The fluorine atom as a halogen bond donor, viz. a positive site†
Pierangelo
Metrangolo
*ab,
Jane S.
Murray
c,
Tullio
Pilati
ad,
Peter
Politzer
c,
Giuseppe
Resnati
*abd and
Giancarlo
Terraneo
ab
aNFMLab, Dept. of Chemistry, Materials and Chemical Engineering, “Giulio Natta”, Politecnico di Milano, Via L. Mancinelli 7, 20131, Milan, Italy. E-mail: pierangelo.metrangolo@polimi.it; giuseppe.resnati@polimi.it; Fax: +39 02 2399 3180; Tel: +39 02 2399 3041/3032
bCNST-IIT@POLIMI, Via Pascoli 70/3, 20133, Milan, Italy
cCleveTheoComp, 1951 W. 26th Street, Suite 409, Cleveland, OH 44113, USA
dISTM-CNR, Università di Milano, Via Venezian 21, 20133, Milan, Italy
First published on 11th July 2011
Abstract
When bound to residues which can work as particularly strong electron withdrawing groups, fluorine can display a region of positive electrostatic potential (positive σ-hole). Fluorine can thus function as a halogen bond donor and form complexes with lone-pair-containing neutral atoms and anions. Examples in the gas, liquid, and solid phases are discussed.
The halogen bond (XB) is the attractive interaction between a region of positive electrostatic potential in a covalently bonded halogen atom and a negative site on an atom or group. XB effectively directs recognition and self-assembly processes in the solid, liquid, and gas phases1 and therefore the full understanding of this interaction is crucial in the development of numerous different fields. This requires identifying which halogens can develop a positive electrostatic potential (positive σ-hole) and which σ-holes can be involved in XBs.
Iodine, bromine, and chlorine atoms in dihalogens, interhalogens, and a wide variety of organohalogen molecules frequently have positive σ-holes which form XBs. Experimental results and theoretical calculations consistently show that the greater the polarizability and the lower the electronegativity of a halogen atom, the more positive is its σ-hole and the stronger is the XB to which it gives rise. The strength of the XBs formed by a halogen derivative with a given electron rich moiety (XB acceptor) thus decreases in the order I > Br > Cl. Fluorine is the least polarizable and the most electronegative halogen and therefore it is least likely to have a positive σ-hole and to function as a XB donor. For instance, a positive potential is present on the bromine of bromo-3,5-difluorobenzene but the fluorines are negative on their entire surface (Fig. 1).
 | ||
| Fig. 1 Computed electrostatic potential (obtained with the Wave Function Analysis-Surface Analysis Suite)12 on the 0.001 au molecular surface of 3,5-difluorobromobenzene. In A the bromine is at the right; in B one of the two fluorines is at the right. Color ranges, in kcal mol−1, are: red, greater than 12; yellow, between 12 and 6; green, between 6 and 0; blue, negative. The bromine atom has a positive region on its outer surface, along the extension of the C–Br bond, with the characteristic negative belt around its lateral sides. The fluorines have completely negative potentials on their surfaces. | ||
There is disagreement as to whether and when fluorine can develop a positive σ-hole and as to whether it can be involved in XB to any significant extent. For example, it has been stated that “XB… is non-existent with organofluorine compounds”3 but it has also been argued that “fluorine atom has the capability of forming halogen bonds”4 and that “the sigma hole will be present on fluorine atom, which now can participate in interactions with the negative electrostatic region of another fluorine/nitrogen/oxygen atom”.5
These issues are of particular relevance to an IUPAC project6 aimed at giving a modern definition of XB by taking a comprehensive look at intermolecular interactions involving halogens as electrophilic species.
In this paper we collect and discuss some experimental findings and theoretical studies proving that fluorine atoms in F2 and some fluoroorganics can indeed develop a positive σ-hole and work as a XB donor, although these capabilities are much less pronounced than for the other halogens. Fluorine XB can also affect recognition and self-assembly processes, but only under specific circumstances.
Like the other dihalogens, but to a lesser extent, molecular fluorine displays a positive σ-hole on the outermost portions of the molecule7 (Fig. 2A). Its presence allows the formation of adducts with Lewis bases and anionic species. Trihalide anions8 X3− are simple halogen-bonded (X-bonded) adducts in which X2 is the XB donor and X− the acceptor. I3− and Br3− are well known, Cl3− is less common and F3− has been observed only under special conditions. It was first reported in argon and neon matrices as [M+][F3−] (M = K, Cs, Rb) salts.9 The F3− anion was later found in the gas phase by mass spectrometry;10 energy-resolved collision-induced dissociation cross-sectional measurements in two tandem mass spectrometers revealed a unimolecular gas-phase F2 elimination value [F3− → F2 + F−] of 98.4 ± 10.6 kJ mol−1.
 | ||
| Fig. 2 Computed electrostatic potentials (using the Wave Function Analysis-Surface Analysis Suite)12 on 0.001 au molecular surfaces of F2 (A) and CF3SO2OCOF (B, the CF3 group is at the right). Color ranges, in kcal mol−1, are: red, greater than 20; yellow, between 20 and 9; green, between 9 and 0; blue, negative. Black hemispheres denote the positions of the most positive potentials associated with the fluorines. | ||
This can be compared to 99 ± 5, 127 ± 7 and 126 ± 6 kJ mol−1 for Cl3−, Br3−, and I3−, respectively. Confirming predictions from modelling10a and consistent with the geometry expected for an X-bonded adduct, IR and Raman spectra showed a linear arrangement of the three fluorine atoms in F3− (linear centrosymmetric D∞h geometry). State-of-the-art quantum chemical calculations indicate that F5− is also a thermodynamically stable species and suggest the V-shaped (C2V) structure commonly observed for pentahalide anions I5− and Br5−.8
Molecular fluorine reacts violently with most organic compounds at ambient temperatures and pressures, so it is no surprise that its adducts with neutral organics have been studied only when the technology of microwave spectrometry allowed for their generation under collisionless conditions. Similar to the analogous complexes of other halogens and interhalogens, in the dimer NH3⋯F2 the fluorine is bound to the nitrogen atom and the axis of the F2 molecule lies along the C3v axis of NH3.11 A C3v symmetry was found also for the adduct CH3CN⋯F2, and the dimer HCN⋯F2 is linear. These geometries as well as those of other F2 adducts (e.g. with H2O, H2S, and (CH2)2O) confirm the general ability of F2 to halogen bond with heteroatoms possessing lone pairs and to give adducts with the expected geometries wherein a positive end of F2 seeks out a non-bonding electron pair. The ratios of the intermolecular separations11 to the sums of the van der Waals radii (Table 1) and also the stretching force constants show that the interactions with F2 are weaker than in the corresponding complexes of larger halogens. The strengths of the interactions for a given XB acceptor correlate, by both geometric and energetic features, with the magnitudes of the positive σ-hole potentials on the XB donors, which are also listed in Table 1.
| Adduct | Ratio of donor⋯acceptor separation to sum of van der Waals radii | Force constant Kσ | σ-Hole potential on XB-donor |
|---|---|---|---|
| H3N⋯F2 | 0.91 | 4.7 | 13.8 |
| H3N⋯Cl2 | 0.83 | 12.7 | 23.8 |
| H3N⋯Br2 | 0.80 | 18.5 | 29.1 |
| H2O⋯F2 | 0.91 | 3.63 | 13.8 |
| H2O⋯ClF | 0.79 | 14.2 | 30.9 |
| H2S⋯F2 | 0.96 | 2.36 | 13.8 |
| H2S⋯Cl2 | 0.92 | 6.23 | 23.8 |
| HCN⋯F2 | 0.93 | 2.61 | 13.8 |
| HCN⋯Cl2 | 0.88 | 6.55 | 23.8 |
Interactions of F2 are stronger than in related van der Waals molecular complexes. For instance, argon gives dimers with both acetonitrile and oxirane and the respective Kσ are 1.92 and 2.18 N m−1;13 the values for the F2 analogues are 2.5 and 3.09 N m−1, respectively. In addition, the geometry of the complex CH3CN⋯F2 reflects that of a typical XB, with the axial nuclei in the order C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯F–F (Fig. 3A), while CH3CN⋯Ar has a T-shaped geometry in which argon lies nearly orthogonal to the C–C bond of acetonitrile (Fig. 3B). These geometric differences suggest that different driving forces are assembling the complexes.
N⋯F–F (Fig. 3A), while CH3CN⋯Ar has a T-shaped geometry in which argon lies nearly orthogonal to the C–C bond of acetonitrile (Fig. 3B). These geometric differences suggest that different driving forces are assembling the complexes.
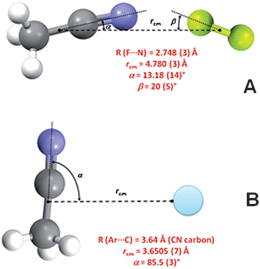 | ||
| Fig. 3 Structure parameters of the X-bonded complex CH3CN⋯F2 (A) and the van der Waals complex CH3CN⋯Ar (B). | ||
A positive σ-hole is found when fluorine is bound to another fluorine (the most electronegative element), Fig. 2A, and sometimes when it is linked to O, N, C or other atoms, if these are components of particularly strong electron withdrawing groups. CF3C(O)OF and (CF3SO2)2NF, well known electrophilic fluorinating reagents,14 are prototypical examples. The fluorines on the oxygen and on the nitrogen have positive potentials of +5.2 and +8.0 kcal mol−1, respectively (B3PW91/6-31G(d,p) calculations). The fluorines on the CF3 groups have maxima of +7.2 to +8.5 in the former and +2.4 to +3.3 kcal mol−1 in the latter molecules. It is noteworthy that if a fluorine is attached to a sufficiently strongly electron withdrawing residue, then there may be no negative belt on its lateral sides (as there is in F2, see Fig. 2A); instead it may have a positive electrostatic potential on its entire surface.15
In CF3SO2OCOF (Fig. 2B and Fig. 4B) the fluorine on the carbonyl residue has a positive cap (2.5 kcal mol−1) while those of the CF3 group have positive potentials on their entire surfaces, as do also the fluorines in fluorotrinitromethane16 and cyanogen fluoride.4 The heavier halogens can behave similarly, but it is less common to have positive potentials on their entire surfaces as, having more electrons, depletion of the characteristic negative belts is more difficult.
 | ||
| Fig. 4 Ball and stick representations of 1D infinite chains formed by 3-nitro-trifluoroacetylbenzene (A), CF3SO2OCOF (B), and (η5-(4-trifluoromethyl-2,3,5,6-tetrafluorophenyl)cyclopentadienyl)-tricarbonyl-manganese (C). The digits close to the XB (dashed lines) are the covalent bond/XB angle around fluorine and the normalized contact (Nc), namely the ratio Nc = Dij/(rvdWi + rvdWj), where Dij is the distance between the atoms i and j and rvdWi and rvdWj are the van der Waals radii for fluorine and oxygen, 147 and 152 pm, respectively. Colour code: grey, carbon; yellow, sulfur; red, oxygen; light green, fluorine; blue, nitrogen; purple, manganese. In A and C hydrogen atoms have been omitted for simplicity. In C also two carbonyl residues and one cyclopentadienyl group were deleted for clarity. | ||
With atoms possessing lone pairs, carbon-bound fluorines can interact attractively in solution and in the solid; in both phases, the adducts formed present the typical features of X-bonded systems. Topological analyses of the electron densities in complexes of simple fluoroorganics with ammonia4 reveal the existence of bond critical points between the fluorine and nitrogen atoms, showing that an N⋯F–C XB is present in these systems. Similar to other haloalkanes,17 perfluoroalkanes and amines in solution show charge transfer (CT) bands in the UV region.18 A linear relationship has been observed, for a given perfluorocarbon, between the energy of the CT band and the known ionization potential of the amine. The measured equilibrium constants (K ≈ 1) and extinction coefficients (ε ≈ 100) were rather small relative to the better known classes of CT complexes. ε decreases as K increases, as is often found for weak complexation. ΔH° and ΔS° were estimated to be in the ranges −0.57/−3.66 kcal mol−1 and −3.6/−12.6 cal mol−1, respectively. These interactions are weaker than similar ones by heavier halogens and are comparable in strength to aromatic stacking and weak hydrogen bonding.
The positive σ-holes in halogens are localized on the extensions of the covalent bonds in which they are involved; this explains the directionality which is a distinctive feature of XB.7 For instance, the Cambridge Structure Database (CSD) gives a histogram of occurrences as a function of the O⋯I–C contact angle which shows a well defined peak (median at 167.6°, see ESI†). This is a likely consequence of the pronounced iodine σ-holes present in iodocarbons and the remarkable strengths of the XBs to which they give rise.19 A similar behaviour is shown by O⋯Br–C and O⋯Cl–C contacts. The corresponding peaks become lower and move to smaller angles as the halogen becomes more electronegative (medians at 164.9 and 162.9° for Br and Cl, respectively), consistent with a correlation between directionality and magnitude of the halogen σ-hole potential. A peak hardly appears in the O⋯F–C histogram, but is present in the histogram O⋯F–C–SO2 (median at 159.3°). This suggests that most fluoroorganics either do not have positive σ-holes or that they are too weak to determine a directional bias in fluorine centred interactions. A positive σ-hole, and the resulting XB, may be a structure-stabilizing factor only in systems where fluorine is close to strongly electron withdrawing groups.
Indeed, many structures can be found in the CSD that meet the standard geometric requirements for fluorine functioning as a XB donor. Examples of one21 and two-dimensional22 networks are presented in Fig. 4 and 5, respectively, but many other structures show short linear contacts (see ESI†), demonstrating the ability of fluorine to work as a XB donor.23 While disorder is frequently present in crystals containing fluorinated residues, those mentioned above show no disorder, so that generalizations drawn from contact distances and angles are reliable.
 | ||
| Fig. 5 Ball and stick representations of the square grid formed by 4-(2,3,5,6-tetrafluoro-4-nitrophenyl)-1,2,3,5-dithiadiazolyl radical. Digits and color code as in Fig. 1. | ||
In conclusion, fluorine is commonly viewed as an electron-rich site with a negative electrostatic potential on its entire surface (Fig. 1). This is true in most cases, but some exceptions exist.24 Both of the fluorines in F2 possess positive σ-holes and the same is found in some compounds in which fluorine is bound to O, N, and/or C atoms if these can function as particularly strong electron-withdrawing moieties.
There is no contradiction here;7 as with chlorine, bromine and iodine, the anisotropic electronic charge distribution around covalently bound fluorine can give rise to a positive σ-hole in its outer portion and a negative (or more weakly positive) belt around its lateral sides. Fluorine can thus work as XB donor and examples have been discussed in the solid, liquid, and gas phases. Anions and neutral lone pair possessing atoms can be the XB acceptors. Calculated and experimental interaction energies for a halocarbon and a XB acceptor site are weakest for fluorine25 and a fluorocarbon moiety can act as XB donor site in crystals when fluorine is close to a very powerful electron withdrawing group and no stronger competing intermolecular interactions control the crystal packing and prevent the XB from showing out. The impact of this newly revealed aspect of fluorine behaviour may extend to several cases in which recognition phenomena of fluorine derivatives play a role.
Acknowledgements
P.M., G.R., and G.T. thank Fondazione Cariplo (“New-Generation Fluorinated Materials as Smart Reporter Agents in 19F MRI”) and MIUR (“Engineering of the Self-assembly of Molecular Functional Materials via Fluorous Interactions”) for financial support.Notes and references
- A. C. Legon, Phys. Chem. Chem. Phys., 2010, 12, 7736–7747 RSC; G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772–3783 RSC; K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC; P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS; P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef.
- F. A. Bulat, A. Toro-Labbé, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS.
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez, J. Benz, J.-M. Plancher, G. Hartmann, D. W. Banner, W. Haap and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 314–318 CrossRef CAS.
- Y.-X. Lu, J.-W. Zou, Q.-S. Yu, Y.-J. Jiang and W.-N. Zhao, Chem. Phys. Lett., 2007, 449, 6–10 CrossRef CAS.
- D. Chopra and T. N. Guru Row, CrystEngComm, 2011, 13, 2175–2186 RSC.
- http://blogs.rsc.org/ce/2010/11/02/defining-the-halogen-bond-iupac-task-group-need-your-input/, Chem. Int., 2010, 32(2)20 Search PubMed; http://www.iupac.org/web/ins/2009-032-1-100; http://www.halogenbonding.eu/.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–196 CrossRef CAS; K. E. Riley, J. S. Murray, M. C. Concha, P. Hobza and P. Politzer, J. Chem. Theory Comput., 2009, 5, 155–163 CrossRef; P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC; J. S. Murray, K. E. Riley, P. Politzer and T. Clark, Aust. J. Chem., 2010, 63, 1598–1607 CrossRef.
- P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649–1684 CrossRef CAS.
- S. Riedel, T. Kochner, X. Wang and L. Andrews, Inorg. Chem., 2010, 49, 7156–7164 CrossRef CAS and references cited therein; B. S. Ault and L. Andrews, J. Am. Chem. Soc., 1976, 98, 1591–1593 CrossRef; B. S. Ault and L. Andrews, Inorg. Chem., 1977, 16, 2024–2028 CrossRef.
- (a) A. A. Tuinman, A. A. Gakh, R. J. Hinde and R. N. Compton, J. Am. Chem. Soc., 1999, 121, 8397–8398 CrossRef CAS; (b) A. Artau, K. E. Nizzi, B. T. Hill, L. S. Sunderlin and P. G. Wenthold, J. Am. Chem. Soc., 2000, 122, 10667–10670 CrossRef CAS.
- A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef and references cited therein.
- F. A. Bulat, A. Toro-Labbé, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS.
- W. Lin and S. E. Novick, J. Mol. Spectrosc., 2007, 243, 32–36 CrossRef CAS; R. A. Collins, A. C. Legon and D. J. Millen, THEOCHEM, 1986, 135, 435–445 CrossRef.
- S. D. Taylor, C. C. Kotoris and G.I. Hum, Tetrahedron, 1999, 55, 12431–12477 CrossRef CAS.
- This can happen also with the larger halogens, but it is harder to accomplish as there are more electrons on them, therefore the characteristic negative belt is normally observed.
- M. Göbel, B. H. Tchitchanov, J. S. Murray, P. Politzer and T. M. Klapötke, Nat. Chem., 2009, 1, 229–235 CrossRef.
- S. V. Rosokha and J. K. Kochi, Struct. Bonding, 2008, 126, 137–160 CrossRef CAS.
- J. Burdeniuc, M. Sanford and R. H. Crabtree, J. Fluorine Chem., 1998, 91, 49–54 CrossRef CAS.
- P. Metrangolo, G. Resnati, T. Pilati and S. Biella, Struct. Bonding, 2008, 126, 105–136 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- L. O. Atovmyan, N. I. Golovina, L. T. Eremenko, N. G. Zhitomirskaya, G. V. Oreshko and M. A. Fadeev, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1984, 33, 504–510 CrossRef CAS (CSD code CUVFAA); C. O. D. Vedova, A. J. Downs, V. P. Novikov, H. Oberhammer, S. Parsons, R. M. Romano and A. Zawadski, Inorg. Chem., 2004, 43, 4064–4071 CrossRef (CSD code AZOYAP); P. A. Deck, B. D. McCauley and C. Slebodnick, J. Organomet. Chem., 2006, 691, 1973–1983 CrossRef (CSD code YELCOG).
- A. Alberola, R. J. Less, C. M. Pask, J. M. Rawson, F. Palacio, P. Oliete, C. Paulsen, A. Yamaguchi, R. D. Farley and D. M. Murphy, Angew. Chem., Int. Ed., 2003, 42, 4782–4785 CrossRef CAS (CSD code ILUKIJ A).
- D. Chopra, V. Thiruvenkatam, S. G. Manjunath and T. N. Guru Row, Cryst. Growth Des., 2007, 7, 868–874 Search PubMed (CSD code SIDLIB); T. A. Hamor and D. J. Watkin, J. Chem. Soc., Perkin Trans. 2, 1974, 140–146 RSC (CSD code BODFNB); O. G. Polyakov, S. M. Ivanova, C. M. Gaudinski, S. M. Miller, O. P. Anderson and S. H. Strauss, Organometallics, 1999, 18, 3769–3771 CrossRef CAS (CSD code QAPHOE); P. Nockemann, B. Thijs, T. N. Parac-Vogt, K. Van Hecke, L. Van Meervelt, B. Tinant, I. Hartenbach, T. Schleid, V. T. Ngan, M. T. Nguyen and K. Binnemans, Inorg. Chem., 2008, 47, 9987–9999 CrossRef (CSD code TOJREQ); P. Nockemann, B. Thijs, S. Pittois, J. Thoen, C. Glorieux, K. Van Hecke, L. Van Meervelt, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2006, 110, 20978–20992 CrossRef (CSD code JESSUL); A. Takahashi, H. Yanai and T. Taguchi, Chem. Commun., 2008, 2385–2387 RSC (CSD code SIZPIB); V. E. Platonov, A. Haas, M. Schelvis, M. Lieb, K. V. Dvornikova, O. I. Osina and Y. V. Gatilov, J. Fluorine Chem., 2001, 109, 131–139 CrossRef (CSD code XIHLUV).
- R. Berger, G. Resnati, P. Metrangolo, E. Weberd and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC.
- P. Politzer, J. S. Murray and M. C. Concha, J. Mol. Model., 2007, 13, 643–650 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scatterplots from CSD. See DOI: 10.1039/c1ce05554b |
| This journal is © The Royal Society of Chemistry 2011 |