Making gold nanoparticles fluorescent for simultaneous absorption and fluorescence detection on the single particle level†
Alexander
Gaiduk
*,
Paul V.
Ruijgrok
,
Mustafa
Yorulmaz
and
Michel
Orrit
*
Institute of Physics, Leiden University, P.O. Box 9504, 2300 RA Leiden, The Netherlands. E-mail: gaiduk@molphys.leidenuniv.nl; orrit@molphys.leidenuniv.nl; Fax: +31 71520 5910
First published on 1st November 2010
Abstract
We demonstrate a simple way of making individual 20 nm gold nanoparticles fluorescent (with a fluorescence quantum yield of about 10−6) in glycerol. Gold NPs prepared in such a way have bright fluorescence for a long time under moderate excitation, and their fluorescence remains when the solvent is exchanged to water. We propose to use these nanoparticles as a calibration standard for simultaneousdetection of fluorescence and absorption (by means of photothermal detection), and experimentally demonstrate the theoretically predicted shift in axial positions of these signals. Simultaneous absorption and fluorescence detection of such stable labels makes them attractive for multidimensional tracking and screening applications.
Introduction
Absorption and fluorescence properties of nanoobjects depend on their chemical structure, composition, dimensions and local environment. These properties determine the use of nanoobjects as labels for optical detection and sensing. Fluorescence microscopy provides a broad range of well-known methods for sensitive fluorescence detection. In absorption microscopy only a few existing methods are capable to directly detect absorption of individual nanoobjects at room temperature.1–3 Recently, room-temperature detection of a single molecule's absorption has been demonstrated in absorption microscopy.4,5 Photothermal microscopy is one of the methods allowing background-free detection of absorbers in the absence of residual absorption of the sample.5–11 Developments of photothermal correlation spectroscopy12–14 and wide-field detection15 open new perspectives for photothermal microscopy in a variety of applications. Combined with fluorescence microscopy it can directly correlate absorption and fluorescence characteristics of single nanoobjects. A simultaneous insight into absorption and fluorescence delivers understanding of the photophysics of labels, provides an additional detection dimension, and expands the observation time and tracking capabilities when labels start to blink. High-throughput molecular and cell sorting,16 tracking and imaging in live cells,17–20 as well as biosensing are a few examples of applications where durable labels and multidimensional detection are beneficial.Most fluorescent labels, such as organic molecules, suffer from blinking and bleaching already at low excitation powers, which limits their use. Semiconductor nanoparticles and nanowires can be a photostable alternative.21,22 Still, different excitation regimes are needed for photothermal and fluorescence detection of a single semiconductor nanoparticle, and complicate their simultaneous registration.
Gold nanoparticles (NPs) of various sizes and shapes are commonly used for the calibration of the signal in photothermal microscopy and for measurements of absorption cross-sections.22 The synthesis of gold NPs is well established, and their absorption properties measured on a single NP level are in good agreement with the results predicted by Mie theory.3,23 The photoluminescence of gold NPs itself is of great interest.24 An efficiency of 10−6 has been reported for 4–80 nm (diameter) NPs in water,25 and different theories have been employed to understand the emission of AuNPs.25,26 Even higher photoluminescence efficiencies of >10−3 have been observed from small (28 atoms) molecular-like gold nanoclusters in ensemble measurements.27 A quantum yield of 3% has been measured for polymer-stabilized gold clusters of 2.2–3.4 nm diameters where authors have systematically varied the polymer to gold ratios in order to tune the properties of fluorescence emission.28 The highest quantum yield of ∼41% has been reported for Au8 NPs stabilized in dendrimer aqueous solution.29 To conclude, the photoluminescence efficiency tends to decrease when the size of NPs increases, and it is argued that clustering, surface modification of NPs, and imbedding/stabilizing by the medium can influence the observed fluorescence signals.28,30 However, there is no experimental evidence gained on single NPs, except for NPs with diameters larger than 100 nm.31
Noble metal nanoparticles decorated with fluorophores (dye molecules and fluorescent proteins) are promising labels.32–34 An enhancement of dye fluorescence is usually observed on ensembles of large NPs or aggregates. Experiments on the single NP level will deliver a better characterization of underlying mechanisms and a reliable control.
In this work we first show a way to make commercial 20 nm diameter gold NPs fluorescent without fluorophores. For the first time (to our knowledge) we demonstrate simultaneous photothermal and fluorescence detection of single gold NPs. We then use these NPs for the calibration of the spatial overlap in simultaneous photothermal and fluorescence detection. After spatial alignment, we experimentally demonstrate the theoretically predicted axial displacement between the maximal photothermal and fluorescence signals. Finally, we shortly discuss the possible origins of the observed fluorescence.
Method
Photothermal microscopy is a method based on interferometric sensing of optical absorption, described in detail elsewhere.7,11 Briefly, the temperature of a nanoobject is modulated by means of a modulated heating laser beam. The absorbed energy which is dissipated into heat gives rise to temperature variations around the nanoabsorber. The maximum temperature rise in steady state on the surface of the particle is ΔTsurf = σabsIheat/(4πκR), where σabs is the absorption cross-section of the NP, Iheat is the intensity (irradiance) of the heating laser, κ is the thermal conductivity of the medium around the NP, R is its radius. A consequent change in refractive index of the media around the nanoabsorber scatters a probe laser, which is spatially overlapped with the heating beam. The scattered light interferes in the far-field with a reference wave, in practice the reflected or transmitted probe laser. The probe light is collected by a microscope objective and is recorded by a photodiode and a lock-in-amplifier. Demodulated small changes of the probe light give the photothermal signal. It is proportional to the heating and probe powers, as well as to the temperature derivative of the refractive index of the medium. The signal-to-noise ratio in photothermal detection under ideal conditions is only limited by the photon noise of the probe laser.Sample preparation
Samples of colloidal suspensions of gold NPs with diameters of 20 nm (British Biocell International, EM.GC20) were prepared by dilution in Milli-Q water at volume ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶8. Approximately 50 μL of the suspension were deposited on the surface of cleaned glass immediately after filtration through a 450 nm porous membrane and spin coated at 2000 rpm for 5 s, followed by drying at 4000 rpm for 90 s.
∶8. Approximately 50 μL of the suspension were deposited on the surface of cleaned glass immediately after filtration through a 450 nm porous membrane and spin coated at 2000 rpm for 5 s, followed by drying at 4000 rpm for 90 s.
Glass coverslides (Menzel, Germany) were cleaned in several steps by sonication for 20 min in 2% Hellmanex (Hellma) solution in water, acetone, ethanol (both 96% purity) and in Milli-Q water after each cleaning step. Experiments were performed in a cell (approx. 50–150 μL volume) made from a rubber o-ring or a top of a plastic Eppendorf tube attached to the coverslip. Glycerol (>99.5%, spectrophotometric grade), pentane and hexane (all AR grade) were used as photothermal transducing fluids for our experiments.
Experimental setup
The experimental setup is described in detail elsewhere.11,51 Briefly, it is based on an inverted optical microscope, Olympus IX71, equipped with Olympus 60× oil immersion objectives (NA = 1.4 or NA = 1.45). The heating beam is either provided by a laser diode at 532 nm (Shanghai Lasers, power 150 mW) or at 514 nm by an Ar-Ion laser (Coherent Innova 300). The heating light passes an acousto-optical modulator which modulates the heating light at Ω = 740 kHz. The probe beam (790 or 800 nm) is produced by a Ti:sapphire laser (Mira, Coherent, or S3900s, Spectra Physics) pumped with a Coherent Verdi V10, or an Ar-Ion laser. Sets of spatial filters and telescopes expand the initial beams to ∼20 mm to overfill the entrance pupil of the microscope objective (∼10 mm). The telescopes also serve to adjust the convergence of the pump and probe beams, and thereby the respective positions of their foci, compensating for residual chromatic aberration in the objective. The beams are overlapped at a dichroic mirror and sent towards the objective. The photothermal signal is collected in a back-scattered mode and detected by a Si photodiode (DHPCA-100-F, Femto). The fluorescence detection is performed in the backwards direction either by splitting the back-scattered light with a 70/30 beamsplitter (70% fluorescence transmission) or by a spectral separation of the fluorescence signal at the dichroic mirror (AHF z532/NIR). The fluorescence signal is spectrally selected by a set of bandpass filters (AHF 615/150 and Omega 595/100) and detected by the avalanche photon counting module (SPCM-AQR-16). The experiment is controlled with home-written LabView software, and the data are collected by a data acquisition card (ADWin Gold). The raster scanning of the samples is performed with a 3-axis piezostage (either NanoCube or MARS II, Physik Instrumente). A spectrograph (USB4000, Ocean Optics) is used to obtain fluorescence spectra.Results and discussion
The results of simultaneous photothermal and fluorescence imaging are shown in Fig. 1(A–I). Time-marks next to each row of images indicate the time since the start of the experiment. Three single 20 nm diameter gold NPs are observed in Fig. 1A, situated more than 500 nm apart from each other. The photothermal signal-to-noise ratios are higher than 300 with 0.26 mW heating power (532 nm) and 40 mW probe power (800 nm) at the sample, and integration time (Δt) of 1 ms. The corresponding temperature elevation at the NP surface caused by the heating light is about 23 K. The absorption of the probe light provides an additional non-modulated temperature rise of 9 K. The full widths at half maximum (FWHM) of the Gaussian fit of the shape of photothermal signal are 220 nm and 260 nm (for vertical and horizontal directions, respectively). The overall performance of the photothermal detection is as reported previously.11 The simultaneously acquired confocal fluorescence image (Fig. 1B) shows only the background signal in glycerol and no detectable fluorescence signal from gold NPs. The vertical lines observed in the fluorescence image (along the fast scan axis) are due to diffusing fluorescent impurities in glycerol. We estimate the lowest detectable fluorescence quantum yield of a 20 nm gold NP in these particular experiments to be 6.4 × 10−9. Calculations are based on the background counts (2.2 kcounts/10 ms) and detection efficiency estimations of 5%.51 The overlap of the simultaneously detected photothermal (A) and fluorescence (B) signal is shown in (C) with the red and green colour representing the photothermal and fluorescence signal, respectively. As a next step, we focus laser beams on the central NP and perform an experiment similar to the one depicted in Fig. 2. The graph shows simultaneously recorded photothermal (red) and fluorescence (green) time-traces normalized to the heating power. The top part of the graph illustrates how the heating power is varied in the experiment. The temperature rise on the surface of the nanoparticles (ΔTsurf) and the photothermal signal depend linearly on the heating power. At approximately 98 s, the heating power has been increased and, with it, the temperature of the NP. The increase of the heating power (514 nm) from 0.26 mW up to 0.85 mW leads to a temperature rise from 17 K up to 57 K on the surface of the NP. At the same time a dramatic increase of the fluorescence signal is observed, as well as fluorescence fluctuations. The fluorescence signal remains after the heating power is reduced back to 0.26 mW. Consequent simultaneous photothermal and fluorescence raster scans reveal no change in the photothermal picture (Fig. 1D). The unaltered absorption of light suggests no fragmentation of the particle has occurred. A fragmentation of a similar size NPs is previously reported with the intense pulsed laser light, where the temperatures rise above 1000 K.35 We use moderate heating with CW lasers with an order of magnitude lower temperature elevation (less than 100 K) at the surface of NPs. The absence of NPs fragmentation is supported in independent experiments by SEM imaging after the studies of the vibrations of individual gold NPs, where temperature elevation about 300 K is used.36,37 In contrast to the unchanged absorption, the confocal fluorescence image (Fig. 1E) shows a clear change. A fluorescent spot appears with a signal-to-background ratio of 1.4, signal-to-noise ratio of more than 50, and a full width at half-maximum (FWHM) of 300 and 340 nm (for vertical and horizontal directions correspondingly). The spatial overlap of the two images is shown in Fig. 1F with the yellow colour encoding the overlap of photothermal and fluorescence signals. The same effect is observed on other NPs in the sample (about 50 NPs tried) and further demonstrated in (G, H, I), where the other two NPs in the image are made fluorescent in the same manner as the middle one. A single gold NP with the calculated absorption cross-section of 630 nm2 in glycerol would absorb 1.5 × 1012photons s−1 with a heating power of 0.26 mW at 532 nm. Thus, the quantum yield of fluorescence is calculated to be about 1.4 ± 0.5 × 10−6, assuming one-photon excitation with the heating beam and with our estimated detection efficiency. The obtained fluorescence persists under illumination for a long time (tested up to 10 min), although its intensity fluctuates. In the particular experiment presented in Fig. 1 fluorescence was also detected as particles were revisited after 148 min. The observed stability of the fluorescence signal provides enough time for the alignment of fluorescence detection, and we demonstrate a good spatial overlap between photothermal and fluorescence signals in the scanning plane (lateral overlap). The brightness of NPs is about 0.9 counts/(J cm−2), which is an order of magnitude less than the brightness of closest in size (diameter of 3.6 nm) fluorescence labels, CdSe/ZnS quantum dots (QD) with fluorescence quantum yield of 40%.38 However, QDs show fluorescence intermittence with 1 ms–10 s characteristic times, have fluorescence saturation intensities of 10–80 kW cm−2, and photobleach.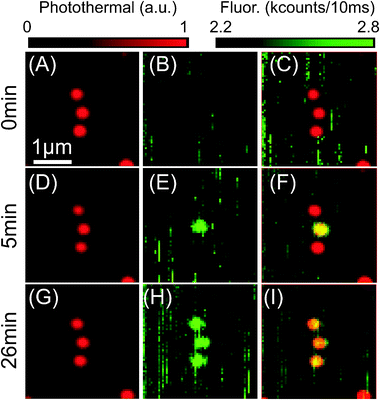 | ||
| Fig. 1 Photothermal (A, D, G) and corresponding fluorescence (B, E, H) raster scan images simultaneously obtained on 20 nm diameter gold NPs on glass surface in glycerol. (C, F, I) shows the overlap of the two signals. Time-marks next to each row indicate the time since the start of the experiment. Vertical lines (along fast scan axis) in fluorescence images originate from diffusing fluorescent impurities in glycerol. There is no detectable fluorescence observed from single NPs in (B), while (E) demonstrates bright fluorescence from the treated central NP (Fig. 2A) and others remain non-fluorescent. Finally all three nanoparticles have been made fluorescent, as shown in (H). Experimental parameters for raster scans are Pheat = 0.26 mW and Pprobe = 40 mW at the sample, Δt = 1 ms, ΔTsurf due to heating light (532 nm) is about 23 K. | ||
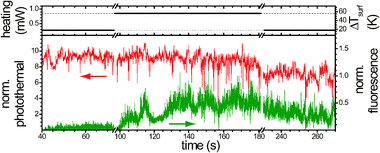 | ||
| Fig. 2 A time trace illustrating the appearance of the fluorescence signal from a single 20 nm gold NP. An increase of the heating power (top, black) at the first axis break (98 s) leads to the temperature rise at the NP. A significant change in fluorescence signal and its fluctuations are observed at the same time (green). At the second axis break (182 s) the heating power is reduced to its starting value. The normalized fluorescence signal remains above its background level at the start. Due to the mechanical drift in the setup (about 20 nm min−1) the photothermal signal (red) is about 20% smaller after 230 s. The wavelength of the heating light is 514 nm. | ||
To probe the spatial overlap in the axial direction, we perform a one dimensional scan on a NP in the direction perpendicular to the sample plane and simultaneously record photothermal and fluorescence signals from the NP (Fig. 3). A single fluorescence maximum and a single photothermal maximum are observed in contrast with the results reported on photothermal measurements of semiconductor wires (optical absorption).22 We take this single maximum of the photothermal signal as a convenient criterion for the proper relative alignment of the heating and probe foci. We report a FWHM of the fluorescence signal along z-axis of 780 nm, which is slightly larger than the FWHM of the photothermal signal (620 nm). These values are in a good agreement with expected diffraction-limited values. The position of the maximum of the fluorescence signal (focused position of the heating beam) and the position of the maximal photothermal signal (the overlap of the heating and probe) are found to be shifted by 250 nm. The shift is well explained by difference in the spatial modes between the incident and scattered fields, which theoretically predicts a discrepancy in z-axis between fluorescence and photothermal signals.10,39–41 With this result we experimentally demonstrate that the model of Hwang and Moerner41 for paraxial rays also holds for large numerical aperture at the focus of an immersion objective.
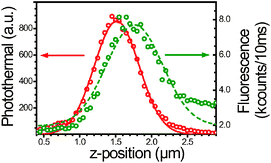 | ||
| Fig. 3 Z-scan (perpendicular to the glass surface) on the fluorescent 20 nm gold particle. A red solid line shows a Gaussian fit to data points of photothermal signal and a green dashed line shows the fit for the fluorescence. Central positions and the width of fits are 1.52 ± 0.62 μm and 1.77 ± 0.78 μm for photothermal and for fluorescence, respectively. A shift (250 nm) between the maxima of two signals is observed. | ||
In order to understand the origin of the fluorescence of NPs we have recorded a fluorescence spectrum of a single NP in the spectral region between 560 nm and 640 nm.51 From the featureless shape of the fluorescence spectrum in the limited spectral range (due to the limitations of the present setup) it is difficult to judge if the spectrum resembles the absorption of gold NP. Thus, we cannot give any solid conclusion as to whether the fluorescence is due to NP's plasmon emission only,25,26 or whether other mechanisms are involved.
The enhancement of fluorescence upon illumination can be explained by the modification of the surface of NPs, caused by thermal processes, photochemical processes,42–44 or a combination of those. The heating can lead to fluorescence due to the modification of the surface electronic properties of gold due to the removal or addition of ligands and the rearrangement of gold atoms at the surface. A temperature-mediated mechanism is supported by our observation of the enhancement of fluorescence after an exposure to only the probe light with a power larger than 150 mW at the sample. In contrast, no enhancement was seen with heating light power kept as low as 0.26 mW for times longer than 200 s, or with the probe light only with powers below 100 mW for times longer than 100 s. We also observe fluorescence appearance in pentane and hexane, organic solvents that are chemically different from glycerol. This fluorescence enhancement effect is not observed in water, although fluorescent gold NPs prepared in glycerol remain fluorescent when glycerol is exchanged with water.51 The simplest temperature-activated chemical reactions in the above-mentioned solvents would first lead to saturated and therefore non-fluorescent products. A high enough temperature elevation at the surface of NPs, however, can result in the formation of more and more complex unsaturated products, which may decorate the NP and generate fluorescence. An additional complication is the possible catalytic role of the gold surface, which may act in more specific ways than just a local “frying pan”.45–47 Hereafter, we speculate about possible surface chemistry on NPs in glycerol, which will have to be tested experimentally in the future: (i) thermal decomposition of glycerol or residual fatty acids in glycerol,48 (ii) formation of a reactive acrylic acid aldehyde (acrolein, H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CHO) obtained in thermal decomposition of glycerol by dehydration in the presence of a catalyst. Recently, reversible and irreversible temperature mediated reactions of glycerol and citric acid49 have been studied with the method of differential scanning calorimetry (DSC) in the temperature range of 20–220 °C. The work reports irreversible esterification and glycerol citrate polyesters formation. Also, Ruppert et al.50 have studied a selective etherification of glycerol to linear, branched and cyclic diglycerols, and triglycerol in the presence of alkaline earth metal oxides.
CH–CHO) obtained in thermal decomposition of glycerol by dehydration in the presence of a catalyst. Recently, reversible and irreversible temperature mediated reactions of glycerol and citric acid49 have been studied with the method of differential scanning calorimetry (DSC) in the temperature range of 20–220 °C. The work reports irreversible esterification and glycerol citrate polyesters formation. Also, Ruppert et al.50 have studied a selective etherification of glycerol to linear, branched and cyclic diglycerols, and triglycerol in the presence of alkaline earth metal oxides.
Conclusions
In this communication we demonstrate a simple way to make commercial 20 nm gold NPs fluorescent, and we observe this effect in different solvents (glycerol, hexane, pentane). The fluorescence enhancement appears after illumination at moderately high power, which leads to a transient temperature elevation at the surface of NPs and modification of their surface properties. We use these NPs as the calibration sample for simultaneous fluorescence and photothermal microscopy, and solve the problem of spatial overlap of two signals. We shortly speculate on the origin of the observed fluorescence in order to stimulate research and suggest that temperature-assisted chemical reactions lead to a complex modification of the surface of gold NPs. More rigorous studies on a single particle level involving Raman spectroscopy or two-photon excited photothermal microscopy20 would allow one to identify and separate between different mechanisms of NPs fluorescence, depending on NP's size, surface modification and environment. Absorption and fluorescence properties of the described fluorescent NPs can be used for various applications where the combination of photostability and brightness of the labels for a long time are required.Acknowledgements
We thank Dr. Leandro Tabares, Alex P. Gaiduk and Prof. J. Lugtenburg for helpful discussions, Ljubiša Babic for lending us the spectrograph, and Dr. Peter Zijlstra for the help with SEM and discussions on the gold fluorescence. We acknowledge financial support by ERC (Advanced Grant SiMoSoMa). AG acknowledges The British Council and Platform Bèta Techniek for financial support. This work is a part of the research program of the “Stichting voor Fundamenteel Onderzoek der Materie”, which is financially supported by the Netherlands Organization for Scientific Research.References
- J. Hwang, M. M. Fejer and W. E. Moerner, Phys. Rev. A, 2006, 73, 021802 CrossRef.
- P. Kukura, M. Celebrano, A. Renn and V. Sandoghdar, Nano Lett., 2009, 9, 926 CrossRef CAS.
- M. A. van Dijk, A. L. Tchebotareva, M. Orrit, M. Lippitz, S. Berciaud, D. Lasne, L. Cognet and B. Lounis, Phys. Chem. Chem. Phys., 2006, 8, 3486 RSC.
- M. Celebrano, P. Kukura, A. Renn, V. Sandoghdar, http://arxiv.org/abs/1007.3457.
- A. Gaiduk, M. Yorulmaz, P. V. Ruijgrok and M. Orrit, Science, 2010, 330, 353 CrossRef CAS.
- D. Boyer, P. Tamarat, A. Maali, B. Lounis and M. Orrit, Science, 2002, 297, 1160 CrossRef CAS.
- S. Berciaud, D. Lasne, G. A. Blab, L. Cognet and B. Lounis, Phys. Rev. B: Condens. Matter, 2006, 73, 045424 CrossRef.
- S. Berciaud, L. Cognet, G. A. Blab and B. Lounis, Phys. Rev. Lett., 2004, 93, 257402 CrossRef.
- M. Tokeshi, M. Uchida, A. Hibara, T. Sawada and T. Kitamori, Anal. Chem., 2001, 73, 2112 CrossRef CAS.
- S. E. Bialkowski, Photothermal Spectroscopy Methods for Chemical Analysis, Wiley, 1996 Search PubMed.
- A. Gaiduk, P. V. Ruijgrok, M. Yorulmaz and M. Orrit, Chem. Sci., 2010, 1, 343 RSC.
- P. M. R. Paulo, A. Gaiduk, F. Kulzer, S. F. G Krens, H. P. Spaink, T. Schmidt and M. Orrit, J. Phys. Chem. C, 2009, 113, 11451 CrossRef CAS.
- R. Radünz, D. Rings, K. Kroy and F. Cichos, J. Phys. Chem. A, 2009, 113, 1674 CrossRef CAS.
- V. Octeau, L. Cognet, L. Duchesne, D. Lasne, N. Schaeffer, D. G. Fernig and B. Lounis, ACS Nano, 2009, 3, 345 CrossRef CAS.
- E. Absil, G. Tessier, M. Gross, M. Atlan, N. Warnasooriya, S. Suck, M. Coppey-Moisan and D. Fournier, Opt. Express, 2010, 18, 780 CrossRef CAS.
- F. Kulzer, N. Laurens, J. Besser, T. Schmidt, M. Orrit and H. P. Spaink, Chem. Phys. Chem., 2008, 9, 1761 CrossRef CAS.
- D. Lasne, G. A. Blab, S. Berciaud, M. Heine, L. Groc, D. Choquet, L. Cognet and B. Lounis, Biophys. J., 2006, 91, 4598 CrossRef CAS.
- D. Lasne, G. A. Blab, F. De Giorgi, F. Ichas, B. Lounis and L. Cognet, Opt. Express, 2007, 15, 14184 CrossRef CAS.
- L. Cognet, C. Tardin, D. Boyer, D. Choquet, P. Tamarat and B. Lounis, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11350 CrossRef CAS.
- S. Lu, W. Min, S. Chong, G. R. Holtom and X. S. Xie, Appl. Phys. Lett., 2010, 96, 113701 CrossRef.
- S. Berciaud, L. Cognet and B. Lounis, Nano Lett., 2005, 5, 2160 CrossRef CAS.
- J. Giblin, M. Syed, M. T. Banning, M. Kuno and G. Hartland, ACS Nano, 2010, 4, 358 CrossRef CAS.
- A. Tcherniak, J. W. Ha, S. Dominguez-Medina, L. S. Slaughter and S. Link, Nano Lett., 2010, 10, 1398 CrossRef CAS.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409 CrossRef CAS.
- E. Dulkeith, T. Niedereichholz, T. A. Klar, J. Feldmann, G. von Plessen, D. I. Gittins, K. S. Mayya and F. Caruso, Phys. Rev. B: Condens. Matter, 2004, 70, 205424 CrossRef.
- O. P. Varnavski, T. Goodson III, M. B. Mohamed and M. A. El-Sayed, Phys. Rev. B: Condens. Matter, 2005, 72, 235405 CrossRef.
- S. Link, A. Beeby, S. Fitzgerald, M. A. El-Sayed, T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2002, 106, 3410 CrossRef CAS.
- N. Schaeffer, B. Tan, C. Dickinson, M. J. Rosseinsky, A. Laromaine, D. W. McComb, M. M. Stevens, Y. Wang, L. Petit, C. Barentin, D. G. Spiller, A. I. Cooper and R. Lévy, Chem. Commun., 2008, 3986 RSC.
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780 CrossRef CAS.
- J. P. Wilcoxon, J. E. Martin, F. Parsapour, B. Wiedenman and D. F. Kelley, J. Chem. Phys., 1998, 108, 9137 CrossRef CAS.
- M. R. Beversluis, A. Bouhelier and L. Novotny, Phys. Rev. B: Condens. Matter, 2003, 68, 115433 CrossRef.
- Y. Fu and J. R. Lakowicz, Laser Photonics Rev., 2009, 3, 221 Search PubMed.
- Y. Fu, J. Zhang and J. R. Lakowicz, J. Am. Chem. Soc., 2010, 132, 5540 CrossRef CAS.
- Y. Fu, J. Zhang and J. R. Lakowicz, Biochem. Biophys. Res. Commun., 2008, 376, 712 CrossRef CAS.
- Z. Peng, T. Walther and K. Kleinermanns, J. Phys. Chem. B, 2005, 109, 15735 CrossRef CAS.
- A. L. Tchebotareva, M. A. van Dijk, P. V. Ruijgrok, V. Fokkema, M. H. S. Hesselberth, M. Lippitz and M. Orrit, ChemPhysChem, 2009, 10, 111 CrossRef CAS.
- G. V. Hartland, Annu. Rev. Phys. Chem., 2006, 57, 403 CrossRef CAS.
- B. Lounis, H. A. Bechtel, D. Gerion, P. Alivisatos and W. E. Moerner, Chem. Phys. Lett., 2000, 329, 399 CrossRef CAS.
- T. Berthoud, N. Delorme and P. Mauchien, Anal. Chem., 1985, 57, 1216 CrossRef CAS.
- M. Tokeshi, J. Yamaguchi, A. Hattori and T. Kitamori, Anal. Chem., 2005, 77, 626 CrossRef CAS.
- J. Hwang and W. E. Moerner, Opt. Commun., 2007, 280, 487 CrossRef CAS.
- K. L. McGilvray, M. R. Decan, D. Wang and J. C. Scaiano, J. Am. Chem. Soc., 2006, 128, 15980 CrossRef CAS.
- M. Sakamoto, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2009, 131, 6 CrossRef CAS.
- M. Sakamoto, T. Tachikawa, M. Fujitsuka and T. Majima, Langmuir, 2009, 25, 13888 CrossRef CAS.
- L. Stehr, C. Hrelescu, R. A. Sperling, G. Raschke, M. Wunderlich, A. Nichtl, D. Heindl, K. Kurzinger, W. J. Parak, T. A. Klar and J. Feldmann, Nano Lett., 2008, 8, 619 CrossRef CAS.
- M. Reismann, J. C. Bretschneider, G. von Plessen and U. Simon, Small, 2008, 4, 607 CrossRef CAS.
- A. S. Urban, M. Fedoruk, M. R. Horton, J. O. Raedler, F. D. Stefani and J. Feldmann, Nano Lett., 2009, 9, 2903 CrossRef CAS.
- Communication with Sigma-Aldrich.
- R. A. Holser, J. Appl. Polym. Sci., 2008, 110, 1498 CrossRef CAS.
- A. M. Ruppert, J. D. Meeldijk, B. W. M. Kuipers, B. H. Erné and B. M. Weckhuysen, Chem.–Eur. J., 2008, 14, 2016 CrossRef CAS.
- See ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental setup; absorption and fluorescence spectra of 20 nm gold NPs solution; SEM, AFM and photothermal imaging of 20 nm gold NPs; calculations of the fluorescence quantum yield; photothermal and fluorescence imaging of 20 nm gold NPs after solvent exchange. See DOI: 10.1039/c0cp01389g |
| This journal is © the Owner Societies 2011 |