydroxyl group using 2-cyanoethyl N,N-diisopropylphosphoramidite chloride in the presence of N,N-diisopropylammonium tetrazolate completed the synthesis (Scheme 13). The overall yield from 44 to 48 was 13%.
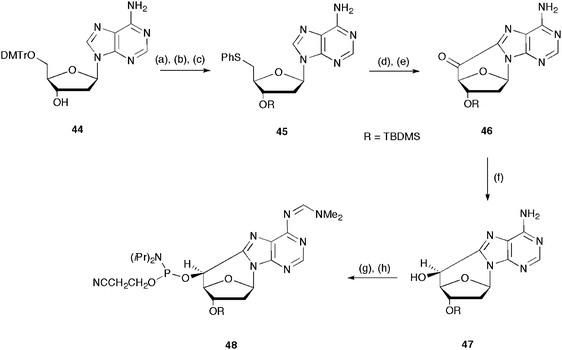 | ||
| Scheme 13 Synthetic steps for the preparation 5′-O-phosphoramidite 48 utilized for the reverse (5′ → 3′) oligonucleotide synthesis (ref. 46). Steps: (a) TBDMSCl, DBU, CH3CN, rt, 73%; (b) I2, MeOH, 30–40 °C, 64%; (c) PhSSPh, (n-Bu)3P, DMF, 95%; (d) hν (254 nm), P(OMe)3, CH3CN, Ar, 50%; (e) SeO2, py, reflux, 5 h, 92%; (f) KBH4, MeOH, 2 h, rt, 72%; (g) DMF dimethyl acetal, MeOH, 14 h, rt, 93%; (h) iPr2NP(Cl)[O(CH2)2CN, DIEA, THF, 0–25 °C, 2.5 h, 95%. | ||
The effect of (5′S)-5′,8-cdAdo on the conformation of di- and trinucleotides was investigated by NMR and DFT calculations.47 The compound 5′-TX-3′ and 5′-TXT-3′ where X = (5′S)-5′,8-cdAdo were synthesized using the classical phosphoramidite strategy reported above. It is reported that the C5′–C8 bond induces an unusual West (0T1) conformation of the furanose ring that is independent of the sugar substituents. It is postulated that the rigid structure of (5′S)-5′,8-cdAdo can strongly influence the global geometry of oligonucleotides.
4. Biological features of (5′S)- and (5′R)-5′,8-cyclo-2′-deoxyadenosine lesions
DNA is protected by a variety of repair systems, including nucleotide excision repair (NER) and base excision repair (BER). These two systems remove the majority of DNA lesions.3,48DNA damage produced by reactive oxygen species, hydrolysis, and methylating agents is removed primarily by the BER pathway. On the other hand, major helix-distorting lesions, like cyclobutane pyrimidine dimers (CPDs) or bulky adducts, are corrected by the NER pathway.We reported above that when DNA is exposed to hydroxyl radical (HO˙), hydrogen abstraction from the C5′ position of the sugar is one of the possible events leading to the formation of purine 5′,8-cyclonucleoside lesions. Are these lesions substrates for BER and/or NER? Due to expected distortion of the DNA structure and the presence of a covalent bond between the C5′ and C8 positions, intuitively the answers is yes for NER and no for BER. Indeed, NER represents the only pathway currently identified for the repair of these lesions.
In 2000, two groups independently reported on the biological aspects of 5′,8-cdAdo lesions,46,49 addressing two main questions: (i) can the purine cyclonucleotide lesions be recognized and repaired? (ii) can DNA synthesis involving polymerases proceed with these lesions?
The first group synthesized plasmid DNA from the modified oligonucleotide sequences containing either the (5′R)-5′,8-cdAdo or (5′S)-5′,8-cdAdo diastereomer lesion,49 and tested for glycosylase and nuclease assays. First, the substrate was tested with HeLa cell protein extract searching for the DNA glycosylase activity. No indication of such hydrolytic activity was obtained. This confirmed that the 5′,8-cdAdo lesions cannot be cleaved at the glycosidic bond, which should have rendered the hydrolysis product eventually processed by nuclease enzymes. It is also worth noting that from these first studies the possibility of a direct cleavage of the 5′,8 bond of cyclonucleotides was ruled out, which renders more intriguing the role of this free radical damage in the biological environment. On the other hand, a 7059bp plasmid DNA containing either the 5′R or 5′S diastereomer of 5′,8-cdAdo was exposed to the nuclease activity of the cellular extract. Human DNA glycosylases active in the base excision repair (BER) pathway were unsuccessful, whereas both diastereomers can be removed by NER, with more efficiency for the 5′R diastereomer than the corresponding 5′S.49 The efficiency was always tested in comparison with the recognition of the other bulky CPD lesion, and experimental evidence indicates that the efficiency of human NER for CPDs and 5′,8-cdA is similar.49
The second group studied more particularly the (5′S)-5′,8-cdAdo lesion inserted in duplex DNA substrates,46 also reporting that this lesion is repaired by NER and not by BER. The presence of a single (5′S)-cdAdo on the transcribed strand of a gene was found to be a strong block to gene expression in Chinese hamster ovary and human cells, with efficiency comparable to CPDs. This effect of gene expression blockage can be meaningful for those patients suffering from xeroderma pigmentosum (XP) and displaying a severe NER defect; indeed it can be hypothesized that the accumulation of the purine cyclonucleoside lesions in an active gene stops the transcription and gives the basis for the neurodegenerative consequences associated to the XP disease. The molecular basis of this working hypothesis was actually tested in human cells transfected with the 5′,8-cdAdo lesion to an active gene, before measuring its activity through a modified luciferase response, as well as directly monitoring the transcriptional activity of cells from a XP patient suffering from neurodegeneration since childhood.46
The fact that there are different polymerase enzymes that can work with modified DNA is another interesting aspect addressed with the purine 5′,8-cyclonucleoside damage. The latter lesions are interesting for modelling the interactions of the different diasteromers and allow gaining interesting insights in the preferred conformational requirements for enzyme recognition. The presence of any of the two diastereomers of 5′,8-cdAdo was found to hamper the primer extension by T7 DNA polymerase and mammalian DNA pol δ.49 In case of the T7 DNA polymerase, a residue opposite to the lesion could be incorporated and a slight read-through activity was seen with the 5′R diastereomer. On the other hand, with DNA pol δ the blockage occurred before the 5′,8-cdAdo lesion and no read-through activity was observed. The role of by-pass polymerases is highlighted because of their ability to overpass bulky damage, as it has been described in the case of CPD lesions.50 Therefore, stereospecific differences between the 5′S and 5′R diastereomer residues can affect not only the relative resistance to exonucleolytic activity, but lead to different efficiencies in translesion synthesis by translesional enzymes, such as human polymerase η.51 As a matter of fact, pol η was able to catalyze translesion synthesis of the 5′R diastereomer containing a DNA strand, whereas the 5′S diastereomer was a blocking lesion, thus preventing chain elongation while allowing incorporation of one nucleotide opposite to the purine cyclonucleoside.
Another biological activity, associated with the blocking effect on gene transcription by purine cyclonucleosides, was explored by examining the blockage of protein binding, which is involved in a transcription factor binding. (5′S)-5′,8-cdAdo containing DNA sequences were used for this purpose and the alteration of the TATA binding protein functionality was studied in vivo; this was achieved using NER-deficient cells which are unable to repair the lesion.52 A single 5′,8-cdAdo residue could strongly disrupt binding of the TATA binding protein to the TATA box. Transfection of the lesion in SV40 transformed fibroblasts from a XP patient, was found to lead to a reduction of the gene expression of ca. 70%.
In a recent study,53 the levels of 5′S and 5′R diastereomers of 5′,8-cdAdo in wild-type, neil1−/−and ogg1−/− mice have been measured (see the next section for details of quantitative determination of these lesions in DNA). The levels of these lesions were shown a significant accumulation in neil1−/−mice suggesting that NEIL1 plays a role in the cellular repair of (5′S)- and (5′R)-5′,8-cdAdo. Since these lesions are repaired by NER and not by BER, it is proposed that NEIL1 is involved in the removal of nucleotides in addition to its function as a DNA glycosylase in BER.
It is worth mentioning that the biological effects of the 5′R and 5′S diastereomers of 5′,8-cdGuo are limited to unpublished results reported in a review article.54 Both diastereomers can be bypassed by the bacterial polymerases during DNA replication, giving rise to a preferential adenine incorporation in front of the lesion indicative of the likely occurrence of G > T transversions.
In his recent reviews, Brooks discussed the evidence that these lesions may be in part responsible for the neurodegeneration that occurs in some XP patients.55,56
5. Evaluation of DNA damage
5.1 Quantitative determination of purine 5′,8-cyclo-2′-deoxynucleosides lesions
One of the first evidence for the presence of purine 5′,8-cyclonucleotides as damage biomarkers in mammalian tissues came from the chromatographic separation and tentative identification of dinucleotides containing the 5′,8-cdAdo (i.e., 5′-AX-3′, 5′-CX-3′, 5′-GX-3′, 5′-TX-3′, where X = 5′,8-cdAdo without addressing the diastereomeric ratios), after digestion and 32P-postlabeling of rat (or mice) genetic material.57 The digestion of DNA to dinucleotides was obtained by micrococcal nuclease plus spleen phosphodiesterase followed by a short treatment with nuclease P1. The 5′,8-cdAdo containing dinucleotides were proposed to be part of the type II–I compounds, which are specifically known to increase in vivo by exposure of mammals to oxidative stress conditions, and to be repaired by NER but not BER. Levels of 5′,8-cdAdo in fetal and postnatal rat liver were in the order of 180–320 lesion/cell, which is 1–2 orders of magnitude lower than 8-oxo-dGuo. Indeed, the 32P-postlabeling approach is a very sensitive methodology allowing the detection of 1–5 lesions in 1010 normal nucleotides in small DNA samples lower than 1 μg, a low-limit detection that cannot be reached even with the modern mass spectroscopic techniques. Levels of hepatic and renal type II–I compounds, including 5′,8-cdAdo containing dinucleotides, were generally higher in NQO1-null than wild-type mice (NQO1 plays an important role in the protection against oxidatively generated genetic damage and carcinogenesis).58 It is worth mentioning that levels of total type II–I compounds in liver and kidney of untreated rats displayed a gradual 2- to 3-fold increase from ages 1 to 24 months.58Attempts to accurately determine the level of these lesions in DNA by enzymatic digestion followed by chromatographic methods coupled to mass spectrometric (MS) analyses are numerous. The reports along the years account for different results, even when identical experimental conditions were used. In Table 2 are collected all the data available in the literature regarding the measurement of 5′,8-cdAdo and 5′,8-cdGuo in gamma-irradiated samples of calf thymus DNA in aqueous solutions. Data are reported in term of frequency of lesions/106nucleosides/Gy, since in most cases a linear dependence of lesions/106nucleosidesvs. dose has been reported. For each experiment, the dose range and degassing conditions for the preparation of the sample are given. The diastereomeric outcomes 5′R/5′Sare also reported when available. As it can be seen, the actual scenario is quite confusing. Most of the results were obtained by Dizdaroglu and co-workers (Table 2, normal font)30,59–64 starting from the middle of the eighties, and very recently by others (Table 2, bold font).23 The latest results are quite consistent with the yield of radiation-induced HO˙ radicals: γ-radiolysis of N2O-saturated aqueous solutions produces twice of HO˙ than in solutions deaerated by a stream of N2. Indeed, the yields of both 5′,8-cdAdo and 5′,8-cdGuo are doubled going from N2- to N2O-saturated solutions. The yield of 5′,8-cdGuo was found to be higher than that of 5′,8-cdAdo, the ratio of formation between 5′,8-cdGuo and 5′,8-cdAdo being ∼1.5. It is worth underlining that a major discrepancy can be found between some previously data reported (normal font) and the most recent one (bold font): ∼20-fold excess of lesions/106nucleosides/Gy for 5′,8-cdAdo and ∼5.5-fold excess for 5′,8-cdGuo have been measured in the latest work.
| Yeara | Methodb | Conditionsc | 5′,8-cdAdo | 5′,8-cdGuo | Ref. | ||
|---|---|---|---|---|---|---|---|
| Lesions/106Nu/Gy | 5′R/5′S | Lesions/106Nu/Gy | 5′R/5′S | ||||
| a Publication year. b In GC-MS the samples were trimethylsilylated prior to the analysis. c Used gas and irradiation dose. d LC/MS and GC-MS yielded similar results. e Only 5′S is reported. | |||||||
| 1986 | GC-MS | N2O, 400 Gy | ∼3:1 |
30 | |||
| 1988 | GC-MS | N2O, 0–40 Gy | ∼0.5 | 0.8:1 |
∼0.4 | 0.8:1 |
59 |
| 2001 | GC-MS | N2O, 0–80 Gy | 0.70 | ∼2:1 |
60 | ||
| 2001 | LC/MS | N2O, 0–80 Gy | 0.65 | ∼2:1 |
60 | ||
| 2002 | GC-MS | N2O, 0–40 Gy | 0.70 | 3.51 | 0.28:1 |
61 | |
| 2002 | LC/MS | N2O, 0–40 Gy | 0.65 | 3.37 | 0.32:1 |
61 | |
| 2003 | LC/MS d | N2O, 0–40 Gy | ∼1.3 | 3:1 |
∼3.9 | 0.24:1 |
62 |
| 2004 | LC/MS | N2O, 2 Gy | 0.80 | e | 63 | ||
| 2009 | LC/MS/MS | N2, 0–60 Gy | 1.23 | 0.45:1 |
64 | ||
| 2010 | LC/MS/MS | N2, 0–60 Gy | 6.5 |
∼4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
11.1 |
∼3:1
|
23 |
| 2010 | LC/MS/MS | N2O, 0–60 Gy | 14.2 |
∼4:1
|
20.1 |
∼3:1
|
23 |
The effect of molecular oxygen on the formation of purine 5′,8-cyclonucleosides in gamma-irradiated samples of calf thymus DNA in aqueous solutions has also been addressed.23Fig. 7 shows the results of the irradiation of DNA aqueous solutions saturated with mixtures of N2/O2 in different proportions. The formation of both 5′,8-cdGuo and 5′,8-cdAdo decreases with the increase of the oxygen concentration, indicating competition paths of the C5′ radical between cyclization and addition to oxygen (Scheme 14). However, the formation of both 5′,8-cdAdo and 5′,8-cdGuo is still operating and significant in all the experiments, even under high oxygen concentration (0.3 mM). This contrasts with early data that excluded the occurrence of cyclization in the presence of oxygen. It is interesting to evaluate that the formation of 5′,8-cdGuo decreases faster than 5′,8-cdAdo with the increase of oxygen concentration, suggesting different rates of cyclization for the corresponding C5′ DNA radicals (Scheme 14).
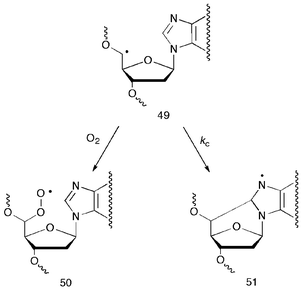 | ||
| Scheme 14 Partition of C5′ DNA radical 49 between addition to oxygen and cyclization to give radicals 50 and 51, respectively. | ||
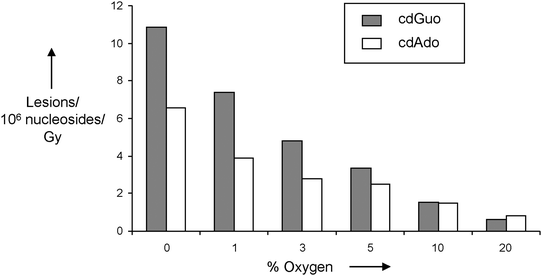 | ||
| Fig. 7 Radiation-induced formation of 5′,8-cdGuo and 5′,8-cdAdo in N2/O2 saturated aqueous solutions of DNA with increasing amounts of oxygen. Taken from ref. 23. | ||
The most incongruent experimental results shown in Table 2 concern the 5′R/5′S ratios, changing from 3:1 to 0.45:1 for 5′,8-cdAdo, and from 3:1 to 0.24:1 for 5′,8-cdGuo within the reported values even from the same laboratory. The conformational restrictions in double-stranded DNA should result in an average diastereomeric ratio of both 5′,8-cdAdo and 5′,8-cdGuo that is expected to be nearly constant for similar experiments. On the other hand, the 5′R/5′S ratio was found to be constant within experimental errors for both purine 5′,8-cyclo-2′-deoxyribonucleosides in all the reported experiments in Fig. 7, with the 5′R diastereomer predominantly formed leading to a ratio 5′R/5′S of ∼3 and ∼4 for 5′,8-cdGuo and 5′,8-cdAdo, respectively.
Many factors are involved in the accurate detection of the cyclonucleoside lesions and their diastereomeric ratios. Further efforts are required to unequivocally perform these measurements. In their paper of 2009, Dizdaroglu and co-workers found a value of 5′,8-cdAdo lesions using LC/MS/MS, which is 4-fold higher than their previous values detected by GC-MS or LC/MS (taking into consideration the HO˙ radical yield).64 Furthermore, this value is not far from that of 6.5 obtained by Cadet and co-workers under similar irradiation conditions and type of analytical tool.23 On the other hand, the discrepancy on the 5′R/5′S ratio (0.45 vs. 4) is more pronounced, suggesting that an important step to be considered concerns the release of the (5′R)-5′,8-cdAdo lesion by one of the enzymatic systems used in the digestion procedure. Indeed, there is some controversy in the literature regarding the ability of different enzymes to release cyclonucleoside lesions.
Early studies used a combination of four enzymes nuclease P1, snake venom phosphodiesterase, calf spleen phosphodiesterase and alkaline phosphatase.30,59–62 Later, it was reported that (5′S)-5′,8-cdAdo containing oligonucleotides do not release completely this lesion as a nucleoside using similar combination of enzymes.44Dinucleotides and oligonucleotides containing (5′S)-5′,8-cdAdo were further used for determining the hydrolysis efficiency with nuclease P1, snake venom phosphodiesterase, and alkaline phosphatase. The dinucleotide 5′-TX-3′ where X = (5′S)-5′,8-cdAdo was the most resistant to hydrolysis which was incomplete (50%) after 48 h of incubation.63 These data suggest that the conditions of release of cyclonucleoside lesions upon enzymatic digestion must be carefully evaluated, since each nucleotide sequence and each diastereomer might follow its specific kinetics, which is not yet known. Therefore the enzymatic digestion tailored for the (5′S)-5′,8-cdAdo moiety may not be the same of other cyclonucleoside lesions and the extrapolation by analogy is likely to be incorrect, thus affecting the quantitative final evaluation of the DNA damage.
An important step is to ascertain the complete release of all four cyclonucleoside lesions by DNA enzymatic digestion, that has been addressed only very recently.23 In this study the digestion was optimized using the diastereomers of 5′,8-cdAdo and 5′,8-cdGuo in oligosequences as reference, and a three step procedure was carried out: (i) digestion started by a mixture of two endonucleases (nuclease P1 and DNAse II) to which a 3′-exonuclease (phosphodiesterase II) was added; (ii) a 5′-exonuclease was then used (phosphodiesterase I) together with alkaline phosphatase in order to convert nucleotides into the corresponding nucleosides, and (iii) the first step using nuclease P1, DNAse II and phosphodiesterase I was repeated. It is important to point out that, treating biological samples by this procedure, the 5′R diastereomers resulted in being the main lesions instead of the 5′S forms,23 in contrast to what was reported previously (Table 2).
In summary, the data of early work in Table 2 should be taken with some precaution, checking the detection methodology together with the quantitative recovery of the nucleosides after digestion. The LC/MS/MS analytic methodology is the most appropriate one, and the liquid chromatography/isotope dilution tandem mass spectrometry methodology, using the 5′R and 5′S diastereomers of [15N5]labeled 5′,8-cdAdo as references, is a highly accurate tool, providing that the processing of the sample has occurred quantitatively. In particular, the hydrolytic pathway for the 5′R diastereomer of 5′,8-cdAdo has not yet been defined, in order to show whether there are differences in the enzyme response.
There are also several articles that report attempts to accurately determine the level of purine cyclonucleoside lesions in DNA in different non-irradiated biological samples. Most of the works deals with the (5′S)-5′,8-cdAdo, although sporadically reports on (5′R)-5′,8-cdAdo and the two diastereomers of 5′,8-cdGuo have also appeared. For example, the levels of (5′S)-5′,8-cdAdo were found to be between 0.26–0.70 lesions/107nucleosides in human blood65 or breast66 and mouse liver67 using the LC/MS technique. Moreover, the levels (lesions/107nucleosides) of 5′S and 5′R diastereomers of cdAdo were shown to be 0.95 and 0.40, respectively, in calf thymus DNA, and 0.48 and 1.13 in mouse liver DNA using LC/MS/MS coupled with isotope dilution methodology.64 Also, the levels of (5′S)-5′,8-cdAdo and both 5′R and 5′S diastereomers of 5′,8-cdGuo were found to be between 0.2–3.0 lesions/106nucleosides in the DNA of human keratinocytes and to increase significantly upon X-rays exposure of 5 Gy.68,69 Indeed the yields of radiation-induced cyclonucleosides per 109 normal nucleosides and per Gy were found to be 14, 100 and 30 for (5′S)-5′,8-cdAdo and the 5′S and 5′R diastereomers of 5′,8-cdGuo, respectively. The radiation-induced yields of cyclonucleosides in cellular DNA have been also recently evaluated by using the effective DNA enzymatic digestion described above.23 Focusing on the 5′R diastereomer of 5′,8-cdAdo by using the isotope dilution technique in combination with LC/MS/MS, the DNA of human monocytes was analyzed prior and after exposure to ionizing radiation. The (5′R)-5′,8-cdAdo in untreated cells was found to be lower than the limit of detection (estimated to be around 0.1 lesion/109nucleosides), whereas its yield after 2 kGy of irradiation, which is biologically irrelevant, was estimated to be 0.2 lesion/109nucleosides/Gy.23 This level of lesions is at least two orders of magnitude lower than the radiation-induction formation yields of both 5′,8-cdAdo and 5′,8-cdGuo that was previously measured68,69 using the LC/MS. This may be explained by the fact that the latter method is not accurate when high sensitivity is required to detect lesions within the subfemtomole range.70 The latest findings of purine cyclonucleosides directly detected in urine samples of healthy volunteers, as free nucleosides identified by LC/MS/MS with isotope dilution, is going to open a large debate on the origin of these lesions and their use as predictive biomarkers for the level of free radical stress in different health conditions.71
5.2 Comparison with 8-oxo-purine lesions
In this context, a comparison with other well-known purine lesions, like 8-oxo-7,8-dihydro-2′-deoxyadenosine (8-oxo-dAdo) and 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dGuo), provides further mechanistic insights. Table 3 shows the data obtained for the various lesions from the irradiation studies of calf thymus DNA.23 The same order 8-oxo-dGuo > 5′,8-cdGuo > 5′,8-cdAdo > 8-oxo-dAdo is observed in N2-deareated or in N2O-saturated aqueous solutions. As expected, the lesions are doubled in N2O because the concentration of HO˙ radicals is double. This behaviour is in accordance with the occurrence of a competition between HO˙ addition at C8 and hydrogen abstraction from C5′, the product ratio being proportional to the relative rate constants. In the presence of molecular oxygen the scenario changes since the formation of purine cyclonucleosides decreases dramatically, whereas the frequency of 8-oxo-7,8-dihydropurine derivatives increases substantially. Indeed, oxygen intercepts the C5′ radical prior to cyclization as reported in Scheme 14, whereas the addition of HO˙ radicals is independent from the presence or absence of oxygen. The presence of oxygen only facilitates the subsequent oxidation to give the 8-oxo-7,8-dihydropurine derivatives (cf.Scheme 11).It is also worth mentioning that the radiation-induced formation of 8-oxo-dGuo and 8-oxo-dAdo,72,73 in the DNA of human monocytes has been determined by LC/MS/MS to be 20 and 4 lesions/109nucleosides/Gy, respectively. These lesions are 1–2 orders of magnitude higher than the amount of (5′R)-cdAdo reported above under similar conditions and fit very well with the data of isolated DNA irradiated in the presence of oxygen shown in Table 3. Moreover, the observed decrease by about three orders of magnitude of the radiation yields of (5′R)-cdAdo, 8-oxo-dGuo and 8-oxo-dAdo in cellular DNA with respect to isolated DNA is not a surprise, due to the cellular content that leads to efficient scavenging of HO˙ and for the so-called non-enzymatic fast repair.74
6. Conclusive remarks
The chemical biology approach used for studying purine 5′,8-cyclonucleoside lesions has brought significant achievements so far. The site-specific generation of sugar radicals has been the key approach for a better understanding of chemical molecular mechanisms occurring at the biological level. The chemistry of the C5′ radicals in the purine nucleosides is fairly well understood and it is now clear that the fate of the C5′ radicals is partitioned between unimolecular processes (cyclizations) and bimolecular processes (reactions with oxygen, thiols, or oxidants). Therefore, the local concentration of these components and pH are extremely important in selecting the preferred pathway.From the biological studies achieved so far, several important aspects emerge: (i) the synthetic access to the lesions with the complete library of purine cyclonucleosides should represent a powerful tool for further development of biophysical, biochemical and analytical methods; (ii) the enzymatic digestion involving all four possible purine cyclonucleoside lesions in synthetic oligonucleotides should be investigated deeply; (iii) the biological importance of these lesions should be mostly linked to a deficiency of the repair systems (such as in XP patients) therefore, further studies should be devoted to prevent accumulation that can be harmful for cell replication, and (iv) it should be advisable to unify the methodology used by different laboratories to identify and quantify properly these lesions in order to fully demonstrate their importance in vivo.
In the previous sections, we critically reviewed the quantitative determination of purine 5′,8-cyclo-2′-deoxyribonucleoside lesions. The underlined discrepancies between earlier and recent data indicate that any speculations on the potential involvement of these lesions in human health and diseases are premature.71,75 In order to assess the role of these lesions in oxidative stress-related diseases it would be useful to develop assays with high sensitivity and easy to perform on biological specimens for clinical screenings. In the middle eighties Fuciarelli et al. reported an enzyme-linked immunosorbent assay (ELISA) with specificity for 5′,8-cycloadenosine-5′-monophosphate (5′,8-cAMP) and its 2′-deoxy analog 5′,8-cdAMP.76 The formation of 5′,8-cdAMP was detected in native DNA. Moreover, the yields of 5′,8-cdAMP was found to be 2–3 times higher in irradiated ds-DNA than in ss-DNA. In this view, efforts devoted to the implementation of immuno assays, based on the antibody response using the purine cyclonucleoside library as antigens, should be potentiated. However this may represent a challenging issue since very high sensitivity is required to detect very low amounts of purine cyclonucleosides.
Acknowledgements
We thank Dr Jean Cadet for helpful discussions. The support and sponsorship by COST Action CM0603 on “Free Radicals in Chemical Biology (CHEMBIORADICAL)” are kindly acknowledged.References
- B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine, University Press, Oxford, 4th edn, 2007 Search PubMed.
- C. C. Witherbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS.
- E. C. Friedberg, G. C. Walker and W. Siede, DNA Repair Mutagenesis, ASM Press, Washington, 1995 Search PubMed.
- J. C. Delaney and J. M. Essigmann, Chem. Res. Toxicol., 2008, 21, 232–252 CrossRef CAS.
- C. von Sonntag, Free-Radical-Induced DNA Damage and Its Repair: A Chemical Persective, Springer-Verlag, Berlin, 2006 Search PubMed.
- P. C. Dedon, Chem. Res. Toxicol., 2008, 21, 206–219 CrossRef.
- D. Sy, C. Savoye, M. Begusova, V. Michalik, M. Charlier and M. Spotheim-Maurizot, Int. J. Radiat. Biol., 1997, 72, 147–155 CrossRef CAS.
- B. Balasubramanian, W. K. Pogozelski and T. D. Tullius, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9738–9743 CrossRef CAS.
- W. Chan, B. Chen, L. Wang, K. Taghizadeh, M. S. Demott and P. C. Dedon, J. Am. Chem. Soc., 2010, 132, 6145–6153 CrossRef CAS.
- B. Aydogan, D. T. Marshall, S. G. Swarts, J. E. Turner, A. J. Boone, N. G. Richards and W. E. Bolch, Radiat. Res., 2002, 157, 38–44 CrossRef CAS.
- C. Chatgilialoglu, in Radical and Radical Ion Reactivity in Nucleic Acid Chemistry, ed. M. M. Greenberg, Wiley, New Jersey, 2009, pp. 99–133 Search PubMed.
- A. C. Bryant-Friedrich, Adv. Mol. Toxicol., 2010, 4C, 127–155 Search PubMed.
- C. von Sonntag, The Chemical Basis of Radiation Biology, Taylor & Francis, Philadelphia, 1987 Search PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CAS.
- K. Keck, Z. Naturforsch., 1968, 23B, 1034–1043 Search PubMed.
- J. A. Raleigh, W. Kremers and R. Whitehouse, Radiat. Res., 1976, 65, 414–422 CrossRef CAS.
- J. A. Raleigh and A. F. Fuciarelli, Radiat. Res., 1985, 102, 165–175 CrossRef CAS.
- T. P. Haromy, J. Raleigh and M. Sundaralingam, Biochemistry, 1980, 19, 1718–1722 CrossRef CAS.
- N. Mariaggi, J. Cadet and R. Téoule, Tetrahedron, 1976, 32, 2385–2387 CrossRef CAS.
- A. Matsuda, M. Tezuka and T. Ueda, Tetrahedron, 1978, 34, 2449–2452 CrossRef CAS.
- C. Chatgilialoglu, M. Guerra and Q. G. Mulazzani, J. Am. Chem. Soc., 2003, 125, 3939–3848.
- C. Chatgilialoglu, R. Bazzanini, L. B. Jimenez and M. A. Miranda, Chem. Res. Toxicol., 2007, 20, 1820–1824 CrossRef CAS.
- N. Belmadoui, F. Boussicault, M. Guerra, J. L. Ravanat, C. Chatgilialoglu and J. Cadet, Org. Biomol. Chem., 2010, 8, 3211–3219 RSC.
- J. A. Theruvathu, P. Jaruga, M. Dizdaroglu and P. J. Brooks, Mech. Ageing Dev., 2007, 128, 494–502 CrossRef CAS.
- B. T. Karwowski, Cent. Eur. J. Chem., 2010, 1, 134–141 Search PubMed.
- L. B. Jimenez, S. Encinas, C. Chatgilialoglu and M. A. Miranda, Org. Biomol. Chem., 2008, 6, 1083–1086 RSC.
- B. T. Karwowski, J. Mol. Struct.-Theochem., 2009, 915, 73–78 CrossRef CAS.
- L. P. Candeias and S. Steenken, Chem. Eur. J., 2000, 6, 475–484 CrossRef CAS.
- C. Chatgilialoglu, M. D'Angelantonio, M. Guerra, P. Kaloudis and Q. G. Mulazzani, Angew. Chem., Int. Ed., 2009, 48, 2214–2217 CrossRef CAS.
- M. Dizdaroglu, Biochem. J., 1986, 238, 247–254 CAS.
- F. Boussicault, P. Kaloudis, C. Caminal, Q. G. Mulazzani and C. Chatgilialoglu, J. Am. Chem. Soc., 2008, 130, 8377–8385 CrossRef CAS.
- L. B. Jimenez, S. Encinas, M. A. Miranda, M. L. Navacchia and C. Chatgilialoglu, Photochem. Photobiol. Sci., 2004, 3, 1042–1046 RSC.
- M. Russo, L. B. Jimenez, Q. G. Mulazzani, M. D'Angelantonio, M. Guerra, M. A. Miranda and C. Chatgilialoglu, Chem. Eur. J., 2006, 12, 7684–7693 CrossRef CAS.
- M. L. Navacchia, A. Manetto, P. Montevecchi and C. Chatgilialoglu, Eur. J. Org. Chem., 2005, 4640–4648 CrossRef CAS.
- C. Chatgilialoglu, Chem. Eur. J., 2008, 14, 2310–2320 CrossRef CAS.
- A. Manetto, D. Georganakis, T. Gimisis, L. Leondiadis, P. Mayer, T. Carell and C. Chatgilialoglu, J. Org. Chem., 2007, 72, 3659–3666 CrossRef CAS.
- M. Newcomb, Tetrahedron, 1993, 49, 1151–1176 CrossRef CAS.
- R. Flyunt, R. Bazzanini, C. Chatgilialoglu and Q. G. Mulazzani, J. Am. Chem. Soc., 2000, 122, 4225–4226 CrossRef CAS.
- C. Chatgilialoglu, M. Duca, C. Ferreri, M. Guerra, M. Ioele, Q. G. Mulazzani, H. Strittmatter and B. Giese, Chem. Eur. J., 2004, 10, 1249–1255 CrossRef CAS.
- C. Chatgilialoglu, M. D'Angelantonio, P. Kaloudis, Q. G. Mulazzani and M. Guerra, J. Phys. Chem. Lett., 2010, 1, 174–177 Search PubMed.
- R. B. Zhang and L. A. Eriksson, Chem. Phys. Lett., 2006, 417, 303–308 CrossRef CAS.
- M. L. Navacchia, C. Chatgilialoglu and P. Montevecchi, J. Org. Chem., 2006, 71, 4445–4452 CrossRef CAS.
- A. Matsuda, M. Tezuka, K. Niizuma, E. Sugiyama and T. Ueda, Tetrahedron, 1978, 34, 2633–2637 CrossRef CAS.
- A. Romieu, D. Gasparutto, D. Molko and J. Cadet, J. Org. Chem., 1998, 63, 5245–5249 CrossRef CAS.
- A. Romieu, D. Gasparutto and J. Cadet, Chem. Res. Toxicol., 1999, 12, 412–421 CrossRef CAS.
- P. J. Brooks, D. S. Wise, D. A. Berry, J. V. Kosmoski, M. J. Smerdon, R. L. Somers, H. Mackie, A. Y. Spoonde, E. J. Ackerman, K. Coleman, R. E. Tarone and J. H. Robbins, J. Biol. Chem., 2000, 275, 22355–22362 CrossRef CAS.
- B. T. Karwowski, J. Gaillard, A. Grand and J. Cadet, Org. Biomol. Chem., 2008, 6, 3408–3413 RSC.
- S. S. Wallace, Radiat. Res., 1998, 150, S60–S79 CrossRef CAS.
- I. Kuraoka, C. Bender, A. Romieu, J. Cadet, R. D. Wood and T. Lindahl, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3832–3837 CrossRef CAS.
- C. Biertümpfel, Y. Zhao, Y. Kondo, S. Ramón-Maiques, M. Gregory, J. Y. Lee, C. Masutani, A. R. Lehmann, F. Hanaoka and W. Yang, Nature, 2010, 465, 1044–1049 CrossRef.
- I. Kuraoka, P. Robins, C. Masutani, F. Hanaoka, D. Gasparutto, J. Cadet, R. D. Wood and T. Lindahl, J. Biol. Chem., 2001, 276, 49283–49288 CrossRef CAS.
- C. Marietta, H. Gulam and P. J. Brooks, DNA Repair, 2002, 1, 967–975 CrossRef CAS.
- P. Jaruga, Y. Xiao, V. Vartanian, R. Stephen Lloyd and M. Dizdaroglu, Biochemistry, 2010, 49, 1053–1055 CrossRef CAS.
- D. Gasparutto, A.-G. Bourdat, C. D'Ham, V. Duarte, A. Romieu and J. Cadet, Biochimie, 2000, 82, 19–24 CrossRef CAS.
- P. J. Brooks, Neuroscience, 2007, 145, 1407–1417 CrossRef CAS.
- P. J. Brooks, DNA Repair, 2008, 7, 1168–1179 CrossRef CAS.
- K. Randerath, G.-D. Zhou, R. L. Somers, J. H. Robbins and P. J. Brooks, J. Biol. Chem., 2001, 276, 36051–36057 CrossRef CAS.
- G.-D. Zhou, K. Randerath, K. C. Donnelly and A. K. Jaiswal, Int. J. Cancer, 2004, 112, 877–883 CrossRef CAS.
- J. M. L. Dirksen, W. F. Blakely, E. Holwitt and M. Dizdaroglu, Int. J. Radiat. Biol., 1988, 54, 195–204 CAS.
- M. Dizdaroglu, P. Jaruga and H. Rodriguez, Free Radical Biol. Med., 2001, 30, 774–784 CrossRef CAS.
- P. Jaruga, M. Birincioglu, H. Rodriguez and M. Dizdaroglu, Biochemistry, 2002, 41, 3703–3711 CrossRef CAS.
- M. Birincioglu, P. Jaruga, G. Chowdhury, H. Rodriguez, M. Dizdaroglu and K. S. Gates, J. Am. Chem. Soc., 2003, 125, 11607–11615 CrossRef CAS.
- P. Jaruga, J. Theruvathu, M. Dizdaroglu and P. J. Brooks, Nucleic Acids Res., 2004, 32, e87 CrossRef.
- P. Jaruga, Y. Xiao, B. C. Nelson and M. Dizdaroglu, Biochem. Biophys. Res. Commun., 2009, 386, 656–660 CrossRef CAS.
- G. Kirkali, M. Tunca, S. Gene, P. Jaruga and M. Dizdaroglu, Free Radical Biol. Med., 2008, 44, 386–393 CrossRef CAS.
- K. M. Anderson, P. Jaruga, C. R. Ramsey, N. K. Gilman, V. M. Green, S. W. Rostad, J. T. Emerman, M. Dizdaroglu and D. C. Malins, Cell Cycle, 2006, 5, 1240–1244 CrossRef CAS.
- G. Kirkali, N. C. de Souza-Pinto, P. Jaruga, V. A. Bohr and M. Dizdaroglu, DNA Repair, 2009, 8, 274–278 CrossRef CAS.
- M. D'Errico, E. Parlanti, M. Teson, B. M. Bernardes de Jesus, P. Degan, A. Calcagnile, P. Jaruga, M. BjØrås, M. Crescenzi, A. M. Pedrini, J.-M. Egly, G. Zambruno, M. Stefanini, M. Dizdaroglu and E. Dogliotti, EMBO J., 2006, 25, 4305–4315 CrossRef.
- M. D'Errico, E. Parlanti, M. Teson, P. Degan, T. Lemma, A. Calcagnile, I. Iavarone, P. Jaruga, M. Ropolo, A. M. Pedrini, D. Orioli, G. Frosina, G. Zambruno, M. Dizdaroglu, M. Stefanini and E. Dogliotti, Oncogene, 2007, 26, 4336–4343 CrossRef.
- J. Cadet and H. Poulsen, Free Radical Biol. Med., 2010, 48, 1457–1459 CrossRef CAS.
- P. Jaruga and M. Dizdaroglu, Biochem. Biophys. Res. Commun., 2010, 397, 48–52 CrossRef CAS.
- J. Cadet, T. Douki, D. Gasparutto and J.-L. Ravanat, Mutat. Res., 2003, 531, 5–23 CrossRef CAS.
- J. Cadet, T. Douki and J.-L. Ravanat, Acc. Chem. Res., 2008, 41, 1075–1083 CrossRef CAS.
- R. Zheng, Y. Shi, Z. Jia, C. Zhao, Q. Zhang and X. Tan, Chem. Soc. Rev., 2010, 39, 2827–2834 RSC.
- P. Jaruga and M. Dizdaroglu, DNA Repair, 2008, 7, 1413–1425 CrossRef CAS.
- A. F. Fuciarelli, G. G. Miller and J. A. Raleigh, Radiat. Res., 1985, 104, 272–283 CAS.
Footnote |
| † Dedicated to the memory of Dr Quinto G. Mulazzani, who drove our group to radiation chemistry. |
| This journal is © The Royal Society of Chemistry 2011 |