Supercritical hydrothermal synthesis of hydrophilic polymer-modified water-dispersible CeO2 nanoparticles†
Minori
Taguchi
*a,
Seiichi
Takami
b,
Tadafumi
Adschiri
c,
Takayuki
Nakane
a,
Koichi
Sato
a and
Takashi
Naka
a
aNational Institute for Materials Science, 1-2-1 Sengen, Tsukuba, 305-0047, Japan. E-mail: taguchi.minori@nims.go.jp
bInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, 980-8577, Japan
cWPI, Advanced Institute for Materials Research, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, 980-8577, Japan
First published on 23rd December 2010
Abstract
We have succeeded in the simple and rapid synthesis of the hydrophilic polymer-modified CeO2 nanoparticles using a supercritical hydrothermal method. To prepare the nanoparticles, Ce(OH)4 as precursor was treated in a batch-type reactor with supercritical water in the presence of either polyvinyl alcohol (PVA) or polyacryl acid (PAA) as surface modifiers. The hydrophilic polymers attached to the surface of the CeO2 nanoparticles by the coordination bond between the functional groups, such as hydroxyl (–OH) or carboxyl (–COOH), of the polymers and the Ce atoms. The amount of the attached polymers on the surface of the CeO2 nanoparticles tended to increase with a decrease in the molecular weight of the polymer. The morphology and the particle size of the nanoparticles were cuboctahedral and about 20 nm, respectively. The nanoparticles were dispersed in water by virtue of the functional groups on the polymers. Notably, the ζ potential of PAA-modified CeO2 nanoparticles did not become zero in the measured pH range between 3 and 11. Interestingly, the surface modification by the polymers controlled the band gap of the nanoparticles, suggesting the possibility of tuning the electronic and the optical properties of the metal oxide nanoparticle by modifying their surface with organic molecules.
Introduction
Organic–inorganic hybrid nanoparticles have attracted great interest.1–14 This is because the incorporation of the inorganic nanoparticles into organic molecules including polymers enables the realization of hybrid nanoparticles with the merits of organic molecules, such as wide tunability, ease of processing, and structural flexibility, coupled with the physical properties of the inorganic nanoparticles such as magnetic, electronic, catalytic, and optical properties. Notably, when polymers are hybridized to inorganic nanoparticles, the properties, such as thermal stability, mechanical strength, transparency, dispersibility, and solubility of the nanoparticles, are tuned by the unique properties of the polymers.7–14 Hydrophilic and/or polyelectrolyte polymer-modified nanoparticles are widely studied to be used for biology and medicine.10–14 Further, functional multilayer thin films and nanoparticles, which enable the application in gas-sensor, permeable membrane, and drug-delivery system, can be prepared using the electrostatic interaction of the polymer in the appropriate solvent.14–16 Thus, polymer-modified nanoparticles enable the design of various functional hybrid nanomaterials, and can be applied to numerous fields. On the other hand, an interesting characteristic on the interface between the organic molecules and the surface of the nanoparticles in the hybrid nanoparticles is often observed.17–19 The properties of the inorganic nanoparticles are controlled by the electrostatic interaction between the organic molecules and the surface of the nanoparticles. Therefore, the investigation of the interface is important for leading to the discovery of novel properties. Hybridization of polymers with inorganic nanoparticles is a critical step in demonstrating these special properties of the polymer-modified nanoparticles. Thus far, several strategies have been developed to synthesize polymer-modified nanoparticles.7–14 However, these processes typically require several experimental steps with long time reaction for the preparation. In most cases, as the first step, surface-modified nanoparticles were prepared, which were then transferred from organic solvent to aqueous solution using a ligand exchange procedure, and finally copolymers were conjugated with various organic molecules. It is therefore essential to develop simple and one-pot methods for synthesizing polymer-modified nanoparticles.To synthesize surface-modified metal oxide nanoparticles, our group has proposed in situ surface modification in a batch-20–22 and a flow-type reactor23,24 under supercritical hydrothermal condition. Supercritical water (SCW) is being increasingly used in materials chemistry, more specifically, nanoparticle synthesis in SCW, because of faster nucleation and crystallization of metal oxides.20–28SCW also possesses a unique characteristic which is larger miscibility with organic ligand molecules. By harnessing these properties, we can prepare various surface-modified metal oxide nanoparticles. Thus far, we have not only synthesized surface-modified metal oxide nanoparticles but also discussed the relationship between the morphology of the nanoparticles and the organic modifier. In particular, we have extensively investigated the relationship between the growth mechanism and the morphology of the CeO2 nanoparticles in the presence of dicarboxylic acids with various chain lengths as modifiers.22 However, polymer-modified metal oxide (CeO2) nanoparticles have not been synthesized using SCW yet. During these studies, CeO2 nanoparticles have been thoroughly investigated as they can be used as three-way catalysts, oxygen ion conductors, polishing agents, gate oxides, and UV-shielding materials.22,23,29–42 Especially, water-dispersible surface-modified CeO2 nanoparticles can be possibly used as cosmetic materials using the UV-shielding property.31
We tried to synthesise hydrophilic polymer-modified CeO2 nanoparticles using SCW. In the present study, Ce(OH)4 as precursor was treated in a batch-type reactor with SCW in the presence of either polyvinyl alcohol (PVA) or polyacryl acid (PAA) as a surface modifier. The experimental results showed that the polymers attached to the surface of the CeO2 nanoparticles through the coordination bond between the functional groups, such as hydroxyl or carboxyl, of the polymers and the Ce atoms. The CeO2 nanoparticles were dispersed in water by virtue of their functional groups. Interestingly, the band gap of the CeO2 nanoparticles was controlled by the polymers through the surface modification.
Experimental section
Synthesis of surface-modified CeO2 nanoparticles
Ce(OH)4 was purchased from Aldrich. Polyvinyl alcohol (PVA500: Mw 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and PVA1000: Mw 44000) and polyacryl acid (PAA: Mw 5000) were purchased from Wako Chemicals.
000 and PVA1000: Mw 44000) and polyacryl acid (PAA: Mw 5000) were purchased from Wako Chemicals.
Each polymer was dissolved in distilled water (1.0 wt%). Ce(OH)4 (0.25 mmol) and each polymer solution (2.5 mL) was transferred to a pressure-resistant Hastelloy C vessel (inner volume: 5.0 mL). A hydrothermal reaction was performed using an electric furnace at 400 °C and 38 MPa for 10 min. The reaction was terminated by submerging the vessel in a water bath at room temperature. After the reaction, unreacted polymers were removed by a combination of repeated centrifugation and decantation, alternately with water and methanol. Finally, the products were dried in air. Hereafter, these products are designated as 1 (PVA500-modified CeO2), 2 (PVA1000-modified CeO2), and 3 (PAA-modified CeO2). Unmodified nanoparticles were also prepared under the same conditions (at 400 °C and 38 MPa for 10 min) without adding the polymers (product 4).
Physical methods
X-Ray diffraction (XRD) patterns were recorded using a RINT-2000 spectrometer (Rigaku) with Cu Kα (λ = 1.542 Å) radiation. According to Scherrer equation, the particle (crystalline) sizes of the products were determined from the full width at half maximum (FWHM) of the XRD patterns, and a shape factor of 0.9. A transmission electron microscope (TEM, JEM-1200EX, JEOL) was used to obtain the magnified image of the products. The particle sizes of the products were evaluated from the TEM images. To estimate the particle size, we used eqn (1) and 2 as follows,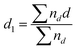 | (1) |
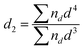 | (2) |
Fourier transform infrared (FT-IR) spectra were recorded using a FT/IR-680 Plus (JASCO) with KBr pellets. The surface state of the products was investigated by the FT-IR measurement to elucidate the surface chemical character of the CeO2 nanoparticles and the attaching polymers. Thermogravimetric analysis (TGA) was performed at a ramp rate of 10 °C min−1 under an argon atmosphere using a DTG-60H (Rigaku). The amount of the polymers in the products was measured by the TGA. We estimated the amount of the polymers attached to the surface of the CeO2 nanoparticles, and that attached to one CeO2 nanoparticle based on the estimated particle size (DTEM) and the density of bulk CeO2 (7.172 g cm−3) using eqn (3)–(5). It is assumed that the entire surface of a particle is modified with the polymers, and the morphology of the particle is spherical.
| N × (4πr3/3) = (m × (100 − a)/100)/7.172 | (3) |
| S = N × 4πr2 | (4) |
| MN = (m × a/100)/Mw × NA | (5) |
Here, N (number) and S (nm2) are the total number and the surface area of the product, respectively. r, m, and a are the radius (nm), the starting weight (g), and the weight loss of the product (%) of TGA measurement, respectively. MN and MW are the total number and the molecular weight of the polymer (modifier), respectively, and NA is Avogadro's number.
ζ Potential analysis was used to measure the surface charge of the polymer-modified CeO2 nanoparticles as a function of pH in water and to determine optimum dispersion conditions. The ζ potentials were measured with a Zetasizer system (MPT-2, Malvern Instruments). The pH of suspensions was adjusted to values ranging from 3 to 11 using appropriate amounts of HCl and KOH. To examine the optical property of the polymer-modified CeO2 nanoparticles, UV-visible absorption spectra of the products dispersed in water were measured using a V-570 spectrophotometer (JASCO). The optical absorption coefficient α was calculated from the obtained spectra in order to determine the band gap energies in the nanoparticles.
| α = (2.303 × 103Aρ)/lc | (6) |
Here, A is the absorbance of the product, ρ is the density of bulk CeO2 (7.172 g cm−3), l is the path length of the quartz cell (1 cm), and c is the concentration of the product in suspensions.32–34
Results and discussion
(1) Structural analysis
The crystal structure of the obtained products was examined by XRD. Fig. 1 shows the XRD patterns of the products. The XRD results indicate that the products were well crystallized and have a cubic structure (space groupFm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) known for bulk CeO2 (JCPDS card no. 43-1002). The lattice constants of the products were larger than that of the bulk, which is possibly due to the oxygen vacancy and/or the valence change of the Ce ion in the products.34–37 The calculated lattice constants and the crystalline (particle) sizes from the Scherrer equation are summarized in Table 1.
m) known for bulk CeO2 (JCPDS card no. 43-1002). The lattice constants of the products were larger than that of the bulk, which is possibly due to the oxygen vacancy and/or the valence change of the Ce ion in the products.34–37 The calculated lattice constants and the crystalline (particle) sizes from the Scherrer equation are summarized in Table 1.
| Products | a/nm | D XRD /nm | D TEM /nm | Morphology | {111}/{200} intensity ratio |
|---|---|---|---|---|---|
| PVA500-CeO2 | 0.5421 | 21.6 ± 1.25 | 17.4 | Cuboctahedral | 3.07 |
| PVA1000-CeO2 | 0.5421 | 21.4 ± 1.37 | 17.9 | Cuboctahedral | 3.11 |
| PAA-CeO2 | 0.5418 | 20.9 ± 1.20 | 17.4 | Cuboctahedral | 3.05 |
| Unmodified CeO2 | 0.5414 | 17.8 ± 1.54 | 15.0 | Truncated octahedral | 3.43 |
| JCPDS43-1002 | 0.5411 | — | — | — | 3.70 |
 | ||
| Fig. 1 XRD patterns of prepared CeO2 nanoparticles: (a) PVA500-modified; (b) PVA1000-modified; (c) PAA-modified; and (d) unmodified. | ||
The detailed morphology of the products was investigated by TEM (Fig. 2). Products 1–3 had a hexagonal shape and therefore were considered cuboctahedral.22,39,40,43 The morphology of product 4 was a truncated octahedral. This result suggests that the presence of polymers during the synthesis of CeO2 influenced the morphology of the CeO2 nanoparticles. The volume weighted averages of the particle diameter for the products were evaluated from the TEM images, whose values were calculated using eqn (1) and 2 in the physical methods. Because there was a distribution in the particle diameter of the products, the volume weighted averages were calculated. These values are shown in Table 1. The arithmetic mean particle diameters and the size distributions of the products are shown in Fig. S1†. The observed particle sizes are close to those determined from the XRD patterns.
 | ||
| Fig. 2 TEM images of prepared CeO2 nanoparticles: (a) PVA500-modified; (b) PVA1000-modified; (c) PAA-modified; and (d) unmodified; scale bar = 20 nm. | ||
(2) Analysis of interfacial properties
 | ||
| Fig. 3 FT-IR spectra of prepared CeO2 nanoparticles: (a) PVA500-modified; (b) PVA1000-modified; (c) PAA-modified; and (d) unmodified. | ||
In product 1, the broad and strong band at 3399 cm−1 is assigned to the stretching vibration of the O–H group in PVA500.44–47 The weak peak at 2961 cm−1 is assigned to the asymmetric (νas) stretching vibration of the –CH3group, and the peaks at 2926 and 2856 cm−1 are assigned to the asymmetric (νas) and symmetric (νs) stretching vibrations of the –CH2– group in PVA500, respectively.44–47 In product 2, these peaks were also observed, but they appeared at 3388 (ν O–H), 2958 (νas –CH3), 2926 (νas –CH2–), and 2857 (νs –CH2–) cm−1 in PVA1000. In a study on the interaction between the hydroxyl (O–H) part in PVA and metal ions, the frequency of O–H stretching shifted towards a lower wavenumber when there are interactions such as coordination bonds between the O–H group and the metal ions.46 In the present study, the stretching vibration of the O–H group of original PVA500 and PVA1000 was shown at 3445 and 3419 cm−1, respectively (Fig. S2†). That is, the PVA attaches with the surface of the CeO2 nanoparticles and becomes a capping agent for the nanoparticles. Further, since the alcohol group is a Lewis base ligand it can be used as the capping agent for metal oxide nanoparticles.48 Although the amount of the O–H groups in the PVA coordinated with the Ce atoms on the surface of the CeO2 nanoparticles cannot be estimated, it is reasonable to believe that the O–H group in PVA attaches to the Ce atoms through the coordination bond in products 1 and 2.
In product 3, the weak peak at 2954 cm−1 is assigned to the asymmetric (νas) stretching vibration of the –CH3group, and the peaks at 2926, 2857 and 1451 cm−1 are assigned to the asymmetric stretching (νas), symmetric stretching (νs), and scissoring bending (δsc) vibrations of the –CH2– in PAA, respectively.44 The weak stretching mode of the –COOH group was observed at 1709 cm−1, thus suggesting the presence of free carboxyl groups in PAA on the surface of the CeO2 nanoparticles.22,44 The strong peaks were observed at 1547 and 1413 cm−1, which are assigned to the asymmetric (νas) and the symmetric (νs) stretching vibration of the –COO−group, respectively.44 These absorption bands indicated that the carboxyl group in PAA is modified to the surface of the CeO2 nanoparticles through the bidentate coordination bonding.10,22,49
Based on these observations, we concluded that the polymers are modified to the surface of the CeO2 nanoparticles through the coordination bond between the functional groups, such as hydroxyl (–OH) or carboxyl (–COOH), of the polymers and the Ce atoms, although FT-IR spectra could not provide quantitative information on its amount on the surface of the nanoparticles.
| Products | Molecular weight (ca.) | Weight loss (%) from TGA | Coverage of polymer/molecule nm−2 | Calculated number of polymers that attached to one CeO2 nanoparticle |
|---|---|---|---|---|
| PVA500-CeO2 | 22000 |
1.69 | 0.008 | 5.47 |
| PVA1000-CeO2 | 44000 |
2.09 | 0.005 | 3.14 |
| PAA-CeO2 | 5000 | 7.55 | 0.151 | 105 |
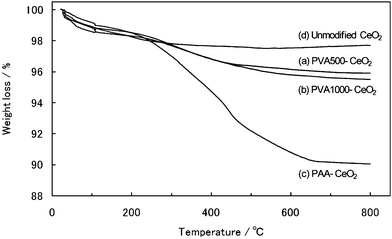 | ||
| Fig. 4 TGA curves of prepared CeO2 nanoparticles: (a) PVA500-modified; (b) PVA1000-modified; (c) PAA-modified; and (d) unmodified. The measurements were conducted at a ramp rate of 10 °C min−1 under an argon atmosphere. | ||
 | ||
| Fig. 5 ζ Potentials of prepared CeO2 nanoparticles: (■) PVA500-modified; (□) original PVA500; (▲) PVA1000-modified; (△) original PVA1000; (●) PAA-modified; (○) original PAA; and (◆) unmodified at various pH values. | ||
(3) Control in the morphology by polymer hybridization
To investigate the growth mechanism of the CeO2 nanoparticles, we have focused on the morphologies of respective products using the TEM images. The morphology in CeO2 depends on the growth ratio of the crystalline planes, which can be estimated from the XRD peak intensity.22,34,39To confirm the morphology from the TEM images, we simply calculated an intensity ratio between the {111} plane and the {200} plane of the XRD pattern (Table 1). In products 1–3, the intensity ratio of the {111} plane to the {200} plane decreased compared to that in product 4 (unmodified). That is, the occurrence ratio of the {111} plane was larger than that of the {200} plane in product 4. As a result, the morphology of unmodified CeO2 nanoparticles was determined to be a truncated octahedral. On the other hand, the morphologies of products 1–3 were identified as a cuboctahedral, which was confirmed by the decreased growth rate of the {200} plane. It is postulated that the growth rate of the {200} plane decreased due to the surface modification with the polymers. Therefore, the polymers might be preferably bound on to the {200} plane of the CeO2 nanoparticles to reduce the growth rate of the {200} plane. Tasker investigated the stability of ionic crystals.53 In the ionic crystals of type MX2 with fluorite structure, the surface energy of the (100) plane is larger than that of the (111) plane in which M and X are metal ion and oxide, respectively. This is true of CeO2 with fluorite structure. The dipole moment in the (100) plane arises toward perpendicular to the plane. Therefore, there may be a strong interaction between the functional groups, such as –OH (–O−) or –COOH (–COO−), and the Ce ion of the {200} plane in the CeO2 nanoparticles. Fig. 6 shows a schematic illustration of the mechanism of the crystal growth of the polymer-modified CeO2 nanoparticles.
 | ||
| Fig. 6 Schematic illustration of the controlled morphology of the CeO2 nanoparticles: (a) Truncated octahedral in the case when the CeO2 nanoparticles were prepared without adding polymers. The morphology is truncated octahedral, because the growth rate of the {200} plane is faster than that of the {111} plane. (b) Polymers as modifier randomly attached to the surface of the CeO2 nanoparticles as there were many reaction parts (functional groups) in the polymers. Since the polymers attached preferentially to the {200} plane of the CeO2 nanoparticles, growth of the {200} planes was slower than in the case of (a). Then, cuboctahedral CeO2 nanoparticles were obtained. | ||
(4) Optical property
UV-visible absorption spectra of the products dispersed in water were shown in Fig. 7a. All products exhibited an absorption edge around 370 nm, which is characteristic of the band gap of the CeO2 nanoparticles. We determined the band gap energies in the polymer-modified CeO2 nanoparticles from the optical absorption coefficient α which was calculated using eqn (6) in the physical methods. To estimate the direct (Ed) and indirect (Ei) band gap energies of the products, we calculated (αhν)2versusphoton energy and α1/2versusphoton energy for the products, respectively. From the intersection of the extrapolated linear portion, the Ed of products 1–4 was determined as 3.61, 3.62, 3.54, and 3.52 eV, respectively (Fig. 7b). The Ei of products 1–4 was determined as 2.99, 3.01, 2.82, and 2.83 eV, respectively (Fig. 7c). The Ed of the products increases compared with that of bulk CeO2 (3.19 eV for Ed and 3.01 eV for Ei).54 However, the Ed to the particle size in the products was still larger than those indicated in other reports.29,32,34,38Fig. 7d is the band gap energies versus the particle size (DTEM) estimated from TEM images for the products, wherein some data points taken from the reference were also included.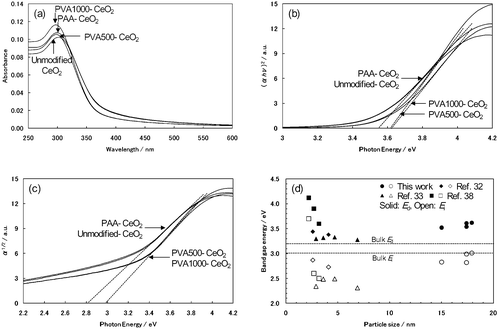 | ||
| Fig. 7 (a) UV-visible absorption spectra of prepared CeO2 nanoparticles in water at room temperature. (b) Plot of (αhν)2vs.Ephoton for prepared CeO2 nanoparticles. (c) Plot of α1/2vs.Ephoton for prepared CeO2 nanoparticles. (d) Plot of band gap energy vs. particle size (DTEM): (●, ○) this work; (◆, ◇) ref. 32; (▲, △) ref. 33; and (■, □) ref. 38. | ||
In general, the UV absorption in CeO2 is brought about by a charge-transfer (CT) transition between O 2p and Ce 4f bands.38 Tsunekawa et al. indicated the increase in the band gap for CeO2−x nanoparticles.38,41,42 According to them, the blue shift of the direct band gap (Ed) in the nanoparticles is due to the valence change of the Ce ion rather than the quantum size effect. The valence change of the Ce ion influences the CT transition between O 2p and Ce 4f bands because the electronic state of the Ce 4f (conduction band) changes with the valence change. The valence of the Ce ion tends to change from 4+ to 3+ with a decrease in the particle size. Thus, the CT transition depends on the particle size. That is, although there is a relationship between the blue shift of the Ed and the particle size, the blue shift of the Ed in the nanoparticles is induced by the valence change of the Ce ion due to a decrease in the particle size. They have also shown a relationship between the Ed and the particle size. The blue shift of the Ed is inversely proportional to the 2.2 power of the particle size.38
To investigate the cause for the increase in the Ed of our products, we focused on the lattice constants of the products. The lattice constants of all products were larger than that of bulk CeO2, which shows the oxygen vacancy and/or the valence change of Ce ion.34–37 When the lattice constant of the products increased, the Ed of the products also tended to expand. This result is consistent with other reports.33,34,38 It was suggested that the blue shift of the Ed in the products is also due to the valence change of the Ce ion. These results were verified compared with other reports.32,33,38Fig. 8a plots the lattice constant against DTEM for the products. Further, based on Tsunekawa's method,38 a log–log plot of ΔEdvs.DTEM for the products was calculated in order to confirm the relationship between the blue shift of the Ed and the particle size (Fig. 8b). Herein, ΔEd is the shift of Ed obtained by the extrapolation in Fig. 7b from the value for the direct transition of bulk CeO2 (3.19 eV). Although the Ed tends to expand with an increase in the lattice constant, the distinctive relationship between the blue shift of the Ed and the particle size like other reports cannot be found in our products from Fig. 8b. Therefore, there might be also other reasons for an increase in the Ed of our products besides the particle size.
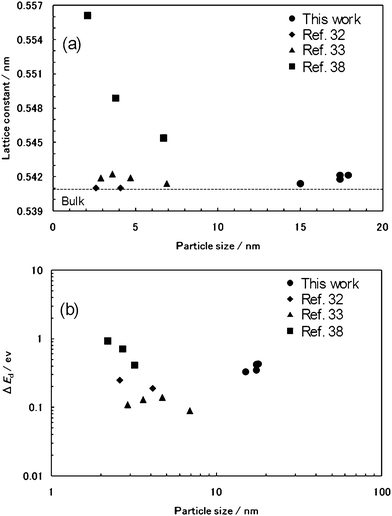 | ||
| Fig. 8 (a) Plot of the lattice constant vs.DTEM. (b) log–log plot of ΔEdvs.DTEM, where ΔEd is the shift in direct band gap from that of bulk crystal: (●) this work; (◆) ref. 32; (▲) ref. 33; and (■) ref. 38. | ||
Zhang et al. noted that the band gap energy in CeO2 nanocrystal changed by the surface condition of the nanocrystal.33 The properties of the inorganic nanoparticles were changed by the electrostatic interaction on the interface between the organic molecules and the surface of the nanoparticles.17–19 In the present study, there might be a strong interaction between the functional groups and the Ce ion of the {200} plane in the CeO2 nanoparticles. The surface modification of the CeO2 nanoparticles might lead to the change in the surface electronic state of the nanoparticles, which caused the blue shift of the Ed. Therefore, the expanded Ed in the products was possibly induced by the electrostatic interaction between the polymer as surface modifier and surface of the CeO2 nanoparticles. In addition, we considered the band gap among products 1–3. The Ed and Ei of products 3 (PAA-modified) were slightly different from those of products 1 and 2 (PVA-modified), despite their particle sizes being similar. This result suggests that the band gap depends on the surface modifier and/or condition. Although the indirect band gap of the CeO2 nanoparticle has not been discussed in detail, the surface condition of the CeO2 nanoparticle should relate to the indirect band gap. These results suggested that the band gap in the CeO2 nanoparticle can be controlled by the functional groups of the surface modifier. On the other hand, the blue shift of the band gap in the products may be derived from the preparation condition or method. That is, the valence of the Ce ion of CeO2 nanoparticles synthesized using SCW differs from that of those synthesized by other reports.
Conclusion
We have succeeded in simply, easily, and rapidly synthesizing hydrophilic polymer-modified CeO2 nanoparticles in a batch-type reactor using SCW. The polymers attached to the surface of the CeO2 nanoparticles through the coordination bond between the functional groups of the hydrophilic polymers and the Ce atoms. A polymer with a small molecular weight was more densely modified to the surface of the CeO2 nanoparticles because of the steric hindrances and the repulsive forces among the hydrophilic parts in the polymers. Further, the presence of polymers during the synthesis of CeO2 nanoparticles influenced the morphology of the CeO2 nanoparticles. The synthesized polymer-modified CeO2 nanoparticles were dispersed in water by virtue of their surface functional groups. In particular, PAA-modified CeO2 nanoparticles (product 3) exhibited negative ζ potentials in the measured pH range between 3 and 11 because of the carboxyl group. Interestingly, the band gaps in the CeO2 nanoparticles were controlled by the attaching polymers on their surface. This phenomenon suggests that the improved properties of CeO2 besides its original properties promise novel applications. These polymer-modified CeO2 nanoparticles enable the design of various functional hybrid nanomaterials since the nanoparticles are water-dispersible and the surface is negatively charged due to the functional groups on the surface of the nanoparticles. Our study offers a simple, rapid, and green chemistry approach for preparing polymer-modified CeO2 nanoparticles using SCW as a reaction medium. Because the procedure is convenient and the chemicals used in this work are readily available, this method is expected to be applicable to various metal oxide nanoparticles.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (S) (KAKENHI) no. 20226015.Reference
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- T. Hyeon, Chem. Commun., 2003, 927–934 RSC.
- C. Sanchez, B. Lebeau, F. Chaput and J. P. Boilot, Adv. Mater., 2003, 15, 1969–1994 CrossRef CAS.
- Z. Chen, H. Chen, H. Hu, M. Yu, F. Li, Q. Zhang, Z. Zhou, T. Yi and C. Huang, J. Am. Chem. Soc., 2008, 130, 3023–3029 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS.
- M. Kasture, S. Singh, P. Patel, P. A. Joy, A. A. Prabhune, C. V. Ramana and B. L. V. Prasad, Langmuir, 2007, 23, 11409–11412 CrossRef CAS.
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107–1110 CrossRef CAS.
- Y. T. Vu and J. E. Mark, Colloid Polym. Sci., 2004, 282, 613–619 CrossRef CAS.
- M. A. Lee, B. J. Park, I.-J. Chin and H. J. Choi, J. Electroceram., 2009, 23, 474–477 CrossRef CAS.
- T. Zhang, J. Ge, Y. Hu and Y. Yin, Nano Lett., 2007, 7, 3203–3207 CrossRef CAS.
- S. S. Banerjee and D.-H. Chen, Chem. Mater., 2007, 19, 6345–6349 CrossRef CAS.
- A. Shkilnyy, M. Soucé, P. Dubois, F. Warmont, M.-L. Saboungi and I. Chourpa, Analyst, 2009, 134, 1868–1872 RSC.
- M. D. Rowe, C.-C. Chang, D. H. Thamm, S. L. Kraft, J. F. Harmon, Jr, A. P. Vogt, B. S. Sumerlin and S. G. Boyes, Langmuir, 2009, 25, 9487–9499 CrossRef CAS.
- S. Chen, Y. Li, C. Guo, J. Wang, J. Ma, X. Liang, L.-R. Yang and H.-Z. Liu, Langmuir, 2007, 23, 12669–12676 CrossRef CAS.
- P. Bertrand, A. Jonas, A. Laschewsky and R. Legras, Macromol. Rapid Commun., 2000, 21, 319–348 CrossRef CAS.
- P. T. Hammond, Adv. Funct. Mater., 2004, 16, 1271–1293 CAS.
- R. Mikami, M. Taguchi, K. Yamada, K. Szuki, O. Sato and Y. Einaga, Angew. Chem., Int. Ed., 2004, 43, 6135–6139 CrossRef CAS.
- M. Taguchi, K. Yamada, K. Szuki, O. Sato and Y. Einaga, Chem. Mater., 2005, 17, 4554–4559 CrossRef.
- M. Suda, N. Kameyama, M. Suzuki, N. Kawamura and Y. Einaga, Angew. Chem., Int. Ed., 2007, 47, 160–163.
- D. Rangappa, T. Naka, A. Kondo, M. Ishii, T. Kobayashi and T. Adschiri, J. Am. Chem. Soc., 2007, 129, 11061–11066 CrossRef CAS.
- J. Zhang, S. Ohara, M. Umetsu, T. Naka, Y. Hatakeyama and T. Adschiri, Adv. Mater., 2007, 19, 203–206 CrossRef CAS.
- M. Taguchi, S. Takami, T. Naka and T. Adschiri, Cryst. Growth Des., 2009, 9, 5297–5303 CrossRef CAS.
- S. Takami, S. Ohara, T. Adschiri, Y. Wakayama and T. Chikyow, Dalton Trans., 2008, 5442–5446 RSC.
- D. Rangappa, S. Ohara, M. Umetsu, T. Naka and T. Adschiri, J. Supercrit. Fluids, 2008, 44, 441–445 CrossRef CAS.
- Y. G. Gogotsi and M. Yoshimura, Nature, 1994, 367, 628–630 CrossRef CAS.
- J. A. Darr and M. Poliakoff, Chem. Rev., 1999, 99, 495–541 CrossRef CAS.
- Y. Arai, T. Sako and Y. Takebayashi, Supercritical Fluids, Springer, Berlin, 2001 Search PubMed.
- Y.-P. Sun, Supercritical Fluid Technology in Materials Science and Engineering, Marcel Dekker, New York, 2002 Search PubMed.
- C. Ho, J. C. Yu, T. Kwong, A. C. Mak and S. Lai, Chem. Mater., 2005, 17, 4514–4522 CrossRef CAS.
- D. Zhang, H. Fu, L. Shi, C. Pan, Q. Li, Y. Chu and W. Yu, Inorg. Chem., 2007, 46, 2446–2451 CrossRef CAS.
- Y. Minamidate, S. Yin and T. Sato, Mater. Chem. Phys., 2010, 123, 516–520 CrossRef CAS.
- T. Masui, K. Fujiwara, K. Machida, G. Adachi, T. Sakata and H. Mori, Chem. Mater., 1997, 9, 2197–2204 CrossRef CAS.
- Y.-W. Zhang, R. Si, C.-S. Liao, C.-H. Yan, C.-X. Xiao and Y. Kou, J. Phys. Chem. B, 2003, 107, 10159–10167 CrossRef CAS.
- H.-I. Chen and H.-Y. Chang, Ceram. Int., 2005, 31, 795–802 CrossRef CAS.
- X.-D. Zhou and W. Huebner, Appl. Phys. Lett., 2001, 79, 3512–3514 CrossRef CAS.
- M. Leoni, R. D. Maggio, S. Polizzi and P. Scardi, J. Am. Ceram. Soc., 2004, 87, 1133–1140 CAS.
- S. Tsunekawa, R. Sahara, Y. Kawazoe and K. Ishikawa, Appl. Surf. Sci., 1999, 152, 53–56 CrossRef CAS.
- S. Tsunekawa, T. Fukuda and A. Kasuya, J. Appl. Phys., 2000, 87, 1318–1321 CrossRef CAS.
- N. B. Kirk and J. V. Wood, J. Mater. Sci., 1995, 30, 2171–2175 CrossRef CAS.
- Z. L. Wang and X. Feng, J. Phys. Chem. B, 2003, 107, 13563–13566 CrossRef CAS.
- S. Tsunekawa, J. T. Wang, Y. Kawazoe and A. Kasuya, J. Appl. Phys., 2003, 94, 3654–3656 CrossRef CAS.
- S. Tsunekawa, J.-T. Wang and Y. Kawazoe, J. Alloys Compd., 2006, 408, 1145–1148 CrossRef.
- Z. L. Wang, J. Phys. Chem. B, 2000, 104, 1153–1175 CrossRef CAS.
- K. Nakanishi, Infrared Absorption Spectroscopy: Practical, Holden-Day, San Francisco, 1962 Search PubMed.
- A. Tawansi, A. H. Oraby, H. M. Zidan and M. E. Dorgham, Physica B (Amsterdam), 1998, 254, 126–133 CrossRef CAS.
- G. N. H. Kumar, J. L. Rao, N. O. Gopal, K. V. Narasimhulu, R. P. S. Chakradhar and A. V. Rajulu, Polymer, 2004, 45, 5407–5415 CrossRef CAS.
- I. S. Elashmawi, N. A. Hakeem and M. S. Selim, Mater. Chem. Phys., 2009, 115, 132–135 CrossRef CAS.
- A. K. Boal, K. Das, M. Gray and V. M. Rotello, Chem. Mater., 2002, 14, 2628–2636 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 1997 Search PubMed.
- L. Shen, P. E. Laibinis and T. A. Hatton, Langmuir, 1999, 15, 447–453 CrossRef CAS.
- L. F. Hakim, J. H. Blackson and A. W. Weimer, Chem. Eng. Sci., 2007, 62, 6199–6211 CrossRef CAS.
- S. L. Kettlewell, A. Schmid, S. Fujii, D. Dupin and S. P. Armes, Langmuir, 2007, 23, 11381–11386 CrossRef CAS.
- P. W. Tasker, J. Phys. C: Solid State Phys., 1979, 12, 4977–4984 CrossRef CAS.
- Z. C. Orel and B. Orel, Phys. Status Solidi B, 1994, 186, K33–K37.
Footnote |
| † Electronic supplementary information (ESI) available: The arithmetic mean particle diameters and the size distributions from TEM images of the surface-modified CeO2 nanoparticles, FT-IR spectra and TGA curves of original hydrophilic polymers, and photographs showing the surface-modified CeO2 nanoparticles dispersed in water. See DOI: 10.1039/c0ce00467g |
| This journal is © The Royal Society of Chemistry 2011 |