Ultrafast excitation transfer and charge stabilization in a newly assembled photosynthetic antenna-reaction center mimic composed of boron dipyrrin, zinc porphyrin and fullerene
Francis
D'Souza
*ab,
Channa A.
Wijesinghe
b,
Mohamed E.
El-Khouly
c,
Jessica
Hudson
b,
Marja
Niemi
d,
Helge
Lemmetyinen
d,
Nikolai V.
Tkachenko
*d,
Melvin E.
Zandler
b and
Shunichi
Fukuzumi
*ce
aDepartment of Chemistry, 1155 Union Circle, #305070, Denton, TX 76203, USA
bDepartment of Chemistry, Wichita State University, Wichita, KS 67260-0051, USA. E-mail: Francis.DSouza@UNT.edu; Tel: +940-369-8832
cGraduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: Fukuzumi@chem.eng.osaka.u.ac.jp; Tel: +81-6-6879-7368
dDepartment Chemistry and Bioengineering, Tampere University of Technology, P.O. Box 541, 33101, Tampere, Finland. E-mail: nikolai.tkachenko@tut.fi
eDepartment of Bioinspired Science, Ewha Womens’ University, Seoul, 120–750, Korea
First published on 16th September 2011
Abstract
A self-assembled supramolecular triad as a model to mimic the light-induced events of the photosynthetic antenna-reaction center, that is, ultrafast excitation transfer followed by electron transfer ultimately generating a long-lived charge-separated state, has been accomplished. Boron dipyrrin (BDP), zinc porphyrin (ZnP) and fullerene (C60), respectively, constitute the energy donor, electron donor and electron acceptor segments of the antenna-reaction center imitation. Unlike in the previous models, the BDP entity was placed between the electron donor, ZnP and electron acceptor, C60 entities. For the construction, benzo-18-crown-6 functionalized BDP was synthesized and subsequently reacted with 3,4-dihydroxyphenyl functionalized ZnP through the central boron atom to form the crown-BDP-ZnP dyad. Next, an alkyl ammonium functionalized fullerene was used to self-assemble the crown ether entity of the dyad via ion–dipole interactions. The newly formed supramolecular triad was fully characterized by spectroscopic, computational and electrochemical methods. Steady-state fluorescence and excitation studies revealed the occurrence of energy transfer upon selective excitation of the BDP in the dyad. Further studies involving the pump–probe technique revealed excitation transfer from the 1BDP* to ZnP to occur in about 7 ps, much faster than that reported for other systems in this series of triads, as a consequence of shorter distance between the entities. Upon forming the supramolecular triad by self-assembling fullerene, the 1ZnP* produced by direct excitation or by energy transfer mechanism resulted in an initial electron transfer to the BDP entity. The charge recombination resulted in the population of the triplet excited state of C60, from where additional electron transfer occurred to produce C60•−:crown-BDP-ZnP•+ ion pair as the final charge-separated species. Nanosecond transient absorption studies revealed the lifetime of the charge-separated state to be ∼100 μs, the longest ever reported for this type of antenna-reaction center mimics, indicating better charge stabilization as a result of the different disposition of the entities of the supramolecular triad.
Introduction
In natural photosynthesis, photon energy is efficiently funneled with the help of light harvesting antenna pigments to the reaction center to accomplish a long lived charge-separated state as a consequence of sequential electron transfer occurring within the energetically well-aligned entities of the reaction center.1 Inspired by this, there have been a number of artificial photosynthetic models in the literature capable of mimicking the energy and electron transfer processes of natural photosynthesis.2–16 Both covalent bonding as well as self-assembled supramolecular bonding approaches have been utilized in the construction of these model systems. However, construction of artificial antenna-reaction center models revealing both ultrafast energy transfer and formation of long-lived charge-separated states have remained a challenge due to the associated synthetic challenges in addition to matching and positioning of the energy states of the different entities.Supramolecular assembly of boron dipyrrin (BDP or BODIPY®) as antenna, zinc porphyrin (ZnP) as primary electron donor, and fullerene (C60) as primary electron acceptor has been one of the successful approaches to build antenna-reaction center models.17–19 In these model compounds excitation of BDP results in efficient excitation transfer to the ZnP entity from where an electron transfer originates to form the ZnP•+–C60•− radical ion-pair. Using this strategy, to-date, we have built model compounds of the type 1–3.17–19 Models 1 and 2 utilized metal–ligand axial coordination approach to link imidazole functionalized fullerene to ZnP of the covalently linked BDP-ZnP dyads having up to four BDP entities.17,18 In model 3, crown ether-alkyl cation binding approach was utilized to assemble fullerene with the crown appended ZnP-BDP dyad.19 In these models the donor and acceptor entities were placed adjacent to each other to facilitate sequential energy and electron-transfer processes. Although charge stabilization to some extent was observed, a long-lived charge stabilized state was difficult to attain. In the present investigation, we have intentionally placed the antenna unit between the electron donor zinc porphyrin and electron acceptor fullerene to increase the distance between them (model 4 in Scheme 1). Based on photo and redox considerations, such a combination would make the BDP entity not only an energy donor but also a primary electron acceptor from the excited ZnP (vide infra). The one-electron reduced BDP thus formed can subsequently undergo electron migration to the fullerene entity or result in the formation of excited triplet state fullerene from where additional electron transfer can take place. The ultimate result of this would be distantly separating the final radical cation (ZnP•+) and radical anion (C60•−) species, a requirement for generating long-lived charge-separated species. This novel concept has been verified in the present study by synthesizing and assembling supramolecular triad, 4.
 | ||
| Scheme 1 Supramolecular antenna-reaction center models built using boron dipyrrin, zinc porphyrin and fullerene entities (1–4) in our laboratories. The crown-BDP and ZnP are control compounds used to probe energy transfer and electron transfer in the newly designed crown-BDP-ZnP dyad binding to alkyl ammonium functionalized C60 (model 4) antenna-reaction center mimic in the present study. | ||
Results and discussion
Syntheses
The synthesis of crown-BDP-ZnP dyad involved a multi-step procedure as outlined in Scheme 2 while the details are given in the experimental section. Briefly, 5-(3,4-dihydroxyphenyl)-10,15,20-triphenylporphyrin 4a was prepared by the reaction of pyrrole and 3,4-dimethoxybenzaldehyde followed by chromatographic separation. The dimethoxyphenyl derivative was converted into dihydroxyphenyl by reacting with BBr3 in CH2Cl2. The BDP derivative, 4b, was prepared by the reaction of 2,4-dimethylpyrrole and 3-formyl-benzo-18-crown-6 in the presence of trifluoroacetic acid and p-chloranil in CH2Cl2 followed by boronation. Finally, the crown-BDP-ZnP dyad was prepared by reacting equimolar amounts of 4a and 4b in CH2Cl2 in the presence of AlCl3 followed by chromatographic separation. Metallation using zinc acetate followed by chromatographic purification yielded 4c. The newly synthesized compound was stored in dark prior performing spectral and photochemical studies. | ||
| Scheme 2 Synthetic procedure adopted for the crown-BDP-ZnP dyad host system in the present study. | ||
Optical absorption and emission studies of the dyad, 4c and triad, 4
The absorption spectrum of crown-BDP-ZnP dyad exhibited bands at 433, 510, 562 and 604 nm revealing the presence of both BDP and ZnP chromophores (Fig. 1a). The BDP band at 510 nm was found to be red shifted by ∼6 nm compared to the control, crown-BDP lacking ZnP while the ZnP Soret was found also to be red shifted by ∼3 nm compared to control, zinc tetratolylporphyrin, ZnP. These observations suggest a fair amount of electronic interactions between the two chromophores perhaps due to the linkage through the BDP core involving the central boron atom.16g,h The emission spectrum of crown-BDP-ZnP when excited at 510 nm corresponding to BDP revealed a weak emission at 526 nm corresponding to BDP along with additional weak emission bands at 607 and 648 nm corresponding to the appended ZnP (Fig. 1b). A comparison of BDP emission intensity in the crown-BDP-ZnP dyad to that of crown-BDP control revealed quenching of the former by over 90%. These results suggest the occurrence of energy or electron transfer from singlet excited BDP to ZnP in the dyad. By using 430 nm excitation light, which selectively excited the ZnP entity (Fig. 1c), the ZnP emission bands of crown-BDP-ZnP dyad at 605 and 653 nm were found to be quenched by ca. 96% as compared with the emission intensity of ZnP. The quenching of 1ZnP* by the attached BDP may involve energy transfer and/or electron transfer processes. Since the energy transfer from the 1ZnP* (2.04 eV) to BDP (2.43 eV) is thermodynamically not feasible, the electron transfer from 1ZnP* to BDP is expected to dominate based on these measurements. | ||
| Fig. 1 (a) Absorption spectra of (i) crown-BDP-ZnP, (ii) ZnP, and (iii) crown-BDP in benzonitrile. (b) Fluorescence spectra of (i) crown BDP-ZnP and (ii) ZnP in benzonitrile; λex = 510 nm. (c) Fluorescence spectra of (i) ZnP and (ii) crown BDP-ZnP in benzonitrile; λex = 430 nm. | ||
The excitation spectrum of the crown-BDP-ZnP was recorded by fixing the emission monochromator to 648 nm and scanning the excitation wavelength. Such spectrum shown in Fig. 2 revealed bands not only of ZnP but also that of BDP suggesting excitation transfer from singlet excited BDP to ZnP in the dyad.20 In order to estimate the efficiency of energy transfer, the intensity ratio of the 526 nm band of BDP to 562 nm band of ZnP was calculated and compared with the ratio of the bands from optical absorption spectrum in Fig. 1a. This ratio from the excitation spectrum was found to be about 25% compared to that obtained from the absorption spectral data. These results suggest occurrence of fairly efficient energy transfer from the 1BDP* to ZnP in the dyad.
 | ||
| Fig. 2 (i) Absorption spectrum of crown ether-BDP-ZnP dyad, and (ii) excitation spectrum of the crown-BDP-ZnP dyad obtained by fixing the emission monochromator wavelength to 648 nm and scanning the excitation wavelength in benzonitrile. | ||
The self-assembly of the crown-BDP-ZnP dyad and alkyl ammonium functionalized fullerene was monitored spectroscopically using optical absorption and emission studies. Fig. 3a shows absorption spectral changes observed during increasing addition of fullerene to the dyad solution in benzonitrile. The bands corresponding to both BDP and ZnP revealed an increase without any appreciable spectral shifts. The fulleropyrrolidine peak appeared at 328 nm. The final spectrum of the triad was a straightforward sum of the spectra of the dyad and fullerene, suggesting a lack of intermolecular interaction between the fullerene and BDP or ZnP entities. Interestingly, the fluorescence spectrum of the dyad revealed additional quenching corresponding to both BDP and ZnP entities (Fig. 3b). Such quenching was negligent when the dyad was titrated with 2-phenyl fulleropyrrolidine lacking the alkyl ammonium cation. These studies suggest alkyl ammonium binding to the crown ether void and subsequent intrasupramolecular quenching.11a The quenching data were analyzed using the Benesi–Hildebrand method21 as shown in Fig. 3b inset. A binding constant of 1.9 × 103 M−1 was obtained suggesting moderately stable complex formation of the alkyl ammonium cation and the crown ether.
 | ||
| Fig. 3 (a) Absorption and (b) emission spectral changes observed for the crown-BDP-ZnP (6 × 10−6 M) on increasing addition of alkyl ammonium functionalized fullerene (0.2 eq. each addition). λex = 502 nm. The Figure inset shows Benesi–Hildebrand plot constructed to obtain the binding constant. I0 and I represent the fluorescence intensity of the BDP emission in the absence and presence of added fullerene, respectively. | ||
Computational studies
Further DFT studies at the B3LYP/3-21G(*) level22,23 were performed to visualize the geometry and electronic structure of the supramolecular triad. Fig. 4a shows the optimized structure on a Born–Oppenheimer potential energy surface in which the fullerene was far from both the BDP and ZnP entities to cause any intermolecular interactions, an observation that readily agrees with the optical absorption data shown in Fig. 3a. In the optimized structure, the BDP and ZnP were coplanar with an orthogonal orientation of the bridging phenyl entity. The crown ether bound fullerene was almost perpendicular to the macrocycle planes. The center-to-center distance between Zn and C60 was found to be 23.6 Å while this distance between zinc and boron atoms of the crown-BDP-ZnP dyad was 9.5 Å. The frontier HOMO and LUMO are shown in Fig. 4b and c, respectively. The HOMO was spread all over the porphyrin π-system with some contribution over the phenyl ring connecting the BDP entity while the LUMO was found to be fully localized on the fullerene entity. The B3LYP/3-21G(*) estimated gas phase HOMO–LUMO gap was found to be 0.72 eV. These results suggest ZnP to be the primary electron donor and fullerene being the terminal electron acceptor in spite of arranging them in a different fashion as shown in Fig. 4. | ||
| Fig. 4 (a) B3LYP/3-21G(*) optimized structure of the supramolecular triad comprised of alkyl ammonium functionalized fullerene bound to the crown-BDP-ZnP dyad. The HOMO and LUMO are shown in Figures (b) and (c) respectively. | ||
Electrochemical studies and energy levels
Cyclic voltammetric studies were performed to arrive at the redox potentials of the newly assembled supramolecular triad, 4. Fig. 5 shows cyclic voltammograms of 4 along with the control compounds during both negative and positive scanning of the potential. The first two reductions of the C60 alkyl ammonium cation were located at E1/2 = −1.03 and −1.45 V vs. Fc/Fc+, respectively (Fig. 5a). The peak-to-peak separation and plots of peak current versus square root of scan rate indicated both reductions to be one-electron reversible processes. The crown ether-BDP-ZnP revealed three oxidations located at E1/2 = 0.25 and 0.58 and 0.78 V vs. Fc/Fc+. Control experiments confirmed that the first two processes involve oxidation of the ZnP macrocycle while the third involves the BDP macrocycle. During reduction, the first reversible reduction located at E1/2 = −1.64 V vs. Fc/Fc+, involved BDP entity while the reduction located at −1.90 involved reduction of the ZnP macrocycle (Fig. 5b). The assembled fullerene:crown-BDP-ZnP triad, 4, revealed redox waves corresponding to the presence of all three entities without significant changes in their potentials compared to the values prior to the assembly formation. As shown in Fig. 5c for 4, the first oxidation of ZnP was located at 0.25 V while the first reduction of C60 was located at −1.03 V vs. Fc/Fc+, respectively, resulting in an electrochemically measured HOMO–LUMO gap of 1.28 V. | ||
| Fig. 5 Cyclic voltammograms of (a) C60 alkyl ammonium cation, (b) crown-BDP-zinc porphyrin, and (c) fullerene:crown-BDP-ZnP triad in benzonitrile containing 0.1 (TBA)ClO4. Scan rate = 0.1 V s−1. The * in panel a represents redox process corresponding to ferrocene oxidation used as an internal standard. | ||
Using the electrochemical data, excited energies and distances between the chromophores, the free-energies of charge-separation (ΔGCS) and charge-recombination (ΔGCR) were calculated using eqn (1) and 2 by Weller's approach.24
| −ΔGCR = e(Eox − Ered) + ΔGS | (1) |
| −ΔGCS = ΔE0–0 − (−ΔGCR) | (2) |
Such calculations revealed a ΔGCS value of −0.76 eV for electron transfer from the singlet excited ZnP to fullerene, and ΔGCR value of −1.28 eV for the charge recombination process. For comparison purpose, the ΔGCS and ΔGCR values for electron transfer within the crown-BDP-ZnP dyad were also calculated. In the case of ZnP-BDP dyad, the ΔGCS and ΔGCR values originated from the 1ZnP* were found to be −0.15 and −1.89 eV, respectively. These studies, in addition to the thermodynamic feasibility of the occurrence of electron transfer, also indicate fullerene being the superior electron acceptor in the supramolecular triad, 4, as predicted by the computational studies (location of the frontier HOMO and LUMO).
Photochemical studies
Femtosecond transient absorption spectra recorded at different delay times after excitation for the crown-BDP-ZnP dyad in PhCN at 430 nm are shown in Fig. 6. Transient features corresponding to both ZnP•+ in the 600–700 nm region and BDP•− in the 450–500 nm region were observed indicating the formation of crown-BDP•−-ZnP•+ as radical ion-pair species from the initial energy transfer product, crown-BDP-1ZnP*.17 From the rise and decay of the transient band of ZnP•+ at 632 nm, the rate of the charge-separation (kCS) process was found to be 1.2 × 1012 s−1 while the rate of charge recombination, kCR was evaluated to be 1.17 × 1010 s−1. From the kCR, the lifetime of charge-separated state (τCS) of crown-BDP•−-ZnP•+via the singlet-excited state of ZnP was found to be 85 ps.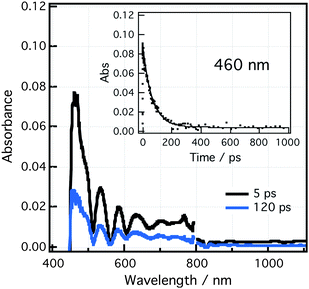 | ||
| Fig. 6 Femtosecond transient absorption spectra of crown-BDP-ZnP dyad in PhCN recorded at 5 and 120 ps after excitation pulse at wavelength of 430 nm. The inset presents a time profile of the transient absorption at 460 nm. | ||
Excitation at 430 nm results in selective generation of the second singlet excited state of the porphyrin chromophore, which decays rapidly to the first singlet excited state. In this case the BDP moiety acts as the electron acceptor only. To investigate the intermolecular energy transfer, the BDP chromophore was directly excited at 510 nm. The results of the transient absorption measurements with excitation at 510 nm are presented in Fig. 7. The decay profiles are more complex with this excitation wavelength since the electron transfer is complemented by the intramolecular energy transfer. The decays were fitted globally with three-exponential model and the decay component spectra are shown in Fig. 7 together with the time resolved spectra right after excitation (at 0 ps) and at 2 ps delay time. As can be expected, the spectrum of the long-lived component is essentially the same as the transient absorption spectrum of the sample excited at 430 nm, and can be attributed to the charge separated state, crown-BDP•−-ZnP•+, due to characteristic absorption of the porphyrin cation in the 600–700 nm range. Right after the excitation (at 0 ps delay) the transient absorption is rather weak in the red part of the spectrum (600–700 nm), as can be expected for BDP excited singlet state. However, the formation of the charge-separated state is at least bi-exponential with time constants of 0.3 and 7 ps. In reality, one can expect three reactions to take place in this time domain: (i) energy transfer from BDP to porphyrin chromophore, crown-1BDP*-ZnP → crown-BDP-1ZnP*, (ii) electron transfer starting from the singlet excited BDP chromophore, crown-1BDP*-ZnP → crown-BDP•−-ZnP•+, and (iii) electron transfer starting from the singlet excited ZnP chromophore, crown-BDP-1ZnP* → crown-BDP•−-ZnP•+. If reaction (ii) was the dominating one, i.e. its rate constant was greater than that of the energy transfer, reaction (i), the formation of the CS state would be essentially mono-exponential, which is not the case. On this basis one can expect a two step process, crown-1BDP*-ZnP → crown-BDP-1ZnP* → crown-BDP•−-ZnP•+, to be the main pathway for the charge-separation. The rate constant of the CS estimated from the measurements with excitation at 430 nm includes two sequential reactions: internal conversion, crown-BDP-2ZnP* → crown-BDP-1ZnP*, and the CS, which means that the rate constant for the CS reaction alone is < 1.2 × 1012 s−1. Therefore the rate constant of CS is likely to be 3 × 1012 s−1, and the rate constant for the energy transfer is 1.4 × 1011 s−1, according to the measurements with excitation at 510 nm. Based on this assumption, the initial fast formation of the CS state is due to partial direct excitation of the ZnP moieties, and the following slower growth of the CS state population is the sequence of two reactions, energy transfer and CS, from which the energy transfer is the time limiting step.
 | ||
| Fig. 7 Transient absorption decay component spectra (symbols with solid lines, the component lifetimes are indicated in the plot) and time resolved spectra (dashed and dotted lines at 0 and 2 ps delay time, respectively) of crown-BDP-ZnP dyad in PhCN at the excitation wavelength of 510 nm. Inset shows the transient absorption decay profile at 630 nm. | ||
Addition of fulleropyrrolidine cation to form the fullerene:crown-BDP-ZnP triad, 4 revealed spectral features not much different from that of the dyad with very weak absorption band of the C60 radical anion at 1000 nm (Fig. 8). The kCR determined by monitoring the decay of BDP•− at 470 nm and ZnP•+ at 637 nm were found to be comparable to those obtained for the crown-BDP-ZnP dyad, being, 1.6 × 1010 s−1 and 1.19 × 1010 s−1, respectively. However, there was some residual featureless absorption left after the charge recombination, and a slow formation of the triplet-excited state of C60 (3C60*) at 700 nm with a rate of 3.60 × 108 s−1 was observed with the decay of the CS state. These results are suggestive of a lack of efficient formation of C60•−-crown BDP-ZnP•+ as charge-separated species. It seems that the C60-crown BDP•−-ZnP•+ undergoes charge recombination to populate the energetically favorable 1C60* which decays to populate the 3C60*. In order to confirm occurrence of such photochemical process and additional electron transfer from the triplet-excited state of C60 (3C60*) in the triad, subsequent nanosecond transient spectral measurements were performed.
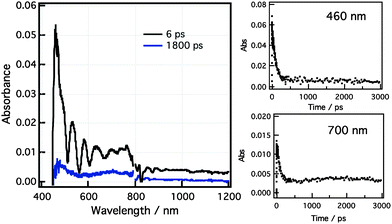 | ||
| Fig. 8 Femtosecond transient absorption spectra of C60:crown-BDP-ZnP triad in PhCN at the excitation wavelength of 430 nm. | ||
Fig. 9 shows the nanosecond transient spectra recorded at different time intervals for the triad 4 at the excitation wavelength of 430 nm. Transient peaks corresponding to 3C60* in the 650–700 nm region, ZnP•+ in the 600 nm region, and C60•− in the 1000 nm region were clearly evident suggesting occurrence of electron transfervia the 3C60* in the triad.7–9 The rates of charge-separation (kCST) and charge-recombination (kCRT) processes via the 3C60* were evaluated by monitoring the rise and decay of C60•−, respectively, as shown in Fig. 9 inset. The kCST and kCRT thus calculated were found to be 1.50 × 105 s−1 and 1.03 × 104 s−1, respectively. The lifetime of the radical ion pair, τCS from kCRT was found to be 100 μs, highest value observed for the triads constructed using BDP, ZnP and fullerene as components (models 1–4 in Scheme 1) to-date.
 | ||
| Fig. 9 Nanosecond transient absorption spectra of the triad 4 at different time intervals in PhCN at the excitation wavelength of 430 nm. The inset shows decay of C60•− at 1000 nm. | ||
Fig. 10 summarizes the photochemical events in the newly assembled supramolecular triad in the present study. The values of different energy levels were calculated from spectral and electrochemical data while the kinetics of different photochemical events leading redox species were arrived from the data using transient spectroscopy both in the femto and nanosecond time scales. The excitation spectrum recorded for the crown-BDP-ZnP dyad revealed energy transfer from 1BDP* to ZnP to populate the 1ZnP*, which in turn donated its electron to the attached BDP forming the crown-BDP•−-ZnP•+. In the supramolecular triad, direct excitation of ZnP resulted in the formation of C60:crown-BDP•−-ZnP•+ as the initial charge-separation product. Although subsequent electron migration to form C60•−:crown-BDP-ZnP•+ seems possible, charge recombination occurs to populate the 1C60* which upon intersystem crossing populates the 3C60*. A relatively slow photoinduced electron transfer subsequently occurs from the ZnP to the triplet-excited state of C60 within the triad resulting into the formation of C60•−:crown-BDP-ZnP•+ as the final charge-separated state with lifetimes of the order of 100 μs.
 | ||
| Fig. 10 Energy level diagram showing the different photochemical events in the investigated supramolecular antenna-reaction center mimic, C60:crown-BDP-ZnP, 4. | ||
A comparison of the photochemical data of the present dyad and triad to the previously reported dyads and triads17–19 deserves special mention, that is, better figures of merit relevant to artificial photosynthesis were obtained for the present systems compared to the previously built BDP-ZnP dyads and BDP-ZnP-C60 triads (1–3). As demonstrated by pump–probe technique, energy transfer from the 1BDP* to ZnP is much faster for the present crown-BDP-ZnP dyad (7 ps) as compared to the rate of excitation transfer in the BDP-ZnP dyads shown in 1–3, as a consequence of shorter distance between the units. In the case of triad 1, where the BDP and ZnP entities were held by a flexible ethylene oxide linkage, upon selective excitation of BDP, efficient singlet-singlet energy transfer (kENTS = 9.2 × 109 s−1) was observed to populate 1ZnP*.17 Subsequent electron transfer to the coordinated C60 (kCSS = 4.7 × 109 s−1) and fairly rapid charge recombination (kCR = 2.0 × 108 s−1) were subsequently observed.17 In the case of triad of the type 2 with varying number of BDP units (1, 2 and 4), the singlet-singlet energy transfer was rather quick and occurred in the time scale ranging between 28 and 48 ps.18 Additionally, a decrease in time constants with increasing the number of BDP units was observed revealing better antenna effect of the dyads having higher number of BDP entities. For the triad 3, having the same antenna, electron donor and electron acceptor but different spatial arrangement of the entities, efficient singlet-singlet energy transfer occurred from the singlet excited state of BDP (10–60 ps time constant depending upon the conformer), a case similar to that observed for the present dyad.19 However, nanosecond transient absorption spectrum of the triad 3 yielded a lifetime for the radical ion pair of about 23 μs, a number that is significantly smaller than the 100 μs lifetime of the radical ion pair observed for the present system 4. These results demonstrate that by careful arrangement of the different entities, it is possible to achieve ultrafast energy transfer and long-lived charge-separated state in model compounds mimicking both antenna and reaction center functionalities.
Summary
A novel photosynthetic ‘antenna-reaction center’ mimic capable of ultrafast excitation transfer followed by electron transfer to generate long-lived charge separated state has been successfully built and studied. For the construction, the antenna mimic, boron dipyrrin, was placed between the electron donor, ZnP and electron acceptor, C60 to modulate the kinetics of the photochemical events. Transient absorption studies using pump–probe technique revealed that singlet energy transfer from the 1BDP* to the ZnP in the crown-BDP-ZnP dyad occurred within 7 ps. Upon forming the supramolecular triad by complexing fullerene to the crown ether void, the 1ZnP* produced either by direct excitation or by energy transfer mechanism resulted in electron transfer to the BDP entity. The charge recombination eventually populated the triplet excited state of C60 from where additional electron transfer occurred to produce C60•−:crown-BDP-ZnP•+ ion pair as the final electron-transfer product. Nanosecond transient absorption studies revealed the lifetime of the charge-separated state to be ∼100 μs, the longest ever reported for these type of antenna-reaction center mimics, indicating better charge stabilization as a result of different arrangement of the antenna, donor and acceptor entities of the supramolecular triad.Experimental
Chemicals
All of the reagents used in the syntheses, and benzonitrile (in sure seal bottle under nitrogen) were obtained from Aldrich Chemicals (Milwaukee, WI). Tetra-n-butylammonium perchlorate, (TBA)ClO4 was obtained from Fluka Chemicals. All the chromatographic materials and solvents were procured from Fisher Scientific and were used as received. The synthesis of alkyl ammonium functionalized fulleropyrrolidine is given elsewhere.25Instruments
The UV-visible spectral measurements were carried out with a Shimadzu Model 2550 double monochromator spectrophotometer. The fluorescence emission was monitored by using a Varian Eclipse spectrometer. A right angle detection method was used. The 1H NMR studies were carried out on a Varian 400 MHz spectrometer. Tetramethylsilane (TMS) was used as an internal standard. Cyclic voltammograms were recorded on an EG&G 263A potentiostat/galvanostat using a three electrode system. A platinum button electrode was used as the working electrode. A platinum wire served as the counter electrode and an Ag/AgCl electrode was used as the reference electrode. A ferrocene/ferrocenium redox couple was used as an internal standard. All the solutions were purged prior to electrochemical and spectral measurements using argon gas. Matrix-assisted laser desorption/ionization time-of-flight mass spectra (MALDI-TOF-MS) were measured on a Kratos Compact MALDI (Shimadzu) for the metal complex in PhCN with dithranol used as a matrix. The computational calculations were performed by DFT B3LYP/3-21(*) method with GAUSSIAN 03 software package22 on high speed PCs. The HOMO and LUMO were generated using the GaussView program.Time resolved spectroscopy
The studied compounds were excited by a Panther OPO pumped by Nd:YAG laser (Continuum, SLII-10, 4–6 ns fwhm) with the powers of 1.5 and 3.0 mJ per pulse. The transient absorption measurements were performed using a continuous xenon lamp (150 W) and an InGaAs-PIN photodiode (Hamamatsu 2949) as a probe light and a detector, respectively. The output from the photodiodes and a photomultiplier tube was recorded with a digitizing oscilloscope (Tektronix, TDS3032, 300 MHz). Femtosecond transient absorption spectroscopy experiments were conducted using an ultrafast source: Integra-C (Quantronix Corp.), an optical parametric amplifier: TOPAS (Light Conversion Ltd.) and a commercially available optical detection system: Helios provided by Ultrafast Systems LLC. The source for the pump and probe pulses were derived from the fundamental output of Integra-C (780 nm, 2 mJ/pulse and fwhm = 130 fs) at a repetition rate of 1 kHz. 75% of the fundamental output of the laser was introduced into TOPAS which has optical frequency mixers resulting in tunable range from 285 nm to 1660 nm, while the rest of the output was used for white light generation. Typically, 2500 excitation pulses were averaged for 5 s to obtain the transient spectrum at a set delay time. Kinetic traces at appropriate wavelengths were assembled from the time-resolved spectral data.The pump–probe instrument used to study transient absorption of the samples with excitation at 510 nm was described elsewhere.18 In brief, the laser system consisted of a primary Ti:sapphire generator (TiF-50, CDP Corp.) pumped by a Nd CW laser (Verdi-6, Coherent Inc.), femtosecond pulse amplifier and optical parametric amplifier (model 2017, CDP Corp.). The time resolved spectra were measured by a CCD detector coupled with a monochromator. Excitation wavelength was 510 nm and overall time resolution of the instrument was ∼200 fs (FWHM).
The nanosecond transient absorption measurements in the near-IR region were measured by laser-flash photolysis; 355 nm light from a Nd:YAG laser (Spectra-Physics and Quanta-Ray GCR-130, 6 ns fwhm) was used as an excitation source. The monitoring lights from a pulsed Xe-lamp were detected via Ge-avalanche photodiode module. The samples were held in a quartz cell (1 × 1 cm) and were deaerated by bubbling argon gas through the solution for 20 min. All measurements were conducted at 298 K. The transient spectra were recorded using fresh solutions in each laser excitation.
Syntheses
Acknowledgements
This work was supported by National Science Foundation (Grant Nos. 0804015 and EPS-0903806) and matching support from the State of Kansas through Kansas Technology Enterprise Corporation, a Grant-in-Aid (Nos. 20108010 and 21750146), the Global COE (center of excellence) program “Global Education and Research Center for Bio-Environmental Chemistry” of Osaka University from Ministry of Education, Culture, Sports, Science and Technology, Japan, KOSEF/MEST through WCU project (R31-2008-000-10010-0) from Korea, and Academy of Finland.References
- (a) Photosynthetic Protein Complexes; A Structural Approach, ed. P. Frome, Wiley-VCH Verlag GmbH & Co., Germany, 2008 Search PubMed; (b) Photosynthetic Light Harvesting, ed. R. Cogdell and C. Mullineaux, Springer, Dordrecht, Netherlands, 2008 Search PubMed; (c) Handbook of Photosynthesis, 2nd edn, ed. M. Pessarakli, CRC Press LLC, Boca Raton, FL, 2005 Search PubMed; (d) Light-Harvesting Antennas in Photosynthesis, ed. B. R. Green and W. W. Parson, Kluwer, Dordrecht, Netherlands, 2003 Search PubMed; (e) R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell Sciences, Oxford, 2002 Search PubMed.
- (a) S. Karrasch, P. A. Bullough and R. Ghosh, EMBO J., 1995, 14, 631 CAS; (b) A. W. Roszak, T. D. Howard, J. Southall, A. T. Gardiner, C. L. Law, N. W. Isaacs and R. J. Cogdell, Science, 2003, 302, 1969 CrossRef CAS; (c) G. McDermott, S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell and N. W. Isaacs, Nature, 1995, 374, 517 CrossRef CAS; (d) The Photosynthetic Reaction Center, ed. J. Deisenhofer and J. R. Norris, Academic Press, San Diego, 1993 Search PubMed; (e) S. Bahatyrova, R. N. Frese, C. A. Siebert, J. D. Olsen, K. O. van der Werf, R. van Grondelle, R. A. Niederman, P. A. Bullough, C. Otto and C. N. Hunter, Nature, 2004, 430, 1058 CrossRef CAS; (f) S. Scheuring, J. N. Sturgis, V. Prima, A. Bernadac, D. Levy and J.-L. Rigaud, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11293 CrossRef CAS; (g) S. Scheuring and J. N. Sturgis, Science, 2005, 309, 484 CrossRef CAS.
- (a) Photochemical Conversion and Storage of Solar Energy, ed. J. S. Connolly, Academic, New York, 1981 Search PubMed; (b) Molecular Level Artificial Photosynthetic Materials, ed. G. J. Meyer, Wiley, New York, 1997 Search PubMed; (c) M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS; (d) J. W. Verhoeven, Adv. Chem. Phys., 1999, 106, 603 CrossRef CAS; (e) A. Osuka, N. Mataga and T. Okada, Pure Appl. Chem., 1997, 69, 797 CrossRef CAS; (f) L. Flamigni, F. Barigelletti, N. Armaroli, J. P. Collin, I. M. Dixon, J.-P. Sauvage and J. A. G. Williams, Coord. Chem. Rev., 1999, 190–192, 671 CrossRef CAS; (g) F. Diederich and M. Gomez-Lopez, Chem. Soc. Rev., 1999, 28, 263 RSC.
- (a) M.-J. Blanco, M. Consuelo Jimenez, J.-C. Chambron, V. Heitz, M. Linke and J.-P. Sauvage, Chem. Soc. Rev., 1999, 28, 293 RSC; (b) V. Balzani, P. Ceroni, A. Juris, M. Venturi, S. Campagna, F. Puntoriero and S. Serroni, Coord. Chem. Rev., 2001, 219–221, 545 CrossRef CAS; (c) V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26 CrossRef CAS.
- (a) M. Bixon, J. Fajer, G. Feher, J. H. Freed, D. Gamliel, A. J. Hoff, H. Levanon, K. Möbius, R. Nechushtai, J. R. Norris, A. Scherz, J. L. Sessler and D. Stehlik, Isr. J. Chem., 1992, 32, 449 Search PubMed; (b) F. D. Lewis, R. L. Letsinger and M. R. Wasielewski, Acc. Chem. Res., 2001, 34, 159 CrossRef CAS.
- (a) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 198 CrossRef CAS; (b) D. Gust and T. A. Moore in The Porphyrin Handbook, ed. K. M. Kadish, K. Smith and R. Guilard, vol. 8, Academic Press, San Diego, 2000, pp. 153–190 Search PubMed; (c) Y. Terazono, G. Kodis, P. A. Liddell, V. Garg, T. A. Moore, A. L. Moore and D. Gust, J. Phys. Chem. B, 2009, 113, 7147 CrossRef CAS.
- (a) S. Fukuzumi and D. M. Guldi, in Electron Transfer in Chemistry, vol. 2., ed. V. Balzani, Wiley-VCH, Weinheim 2001, pp. 270–337 Search PubMed; (b) S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609–620 RSC; (c) S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283 RSC; (d) S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 177–195 CrossRef CAS; (e) S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427–1439 RSC.
- (a) Y. Sakata, H. Imahori, H. Tsue, S. Higashida, T. Akiyama, E. Yoshizawa, M. Aoki, K. Yamada, K. Hagiwara, S. Taniguchi and T. Okada, Pure Appl. Chem., 1997, 69, 1951 CrossRef CAS; (b) H. Imahori and Y. Sakata, Eur. J. Org. Chem., 1999, 2445 CrossRef CAS; (c) H. Imahori, K. Tamaki, Y. Araki, Y. Sekiguchi, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2002, 124, 5165 CrossRef CAS; (d) T. Umeyama and H. Imahori, Energy Environ. Sci., 2008, 1, 120 RSC.
- (a) D. M. Guldi, Chem. Commun., 2000, 321 RSC; (b) D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC; (c) V. Sgobba and D. M. Guldi, Chem. Soc. Rev., 2009, 38, 165 RSC; (d) A. Mateo-Alonso, D. M. Guldi, F. Paolucci and M. Prato, Angew. Chem., Int. Ed., 2007, 46, 8120 CrossRef CAS; (e) D. M. Guldi, Phys. Chem. Chem. Phys., 2007, 9, 1400 RSC; (f) L. Sanchez, M. Nazario and D. M. Guldi, Angew. Chem., Int. Ed., 2005, 44, 5374 CrossRef CAS; (g) G. Bottair, V. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768 CrossRef; (h) M. Martinez-Diaz, V. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090 RSC.
- J. S. Sessler, B. Wang, S. L. Springs and C. T. Brown, in Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Vögtle, ch. 9, Pergamon, 1996 Search PubMed.
- (a) M. E. El-Khouly, O. Ito, P. M. Smith and F. D'Souza, J. Photochem. Photobiol., C, 2004, 5, 79 CrossRef CAS; (b) F. D'Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410 CrossRef CAS; (c) F. D'Souza, O. Ito, in Handbook of Organic Electronics and Photonics, vol. 1, ed. H. R. Nalwa, American Scientific Publishers, 2008, ch. 13, pp. 485–521 Search PubMed; (d) R. Chitta and F. D'Souza, J. Mater. Chem., 2008, 18, 1440 RSC; (e) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC; (f) F. D'Souza and O. Ito in Handbook of Porphyrin Science, Vol. 1 ed. K. M. Kadish, R. Guilard and K. M. Smith, World Science, Singapore, 2010, ch. 4, p. 307 Search PubMed.
- (a) Introduction of Molecular Electronics, ed. M. C. Petty, M. R. Bryce and D. Bloor, Oxford University Press, New York, 1995 Search PubMed; (b) Molecular Electronics: Science and Technology, ed. A. Aviram and M. Ratner: Ann. NY Acad. Sci. 1998, p. 852 Search PubMed; (c) Molecular Switches, ed. B. L. Feringa, Wiley-VCH GmbH, Weinheim, 2001 Search PubMed; (d) D. Gust, T. A. Moore and A. L. Moore, Chem. Commun., 2006, 1169 RSC; (e) V. Balzani, A. Credi and M. Venturi, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, 2008, p. 1–31 Search PubMed; (f) Energy Harvesting Materials, ed. D. L. Andrews, World Scientific, Singapore, 2005 Search PubMed; (g) J. M. Tour, Molecular Electronics; Commercial Insights, Chemistry, Devices, Architectures and Programming, World Scientific, River Edge, NJ, 2003 Search PubMed.
- (a) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2007, 46, 2 CrossRef; (b) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS; (c) A. C. Benniston and G. Copley, Phys. Chem. Chem. Phys., 2009, 11, 4124 RSC; (d) J. D. Megiatto and D. I. Schuster, in Handbook of Carbon Nanomaterials, vol. 1, ed. F. D'Souza and K. M. Kadish, ch. 7, pp. 207–244, Work Scientific Publishing, Singapore, 2011 Search PubMed.
- (a) R. W. Wagner, J. S. Lindsey, J. Seth, V. Palaniappan and D. F. Bocian, J. Am. Chem. Soc., 1996, 118, 3996 CrossRef CAS; (b) R. K. Lammi, R. W. Wagner, A. Ambroise, J. R. Diers, D. F. Bocian, D. Holten and J. S. Lindsey, J. Phys. Chem. B, 2001, 105, 5341 CrossRef CAS; (c) F. Li, S. I. Yang, Y. Ciringh, J. Seth, C. H. Martin, D. L. Singh, D. Kim, R. R. Birge, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1998, 120, 10001 CrossRef CAS; (d) R. W. Wagner and J. S. Lindsey, Pure Appl. Chem., 1996, 68, 1373 CrossRef CAS; (e) R. W. Wagner and J. S. Lindsey, J. Am. Chem. Soc., 1994, 116, 9759 CrossRef CAS; (f) H. L. Kee, C. Kirmaier, L. Yu, P. Thamyongkit, W. J. Youngblood, J. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheidt, R. R. Birge, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 109, 20433 CrossRef CAS.
- (a) H. Imahori, H. Norieda, H. Yamada, Y. Nishimura, I. Yamazaki, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 100 CrossRef CAS; (b) Y. Terazono, G. Kodis, P. A. Liddell, V. Garg, T. A. Moore, A. L. Moore and D. Gust, J. Phys. Chem. B, 2009, 113, 7147 CrossRef CAS; (c) A. Harriman, L. J. Mallon, K. J. Elliot, A. Haefele, G. Ulrich and R. Ziessel, J. Am. Chem. Soc., 2009, 131, 13375 CrossRef CAS; (d) A. Harriman, L. J. Mallon, S. Goeb, G. Ulrich and R. Ziessel, Chem.–Eur. J., 2009, 15, 4553 CrossRef CAS; (e) S. Diring, F. Putoriero, F. Nastasi, S. Campagna and R. Ziessel, J. Am. Chem. Soc., 2009, 131, 6108 CrossRef CAS; (f) J.-Y. Liu, E. A. Ermilov, B. Roder and D. K. P. Ng, Chem. Commun., 2009, 1527 Search PubMed; (g) J.-Y. Liu, H.-S. Yeung, W. Xu, X. Li and D. K. P. Ng, Org. Lett., 2008, 10, 5421 CrossRef CAS; (h) X. Zhang, Y. Xiao and X. Zian, Org. Lett., 2008, 10, 29 CrossRef CAS; (i) R. Ziessel, B. D. Allen, D. B. Rewinska and A. Harriman, Chem.–Eur. J., 2009, 15, 7382 CrossRef CAS; (j) D. Kumaresan, A. Datta and M. Ravikanth, Chem. Phys. Lett., 2004, 395, 87 CrossRef CAS; (k) A. Harriman, J. P. Cesqario, G. Ulrich and R. Ziessel, J. Phys. Chem. A, 2006, 110, 7994 CrossRef CAS.
- (a) M. D. Yilmaz, O. A. Bozdemir and E. U. Akkaya, Org. Lett., 2006, 8, 2871 CrossRef CAS; (b) A. Herrmann, T. Weil, V. Sinigersky, U.-M. Wiesler, T. Vosch, J. Hofkens, F. C. De Schryver and K. Mullen, Chem.–Eur. J., 2001, 7, 4844 CrossRef CAS; (c) J. M. Serin, D. W. Brousmiche and J. M. J. Fréchet, Chem. Commun., 2002, 2605 RSC; (d) J. M. Serin, D. W. Brousmiche and J. M. J. Fréchet, J. Am. Chem. Soc., 2002, 124, 11848 CrossRef CAS; (e) S. Hattori, K. Ohkubo, Y. Urano, H. Sunahara, T. Nagano, Y. Wada, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, J. Phys. Chem. B, 2005, 109, 15368 CrossRef CAS; (f) M. Kollmannsberger, K. Rurack, U. Resch-Genger and J. Daub, J. Phys. Chem. B, 1998, 102, 10211 CrossRef CAS; (g) C. A. Wijesinghe, M. E. El-Khouly, J. D. Blakemore, M. E. Zandler, S. Fukuzumi and F. D'Souza, Chem. Commun., 2010, 46, 3301 RSC; (h) C. A. Wijesinghe, M. E. El-Khouly, N. K. Subbaiyan, M. Supur, M. E. Zandler, K. Ohkubo, S. Fukuzumi and F. D'Souza, Chem.–Eur. J., 2011, 17, 3147 CrossRef CAS.
- F. D'Souza, P. M. Smith, M. E. Zandler, A. L. McCarty, M. Itou, Y. Araki and O. Ito, J. Am. Chem. Soc., 2004, 126, 7898 CrossRef CAS.
- E. Maligaspe, T. Kumpulainen, N. K. Subbaiyan, M. E. Zandler, H. Lemmetyinen, N. V. Tkachenko and F. D'Souza, Phys. Chem. Chem. Phys., 2010, 12, 7434 RSC.
- E. Maligaspe, N. V. Tkachenko, N. K. Subbaiyan, R. Chitta, M. E. Zandler, H. Lemmetyinen and F. D'Souza, J. Phys. Chem. A, 2009, 113, 8478 CrossRef CAS.
- Principles of Fluorescence Spectroscopy, 3rd ed. By J. R. Lakowicz, 2006, Springer, Singapore Search PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
- For additional examples using this basis set, see: M. E. Zandler and F. D'Souza, C. R. Chim., 2006, 9, 960 CrossRef CAS.
- A. Weller, Z. Phys. Chem., 1982, 132, 93 Search PubMed.
- F. D'Souza, R. Chitta, S. Gadde, A. L. McCarty, P. A. Karr, M. E. Zandler, A. S. D. Sankanayaka, Y. Araki and O. Ito, J. Phys. Chem. B, 2006, 110, 5905 CrossRef CAS.
- C. H. Lee and J. S. Lindsey, Tetrahedron, 1994, 50, 11427 CrossRef CAS.
- G. R. Deviprasad, B. Keshavan and F. D'Souza, J. Chem. Soc., Perkin Trans. 1, 1998, 3133 RSC.
- C. Luo, D. M Guldi, H. Imahori, K. Tamaki and Y. Sakata, J. Am. Chem. Soc., 2000, 122, 6535 CrossRef CAS.
- C. Tahtaoui, C. Thomas, F. Rohmer, P. Klotz, G. Duportail, Y. Mely, D. Bonnet and M. Hibert, J. Org. Chem., 2007, 72, 269 CrossRef CAS.
| This journal is © the Owner Societies 2011 |