 Open Access Article
Open Access ArticleDefinitive solid-state 185/187Re NMR spectral evidence for and analysis of the origin of high-order quadrupole-induced effects for I = 5/2†
Cory M.
Widdifield
a,
Alex D.
Bain
b and
David L.
Bryce
*a
aDepartment of Chemistry and Centre for Catalysis Research and Innovation, University of Ottawa, 10 Marie Curie Pvt., Ottawa, Ontario, Canada. E-mail: dbryce@uottawa.ca; Fax: +1 613 562 5170; Tel: +1 613 562 5800 ext. 2018
bDepartment of Chemistry and Chemical Biology, McMaster University, 1280 Main St. W., Hamilton, Ontario, Canada. E-mail: bain@mcmaster.ca for software enquiries
First published on 1st June 2011
Abstract
Rhenium-185/187 solid-state nuclear magnetic resonance (SSNMR) experiments using NaReO4 and NH4ReO4 powders provide unambiguous evidence for the existence of high-order quadrupole-induced effects (HOQIE) in SSNMR spectra. Fine structure, not predicted by second-order perturbation theory, has been observed in the 185/187Re SSNMR spectrum of NaReO4 at 11.75 T, where the ratio of the Larmor frequency (ν0) to the quadrupole frequency (νQ) is ∼2.6. This is the first experimental observation that under static conditions, HOQIE can directly manifest in SSNMR powder patterns as additional fine structure. Using NMR simulation software which includes the quadrupole interaction (QI) exactly, extremely large 185/187Re nuclear quadrupole coupling constants (CQ) are accurately determined. QI parameters are confirmed independently using solid-state 185/187Re nuclear quadrupole resonance (NQR). We explain the spectral origin of the HOQIE and provide general guidelines that may be used to assess when HOQIE may impact the interpretation of the SSNMR powder pattern of any spin-5/2 nucleus in a large, axially symmetric electric field gradient (EFG). We also quantify the errors incurred when modeling SSNMR spectra for any spin-5/2 nucleus within an axial EFG using second-order perturbation theory. Lastly, we measure rhenium chemical shifts in the solid state for the first time.
Introduction
All quadrupolar nuclei (i.e., I > 1/2) possess a nuclear electric quadrupole moment (Q), which will couple with the electric field gradient (EFG) at the nucleus.1 The coupling between Q and the EFG, known as the quadrupole interaction (QI), provides information that can be used to complement other solid-state nuclear magnetic resonance (SSNMR) observables, such as the isotropic chemical shift (CS). Unfortunately, the QI may also drastically broaden the SSNMR signal in powdered samples, sometimes to the extent that the experiment becomes impractical. Despite this potential drawback, SSNMR experiments using quadrupolar nuclei are valuable, as these nuclei can be found in many important areas of chemical research, including biochemistry (e.g., 14N, 17O, 23Na, 25Mg, 43Ca, 67Zn), and materials science (e.g., 6/7Li, 11B, 17O, 27Al).2 The precise determination of the QI is therefore of critical importance to correctly characterize a wide variety of systems using SSNMR spectroscopy. Until now, it was most common to use second-order perturbation theory to model SSNMR line shapes (to see how first- and second-order perturbation theory modifies the Zeeman eigenstates, see the ESI, Fig. S1a†); however, as the sensitivity of SSNMR experiments continues to increase, experiments using previously “inaccessible” nuclei are becoming more common and additional care needs to be taken when analyzing the SSNMR spectra of quadrupolar nuclei that experience a very large QI.Rhenium, a group 7 transition metal, was first detected in 1925, occurs naturally within molybdenum sulfide ores in the earth's crust (∼10−7% abundance), and may exist in at least nine oxidation states (ranging from −1 to +7).3,4 Compounds containing rhenium in a relatively reduced oxidation state, such as Re(III), exhibit metal–metal bonding interactions: for example, K2Re2Cl8 is recognized as containing the first example of a metal–metal quadruple bond.5Rhenium metal possesses very high thermal stability, and is present within the high-temperature alloys used to make jet engine parts.4 In addition, rhenium-containing compounds have been used as catalysts in many types of organic reactions.6–8 The nuclei of the two stable isotopes of rhenium (185Re/187Re) are NMR-active, are present in high natural abundance (37.398(16)% and 62.602(16)% for 185Re and 187Re, respectively),9 and are quadrupolar (I(185/187Re) = 5/2). In addition, they possess relatively high magnetogyric ratios (γ(185Re) = 6.1057 × 107 rad s−1 T−1; γ(187Re) = 6.1682 × 107 rad s−1 T−1).10 Despite the potential wealth of diagnostic information that could be extracted using 185/187Re SSNMR experiments, very few literature reports exist, and they are nearly exclusively restricted to compounds that exhibit very high symmetry,11–13 or are from experiments carried out at liquid helium temperatures.14–16 The paucity of 185/187Re SSNMR studies may be attributed to the very large Q for both NMR-active nuclides (Q(185Re) = 2180(20) mb; Q(187Re) = 2070(20) mb).17 In fact, the line-width factor for 185Re is the highest of the stable elements (1.5 × 104 relative to 1H).10 As such, a very small EFG can result in a rhenium QI that broadens the SSNMR powder pattern to the point that it is undetectable. A partial remedy to this problem is to perform the SSNMR experiments within as high an applied magnetic field (B0) as possible.
As very high magnetic fields (i.e., B0 > 18.8 T) are becoming increasingly available, SSNMR experiments on previously inaccessible nuclei are now potentially feasible, but remain technically challenging as they often require sensitivity-enhancing pulse sequences18,19 and/or variable offset cumulative spectrum (VOCS) data acquisition.20–22 Recently, we have shown that subtle “high-order” (i.e., greater than second-order) quadrupole-induced effects (HOQIE) are present in the SSNMR spectra for 127I (I = 5/2) at B0 = 11.75 T and 21.1 T for some alkaline earth iodides.23 In those cases, the observed HOQIE manifested as a non-uniform frequency-dependent shift of the 127I SSNMR spectrum. While high-order perturbation theory may have been useful for modeling these 127I SSNMR line shapes, we used a simulation code that included Zeeman and QI effects exactly,24,25 as well as 127I NQR experiments, to precisely measure the EFG tensor magnitude, as well as the isotropic iodine chemical shift.
As part of an effort to more generally and completely understand the origin and ramifications of HOQIE on SSNMR spectra for I = 5/2 nuclides, we report here 185/187Re SSNMR spectra for NH4ReO4 and NaReO4 in standard and ultrahigh B0. Prior 185/187Re SSNMR measurements on these two compounds highlighted some of the largest QIs ever measured using NMR, although quantitative EFG tensor information could not be extracted for NaReO4.22 While repeating the prior 185/187Re SSNMR measurements on NaReO4 at 11.75 T, we observed previously undetected fine structure. Using both second-order perturbation theory and exact QI simulations, we comment upon the origin of the fine structure and also outline some guidelines that are generally applicable when modeling the SSNMR spectra for any I = 5/2 nucleus which experiences a large, axially symmetric QI.
Experimental
1 Sample preparation
Both NaReO4 (99.99%) and NH4ReO4 (99.999%) were purchased from Sigma-Aldrich and were received as powders. Sample purity was confirmed by the manufacturer (ESI, Additional Experimental†). Both compounds are stable under normal conditions. All samples were tightly packed into 4 mm o.d. Bruker magic angle spinning (MAS) ZrO2 rotors.2 Solid-state 185/187Re NMR
Experimental data were acquired at the National Ultrahigh-field NMR Facility for Solids in Ottawa and at the University of Ottawa. The Ultrahigh-field Facility experiments used a standard bore Bruker AVANCE II spectrometer, which operates at B0 = 21.1 T (ν0(1H) ≈ 900.08 MHz, ν0(185Re) = 202.738 MHz, and ν0(187Re) = 204.781 MHz), and a 4 mm Bruker HX MAS probe. The experiments performed at the University of Ottawa used a wide bore Bruker AVANCE spectrometer, which operates at B0 = 11.75 T (ν0(1H) ≈ 500.13 MHz, ν0(185Re) = 112.652 MHz, and ν0(187Re) = 113.787 MHz), and a 4 mm Bruker HXY MAS probe. The 185/187Re SSNMR signals were referenced to a 0.1 mol dm−3 solution of NaReO4 in D2O at 0.0 ppm. The 185/187Re pulse lengths used for experiments on NH4ReO4 were established using the solution reference, and include a scaling of the optimized solution pulse by 1/[I + 1/2] = 1/3 to selectively excite the central transition (mI = 1/2 ↔ −1/2; CT) of the solid. Due to the excessive width of the 185/187Re SSNMR signals of NaReO4 (vide infra), the high- and low-frequency pulse lengths were calibrated using the high- and low-frequency CT and satellite transition (ST) discontinuities of the actual powder sample. For further details on the frequency-dependence of the pulse lengths used to acquire the 185/187Re SSNMR signals of NaReO4, see the ESI, Table S1.†The 185/187Re SSNMR signals were acquired using either Solomon (i.e., “solid”) echo (i.e., π/2–τ1–π/2–τ2–acq)26–28 or Hahn echo (i.e., π/2–τ1–π–τ2–acq)29pulse sequences (see also the ESI†). Typical parameters were as follows: π/2 = 1.1 to 1.7 μs; spectral window = 2 MHz; τ1 = 12.75 to 13.8 μs; recycle delay ≈ 100 ms, and between 512 and 1024 complex time-domain data points were collected per scan. All final SSNMR spectra were prepared using VOCS data acquisition methods.20–22 This involves stepping the radiofrequency transmitter at uniform offset values, with the acquisition of a ‘sub-spectrum’ at each step. The offsets used here were 200 and 300 kHz for Hahn and Solomon echo experiments, respectively. For each transmitter setting, between 8192 and 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 transients were collected. Each processed sub-spectrum was combined in the frequency domain via co-addition to produce the final spectrum. Due to the temperature dependence of the 185/187Re QI for these compounds, all experiments were performed at T = 291.8(0.2) K, as monitored via a Bruker ‘type-T’ thermocouple and regulated using a standard Bruker variable temperature unit. For full experimental details, see the ESI, Table S1.†
500 transients were collected. Each processed sub-spectrum was combined in the frequency domain via co-addition to produce the final spectrum. Due to the temperature dependence of the 185/187Re QI for these compounds, all experiments were performed at T = 291.8(0.2) K, as monitored via a Bruker ‘type-T’ thermocouple and regulated using a standard Bruker variable temperature unit. For full experimental details, see the ESI, Table S1.†
3 Solid-State 185/187Re NQR
All experiments were carried out at the University of Ottawa using the AVANCE spectrometer outlined above. In addition, NQR experiments used a 4 mm Bruker HX MAS probe, and all spectra were acquired using the Hahn echo pulse sequence at T = 291.8(0.2) K. Non-optimized, short (≤ 1.6 μs), high-powered pulses were used as the radiofrequency was varied until a particular resonance was detected. The offset used while searching for 185/187Re NQR signals was 200 kHz. For further details, see the ESI, Table S1.†Results and discussion
1 Rhenium-185/187 solid-state NMR
| Compound | ν 1(185Re)/MHz | ν 2(185Re)/MHz | ν 1(187Re)/MHz | ν 2(187Re)/MHz | |CQ(185Re)| b/MHz | |CQ(187Re)|/MHz | η Q | δ iso c/ppm |
|---|---|---|---|---|---|---|---|---|
| a Measurement errors are within parentheses and parameter definitions are as follows: CQ = eQV33/h; ηQ = (V11–V22)/V33, where |V11| ≤ |V22| ≤|V33|; δiso = (δ11 + δ22 + δ33)/3, where δ33 ≤ δ22 ≤ δ11. The frequencies ν1 and ν2 correspond to the doubly-degenerate single quantum NQR resonance frequencies, which for I = 5/2 and ηQ = 0 can be defined as: ν1 = νQ = 3CQ/20 and ν2 = 2νQ = 3CQ/10. All measurements were carried out at T = 291.8(0.2) K. SSNMR line shape simulations were performed using exact theory.24 EFG tensor parameters using NQR data were determined using the procedure outlined by Semin.54 b While CQ may take any real value, |CQ| is measured experimentally using NQR/SSNMR. On the basis of our data, we find that the maximum possible value for the 185Re nuclear electric hexadecapole interaction in NaReO4 is ca. 750 Hz.35 c Rhenium chemical shifts are relative to 0.1 mol dm−3NaReO4 in D2O (δiso(185/187Re) = 0 ppm). | ||||||||
| NaReO4 | 45.102(0.006) | 90.204(0.009) | 42.681(0.005) | 85.362(0.008) | 300.68(0.02) | 284.54(0.02) | <0.003 | 70(40) |
| NH4ReO4 | — | 35.068(0.010) | — | 33.186(0.008) | 116.90(0.04) | 110.62(0.03) | <0.003 | 0(40) |
Rhenium-185/187 NQR experiments were performed (ESI, Fig. S2†) and the resulting transition frequencies (Table 1) allow us to confirm that ηQ(185/187Re) = 0. As the ratio between the mI = ±3/2 ↔ ±5/2 and mI = ±1/2 ↔ ±3/2 transition frequencies (|ΔmI| = 1) is exactly 2 within experimental error, we may conclude that there is no evidence of a nuclear electric hexadecapole interaction.35 Using our new NQR data, we establish that CQ(185Re)=300.68(0.02) MHz and CQ(187Re) = 284.54(0.02) MHz at 291.8 K. The slight discrepancy between our CQ(185/187Re) values and those measured earlier by NQR can be fully attributed to the difference in the respective measurement temperatures.36
To measure the rhenium EFG and CS tensors for this sample using SSNMR, we carried out 185/187Re SSNMR experiments on powdered NaReO4 at B0 = 11.75 and 21.1 T (Fig. 1 and 2). In the present study, the 185/187Re SSNMR signals typically overlap one another due to their similar Larmor frequencies. After careful line shape analysis using exact QI simulation software24 (it is noted here that other exact QI models exist),37–40 quantitative agreement between the rhenium EFG tensor parameters determined using our 185/187Re NQR and NMR measurements was established. In addition, we were able to measure for the first time an isotropic rhenium CS in a solid sample (δiso = 70(40) ppm relative to 0.1 mol dm−3NaReO4 in D2O). This opens up the possibility that 185/187Re SSNMR experiments could be used to report on the local rhenium bonding environment or the oxidation state (under favorable conditions) in solid samples. Indeed, the sparse solution 185/187Re NMR literature data highlight a chemical shift range of ∼6800 ppm.41,42 We were not able to measure rhenium chemical shift anisotropy (CSA) for this sample.
 | ||
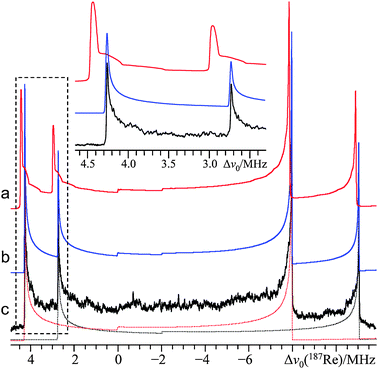
| Fig. 1 Second-order perturbation theory simulation (a, d), exact simulation (b, e), and experimental static VOCS Hahn echo (c, f) 185/187Re SSNMR spectrum of powdered NaReO4, acquired at B0 = 11.75 T (ν0(187Re) = 113.787 MHz; ν0(185Re) = 112.652 MHz) and T = 291.8 K. The second-order perturbation theory spectrum includes only the 187Re signal, to enhance clarity. The high frequency region is depicted in (a, b, c) and the low frequency region is in (d, e, f). Please note that the horizontal scaling is not equivalent between (a, b, c) and (d, e, f): in the inset, the experimental spectrum is displayed using equivalent horizontal scaling for both regions. Below (c and f), the exact simulation line shapes associated with each of 185Re and 187Re are deconvoluted: the long dashed red trace is 187Re, while the short dashed black trace is 185Re. Low-frequency splittings (denoted by double-headed arrows and guide lines) are not predicted by second-order perturbation theory. The discontinuities due to the mI = 1/2 ↔ 3/2 STs are marked by “*”, while the remainder of the discontinuities are due to the mI = 1/2 ↔ −1/2 transition. All simulations use identical EFG tensor parameters, which were also measured independently using 185/187Re NQR experiments. Minor discontinuities in the slope of trace (a) are due to the POWDER algorithm55 used for powder averaging. | ||
 | ||
| Fig. 2 Second-order perturbation theory simulation (a), exact simulation (b), and experimental static VOCS Solomon echo (c) 185/187Re SSNMR spectra of powdered NaReO4, acquired at B0 = 21.1 T (ν0(187Re) = 204.781 MHz; ν0(185Re) = 202.738 MHz) and T = 291.8 K. Below c, the exact simulation signals associated with each of 185Re and 187Re are deconvoluted: the dotted red trace is 187Re, while the dotted black trace is 185Re. Low-frequency splittings are not observed; however, the expected positions of the discontinuities in the analytical simulation are subject to a non-uniform, frequency-dependent shift, which is evidence of HOQIE. All simulations used identical EFG tensor parameters, which were also measured independently using 185/187Re NQR experiments. The inset (top, middle) corresponds to the region within the dashed line box, and is meant to highlight the significant difference between the exact and second-order perturbation theory models. For the inset, the deconvolution traces have been omitted to enhance clarity. | ||
Upon inspection of Fig. 1 and 2, it is immediately clear that the predominantly CT line shapes at both applied fields are very broad (∼16 and 26 MHz for the spectra acquired at B0 = 21.1 and 11.75 T, respectively), but the most striking aspect of the spectrum at 11.75 T is the presence of unexpected high-intensity discontinuities (i.e., not predicted using second-order perturbation theory; Fig. 1f). It is intriguing that the extra discontinuities do not appear in the 185/187Re SSNMR spectrum acquired at 21.1 T. This B0-dependent system response is consistent with the expected behavior of a second-order (or greater) quadrupole-induced effect and can be fully attributed to HOQIE (vide infra). While it is perhaps clear that up to two additional low-frequency discontinuities could be due to the inner (mI = 1/2 ↔ 3/2) ST (one from each of 185Re and 187Re), second-order perturbation theory still fails rather spectacularly when one applies the correct 187Re EFG tensor parameters (from NQR and exact line shape simulations) and includes the STs within the model (Fig. 1a and d). The high-frequency 187Re CT discontinuity position is overestimated by ∼1 MHz, as are both the position of the corresponding low-frequency CT discontinuity and that of the mI = 1/2 ↔ 3/2 ST. Line shape modeling using exact QI software leads to both the correct number and frequency positions for all eight observed discontinuities for NaReO4 using the parameters in Table 1. Most importantly, the exact QI model predicts the experimentally observed fine structure in the low-frequency spectral region for both 185Re and 187Re (Fig. 1f). These additional features are field-dependent, are attributed to the mI = 1/2 ↔ 3/2 ST transition (vide infra), and do not interfere with the CT 185/187Re SSNMR spectra at 21.1 T (Fig. 2).
Although second-order perturbation theory does not predict the correct placement of the CT discontinuities for NaReO4, even at 21.1 T (Fig. 2a), it does predict the correct number of discontinuities at the higher applied field. At 21.1 T, the additional fine structure observed at 11.75 T is not present and it appears that HOQIE manifest as a non-uniform frequency-dependent shift in the positions of the discontinuities (with a notable bias towards a positive-frequency shift; the effective parameters determined using second-order perturbation theory are summarized in the ESI, Table S2†). It is therefore clear that the unusual B0-dependent behavior is due to HOQIE, and that the fine structure observed at 11.75 T must be due to a 3rd-order QI effect on the ST and/or a 4th-order QI effect on the CT and/or ST (as 3rd-order effects on the CT are known to be zero).43 Beyond 4th-order effects are also potentially significant, but are expected to be much smaller than the leading-order contributions. It is possible that fourth-order perturbation theory may be able to produce accurate line shape models in the regime where the value of νQ becomes somewhat comparable to the Larmor frequency (ν0). Overall, it is seen that second-order perturbation theory cannot lead to the correct values for either δiso or CQ(185/187Re) under these conditions and in fact both quantities will be underestimated relative to their true values (vide infra).
To provide a second independent measure of the 185/187Re EFG tensor parameters in NH4ReO4, rhenium NQR experiments were performed (see ESI, Fig. S3†) and the measured NQR transition frequencies are summarized in Table 1. Using the rhenium NQR data in tandem with the multiple field 185/187Re SSNMR data (Fig. 3) (SSNMR data analyzed using exact QI line shape modeling), it is observed that CQ(185Re) = 116.90(0.04) MHz, CQ(187Re) = 110.62(0.03) MHz, and ηQ(185/187Re) = 0 at T = 291.8 K for this sample. After adjusting for the well-known temperature dependence of the rhenium QI in NH4ReO4, these measurements are fully consistent with prior NQR findings. Unlike the 185/187Re SSNMR spectra of NaReO4 recorded at 11.75 T, there is no evidence of unexpected additional fine structure in the 185/187Re CT SSNMR spectrum of NH4ReO4. Using exact QI simulation software, we were able to measure the isotropic rhenium chemical shift for this sample as 0(40) ppm. At the lower applied field, the extracted isotropic chemical shifts using second-order perturbation theory and exact theory do not agree with one another. As was the case in our prior study using 127I SSNMR data,23 and with the 185/187Re SSNMR spectral simulations for NaReO4 at 21.1 T, the second-order simulation produces a chemical shift that is smaller than the true value (for the values extracted using second-order perturbation theory, see the ESI, Table S2†). Unlike the rhenium SSNMR line shape simulations for NaReO4, however, the rhenium EFG tensor parameters extracted for NH4ReO4 using both second-order perturbation theory and exact theory match, within experimental error, when modeling the spectrum acquired at 11.75 T. At 21.1 T, quantitative agreement is found between second-order and exact theory for all the reported parameters in Table 1. Hence, there is no evidence of HOQIE in the spectra for NH4ReO4 at 21.1 T and second-order perturbation theory would be sufficient to model this SSNMR line shape. The quantitative agreement for this final case is sensible, as at 21.1 T, the ratio between ν0 and νQ for NH4ReO4 exceeds 10 to 1, which is a regime where HOQIE are not expected to be significant (i.e., the high-field approximation is valid).
 | ||
| Fig. 3 Exact simulation (a, c), experimental static VOCS Hahn echo (b), and static VOCS Solomon echo (d) 185/187Re SSNMR spectra of powdered NH4ReO4, acquired at (b) B0 = 11.75 T and (d) B0 = 21.1 T. Below b and d, the exact simulation signals associated with each of 187Re (dotted red trace) and 185Re (dashed black trace) are deconvoluted. All experiments were performed at T = 291.8 K. | ||
As the two CT 185/187Re signals for NH4ReO4 are separated at 21.1 T (Fig. 3d), and as the central “step” discontinuity is clearly defined for both isotopes, we attempted to include rhenium CSA in the line shape models for this sample. Unfortunately, the rhenium CS tensor span was too small to be measured and it is noted that the span must be less than ca. 80 ppm. A small tensor span is consistent with the nearly tetrahedral local symmetry about the rhenium atoms.
In Fig. 4, line shape simulations are presented which were generated using the exact simulation software. We examined the regime where the ν0/νQ ratio value ranges from 4.0 to 2.0 in steps of −0.2. As we wish to comment upon the origin of the fine structure for this particular case, we have set ν0 to 113.787 MHz (the value of the NMR resonance condition for the 187Re solution standard at 11.75 T). At ν0/νQ = 4, it is noted that the inner ST discontinuity (i.e., mI = 1/2 ↔ 3/2) is expected to have a relatively high intensity, and there is no additional fine structure. However, as the ν0/νQ value is further decreased, the inner low-frequency ST splits into two discontinuities. Eventually, the discontinuities shift to such an extent that they will both occur within the spectral region which is normally (using second-order perturbation theory) attributed to the CT. For the experimental case of 185/187Re SSNMR of NaReO4 at 11.75 T, the ν0/νQ value is roughly 2.6:1, which closely resembles the ratio used to generate the trace indicated using a dagger in Fig. 4. Hence, we may conclude that the experimentally observed fine structure is due to a high-order splitting in the mI =1/2 ↔ 3/2 ST for each of the 185Re and 187Re nuclides. One can expect this type of fine structure to exist (although it will not likely interfere with the CT signal until ν0/νQ < 3) once ν0/νQ becomes less than 4. This effect on the ST appears to be similar in nature to the splittings observed in certain STMAS experiments, which were attributed to a third-order quadrupole-induced effect.51 Fine structure due to third-order effects is also predicted to arise in 14N (I = 1) MAS NMR spectra when the value of CQ becomes large, although it appears that this has not been experimentally validated.52
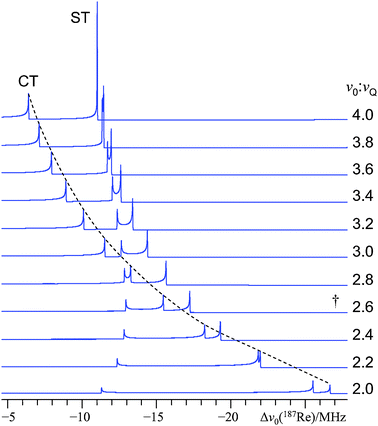 | ||
| Fig. 4 Exact simulations of the low-frequency spectral region (part of the CT and mI = 1/2 ↔ 3/2 ST discontinuities only), which highlight the onset and origin of the HOQIE fine structure for NaReO4. For this particular simulation, the 187Re nucleus at 11.75 T has been assumed; hence, ν0 = 113.787 MHz. By adjusting νQ, the ν0/νQ ratio is varied from 4.0 (top trace) to 2.0 (bottom trace) in steps of −0.2. The dashed line trace clarifies the evolution of the low-frequency CT discontinuity as a function of ν0/νQ. The spectrum corresponding closely to the best-fit spectrum in Fig. 1 is demarked with a dagger above it. | ||
2 General guidelines for NMR spectral analysis when any I = 5/2 nucleus experiences a very large, axial QI
It is well known that second-order perturbation theory is a valid method to model many SSNMR line shapes associated with half-integer quadrupolar nuclei; however, care must be taken to ensure that the high-field condition is satisfied (often taken as ν0 > 10νQ). For a large enough QI (i.e., ν0 < 4νQ), it was established above, using experiment and theory, that additional fine structure is present in the SSNMR spectrum. Under these conditions, second-order perturbation theory does not even predict the correct number of discontinuities, and it is not meaningful to quantify the differences in the extracted NMR parameters between perturbation theory and exact theory. Between these two cases, therefore, there exists a region where the high field approximation is not clearly valid, but the additional fine structure is not observable. We comment here upon the errors in the SSNMR parameters extracted (namely CQ and δiso) in this intermediate regime when using second-order perturbation theory as compared to exact theory. Methodology details for this section can be found in the ESI, Additional Experimental.†We consider here the case where a nuclear spin having I = 5/2 is subjected to an axial EFG and B0. The inclusion of additional effects, such as ηQ ≠ 0, CSA, dipole–dipole, etc., is beyond the scope of the current study. For cases where the high-field approximation is traditionally viewed as being valid (ν0 > 10νQ), it is found that the error in the extracted CQ value is at most ca. 0.1% (Fig. 5a). However, the error in the isotropic chemical shift, even within the high-field condition, can be as large as 40 ppm (Fig. 5b). For a fictitious example where ν0 = 100 MHz and νQ = 10.0 MHz (i.e., CQ = 66.7 MHz), this would mean an error in the CQ value of ∼67 kHz, while the error in the shift would be 4.0 kHz. As the value of the ratio between ν0 and νQ is decreased (i.e., increasing CQ or decreasing ν0), a point is reached where the extracted CQ and δiso values exceed typical experimental measurement errors. The point at which this would occur is of course highly dependent upon the measurement conditions and the sample. As an additional example, for the case where ν0 = 4νQ, the error in the chemical shift extracted using second-order perturbation theory exceeds 1500 ppm (Fig. 5b). At the onset of the additional fine structure (ν0 ≈ 4νQ), the error in the CQ value determined using second-order perturbation theory will be slightly in excess of 0.6% (Fig. 5a). These findings echo the observations noted previously for spin-5/2 nuclei (although not restricted to ηQ = 0): when using second-order perturbation theory to model the SSNMR line shapes, the error in the chemical shift becomes detectable at a relatively greater ν0/νQ value than the error in CQ.23,53 Importantly, it is observed and calculated that the errors in both parameters will always be such that second-order perturbation theory underestimates the true value of the parameter.
 | ||
| Fig. 5 Illustrations of the errors associated with using second-order perturbation theory to model SSNMR line shapes for the case where I = 5/2 and ηQ = 0, relative to an exact simulation. (a) Error in the CQ value as a function of the ν0/νQ ratio, and (b) error in the isotropic chemical shift value as a function of the ν0/νQ ratio. High field conditions are traditionally assumed to be satisfied if ν0/νQ > 10. The lines connecting the data points are guides for the eyes only. | ||
Conclusions
We have presented unambiguous evidence of HOQIE in SSNMR spectra and show that they can manifest in unexpected ways (i.e., not always as simple shifts in the frequencies of the spectral discontinuities). The 185/187Re NMR spectrum of NaReO4 at B0 = 11.75 T displays additional fine structure in the low-frequency region, which is not predicted by second-order perturbation theory, but which is predicted using an exact QI model. The fine structure is not observed experimentally at 21.1 T, and is not predicted to be present at this field using exact QI line shape simulations, which is in accord with the expected behavior of a quadrupole-induced effect on a SSNMR line shape (i.e., higher B0 leads to smaller QI spectral effects). We confirm our NMR QI parameters, and rule out (within experimental error) hexadecapole interaction effects by performing 185/187Re NQR experiments for both samples. We use exact QI simulations to establish that the fine structure will potentially become observable when the ν0/νQ value drops below 4, and that the fine structure originates from the mI = 1/2 ↔ 3/2 ST, which also happens to overlap with the CT. For ν0/νQ values greater than 4, we find that the true values of δiso and CQ will be underestimated when the spectra are modeled using second-order perturbation theory. Knowledge of HOQIE may be of critical importance for the accurate line shape analysis of SSNMR spectra of many quadrupolar nuclei that may experience large QIs, including 63/65Cu, 67Zn, 75As, 79/81Br, 91Zr, 105Pd, 115In, 127I, 209Bi, and others.Acknowledgements
D.L.B. thanks the Natural Sciences and Engineering Research Council (NSERC) of Canada for funding. C.M.W. thanks NSERC for an Alexander Graham Bell CGS D2 scholarship. We are grateful to Dr Victor Terskikh and Dr Eric Ye for technical support. Access to the 900 MHz NMR spectrometer was provided by the National Ultrahigh-Field NMR Facility for Solids (Ottawa, Canada), a national research facility funded by the Canada Foundation for Innovation, the Ontario Innovation Trust, Recherche Québec, the National Research Council Canada, and Bruker Biospin and managed by the University of Ottawa (www.nmr900.ca). NSERC is acknowledged for a Major Resources Support grant.References
- C. P. Slichter, Principles of Magnetic Resonance, ed. M. Cardona, P. Fulde, K. von Klitzing, H. J. Queisser and H. K. V. Lotsch, Springer-Verlag, New York, 3rd edn, 1990, pp. 485–502 Search PubMed.
- S. E. Ashbrook, Phys. Chem. Chem. Phys., 2009, 11, 6892 RSC.
- F. A. Cotton and G. Wilkinson, in Advanced Inorganic Chemistry: A Comprehensive Text, Wiley, Toronto, 4th edn, 1980, p. 883 Search PubMed.
- A. V. Naumov, Russ. J. Non-Ferrous Metals, 2007, 48, 418 Search PubMed.
- F. A. Cotton, Acc. Chem. Res., 1969, 2, 240 CrossRef CAS.
- Y. Kuninobu and K. Takai, Chem. Rev., 2011, 111, 1938 Search PubMed.
- R. Hua and J.-L. Jiang, Curr. Org. Synth., 2007, 4, 151 Search PubMed.
- A. T. Herrmann, T. Saito, C. E. Stivala, J. Tom and A. Zakarian, J. Am. Chem. Soc., 2010, 132, 5962 CrossRef CAS.
- I. L. Barnes, T. L. Chang, P. De Bièvre, J. W. Gramlich, R. J. C. Hageman, N. E. Holden, T. J. Murphy, K. J. R. Rosman and M. Shima, Pure Appl. Chem., 1991, 63, 991.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795 CrossRef CAS.
- M. Bernasson, P. Descouts and G. A. Styles, Helv. Phys. Acta, 1970, 43, 393 Search PubMed.
- J. E. Schirber, L. J. Azevedo and A. Narath, Phys. Rev. B, 1979, 20, 4746 Search PubMed.
- D. G. Klobasa and P. K. Burkert, Magn. Reson. Chem., 1987, 25, 154 Search PubMed.
- A. Narath and D. C. Barham, Phys. Rev., 1968, 176, 479 Search PubMed.
- S. Wada and K. Asayama, J. Phys. Soc. Jpn., 1973, 34, 1163 Search PubMed.
- Y. Nishihara, Y. Yamaguchi, S. Waki and T. Kohara, J. Phys. Soc. Jpn., 1983, 52, 2301 Search PubMed.
- P. Pyykkö, Mol. Phys., 2008, 106, 1965 CrossRef CAS.
- R. Siegel, T. T. Nakashima and R. E. Wasylishen, Concepts Magn. Reson. A, 2005, 26A, 62 Search PubMed.
- R. Siegel, T. T. Nakashima and R. E. Wasylishen, Concepts Magn. Reson. A, 2005, 26A, 47 Search PubMed.
- D. Massiot, I. Farnan, N. Gautier, D. Trumeau, A. Trokiner and J. P. Coutures, Solid State Nucl. Magn. Reson., 1995, 4, 241 CrossRef CAS.
- A. Medek, V. Frydman and L. Frydman, J. Phys. Chem. A, 1999, 103, 4830 CrossRef CAS.
- R. W. Schurko, S. Wi and L. Frydman, J. Phys. Chem. A, 2002, 106, 51 CrossRef CAS.
- C. M. Widdifield and D. L. Bryce, J. Phys. Chem. A, 2010, 114, 10810 Search PubMed.
- A. D. Bain, Mol. Phys., 2003, 101, 3163 CrossRef CAS.
- A. D. Bain and B. Berno, Prog. Nucl. Magn. Reson. Spectrosc., 2011 DOI:10.1016/j.pnmrs.2010.12.002.
- I. Solomon, Phys. Rev., 1958, 110, 61 CrossRef CAS.
- I. D. Weisman and L. H. Bennett, Phys. Rev., 1969, 181, 1341 Search PubMed.
- A. C. Kunwar, G. L. Turner and E. Oldfield, J. Magn. Reson., 1986, 69, 124 CAS.
- E. L. Hahn, Phys. Rev., 1950, 80, 580 CrossRef.
- A. Atzesdorfer and K.-J. Range, Z. Naturforsch. B: Chem. Sci., 1995, 50, 1417 Search PubMed.
- J. Spitaler, C. Ambrosch-Draxl, E. Nachbaur, F. Belaj, H. Gomm and F. Netzer, Phys. Rev. B: Condens. Matter, 2003, 67, 115127 Search PubMed.
- A. A. Boguslavskii, R. S. Lotfullin, R. V. Magera and V. V. Pechenov, Fiz. Tverd. Tela, 1974, 16, 2453 Search PubMed.
- R. A. Johnson and M. T. Rogers, in Advances in Nuclear Quadrupole Resonance: Papers Presented at the International Symposium on Nuclear Quadrupole Resonance, Sept. 28-–29, 1972, Queen Elizabeth College, University of London, England, ed. J. A. S. Smith, 1974, 297 Search PubMed.
- M. T. Rogers and K. V. S. R. Rao, J. Chem. Phys., 1973, 58, 3233 Search PubMed.
- S. L. Segel, J. Chem. Phys., 1978, 69, 2434 Search PubMed.
- S. Günther, O. Lutz, A. Nolle and P. G. Schrade, Z. Naturforsch., 1978, 33a, 1018 Search PubMed.
- G. M. Muha, J. Magn. Reson., 1983, 53, 85 CAS.
- R. B. Creel and D. A. Drabold, J. Mol. Struct., 1983, 111, 85 Search PubMed.
- R. B. Creel, J. Magn. Reson., 1983, 52, 515 Search PubMed.
- B. C. Sanctuary, T. K. Halstead and P. A. Osment, Mol. Phys., 1983, 49, 753 Search PubMed.
- A. Müller, E. Krickemeyer, H. Bögge, M. Penk and D. Rehder, Chimia, 1986, 40, 50.
- Y. Do, E. D. Simhon and R. H. Holm, Inorg. Chem., 1985, 24, 4635 CrossRef CAS.
- A. D. Bain, J. Magn. Reson., 2006, 179, 308 Search PubMed.
- I. P. Swainson and R. J. C. Brown, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 76 Search PubMed.
- K. V. S. R. Rao and M. T. Rogers, J. Magn. Reson., 1972, 8, 392 Search PubMed.
- P. K. Burkert and M. F. Eckel, Z. Naturforsch., 1973, 28b, 379 Search PubMed.
- R. A. Johnson and M. T. Rogers, J. Magn. Reson., 1974, 15, 584 Search PubMed.
- R. J. C. Brown, J. Magn. Reson., 1975, 18, 558 Search PubMed.
- R. J. C. Brown, J. G. Smeltzer and R. D. Heyding, J. Magn. Reson., 1976, 24, 269 Search PubMed.
- R. J. C. Brown and S. L. Segel, J. Chem. Phys., 1977, 67, 3163 Search PubMed.
- S. E. Ashbrook and S. Wimperis, Prog. Nucl. Magn. Reson. Spectrosc., 2004, 45, 53 CrossRef CAS.
- S. Cavadini, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 56, 46 CrossRef CAS.
- C. M. Widdifield, R. P. Chapman and D. L. Bryce, in Annu. Rep. Nucl. Magn. Reson. Spectrosc, ed. G. A. Webb, 2009, vol. 66, 195–326 Search PubMed.
- G. K. Semin, Russ. J. Phys. Chem. A, 2007, 81, 38 Search PubMed.
- D. W. Alderman, M. S. Solum and D. M. Grant, J. Chem. Phys., 1986, 84, 3717 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental; detailed 185/187Re NQR/SSNMR experimental acquisition parameters; energy level diagrams for I = 5/2 under various relative Zeeman/QI strengths; experimental 185/187Re NQR spectra of NaReO4 and NH4ReO4; experimental 185/187Re EFG/CS parameters obtained via second-order perturbation theory; V33/B0 angle corresponding to the low-frequency CT discontinuity using exact QI theory; table of values used to constructFig. 5. See DOI: 10.1039/c1cp20572b |
| This journal is © the Owner Societies 2011 |