Crystal engineering: origins, early adventures and some current trends
Sir
John Meurig
Thomas
*
Dept. of Materials Science, Cambridge, CB2 3QZ. E-mail: jmt2@cam.ac.uk
Occasionally, however, their predictions and expectations as to which product would be formed within a crystal subjected to photoirradiation turned out to be wrong. 9-cyanoanthracene was a case in point. Instead of forming the cis dimer of the diparaanthracene, the trans dimer appeared on irradiating crystals of the monomer, in which molecules were stacked in columns with “incipient cis” orientation. This and other examples proved enigmatic.
In the 1960s, my students and I at Bangor had found that dislocations in crystalline solids often exerted a profound influence on the reactivity of both inorganic and organic materials. Two particular examples were the thermally-stimulated decomposition of calcite (CaCO3)1 and the photodimerization of anthracene.2 My work led Cohen and Schmidt to invite me to their laboratory in 1969, to lecture on crystalline defects and to do joint research there, all focused on the chemical consequences of imperfections in solids. One of the consequences was the discovery that acenaphthylene (Scheme 1), under irradiation with 300 nm light, dimerized preferentially at dislocation cores.3
 | ||
| Scheme 1 Solid-state photodimerization of acenaphthylene.3 | ||
Upon my translation from Bangor to Aberystwyth in 1969, I embarked on a systematic study of how imperfections of one kind or another in organic molecular crystals influenced their excitonic, electronic, photo-physical and photo-chemical behaviour. With Cohen and the late J. O. Williams, we showed how the trans dimer of 9-cyanoanthracene could be formed preferentially at stacking faults within the crystal (Figure 1).4 We also showed5 that excitons, from measurements of their lifetimes as well as prompt and delayed fluorescence, could be used to probe the energetics of structural imperfections in crystalline anthracene. By the time Bill Jones had finished his PhD at Aberystwyth in 1974, he was an expert in the electron microscopy of beam-sensitive materials, a skill which he now utilizes in his pioneering investigations of pharmaceutical materials.6 He mastered dislocation theory and, inter alia, he discovered the ease with which certain organic molecular crystals underwent martensitic transformations.7
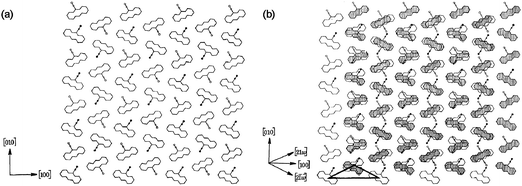 | ||
| Fig. 1 Schematic illustration of (a) the ideal structure of 9-cyanoanthracene projected onto the (001) plane and (b) the nature of the stacking fault that leads to the formation of centrosymmetric dimers in the crystals of 9-cyanoanthracene. This image has been reproduced by permission of the Royal Society.4 | ||
In 1974, having delved into what had been the early pioneering work of Kohlschütter in Germany and Kitaigorodsky in the Soviet Union, I opted, when invited by the Royal Society to present a review lecture, to talk on topography and topology in solid-state chemistry.8 This drew my attention to the work of Hasegawa et al. in Japan and Wegner et al. in Germany, each on photopolymerization within organic crystals, the former in 2,5-distyrylpyrazine (DSP) (Figure 2), the latter in substituted diacetylene monomers (Figure 3). This review also enabled me to explain, inter alia, the distinction between topochemical and topotactic reactions. It also had the added bonus of attracting one of Hasegawa's associates, Hachiro Nakanishi, to join Bill Jones and me, when I moved to Cambridge in the late 1970s.
![Overlay of two molecules in the crystal of 2,5-distyrylpyrazine (green), with the polymer resulting from their solid-state [2 + 2] photopolymerization (orange). This image has been reproduced by permission of the American Chemical Society.9](/image/article/2011/CE/c1ce90016a/c1ce90016a-f2.gif) | ||
| Fig. 2 Overlay of two molecules in the crystal of 2,5-distyrylpyrazine (green), with the polymer resulting from their solid-state [2 + 2] photopolymerization (orange). This image has been reproduced by permission of the American Chemical Society.9 | ||
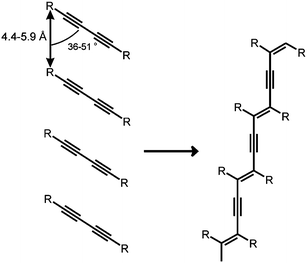 | ||
| Fig. 3 Topochemical polymerization of a diacetylene. | ||
Progress in organic solid state chemistry at the Department of Physical Chemistry, Cambridge in the subsequent years proved rewarding. We were able to determine, by a combination of low-temperature electron microscopy and atom-atom potentials, which Kitaigorodsky had been using some decades earlier, that Simonetta in Milan had refined, in a sophisticated manner, the crystal structure of a metastable phase of anthracene and to predict its phonon spectrum (which was later confirmed experimentally).10 A baffling observation, the fact that crystals of hexahelicene (space group P212121) display very little optical activity, also yielded to our studies. Atom–atom potentials enabled us to explain why this is so. The crystals contain recurrent intergrowths of the right and left-handed regions,11 it was not homochiral. The atom–atom approach also enabled the determination, with the aid of low-temperature electron diffraction by selected area studies in electron microscopy, of the structure of a new phase of pyrene.12
With Nakanishi, Bill Jones and I were fortunate to unearth a rich vein of related organic monomers, based on 2-benzyl-5-benzylidenecyclopentanones (Figure 4). The delicate interdependence of the crystal structure and reactivity in this family of related molecules, derived by various substitutions for X, Y and Z in the rings A, B and C, could be carefully traced.13 One of the most satisfying consequences of this facet of our work was that we could genuinely engineer organic crystals so as to control the photoreactivity of the reactants and crystallinity of the products.14 Moreover, not only could comparisons be made between the various substituents (e.g. chloro and methyl groups) in steering the engineering process towards desirable photoactive reactants,15,16 we were also able to track, with M. B. Hursthouse, the subtle atomic displacements associated with the conversion of two juxtaposed monomers into a dimer occupying essentially the same volume as the original pair of monomers.17
 | ||
| Fig. 4 (a) Schematic diagram of a 2-benzyl-5-benzylidenecyclopentanone molecule and (b) the two neighbouring molecules of its monomer in the crystal (in which bonds are drawn with full lines) compared to the molecule of the dimer (in which bonds are drawn with dotted lines), which is generated from them when the crystal is exposed to ultraviolet radiation. | ||
The topic of crystal engineering is still flourishing: it is, in effect, the rational design of functional molecular solids and it has received voluminous attention. The literature up until 1988 was summarized and addressed in the monograph by Desiraju,18 who has more recently19 reviewed the subject and many of its myriad ramifications up to 2007. One facet of this vast subject, which continues to elicit much attention, devolves upon the importance of co-crystallization.
Co-crystallization was used20 by this author as a stratagem to bury molecules of H2O2 in a readily soluble solid so as to be able to release the peroxide on demand for use as a bleaching agent.20 It was the large number of weak hydrogen bonds involving N–H⋯O and O–H⋯O that would be formed between the guanidinium oxalate dihydrate host and the H2O2 guest that constituted the element of crystal engineering, in my thinking.
In the recent work of Jones et al., which focused on cocrystal architecture and the occurrence of chiral and racemic structures21 and cognate topics, terahertz (i.e. far infrared) time-domain spectroscopy has been used22 to probe cocrystal formation, leading to worthwhile insights.23 One very powerful recent technique involving chiral crystals, which could offer penetrating insights into the mechanism of (in situ) co-crystal formation, is that of vibrational optical activity, pioneered and recently reviewed by Barron and Buckingham.24
It is invidious for anyone, like me, who is no longer active in the field of crystal engineering, to predict possible future developments; but it is surely uncontentious to remark that such is the rapidity with which the structures of solids may now be retrieved from both single-crystal and powdered25 specimens, that it is the arrival of yet far more powerful techniques26 for in situ studies of crystallization (and co-crystallization) that may hold the key to deeper understanding in future investigations.27 So far as the dynamics of change in organic crystalline solids is concerned, there will doubtless be major advances made, especially for those molecular crystals that are not too vulnerable to electron-beam damage, from the application of 4-D electron microscopy (4DEM), where time-resolution of femtoseconds to the bond-breaking and bond-making durations are possible.28,29 Other exciting prospects may emerge from the dramatic success recently reported30,31 from Stanford, whereby femtosecond X-ray protein nanocrystallography is described using the free-electron X-ray laser. One of the penalties that has to be paid in the X-ray laser work is that the crystal under study is destroyed almost immediately after it yields its diffraction pattern! There is no doubt that 4DEM and X-ray laser crystallography together will, and already have, enormously increase our knowledge of the dynamics of change in the solid state.
I am grateful to Professor K. D. M. Harris and Dr T. Friščić for useful discussions.
References
- G. D. Renshaw and J. M. Thomas, Topographical studies of decomposition of single crystals of calcite, Nature, 1966, 209, 1196 CrossRef CAS.
- J. M. Thomas and J. O. Williams, Photochemical transformations in crystalline anthracene: The importance of crystal defects, Chem. Commun. (London), 1967, 432 RSC.
- M. D. Cohen, I. Ron, G. M. J. Schmidt and J. M. Thomas, Photochemical decoration of dislocations inside crystals of acenaphthalene, Nature, 1969, 224, 167 CAS.
- M. D. Cohen, L. Ludmer, J. M. Thomas and J. O. Williams, The role of structural imperfections in the photodimerization of 9-cyanoanthracene, Proc. R. Soc. London, Ser. A, 1971, 324, 459 CAS.
- D. Goode, V. Lupien, W. Siebrand, D. F. Williams, J. M. Thomas and J. O. Williams, Triplet excitons as probes for structural imperfections in crystalline anthracene, Chem. Phys. Lett., 1974, 25, 308 CAS.
- M. Eddleston, E. Bithell and W. Jones, Transmission electron microscopy of pharmaceutical materials, J. Pharma., 2010, 99, 4072 Search PubMed.
- W. Jones, J. M. Thomas and J. O. Williams, Electron and optical microscopic studies of a stress-induced phase transition in 1,8 dichloro-10- methyl anthracene, Philos. Mag., 1975, 32, 1 CAS.
- J. M. Thomas, Topography and topology in solid-state chemistry, Philos. Trans. R. Soc. London, Ser. A, 1974, 277, 251 CAS.
- F. Guo, J. Martí-Rujas, Z. Pan, C. E. Hughes and K. D. M. Harris, Direct structural understanding of a topochemical solid-state photopolymerization reaction, J. Phys. Chem. C, 2008, 112, 19793 CrossRef CAS.
- S. Ramdas, G. M. Parkinson, J. M. Thomas, C. M. Gramaccioli, G. Filippini, M. Simonetta and M. J. Goringe, Determination of crystal structure of metastable anthracene by a novel method, Nature, 1980, 284, 153 CrossRef CAS.
- S. Ramdas, J. M. Thomas, M. E. Jordan and C. J. Eckhardt, Enantiomeric intergrowths in hexahelicenes, J. Phys. Chem., 1981, 85, 2421 CrossRef CAS.
- W. Jones, S. Ramdas and J. M. Thomas, Novel approach to the determination of the crystal structures of organic molecular crystals: Low temperature form of pyrene, Chem. Phys. Lett., 1978, 54, 490 CrossRef CAS.
- J. M. Thomas, Diffusionless reactions and crystal engineering, Nature, 1981, 289, 633.
- W. Jones, H. Nakanishi, C. R. Theocharis and J. M. Thomas, Engineering organic crystals so as to control the photoreactivity of the reactants and the crystallinity of the products, J. Chem. Soc., Chem. Commun., 1980, 610 RSC.
- W. Jones, S. Ramdas, C. R. Theocharis, J. M. Thomas and N. W. Thomas, Crystal engineering of photodimerizable cyclopentanones: Comparison of chloro and methyl substitution as solid-state steering groups, J. Phys. Chem., 1981, 85, 2594 CrossRef CAS . This topic still elicits considerable interest, see ref. 16.
- M. R. Edwards, W. Jones, W. D. S. Motherwell and G. P. Shields, Crystal engineering and chloro-methyl interchange: A CSD analysis, Mol. Cryst. Liq. Cryst., 2001, 356, 337 CrossRef CAS.
- H. Nakanishi, W. Jones, J. M. Thomas, M. B. Hursthouse and M. Motevalli, Static and dynamic single crystal X-ray diffraction studies of some solid-state photodimerization reactions, J. Phys. Chem., 1981, 85, 3636 CrossRef CAS.
- G. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed.
- G. Desiraju, Crystal Engineering: A holistic view, Angew. Chem., Int. Ed., 2007, 46, 8342 CrossRef CAS.
- J. M. Adams, R. G. Pritchard and J. M. Thomas, Preparation and X-ray crystal structure of guanidinium oxalate dihydrate monoperhydrate: A novel example of crystal engineering, J. Chem. Soc., Chem. Commun., 1976, 358–359 RSC.
- T. Friščić and W. Jones, Cocrystal architechture and properties: design and building of chiral and racemic structures by solid-state reactions, Faraday Discuss., 2007, 136, 167 RSC.
- T. Friščić and W. Jones, Recent advances in understanding the mechanism of cocrystal formation via grinding, Cryst. Growth Des., 2009, 9, 1621 CrossRef CAS.
- K. L. Nguyen, T. Friščić, G. M. Day, L. F. Gladden and W. Jones, Terahertz time-domain spectroscopy and the quantitative monitoring of mechanochemical cocrystal formation, Nat. Mater., 2007, 6, 206 CrossRef CAS.
- L. D. Barron and A. D. Buckingham, Vibrational optical activity, Chem. Phys. Lett., 2010, 492, 199 CrossRef CAS.
- K. D. M. Harris, M. Tremayne, P. Lightfoot and P. G. Bruce, Crystal structure determination from powder diffraction data by Monte Carlo methods, J. Am. Chem. Soc., 1994, 116, 3543 CrossRef CAS.
- C. E. Hughes and K. D. M. Harris, Direct observation of a transient polymorph during crystallisation, Chem. Commun., 2010, 46, 4982 RSC.
- B. A. Palmer, K. D. M. Harris and F. Guillaume, A strategy for retrospective mapping the growth history of a crystal, Angew. Chem., Int. Ed., 2010, 49, 5096 CAS . (This paper was highlighted in Chem. Eng. News, 28 June 2010).
- A. H. Zewail and J. M. Thomas,4D Electron microscopy: Imaging in space and time, Imperial College Press, London , 2010 Search PubMed.
- A. H. Zewail and Oh-Hoon Kwon, 4D Electron tomography, Science, 2010, 328, 1668 CrossRef.
- H. N. Chapman, et al., Femtosecond X-ray protein nanocrystallography, Nature, 2011, 470, 73 CrossRef CAS.
- M. M. Seibert, et al., Single mimivirus particles intercepted and imaged with an X-ray laser, Nature, 2011, 470, 78 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |