 Open Access Article
Open Access ArticleSupramolecular architectures of conformationally controlled 1,3-phenyl-dioxalamic molecular clefts through hydrogen bonding and steric restraints†
Juan Saulo
González-González
a,
Francisco Javier
Martínez-Martínez
a,
Ana Lilia
Peraza Campos
a,
Maria
de Jesus Rosales-Hoz
b,
Efrén V.
García-Báez
c and
Itzia I.
Padilla-Martínez
*c
aFacultad de Ciencias Químicas, Universidad de Colima, Km. 9 Carretera Colima-Coquimatlán, C.P. 28400, Coquimatlán, Colima, Mexico
bDepartamento de Química, CINVESTAV-IPN, Av. Instituto Politécnico Nacional 2508, Col. San Pedro Zacatenco, Apartado postal: 14-740, 07000, C.P. 07360, México, D.F., Mexico
cDepartamento de Ciencias Básicas, Unidad Profesional Interdisciplinaria de Biotecnología del Instituto Politécnico Nacional, Av. Acueducto s/n Barrio la Laguna Ticomán, 07340, México, D. F., Mexico
First published on 31st May 2011
Abstract
In this contribution the supramolecular architecture of a series of six 1,3-phenyl-dioxalamic molecular clefts is described. The conformation was controlled by the use of Me and OMe group substitution in the phenyl spacer. The structural and conformational study was carried out by X-ray diffraction analysis, DFT calculations at PBEPBE 6-31+G (3df, 3pd) theory level and variable temperature 1H NMR in solution. The C2-Me group exerts a dual influence on the conformation adopting the endo(sc) or exo(ac) conformations in the oxalamic arms, meanwhile the C2-OMe group leads to the adoption of the exo(ap) conformation. DFT study results showed that the exo(ap)–exo(ap) conformation is more stable than the other conformations due to the conjugation that stabilizes the molecule and minimizes the conformational energy. Supramolecular arrays in oxalamate/oxalamide derivatives of 1,3-diaminobenzene, 2-methyl-benzene-1,3-diamine and 2,4,6-trimethyl-benzene-1,3-diamine are directed by self-complementary N–H⋯O hydrogen bonding interactions, whose organization in the crystal depends on the twist of the oxalamic arms, meanwhile in oxalamate/oxalamide derivatives of 5-tert-butyl-2,6-diamineanisol with an exo(ap)–exo(ap) conformation, the supramolecular arrays are directed by π-stacking, dipolar carbonyl–carbonyl interactions and C–H⋯O soft contacts. N1,N1′-(1,3-(2,4,6-Trimethyl)-phenyl)-bis-(N2-(2-(2-hydroxyethoxy)ethyl)oxalamide) adopts the form of a supramolecular meso-helix, which is the first example of helical 1,3-phenyl-dioxalamide.
1. Introduction
Non-covalent interactions are recognized to be involved in crystal engineering and molecular recognition.1 They have been increasingly used as a powerful tool for construction of many supramolecular architectures, devices and nanostructured materials.2 Among them, hydrogen bonding is particularly important from the biological point of view, because of its participation in several biological processes such as: the stabilization of the double helix of DNA,3enzyme–substrate interactions,4 recognition among proteins5 and drug–acceptor interactions.6Hydrogen bonding is the most important element in the design of self-assembled organic molecules, sometimes with the participation of other intermolecular forces.7 Inter- and intra-molecular hydrogen bonding has been extensively used in the production of complex organized systems due to the reversibility, specificity, directionality and cooperativity of such interactions.Oxalamates are composed of an amide and an ester group directly connected to each other. Thus the functionalities N–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–R allow them to form one dimensional (1-D) networks through intermolecular hydrogen bonding,8 but they also can form two (2-D) and three dimensional (3-D) networks whose architecture in the solid state will depend on the side-arm substituent.
O and O–R allow them to form one dimensional (1-D) networks through intermolecular hydrogen bonding,8 but they also can form two (2-D) and three dimensional (3-D) networks whose architecture in the solid state will depend on the side-arm substituent.
Oxalamides, which are mono-substituted diamides of oxalic acid, represent construction units with high in-plane hydrogen bonding potential; they are self-complementary and capable of unidirectional hydrogen bonding. In many cases, their self-assembly produces robust one-dimensional hydrogen-bonded chains, although, depending on the substitution and presence of additional hydrogen bonding units, a variety of more complex structures might be observed. The NH and amide carbonyl are usually in a trans disposition as also are amide and ester carbonyls or both carbonyl amides, in oxalamate and oxalamide, respectively, thus the most common conformation is trans–anti (t,a) in oxalamates and trans–anti–trans (t,a,t) in oxalamides.9

1,3-Phenyl-dioxalamates and 1,3-phenyl-dioxalamides are good candidates to be used in molecular recognition processes, because of the cavity formed in between the two side arms with a bent conformation and the open possibility of π-stacking between the phenyl spacer. The relative disposition of both arms in relation to the 2-substituted phenyl spacer and among them gives rise to several conformations.
In this contribution the molecular and supramolecular structure of 1,3-phenyl-dioxalamate 1 is revisited, and those corresponding to three 2-substituted 1,3-phenyl-dioxalamates 2–4 are reported. This series of compounds was synthesized starting from 1,3-diaminobenzene C-2 substituted with Me or OMe groups. The presence of these groups bisecting the central phenyl ring was used to tilt the oxalyl side arms out of the plane and thus favor intermolecular hydrogen bonding. In order to test the utility of the hydroxyl group to increase the self-assembling capabilities of the oxalamide moiety into predictable structural patterns, oxalamides with side-arm substituents derived from ethanolamine 5–7 were prepared. Furthermore, the molecular and supramolecular 1,3-phenyl-dioxalamides 5–7 are also analyzed and compared with their parent dioxalamates 2–4. To the best of our knowledge there are recent reports on the topological control10a of their dimensionality in the solid state, but there are no reports about the use of steric effects to control the conformation and so the hydrogen bond directed self-assembly in the solid state of these molecules. Results were supported by DFT calculations and 1H NMR measurements.

2. Results
The relative disposition of the carbonyl group of the phenylamide moiety (PhNHCO) regarding the 2-substituent in the phenyl ring, as a plane of reference, gives rise to several conformers: antiperiplanar (ap), anticlinal (ac), synclinal (sc) and synperiplanar (sp), in agreement with the torsion angle C2–C1–N7–C8 values of ±180–150°, ±150–90°, ±90–30° and ±30–0°, respectively. In Fig. 1 are shown the Newman projections along the C1–N7 bond.11 | ||
| Fig. 1 Newman projections of conformers derived from the C2–C1–N7–C8 torsion angle, viewed along the C1–N7 bond. | ||
In both syn-conformers, the PhNHCO carbonyl is pointing towards the 2-substituent, thus endo positioned with regard to the cavity, whereas the anti-conformers are exo positioned. Taking into account the two arms, ten combinations are possible: three exo–exo (ap–ap, ac–ac, ap–ac), four exo–endo (ap–sc, ap–sp, ac–sc, ac–sp) and three endo–endo (sc–sc, sc–sp, sp–sp).
There are two reported polymorphs of diethyl 1,3-phenyl-dioxalamate 110b and 1′.10c In the conformation exhibited by polymorph 1, both amide carbonyls are located pointing endo to the cavity and syn (sc–sp) positioned regarding to the phenyl ring with C2–C1–N7–C8 and C1–N7–C8–O8 torsion angles of 37.0(3)° and −4.1(3)°, respectively. This conformation allows a full intramolecular hydrogen bonding scheme combining one O⋯H2⋯O three centred and two NH⋯O hydrogen bonding interactions, respectively. Thus a set of four adjacent intramolecular rings are formed, which are described by the graph set notation12 as [S(5)S(6)S(5)S(6)] motif. Meanwhile in 1′, the torsion angles C6–C1–N7–C8 and C1–N7–C8–O8 take values of 32.6(5)° and 179.3(3)°, respectively, in agreement with an exo(ac)–exo(ac) conformation of both oxalamate arms. This conformation favors the formation of two sets of intramolecular six and five membered hydrogen bond motifs [S(6)S(5)]. A common feature of conformers 1 and 1′ is the twist of both oxalamate side arms, one of them tilted up and the other tilted down with regard to the mean plane of the phenyl ring. The out-of-plane conformation is characteristic of disubstituted ethylo- and p-phenyl-bis-oxalamates,13 in contrast with the reported planar conformation of monosubstituted ethyl N-phenyl oxalamate.14
2.1 Intramolecular hydrogen bonding and molecular structure
C8–C9O9 and O18C18–C19O19 torsion angle values between ±165 and ±178°. The N7–C8–C9–O10 and N17–C18–C19–O20 torsion angle values are near to 180°, in close agreement with those values reported for diethyl phenyl-dioxalamates.10
| Compounds | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|
| a X = O in 2–4 and X = N in 5–7. | ||||||
| Atoms | Bond lengths/Å | |||||
| C1–N7 | 1.431(3) | 1.417(5) | 1.402(4) | 1.440(6) | 1.441(3) | 1.409(3) |
| C3–N17 | 1.433(3) | 1.429(5) | 1.413(4) | 1.415(6) | 1.397(3) | |
| N7–C8 | 1.335(3) | 1.344(5) | 1.345(4) | 1.324(6) | 1.317(4) | 1.344(3) |
| N10–C9 | 1.317(6) | 1.313(5) | 1.319(3) | |||
| N17–C18 | 1.335(3) | 1.331(5) | 1.342(4) | 1.338(6) | 1.353(3) | |
| N20–C19 | 1.337(6) | 1.330(4) | ||||
| C8–C9 | 1.535(3) | 1.532(6) | 1.529(4) | 1.505(7) | 1.525(5) | 1.538(4) |
| C18–C19 | 1.543(3) | 1.524(6) | 1.543(5) | 1.517(6) | 1.527(4) | |
| Atoms | Torsion angles/° | |||||
| C2–C1–N7–C8 | 64.4(3) | −60.4(5) | 179.5(3) | −126.5(5) | −72.4(4) | −176.1(3) |
| C2–C3–N17–C18 | 67.4(3) | −103.5(4) | −178.4(3) | −144.5(4) | −177.6(3) | |
| C6–C1–N7–C8 | −117.0(2) | 123.4(4) | 0.3(5) | 55.4(7) | 108.0(3) | −4.9(5) |
| C1–N7–C8–O8 | −0.2(4) | −9.5(6) | −3.6(5) | −1.2(9) | 10.3(6) | −2.3(5) |
| C4–C3–N17–C18 | −113.7(2) | 76.5(4) | 0.8(5) | 38.5(7) | −1.6(5) | |
| C3–N17–C18–O18 | −0.8(3) | −2.1(6) | −4.5(5) | 1.8(8) | 2.5(5) | |
| O8–C8–C9–O9 | −167.9(2) | 165.0(4) | −176.1(3) | −169.1(5) | 179.1(3) | 179.7(3) |
| O18–C18–C19–O19 | −177.3(2) | 168.6(4) | −177.4(4) | −179.6(4) | 178.0(3) | |
| N7–C8–C9–X10 | −169.14(18) | 170.0(3) | −178.6(3) | −165.1(5) | 176.3(3) | −179.7(2) |
| N17–C18–C19–X20 | 179.97(18) | 169.9(3) | −178.1(3) | −177.2(4) | 177.7(2) | |
| N10–C11–C12–O13 | −177.3(3) | −59.5(3) | ||||
| N20–C21–C22–O23 | 66.2(3) | |||||
| O13–C14–C15–O16 | 69.1(4) | |||||
 | ||
| Fig. 2 Molecular structure at 30% probability level and intramolecular interactions of dioxalamates 2–4. | ||
The introduction of a CH3 group in the C-2 position, compound 2, increases the twisting out of the ethyl oxalamate arms from 37.0(3)° in polymorph 1 to 64.4(3) and 67.4(3)° (C2–C1–N7–C8 and C2–C3–N17–C18 torsion angles, respectively), as reported for diamides derived from 2-methyl-benzene-1,3-diamine.15 Thus both oxalamate arms are toward one another, one above the plane of the phenyl ring and the other below this plane with both amide carbonyls in synclinal (sc) disposition in relation to the phenyl ring, but even endo to the cavity. This conformation allows the formation of a set of four adjacent intramolecular rings [S(5)S(7)S(7)S(5)] with the participation of NH and C2–CH3 hydrogen atoms as donors and both carbonyls as acceptors: N7–H7⋯O9, N17–H17⋯O19, C23–H23A⋯O18 and C23–H23B⋯O18, Fig. 2(a). It is worthy to note that, in this compound and in related diamides, the methyl group is pointing towards the amide carbonyl thus favoring the S(7) ring, whereas in monoamides derived from o-tolylamine16 it is always in the opposite direction. In spite of the steric requirements of the C2–CH3 group, this conformer strongly resembles that of polymorph 1, where the central S(6) intramolecular hydrogen bonded rings are substituted by larger S(7) ring motifs. In Table 2 the geometric parameters associated with intramolecular hydrogen bonding of compounds 2–4 are summarized. The interaction lengths are shorter than the sum of the van der Waals radii of the atoms involved (H = 1.20, C = 1.70, N = 1.55, O = 1.50 Å).17
| Comp. | D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | D–H⋯A/° | HB motif |
|---|---|---|---|---|---|---|
| a Σ∠N7H7/° = 359 (a), 360 (c); Σ∠N17H17/° = 360 (b), 359 (d). | ||||||
| 2 | N7–H7⋯O9 | 0.86 | 2.31 | 2.696(3) | 107 | S(5) |
| N17–H17⋯O19 | 0.86 | 2.31 | 2.693(2) | 107 | S(5) | |
| C23–H23A⋯O18 | 0.96 | 2.44 | 3.156(3) | 131 | S(7) | |
| C23–H23B⋯O8 | 0.96 | 2.58 | 3.115(3) | 115 | S(7) | |
| 3 | N7–H7⋯O9 | 0.86 | 2.39 | 2.727(4) | 104 | S(5) |
| N17–H17⋯O19 | 0.86 | 2.37 | 2.728(4) | 105 | S(5) | |
| 4 | N7–H7a⋯O9 | 0.86 | 2.20 | 2.640(4) | 111 | S(5) |
| N7–H7a⋯O23 | 0.86 | 2.27 | 2.694(3) | 110 | S(5) | |
| N17–H17b⋯O19 | 0.86 | 2.25 | 2.678(4) | 111 | S(5) | |
| N17–H17b⋯O23 | 0.86 | 2.25 | 2.703(4) | 110 | S(5) | |
| C4–H4⋯O18 | 0.93 | 2.34 | 2.940(4) | 122 | S(6) | |
| C6–H6⋯O8 | 0.93 | 2.37 | 2.967(4) | 121 | S(6) | |
| 5 | N7–H7⋯O9 | 0.86 | 2.26 | 2.659(5) | 109 | S(5) |
| N10–H10⋯O8 | 0.86 | 2.39 | 2.749(5) | 106 | S(5) | |
| N17–H17⋯O19 | 0.86 | 2.25 | 2.670(5) | 110 | S(5) | |
| N20–H20⋯O18 | 0.86 | 2.36 | 2.727(5) | 106 | S(5) | |
| C4–H4⋯O18 | 0.93 | 2.51 | 2.977(6) | 111 | S(6) | |
| 6 | N7–H7⋯O9 | 0.86 | 2.29 | 2.675(3) | 107 | S(5) |
| N10–H10⋯O8 | 0.86 | 2.32 | 2.701(4) | 107 | S(5) | |
| 7 | N7–H7⋯O9c | 0.86 | 2.20 | 2.647(3) | 112 | S(5) |
| N7–H7⋯O24c | 0.86 | 2.27 | 2.692(3) | 110 | S(5) | |
| N10–H10⋯O8 | 0.86 | 2.33 | 2.714(3) | 107 | S(5) | |
| N17–H17⋯O19d | 0.86 | 2.24 | 2.665(3) | 110 | S(5) | |
| N17–H17⋯O24d | 0.86 | 2.30 | 2.704(3) | 109 | S(5) | |
| N20–H20⋯O18 | 0.86 | 2.32 | 2.693(3) | 106 | S(5) | |
| C4–H4⋯O18 | 0.93 | 2.30 | 2.912(3) | 123 | S(6) | |
| C6–H6⋯O8 | 0.93 | 2.38 | 2.974(3) | 121 | S(6) |
Further increasing the steric restraints with CH3 substituent groups in C2, C4 and C6, compound 3, the oxalamate arms become twisted by −60.4(5)° and −103.5(4)° for C2–C1–N7–C8 and C2–C3–N17–C18 torsion angles, respectively. This result is in agreement with the mean value of ca. 70° reported for 2,4,6-trimethyl-benzene-1,3-diamine.18 The oxalamate arms are also up and down, adopting a synclinal (sc) and anticlinal (ac) disposition, in relation to the mean plane of the phenyl ring, and endo–exo positioned to the cavity. Intramolecular hydrogen bonding is reduced to only two S(5) ring motifs, formed by N7–H7⋯O9 and N17–H17⋯O19 interactions, Fig. 2(b).
In compound 4, the bulkiest OCH3 substituent in C2 leads to the full rotation of both oxalamate side arms to locate them almost in the same plane of the phenyl ring, C2–C1–N7–C8 and C2–C3–N17–C18 torsion angles of 179.5(3)° and −178.4(3)°, as found in diamides derived from 2-methoxy-5-methyl-benzene-1,3-diamine,19 both adopting an antiperiplanar (ap) disposition, in relation to the mean plane of the phenyl ring and exo–exo positioned to the cavity. In this molecule the t-Bu substituent is far away from the cavity, thus the steric effect is mainly exerted by the methoxy group. The OMe group is almost perpendicular to the phenyl ring (C1–C2–O23–C24 = 91.3(3)°) leading the oxygen atom in the same plane. This conformation is probably assisted by cooperative intramolecular hydrogen bonding.20 The amide hydrogen atoms are involved in a three-centred hydrogen bonding with both O-atoms from the ester carbonyl and methoxy groups (O9⋯H7⋯O23 and O19⋯H17⋯O23), an important feature of this interaction is that the sum of angles around the amide H-atom (Σ∠NH) is close to 360°.21 In the opposite side, the amide carbonyls are also interacting with the aromatic hydrogen atoms H4 and H6 (C4–H4⋯O18 and C6–H6⋯O8). The full system is composed of six adjacent rings described by the graph set notation as an [S(5)S(6)S(5)S(5)S(6)S(5)] ring system, Fig. 2(c).
 | ||
| Fig. 3 Molecular structure at 30% probability level and intramolecular interactions of dioxalamides 5–7. | ||
In compound 5, both oxalamide side arms are almost planar with C1–N7–C8–O8 and C3–N17–C18–O18 torsion angles close to 0°. Phenylamide carbonyls (C8O8 and C18O18) are both exo positioned to the cavity and anticlinal (ac) to the mean plane of the phenyl ring, one of them above and the other below the phenyl ring plane with C2–C1–N7–C8 and C2–C3–N17–C18 torsion angles of −126.5(5)° and −144.5(4)°, respectively. This last conformation brings about the formation of intramolecular C4–H4⋯O18 HB, S(6) ring, at the expense of two S(7) rings in the parent oxalamate 2. In addition N7–H7⋯O9, N10–H10⋯O8, N17–H17⋯O19 and N20–H20⋯O18 hydrogen bonding, typical of oxalamides, are observed. Therefore, the intramolecular hydrogen bonding pattern is depicted by two sets of adjacent HB ring systems [S(5)S(5)S(6)] and [S(5)S(5)], Fig. 3(a).
Compound 6 has a crystallographic plane of symmetry and thus only one half of the molecule is observed. The fragment N–CO–CO–N of the oxalamide arms is almost planar with C1–N7–C8–O8 and N10–C11–C12–O13 torsion angles of 10.3(6)° and −177.3(3)°, respectively. The OH group at the end of the arm is in a gauche conformation with O13–C14–C15–O16 torsion angle of 69.1(4)°. Both arms are synclinarly (sc) positioned with respect to the mean plane of the phenyl ring, with C2–C1–N7–C8 torsion angle of −72.4(4)° and the phenylamide carbonyl pointing towards the cavity. The intramolecular HB scheme is described as two sets of [S(5)S(5)] ring systems, Fig. 3(b).
The conformation of oxalamide 7 strongly resembles the conformation observed for the parent oxalamate 4: the oxalamide arms are ap to the phenyl ring (C2–C1–N7–C8 = −176.1(3)° and C2–C3–N17–C18 = −177.6(3)°), both are exo disposed to the cavity and the OMe group is almost perpendicular to the phenyl ring (C1–C2–O24–C25 = 88.6(3)°). The OH group at the end of the arm is in a gauche conformation with N10–C11–C12–O13 and N20–C21–C22–O23 torsion angles of −59.5(3)° and 66.2(3)°, respectively, Fig. 3(c).
The substitution of EtO in oxalamates 2–4 for RNH in oxalamides 5–7 increases the intramolecular HB, allowing the formation of an additional S(5) ring in each arm due to the (t,a,t) conformation of the dioxalamide RHN–CO–CO–NHPh fragment. Therefore, the following patterns are observed: [S(5)S(5)S(6)] and [S(5)S(5)] for 5, two sets of [S(5)S(5)] rings for 6 and eight adjacent rings [S(5)S(5)S(6)S(5)S(5)S(6)S(5)S(5)] for 7. At this point it is worthy to note that the C1–N7/C3–N17 distances enlarge when the oxalyl arm is out of the mean plane of the phenyl ring. This result is consistent with the C1–N7 distance value of 1.414 Å measured in planar N,N'-bis(3,5-dimethyl-phenyl)oxalamide,13 similar to the C1–N7/C3–N17 distance values observed in the corresponding in plane arm of compounds 3–5 and 7. In contrast, a C1–N7 distance value of 1.435 Å has been measured in the almost perpendicular N,N'-bis-(2,4,6-trimethyl-phenyl)oxalamide,13 in agreement with C1–N7/C3–N17 distance values observed in the corresponding out of plane arm of compounds 2, 3, 5 and 6.
When the steric effect is increased by substitution in C4 and C6 positions, compounds 3 and 6, both oxalamate groups are tilted out of the phenyl mean plane to give conformers very similar to those found by X-ray. In compound 3, the conformers endo(sc)–endo(sc), exo(ac)–exo(ac) and exo(ac)–endo(sc) (from XRD) were tested, this last one is more stable than the other two by 1.8 and 1.1 kcal mol−1, respectively. In the case of oxalamide 6, the endo(sc)–endo(sc) conformer was the predicted one, in close agreement with the XRD structure. The calculated planar exo(ap)–exo(ap) conformers of 4 and 7 are in close agreement with XRD structures. In both cases, the N atoms bear a positive formal charge as a consequence of the conjugation of the lone pair of electrons into the phenyl ring. The optimized structures and formal charges are shown in Fig. 4. The exo(ap)–exo(ap) conformation is more stable than the other conformations due to the conjugation that stabilizes the molecule and minimizes the conformational energy.
 | ||
| Fig. 4 Molecular structures and charges of the most stable optimized conformers of compounds 2 (a), 3 (b), 4 (c) and 6 (d) at DFT/PBEPBE 6-31+G (3df, 3pd) level of theory. | ||
| Parameter | Compound | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| a δ 1HDMSO-d6 − δ1HCDCl3 available only for 1–4 for solubility restrictions. | |||||||
| δNHPh | 10.82 | 10.48 | 10.37 | 10.04 | 10.29 | 10.25 | 9.90 |
| ΔδNHPha | 1.89 | 1.62 | 1.90 | 0.79 | |||
| ΔδNHPh/ΔT | −5.50 | −4.70 | −5.21 | −4.20 | −9.30 | −5.39 | −2.00 |
| δNHR | 8.81 | 8.80 | 8.97 | ||||
| ΔδNHR/ΔT | −4.96 | −5.50 | −4.9 | ||||
| δOH | n.o. | 4.65 | n.o. | ||||
| ΔδOH/ΔT | −5.43 | ||||||
2.3 Crystal packing and supramolecular arrangement
Compounds 2 and 3 crystallize as monoclinic systems, space groupP21/c with four molecules in the unit cell, compounds 4 and 5 as triclinic, space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules in the unit cell, whereas compounds 6 and 7 crystallize as monoclinic, space groupC2/c with four molecules in the unit cell. Compound 7 is a special case in which the monoclinic structure has a β angle indistinguishable from 90.0°. The geometric parameters associated with intermolecular non-covalent interactions D–H⋯A are summarized in Table 4. In order to avoid data duplication, the reader is referred to the corresponding entry. Classic hydrogen bonding26 and C–H⋯O27 or C–H⋯π28 interactions are in agreement with accepted criteria. Classification and geometry of CO⋯CO interactions29 and π-stacking30 are in accordance with accepted values.
with two molecules in the unit cell, whereas compounds 6 and 7 crystallize as monoclinic, space groupC2/c with four molecules in the unit cell. Compound 7 is a special case in which the monoclinic structure has a β angle indistinguishable from 90.0°. The geometric parameters associated with intermolecular non-covalent interactions D–H⋯A are summarized in Table 4. In order to avoid data duplication, the reader is referred to the corresponding entry. Classic hydrogen bonding26 and C–H⋯O27 or C–H⋯π28 interactions are in agreement with accepted criteria. Classification and geometry of CO⋯CO interactions29 and π-stacking30 are in accordance with accepted values.
| Comp. | D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | D–H⋯A/° | HB motif |
|---|---|---|---|---|---|---|
| a Σ∠N7H7/° = 332 (a), 351 (c), 355 (e), 349 (h); Σ∠N17H17/° = 332 (b), 348 (d), 354 (g); Σ∠N10H10/° = 356 (f), 360 (i), 342 (j); Σ∠N20H20/° = 357 (k). Symmetry codes: (i) = 2 − x, −y, 1 − z; (ii) = 1 − x, −y, 1 − z; (iii) = 2 − x, −y, 2 − z; (iv) = x, ½ − y,−½ + z; (v) = 1 − x, −y, −z; (vi) = 2 − x, −y, −z; (vii) = −x, 1 − y, 1 − z; (viii) = −x, −y, 1 − z; (ix) = −x, 1 − y, −z; (x) = −1 − x, −y, 1 − z; (xi) = ½ + x, ½ − y, ½ + z; (xii) = ½ − x, ½ − y, −z; (xiii) = −½ − x, ½ − y, −z; (xiv) = −x, −y, −z; (xv) = −½ + x, ½ + y, z; (xvi) = −½ − x, ½ + y, ½ − z; (xvii) = −x, y, ½ − z. | ||||||
| 2 | N7–H7a⋯O18i | 0.86 | 2.39 | 3.074(2) | 137 | R 2 2(16) |
| N7–H7⋯πi | 0.86 | 3.18 | 3.710(2) | 123 | R 2 2(6) | |
| N17–H17b⋯O8ii | 0.86 | 2.32 | 2.995(2) | 136 | R 2 2(16) | |
| C11–H11A⋯O18iii | 0.97 | 2.59 | 3.433(3) | 145 | R 2 2(24) | |
| 3 | N7–H7c⋯O18iv | 0.86 | 2.24 | 3.060(4) | 160 | C(8) |
| C24–H24C⋯O18iv | 0.96 | 2.52 | 3.281(4) | 136 | C(9) | |
| N17–H17d⋯O8v | 0.86 | 2.15 | 2.903(4) | 149 | R 2 2(16) | |
| 4 | C24–H24C⋯O9vii | 0.96 | 2.41 | 3.348(5) | 167 | R 2 2(18) |
| C21–H21B⋯πii | 0.96 | 2.85 | 3.668(4) | 142 | R 2 2(14) | |
| 5 | N7–H7e⋯O18viii | 0.86 | 2.18 | 2.937(5) | 146 | R 2 2(16) |
| N10–H10f⋯O8ix | 0.86 | 2.19 | 2.969(6) | 151 | R 2 2(10) | |
| N17–H17g⋯O19x | 0.86 | 2.20 | 2.932(5) | 142 | R 2 2(10) | |
| N20–H20⋯O9viii | 0.86 | 2.49 | 3.306(5) | 159 | R 2 2(24) | |
| 6 | N7–H7h⋯O16xi | 0.86 | 2.12 | 2.928(4) | 157 | R 2 2(30) |
| N10–H10i⋯O8xii | 0.86 | 2.07 | 2.888(4) | 158 | R 2 2(10) | |
| O16–H16⋯O9xiii | 0.84 | 1.97 | 2.798(3) | 167 | R 2 2(34) | |
| 7 | N10–H10j⋯O13xiv | 0.86 | 2.40 | 3.139(3) | 144 | R 2 2(10) |
| O13–H13⋯O19xiii | 0.84 | 2.08 | 2.893(3) | 164 | R 2 2(30) | |
| C25–H25A⋯O9xiii | 0.96 | 2.53 | 3.274(4) | 134 | R 2 2(18) | |
| C22–H22A⋯O8xv | 0.97 | 2.46 | 3.279(3) | 142 | C(13) | |
| O23–H23⋯O9xvi | 0.84 | 2.03 | 2.817(3) | 156 | C(15) | |
| N20–H20k⋯O23xvii | 0.86 | 2.14 | 2.922(3) | 150 | R 2 2(10) | |
| C27–H27B⋯O24xvii | 0.96 | 2.55 | 3.377(4) | 144 | R 2 2(16) |
 | ||
| Fig. 5 1-D columns along the a axis formed by N–H⋯O, N–H⋯π and pd–π-stacking interactions in compound 2. | ||
Compound 3 also forms amide-linked centrosymmetric dimers, as in 2, through the ac-oxalamate side arm N17–H17⋯O8v (v: 1 − x, −y, −z) to define a R22(16) ring motif. In this case each dimer is connected with four other dimers through N7–H7⋯O18ivhydrogen bonding and C24–H24C⋯O18iv interaction, to form R21(7)[C(8)C(9)] second level ring motif (iv: x, ½ − y,−½ + z). This arrangement, viewed in the bc plane, looks like a brick wall-sheet of alternating squared R22(16) and hexagon R66(32) ring motifs, Fig. 6. The R22(16) dimers are pd–π-stackedvi as the intercentroid and interplanar distances of 4.540(2) and 3.6204(13) Å (vi: 2 − x, −y, −z), respectively, indicate.
 | ||
| Fig. 6 View in the bc plane of a brick wall-sheet of alternating squared R22(16) and hexagon R66(32) like ring motifs, formed by N–H⋯O hydrogen bonding interactions in compound 3. | ||
The extended embracement disposition of 4 and the steric hindrance of the C2-OMe group do not allow intermolecular hydrogen bonding. However, dipolar carbonyl–carbonyl and π-stacking interactions direct the formation of centrosymmetric pairs. Two self-complementary sheared parallel carbonyl–carbonyl interactions are formed: C8O8⋯C19viii [O8⋯C19 = 3.270(4) Å, C8O8⋯C19 = 104.7(3)°] and C18O18⋯C9viii [O18⋯C9 = 3.144(4) Å, C18O18⋯C9 = 101.9(3)°]. Intercentroid and interplanar distances of 3.7950(18) Å and 3.4580(13) Å, respectively, are in agreement with face to face π-stackingviii (viii: −x, −y, 1 − z), Fig. 7(a). These pairs are linked through weak C24–H24C⋯O9vii interactions with the participation of the methyl group from OMe as the donor and ester carbonyl as the acceptor (vii: −x, 1−y, 1 − z). Thus R22(18) ring motifs are outlined developing 1-D zig-zagging tapes along the direction of the a axis. If intramolecular HB is included, [S(5)S(5)] motifs, chair like H4O4, octagons are also observed, Fig. 7(b). T-Shaped C21–H21B⋯πii interactions, R22(14) motif, are responsible to generate 2-D walls in the direction of the c axis.
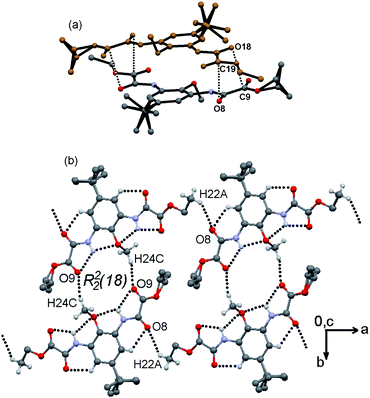 | ||
| Fig. 7 Supramolecular structure of compound 4. (a) Centrosymmetric pair linked by CO⋯CO dipolar and pd–π-stacking interactions (hydrogen atoms and t-Bu group are omitted for clarity). (b) 1-D zig-zagging tape linked through weak C24–H24C⋯O interactions along the a axis, chair like H4O4 octagons are also included. | ||
![(a) Partial view of the rough complex sheet of compound 5, developed by N–H⋯O and O–H⋯O hydrogen bonding interactions in the [2 8 11] mean plane (hydrogen atoms not involved in HB have been omitted for clarity). OxalamideR22(10) and R22(16) ring motifs are clearly appreciated as well as disordered ethanolamine fragment C11–C12–O13–H13. (b) A detail of the disordered hydroxyl group O23–H23 showing the R22(8) ring motif through O–H⋯O interactions.](/image/article/2011/CE/c1ce05302g/c1ce05302g-f8.gif) | ||
| Fig. 8 (a) Partial view of the rough complex sheet of compound 5, developed by N–H⋯O and O–H⋯O hydrogen bonding interactions in the [2 8 11] mean plane (hydrogen atoms not involved in HB have been omitted for clarity). OxalamideR22(10) and R22(16) ring motifs are clearly appreciated as well as disordered ethanolamine fragment C11–C12–O13–H13. (b) A detail of the disordered hydroxyl group O23–H23 showing the R22(8) ring motif through O–H⋯O interactions. | ||
Molecule 6 is linked to other molecule, forming centrosymmetric pairs, by N10–H10⋯O8xii (xii: ½ − x, ½ − y, −z), N7–H7⋯O16xi (xi: ½ + x, ½ − y, ½ + z) and O16–H16⋯O9xiiihydrogen bonding to form R22(16)[R22(10)R22(36)] ring motif. The intermolecular spacing along the oxalamideR22(10) motif is 5.060(3) Å. The propagation of these hydrogen bonding interactions along the (2 0 −2) direction gives rise to a meso-helix. The turn of the helix measures 18.0 Å between two phenyl rings, longer than 15.0 Å measured in the polymorph 1′.10c The meso-helix can be regarded as the alternate propagation of helical conformers of opposite chirality (M and P) around the inversion centre thus the twist sense changes every half-turn of the helix, Fig. 9(a). To the best of our knowledge compound 6 is the first example of helical 1,3-phenyl-dioxalamide. An infinite number of helixes are interlinked by O16–H16⋯O9xiii (xiii: −½ − x, ½ − y, −z) interactions through the ending ethanolamine OH and the ethanolamide CO, forming R22(34) ring motifs, which combined with the above mentioned N7–H7⋯O16 interactions depict a R22(7)[R22(30) R22(34)] second level ring motif. A view of the full set of hydrogen bonding motifs in the ac plane is shown in Fig. 9(b). H2O2 squares and H4O4 boat like octagons appear as intrahelix and interhelix motifs, when three-centred HB is considered: O8⋯H10⋯O8 (Σ∠H10 = 360°) and O9⋯H7⋯O16 (Σ∠H7 = 349°), respectively. Better appreciated is the 20-membered ring in the top view of the helix shown in Fig. 9(c). The whole 2-D architecture is composed of independent-weaving sheets propagating in the (2 0 −2) direction.
 | ||
| Fig. 9 (a) View of supramolecular meso-helix of compound 6, hydrogen bonding interactions lie in the plane. (b) View of the full set of hydrogen bonding motifs in the ac plane: N10–H10⋯O8 (R22(10)), N7–H7⋯O16 and O16–H16⋯O9, intrahelix H2O2 squares and interhelix H4O4 boat like octagons are highlighted. (c) Top view of R22(20) interhelical ring motif. | ||
In molecule 7, where the adopted conformation maximizes the intramolecular hydrogen bonding, also centrosymmetric dimers are formed by hydrogen bonding between one ending hydroxyl group and RNHCO amide carbonyl O13–H13⋯O19xiii, and also by C25–H25A⋯O9xiii soft interactions to form R22(16)[R22(30) R22(18)] second level ring motif. One dimension is generated by the propagation of this motif along the direction of the b axis through N10–H10⋯O13xiv (xiv: −x, −y, −z) hydrogen bonding to form R22(10) ring motifs and C22–H22A⋯O8xv (xv: −½ + x, ½ + y, z) forming C(13) chains, Fig. 10(a). The curvature of the assembly is inherently imprinted by the exo–exo symmetric disposition of the oxalamide arms. The remaining OH, NH and CO groups link the curved chains through O23–H23⋯O9xvi (xvi: −½ − x, ½ + y, ½ − z) and N20–H20⋯O23xvii (xvii: −x, y, ½ − z) interactions, to form C(15) and R22(10) ring motifs. The second dimension is strengthened by the soft contact C27–H27B⋯O24xvii between t-Bu and OMe groups, dipolar carbonyl–carbonyl and π-stacking interactions to form π-stacked pairs. Both π-stacked molecules are related by C2 symmetry axis, in close similarity with the parent oxalamate 4: two self-complementary sheared parallel carbonyl–carbonyl interactions C8O8⋯C19xvii [O8⋯C19 = 3.235(4) Å, C8O8⋯C19 = 105.2(2)°], C18O18⋯C9xvii [O18⋯C9 = 3.284(4) Å, C18O18⋯C9 = 100.4(2)°] and a face-to-face π-stackingxvii with intercentroid and interplanar distances of 3.6756(17) and 3.3608(11) Å, respectively, Fig. 10(b). Thus the full architecture is seen, in the bc plane, as a wall formed by lines of molecules alternately linked only by HB and then by a combination of HB and π-stacking interactions, running along the (−1 0 14) direction, Fig. 10(c). Hydrophobic contacts among these blocks along the direction of the a axis are given between tert-butyl groups with C29⋯C29 distance of 4.184(4) Å.
 | ||
| Fig. 10 (a) Supramolecular 1-D chains formed by O–H⋯O, N–H⋯O and C–H⋯O interactions along the b axis in compound 7. (b) Molecular pair formed by CO⋯CO dipolar and face-to-face π-stacking interactions. (c) Full supramolecular architecture shown as a wall formed by lines of molecules alternately linked only by HB and then by a combination of HB and π-stacking interactions, running along the (−1 0 14) direction. | ||
3. Discussion
The lengthening of the C1–N7/C3–N17 bonds suggests that the twisted conformation breaks the conjugation between the phenyl ring and the amide bond and directs the two oxalyl arms one above and the other below the plane of the phenyl ring. The XRD conformations exhibited by compounds 2–7 are the result of maximization of intramolecular hydrogen bonding and C–H⋯OC interactions, both assisted by dipole–dipole interactions coming from favorable antiparallel arrangements of the local N–H⋯OC dipoles. The conformation of oxalamates 3 and 4 is reliable; it is almost the same in oxalamides 6 and 7, whereas the conformation of oxalamate 2 does not remain in the corresponding oxalamide 5. The transformation of oxalamate into the corresponding oxalamide is accompanied with an increase in the hydrogen bonding capabilities. Intramolecular hydrogen bonding is increased by one S(5) ring motif per arm. The hydrogen bonding nature of the intramolecular O9⋯H7⋯O23⋯H17⋯O19 interactions in compound 7 was confirmed by ΔδNHPh/ΔT measurements, with a value of −2.0 ppm K−1.Theoretical calculations predict the exo(ap)–exo(ap) conformer as the most stable in compounds 2, 4, 5 and 7 and conformer endo(sc)–endo(sc) in compounds 3 and 6 in agreement with the steric effects exerted by the substituents in the phenyl ring. Differences between theoretical predictions and XRD structures in compounds 2 and 5 can be attributed to intramolecular C–H⋯OC interactions which are strong enough to overcome the small energy difference between both conformers.
There is a clear trend, in the solid state, to adopt an exo(ap)–exo(ap) conformation by increasing the steric requirements of the C2-substituent. Further substitution in C4 and C6 leads to both oxalyl arms almost perpendicularly positioned to the phenyl ring resulting in a very reliable conformation also observed in diamides derived from 2,4,6-trimethyl-benzene-1,3-diamine.18
Intermolecular NH⋯OC(amide) hydrogen bonds are preferred over NH⋯OC(ester) as supramolecular synthons in the formation of crystal networks by dioxalamates 2 and 3. This last interaction is exclusively used for intramolecular hydrogen bonding. Carbonyl amide groups are also differentially used for hydrogen bonding in oxalamides, NH⋯OC amide dislike interactions are mainly used for intramolecular HB in oxalamides 5–7 and for supramolecular synthons only when also involved in intramolecular three-centred hydrogen bonding. NH⋯OC amide-like interactions are exclusively used for intermolecular hydrogen bonding in compound 5.
This set of compounds appears as centrosymmetric pairs in the 0-D which further develop 1-D as π-stacked columns, tapes, helix or chains. Walls or zig-zagging, rough or weaving sheets give shape to the 2-D. It is worthy to note that 0-D and 1-D arrays are directed by strong amide N–H⋯O self-complementary hydrogen bonding in dioxalamates 2 and 3 whereas the 2-D is structured through weak C–H⋯O interactions. Hydroxyl combined with amide moieties are not enough to develop a strong 3-D in oxalamides 5 and 6, although they show more complex networks in comparison with their parent oxalamates.
Instead of the characteristic R22(10) ring motif frequently observed in oxalamide and oxalamate supramolecular architectures, the R22(16) ring motif is recurrent in oxalamates 2 and 3, and in oxalamide 5. This motif appears associated with pd–π-stacking interactions between the phenyl spacers in oxalamates 2 and 3, but not in 5. This motif is also observed in diamides derived from 2,4,6-trimethyl-benzene-1,3-diamine18 and strongly resembles the supramolecular staircase 1-D motif shown by polymorph 1.
In spite of the presence of sterically demanding groups in 5 and 6, the R22(10) (NH⋯OC) oxalamide supramolecular motif persists, in contrast with CCDB searches which suggest that in most of the cases when this motif was not present this was due to severe derivatization or the presence of sterically demanding groups.31 The intermolecular spacing along the oxalamideR22(10) tape, in 5 and 6, is in the reported range for other oxalamides.7e Among the set of compounds analyzed, compound 5 shows both R22(10) and R22(16) ring motifs, however this last is not associated with π-stacking interactions, mainly because of the length of the oxalamide arm whose involvement in R22(10) hydrogen bonding motif slips the phenyl planes beyond the limits for such interaction. Whereas the 1-D meso-helix arrangement of oxalamide 6 is similar to that reported for polymorph 1′.
On the other hand, the PhNHCO groups are exclusively engaged in intramolecular three-centred hydrogen bonding, in compounds 4 and 7, and thus are not available for intermolecular interactions. Extensive intramolecular hydrogen bonding, especially when at least four adjacent rings of six or five members are formed, avoids the participation of the amide group in intermolecular hydrogen bonding as has also been observed for oxalamides derived from amino acids.7e Such is the case of compounds 4 and 7. However, in contrast with similar bis-aryloxalamides/oxalamates o-substituted with groups capable of HB, which are characterized for their lack of a supramolecular structure,22 compounds 4 and 7 form well defined 2-D crystal networks built by the concurrence of more varied interactions: CO⋯CO, C–H⋯A (AO, π) and π-stacking interactions, additionally strengthened by NH⋯OH, OH⋯OC HB in oxalamide 7. Among them, CO⋯CO interactions play an important role since they are comparable with hydrogen bonding.28
Blocking 4 and 6 positions is required to maintain a reliable out of-the-plane conformation on the transformation from oxalamates to oxalamides. Nevertheless, the twisted conformation is not enough for intermolecular HB as reported in bis-oxalamides. Neither twisted nor planar conformation forms homomeric intermolecular hydrogen bonding, since bis-oxalamides are discrete units in the solid state.13 The increase in the number of moieties per molecule capable of forming non-covalent interactions plays a decisive role. Thus in 1,3-phenyl-dioxalamates/dioxalamides both twisted and planar conformations are capable to develop 2-D networks.
4. Conclusions
Conformational control of the oxalamate/oxalamide cavities was demonstrated through the use of substituents in the C-2 position. The bulky methyl group favours the out-of-plane conformation in compounds 2 and 5. Whereas the participation of the methyl group in C–H⋯OC interactions favors the endo conformation of the phenylamide carbonyl. Further increasing steric strain with methyl groups in C–4 and C–6 permanently fixes the twisted conformation in compounds 3 and 6. Meanwhile the cooperative participation of the bulky OMe group and its hydrogen-bonding accepting nature leads to the exo conformation in compounds 4 and 7. Particularly the role of intramolecular three-centred hydrogen bonding interactions in the stabilization of the exo(ap)–exo(ap) conformation was demonstrated by variable temperature 1H NMR experiments and IR spectra.Intramolecular hydrogen-bonding interactions are determinant in the adopted conformation and exert a strong influence on the nature of non-covalent interactions that rule the organization at the supramolecular level. More than four adjacent S(n) rings (n = 5, 6), in the same molecule, avoid intermolecular hydrogen bonding leading to CO⋯CO, C–H⋯A (AO, π) and π-stacking interactions to direct the crystal organization in compound 4 and 7, strengthened by NH⋯OH, OH⋯OC hydrogen bonding in this last.
The twisted conformation of the oxalamate moiety, in compounds 2 and 3, allowed the formation of R22(16) NH⋯OC motif which appears associated with pd–π-stacking interactions between the phenyl spacers. This motif strongly resembles the reported one for polymorph 1. In contrast, the twisted conformation of the oxalamide moiety, in compound 6, forms the robust oxalamide tapes characterized by the occurrence of the R22(10) NH⋯OC motif, developing meso-helixes in 1-D, in close similarity with the reported structure of polymorph 1′. Oxalamide 5 shows both R22(16) and R22(10) motifs forming rough 2-D sheets.
Supramolecular versatility of phenyl-oxalamate/oxalamide moieties was demonstrated by means of the formation of dimers (0-D), π-stacked columns, tapes, chains, or helixes (1-D) and sheets or walls (2-D). The lengthening of the oxalyl arm in oxalamides in comparison with oxalamates, slips the phenyl rings beyond π-stacking interactions, in compound 5, and enlarges the turn of the helix in compound 6. Dimensionality is not increased on going from oxalamates 2–4 to oxalamides 5–7, just the 2-D is strengthened showing more complex networks.
The conformations of amide/oxalamate/oxalamide derivatives of the parent 2,4,6-trimethyl-benzene-1,3-diamine and 5-tert-butyl-2,6-diamineanisol are reliable, the former is twisted up and down from the phenyl spacer and the latter is planar and exo to the cavity. In contrast, amide/oxalamate/oxalamide derivatives of both 1,3-diaminobenzene and 2-methyl-benzene-1,3-diamine are more versatile, their final conformation will depend on the forces that bind the crystal lattice.
Among the set of studied compounds, oxalamide derivatives of 2,4,6-trimethyl-benzene-1,3-diamine are prone to form 1-D meso-helix supramolecular architectures. The turn of the helix could be modulated by the pendant RNHCO residue.
5. Experimental
5.1 General methods
All chemicals and solvents were of reagent grade and used as received. Melting points were measured on an Electrothermal IA 9100 apparatus and were uncorrected. IR spectra were recorded neat using a Varian 3100 FT-IR with ATR system Excalibur Series spectrophotometer. Mass spectra were obtained in a Bruker Esquire 6000 spectrometer with an electron ionization mode. 1H and 13C NMR spectra were recorded on a Varian Mercury 300 (1H, 300.08; 13C, 75.46 MHz) instrument in CDCl3 solutions (2–4) or DMSO-d6 (5–7), measured with SiMe4 as the internal reference, chemical shifts are in ppm and nJ(H–H) in hertz. Variable temperature experiments were performed in the same apparatus equipped with a temperature controller to keep the temperature constant within 0.3 °C, from 20–120 °C in 10 °C increments with a delay of 5 min for temperature stabilization. Each spectrum was recorded with 16 scans.| Parameter | Compound | |||||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | 7 | |
| CCDC no. | 795133 | 801211/795129 | 795131 | 801212 | 801213 | 801214 |
| Formulae | C15H18N2O6 | C17H22N2O6 | C19H26N2O7 | C15H20N4O6 | C21H32N4O8 | C19H28N4O7 |
| Form. Wt. | 322.31 | 350.37 | 394.42 | 352.35 | 468.5 | 424.5 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
P
|
P
|
C2/c | C2/c |
| a/Å | 8.2302(12) | 8.0352(7) | 9.9640(3) | 9.0080(18) | 11.4567(6) | 13.0660(30) |
| b/Å | 23.218(3) | 16.0173(14) | 10.2180(3) | 9.5510(19) | 12.9050(7) | 24.3840(50) |
| c/Å | 8.1795(12) | 14.3980(14) | 10.5790(4) | 10.5930(19) | 16.4558(10) | 13.3030(30) |
| α/° | 90 | 90 | 80.969(2) | 94.800(20) | 90.000(0) | 90.000(0) |
| β/° | 94.665(2) | 102.681(2) | 87.691(2) | 99.010(30) | 101.828(2) | 90.000(30) |
| γ/° | 90 | 90 | 87.649(2) | 109.340(30) | 90.000(0) | 90.000(0) |
| V/Å3 | 1557.8(4) | 1807.9(3) | 1062.20 | 840.3(3) | 2381.31(10) | 4238.35(16) |
| Z | 4 | 4 | 2 | 2 | 4 | 8 |
| Measured reflections | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 126 126 |
10973 |
11563 |
5214 | 7143 | 11757 |
| Independent reflections | 2745 | 3184 | 3246 | 2956 | 2096 | 3613 |
| GOOF | 1.160 | 1.050 | 1.157 | 0.960 | 0.820 | 1.121 |
| R int | 0.030 | 0.060 | 0.125 | 0.033 | 0.050 | 0.071 |
| R[F2 >2σ(F2)] | 0.054 | 0.052 | 0.082 | 0.080 | 0.068 | 0.055 |
| wR(F2) | 0.125 | 0.103 | 0.215 | 0.243 | 0.170 | 0.186 |
5.2 Synthesis of compounds
Compounds 2 and 3 were prepared as reported.38O). 1H NMR: 8.86 (s, 2H, NH), 7.70 (d, 2H, 3J = 8.2), 7.28 (t, 1H, 3J = 8.2), 4.42 (q, 4H, O–CH2, 3J = 7.0), 2.20 (s, 3H, CH3), 1.42 (t, 6H, CH2–CH3, 3J = 7.0). 13C NMR: 161.1 (C9), 154.5 (C8), 134.9 (C1, C3), 127.4 (C5), 123.0 (C2), 121.4 (C4, C6), 64.1 (C11), 14.2 (C12), 12.3 (Me). ESI MS [M + Na]+ = 344.8 m/z. Anal. Calcd for C15H18N2O6 (%): C 55.90, H 5.63, N 8.69. Found: C 55.58, H 5.75, N 9.09.
O). 1H NMR: 8.47 (s, 2H, NH), 7.00 (s, 1H, Ph), 4.39 (q, 4H, O–CH2, 3J = 7.1), 2.17 (s, 6H, 2CH3), 2.06 (s, 3H, CH3), 1.41 (t, 6H, CH2–CH3, 3J = 7.1). 13C NMR: 160.9 (C9), 155.1 (C8), 135.3 (C1, C3), 132.7 (C4, C6), 130.8 (C5), 130.4 (C2), 63.9 (C11), 18.5 (2Me), 14.3 (Me), 14.1 (C12). ESI MS [M + Na]+ = 372.8 m/z. Anal. Calcd for C17H22N2O6 (%): C 58.28, H 6.33, N 8.00. Found C 58.03, H 6.25, N 10.56.
O). 1H NMR: 9.25 (s, 2H, NH), 8.21 (s, 2H, Ph), 4.40 (q, 4H, O–CH2, 3J = 7.1), 3.81 (s, 3H, O–CH3), 1.38 (t, 6H, –CH2–CH3, 3J = 7.1), 1.28 (s, 9H, 3CH3). 13C NMR: 160.8 (C9), 154.1 (C8), 149.2 (C2), 137.0 (C5), 129.4 (C1, C3), 114.7 (C4, C6), 63.9 (C11), 61.7 (MeO), 35.3 (C(Me)3), 31.3 (3Me), 14.3 (C12). ESI MS [M + Na]+ = 416.9 m/z. Anal. Calcd for C19H26N2O7 (%): C 57.86, H 6.64, N 7.10. Found (%): C 57.80, H 6.60, N 7.44.
O) cm−1. 1H NMR: 10.28 (s, 2H, N7–H), 8.78 (t, 2H, N10–H), 7.18–7.29 (m, 3H, Ph), 4.70 (b, 2H, OH), 3.48 (t, 4H, OCH2), 3.26 (q, 4H, NCH2), 2.01 (s, 3H, Ar–CH3). 13C NMR: 160.2 (C9), 159.4 (C8), 136.4 (C1), 129.2 (C5), 126.3 (C2), 124.3 (C4), 59.8 (C12), 42.5 (C11), 13.4 (Me). ESI MS [M + Na]+ = 374.8 m/z. Anal. Calcd for C15H20N4O6 (%): C 51.13, H 5.72, N 15.90. Found (%): C 51.03, H 6.00, N 15.90.
O). 1H NMR: 10.20 (s, broad 2H, N7–H), 8.73 (t, 2H, N10–H), 7.00 (s, 1H, Ph), 4.20 (broad, 2H, OH), 3.53 (m, 4H, CH2OH), 3.48 (m, 4H, CH2O–CH2), 3.46 (m, 4H, CH2–OCH2), 3.36 (m, 4H, NCH2), 2.09 (s, 6H, 2CH3), 1.91 (s, 3H, CH3). 13C NMR (δ ppm DMSO-d6): 160.6 (C9), 159.5 (C8), 134.5 (C4), 133.4 (C1), 132.9 (C5), 129.4 (C2), 72.7 (C12), 68.9 (C15), 60.8 (C14), 39.7 (C11), 18.5 (2Me), 14.1 (Me). Anal. Calcd for C22H34N4O7 (%): C 53.84, H 6.88, N 11.96. Found (%): C 53.60, H 7.20, N 11.19.
O). 1H NMR: 9.86 (s, 2H, N7–H), 8.91 (t, 2H, N10–H), 7.88 (s, 2H, Ph), 4.83 (t, 2H, OH), 3.76 (s, 3H O–CH3), 3.50 (t, 4H, OCH2), 3.25 (q, 4H, NCH2), 1.24 (s, 9H, 3CH3). 13C NMR: 160.3 (C9), 158.6 (C8), 147.5 (C2), 140.3 (C5), 130.0 (C1, C3), 116.1 (C4, C6), 59.7 (3CH3), 61.7 (O–Me), 42.7 (C–(CH3)3). Anal. Calcd for C19H28N4O7 (%): C 53.76, H 6.65, N 13.20. Found (%): C 53.73, H 6.71, N 12.78.
Acknowledgements
This work was supported by CONACYT grant 83378, SIP-IPN (Secretaría de Investigación y Postgrado del Instituto Politécnico Nacional), CGIC-UC (Coordinación General de Investigación Científica de la Universidad de Colima) and PROMEP-SEP.References
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd., UK, 2nd edn, 2009 Search PubMed.
- (a) T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401 CrossRef CAS; (b) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS; (c) J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- E. T. Kool, Chem. Rev., 1997, 97, 1473 CrossRef CAS.
- T. Bugg, Introduction to Enzyme and Coenzyme Chemistry, Blackwell Publishing Ltd, Oxford, UK, 2004 Search PubMed.
- O. Keskin, A. Gursoy, B. Ma and R. Nussinov, Chem. Rev., 2008, 108, 1225 CrossRef CAS.
- S. D. Sarker and L. Nahar, Chemistry for Pharmacy Students: General, Organic and Natural Product Chemistry, John Wiley & Sons Ltd, London, England, 2007 Search PubMed.
- (a) P. W. Baures, J. R. Rush, A. V. Wiznycia, J. Desper, B. A. Helfrich and A. M. Beatty, Cryst. Growth Des., 2002, 6, 653 Search PubMed; (b) O. Ugono, N. P. Rath and A. M. Beatty, Cryst. Growth Des., 2009, 9, 4595 CrossRef CAS; (c) C. B. Aakeroy, J. Desper and J. F. Urbina, Chem. Commun., 2005, 2820 RSC; (d) C. B. Aakeroy, J. Desper and I. Hussain, Cryst. Growth Des., 2006, 6, 474 CrossRef; (e) T. L. Nguyen, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 2001, 123, 11057 CrossRef CAS.
- J. W. Lauher, ACA Trans., 2004, 39, 31 Search PubMed.
- (a) R. Marx, S. J. Glasera and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o1425 Search PubMed; (b) F. J. Martínez-Martínez, P. Maya-Lugardo, E. V. García-Báez, H. Höpfl, J. Hernández-Díaz and I. I. Padilla-Martínez, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o2994 Search PubMed; (c) S. Coe, J. J. Kane, T. L. Nguyen, L. M. Toledo, E. Wininger, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 1997, 119, 86 CrossRef CAS; (d) T. L. Nguyen, A. Scott, B. Dinkelmeyer, F. W. Fowler and J. W. Lauher, New J. Chem., 1998, 129 RSC.
- (a) M. C. Muñoz, G. Blay, I. Fernández, J. R. Pedro, R. Carrasco, M. Castellano, R. Ruiz-García and J. Cano, CrystEngComm, 2010, 12, 2473 RSC; (b) I. I. Padilla-Martínez, M. Chaparro-Huerta, F. J. Martínez-Martínez, H. Höpfl and V. García-Báez, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o825 CrossRef; (c) G. Blay, I. Fernandez, J. R. Pedro, R. Ruiz-García, M. C. Muñoz, J. Cano and R. Carrasco, Eur. J. Org. Chem., 2003, 1627 CrossRef CAS; (d) G. Francese, A. Neels, H. Stoeckli-Evans and S. Decurtins, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, o1858 Search PubMed.
- A. D. McNaught and A. Wilkinson, IUPAC. Compendium of Chemical Terminology (the “Gold Book”), Blackwell Scientific Publications, Oxford, UK, 2nd edn, 1997 Search PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- B. Piotrkowska, M. Gdaniec, M. J. Milewska and T. Połonski, CrystEngComm, 2007, 9, 868 RSC.
- E. V. Garcia-Baez, C. Z. Gomez-Castro, H. Hopfl, F. J. Martinez-Martinez and I. I. Padilla-Martinez, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, o541 Search PubMed.
- G. Pickaert, M. Cesario and R. Ziessel, J. Org. Chem., 2004, 69, 5335 CrossRef CAS.
- (a) P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405 CrossRef; (b) A. Saeed, S. Hussain and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o521 Search PubMed; (c) C. P. Price, A. L. Grzesiak, J. W. Kampf and A. J. Matzger, Cryst. Growth Des., 2003, 3, 1021 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- H. Allouchi, M. Cotrait and J. Malthěte, Mol. Cryst. Liq. Cryst., 2001, 362, 101 Search PubMed.
- Y.-Y. Zhu, J. Wu, C. Li, J. Zhu, J.-L. Hou, C.-Z. Li, X.-K. Jiang and Z.-T. Li, Cryst. Growth Des., 2007, 7, 1490 CrossRef CAS.
- (a) R. Wieczorek and J. J. Dannenberg, J. Am. Chem. Soc., 2003, 125, 14065 CrossRef CAS; (b) R. Viswanathan, A. Asensio and J. J. Dannenberg, J. Phys. Chem. A, 2004, 108, 9205 CrossRef CAS.
- R. Taylor, O. Kennard and W. Versichel, J. Am. Chem. Soc., 1984, 106, 244 CrossRef CAS.
- F. J. Martínez-Martínez, I. I. Padilla-Martínez, M. A. Brito, E. D. Geniz, R. C. Rojas, J. B. R. Saavedra, H. Höpfl, M. Tlahuextl and R. Contreras, J. Chem. Soc., Perkin Trans. 2, 1998, 401 RSC.
- I. I. Padilla-Martínez, F. J. Martínez-Martínez, C. I. Guillén-Hernández, M. Chaparro-Huerta, L. C. Cabrera-Pérez, C. Z. Gómez-Castro, B. A. López-Romero and E. V. García-Báez, ARKIVOC, 2005, 401 Search PubMed.
- H. O. Desseyn, S. P. Perlepes, K. Clou, N. Blaton, B. J. V.d. Veken, R. Dommisse and P. E. Hansen, J. Phys. Chem. A, 2004, 108, 5175 CrossRef CAS.
- I. I. Padilla-Martínez, F. J. Martínez-Martínez, E. V. García-Báez, J. M. Torres-Valencia, S. Rojas-Lima and H. Höpfl, J. Chem. Soc., Perkin Trans. 2, 2001, 1817 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441 CrossRef CAS.
- M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
- F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320 CrossRef.
- (a) J. H. Williams, Acc. Chem. Res., 1993, 26, 593 CrossRef CAS; (b) I. Dance and M. Scudder, Chem.–Eur. J., 1996, 2, 481 CrossRef CAS.
- M. Hoffman, U. Rychlewska and B. Warżajtis, CrystEngComm, 2005, 7, 260 RSC.
- Bruker, APEX II, SAINT, SADABS and SHELXTL, Bruker AXS Inc., Madison, Winsconsin, USA, 2004 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, Jr, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Peterson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98, Revision A.9, Gaussian, Inc., Pittsburgh, PA 1998 Search PubMed.
- (a) J. B. Wright, C. M. Hall and H. G. Johnson, J. Med. Chem., 1978, 21, 930 Search PubMed; (b) E. Pardo, R. Ruiz-García, F. Lloret, M. Julve, J. Cano, J. Pasan, C. Ruiz-Perez, Y. Filali, L.-M. Chamoreau and Y. Journaux, Inorg. Chem., 2007, 46, 4504 CrossRef CAS.
Footnote |
| † CCDC reference numbers 2795133, 3801211/795129, 4795131, 5801212, 6801213, and 7801214. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce05302g |
| This journal is © The Royal Society of Chemistry 2011 |