Dendrimers and miktoarm polymers based multivalent nanocarriers for efficient and targeted drug delivery
Ghareb M.
Soliman†
abc,
Anjali
Sharma†
a,
Dusica
Maysinger
*b and
Ashok
Kakkar
*a
aDepartment of Chemistry, McGill University, 801 Sherbrooke St. West, Montreal, Quebec, H3A 2K6, Canada. E-mail: ashok.kakkar@mcgill.ca; Fax: +1 514-398-3797; Tel: +1 514-398-6912
bDepartment of Pharmacology and Therapeutics, McGill University, 3655 Promenade Sir-William-Osler, Montreal, Quebec, H3G 1Y6, Canada. E-mail: dusica.maysinger@mcgill.ca; Fax: +1 514-398-6690; Tel: +1 514-398-1264
cDepartment of Pharmaceutics, Faculty of Pharmacy, Assiut University, Assiut, Egypt
First published on 9th June 2011
Abstract
The delivery of biologically active agents to the desired site in the body and intracellular organelles is still a big challenge despite efforts made for more than five decades. With the elaboration of synthetic methodologies to branched and hyperbranched macromolecules such as miktoarm stars and dendrimers, the focus has shifted to nanocarriers able to release and direct drug molecules to a desired location in a controlled manner. We present here recent developments in the field of targeted drug delivery with a focus on two specific macromolecular nanocarriers, dendrimers and miktoarm stars, and provide examples of these nanocarriers tested in different biological systems. A particular attraction of miktoarm stars is their versatility in achieving superior drug loading within their self-assembled structures. Advantages of dendrimers over linear polymers are that the former provide a platform for development of multivalent and multifunctional nanoconjugates, in addition to their ability to accommodate a large number of molecules inside, or at their surfaces.
 Ghareb M. Soliman | Ghareb Soliman received his BSc in Pharmaceutical Sciences and his MSc in Pharmaceutics from Assiut University, Egypt. In 2010, he obtained his PhD in Pharmaceutical Sciences from the Faculty of Pharmacy, University of Montreal, Canada under the supervision of Prof. Françoise Winnik. He subsequently joined the groups of Professors Dusica Maysinger and Ashok Kakkar in the Departments of Pharmacology and Therapeutics, and Chemistry, McGill University, Montreal, Canada, as a postdoctoral fellow, working on the rational development of optimized nano-delivery systems that enhance the efficacy and minimize toxicity of selected neuroprotective drugs. His current research interests focus on designing multifunctional dendrimers and miktoarm polymers for efficient and targeted drug and gene delivery. |
 Anjali Sharma | Anjali Sharma received her MSc in Applied Chemistry and Pharmaceutical Sciences in 2003 from Guru Nanak Dev University, India. From 2003–2008, she worked as a Research Associate in new drug discovery units of leading pharmaceutical companies (Dr. Reddy’s Laboratories and Panacea Biotec Ltd.) in India. In 2009, she joined McGill University, Montreal, Canada, as a PhD candidate under the supervision of Dr Ashok Kakkar. Her research work includes design and synthesis of multifunctional dendrimers and miktoarm polymers using Cu(I) catalyzed alkyne–azide click chemistry for application in drug delivery. |
 Dusica Maysinger | Dr Dusica Maysinger earned her PhD at the University of Southern California, Los Angeles. She did her postdoctoral training at Max Planck Institute, Karolinska Institute and Oxford University. Her research activities focus on investigating mechanisms of drug actions in diabetes and neurodegenerative disorders. She employs real time imaging of fluorescent nanostructures (e.g. quantum dots, gold nanoparticles, fluorescent dendrimers and miktoarms) in living cells and whole animals in order to understand the nature of cell–nanomaterial interactions and how they activate signal transduction pathways in cells. She has published more than 150 papers in scientific journals. |
 Ashok Kakkar | Dr Ashok Kakkar is an Associate Professor in the Department of Chemistry at McGill University, Montreal, Canada. He obtained training in Chemistry under the directions of Professor Todd B. Marder (PhD University of Waterloo), Professor The Lord Lewis (NSERC Post-doctoral Fellow, University of Cambridge) and Professor Tobin Marks (NSERC Post-doctoral Fellow, Northwestern University). His research interests include developing methodologies to complex architectures (hyperbranched macromolecules including dendrimers, miktoarm polymers, metal nanoparticles), and studying their efficacy in drug delivery. |
1. Introduction
Some of the current challenges facing theranostics (i.e., combining therapy and diagnostics) include preparing drug carriers which are biologically inert, internalise easily into the cells and remain intact until they reach their intended target with high specificity.1,2 Premature drug release and the inability to monitor the fate of drug carriers in vivo are key issues that need to be addressed in developing drug delivery nanotechnology.3,4 Nanocarriers based on biocompatible and/or biodegradable polymers have shown tremendous potential in addressing these unmet needs, and a significant effort has been devoted in the past to incorporate therapeutic agents into such polymeric nanoparticles using hydrophobic and/or electrostatic interactions.5–10 Some of the nanoparticles based on linear block copolymers have advanced well beyond experimental/conceptual phases and many are now in clinical trials, especially for cancer chemotherapy.7,11,12 However, there are still several practical aspects of this technology that need to be addressed before any clinical implementation. For instance, micelles of linear block copolymers have low drug loading efficacy (ca. 5 weight%), insufficient stability in vitro and in vivo and rapid drug release in vivo.13–15 In addition, it is becoming increasingly evident that multitasking using a single particle is essential to enhance their efficacy, a feature that is lacking in linear block copolymers. In this regard dendrimers and miktoarm polymers provide a highly useful platform to achieve these goals.16Branched and hyperbranched macromolecules such as miktoarm stars and dendrimers17–22 offer distinct structural features, but share a common thread in which arms emanate from a central core (Fig. 1). Dendrimers are hyperbranched and globular macromolecules with a well-defined core, backbone and multivalent periphery.19,21 The synthetic methodology to construct dendrimers was introduced by Vögtle and coworkers in 1978.23 However, the first series of dendrimers prepared using a well established method appeared in 1985 with the introduction of poly(amidoamine) (PAMAM) dendrimers by Tomalia et al.24 There has been considerable effort in the recent past that has led to synthetic elaboration of dendrimers, and numerous elegant methodologies now exist to construct these hyperbranched macromolecules with a tailor made architecture.25,26 Miktoarm polymers that are sometimes also referred to as asymmetric polymers or heteroarm polymers are star-shaped macromolecules in which a variety of polymeric arms varying in chemical identity and/or molecular weight branch out from the core.27
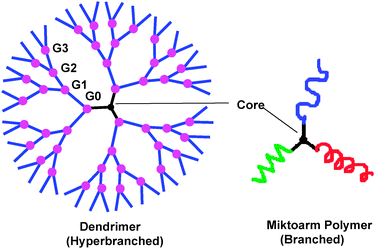 | ||
| Fig. 1 Dendrimer and miktoarm polymer architecture. | ||
The intense interest of the scientific community in exploring the potential of dendrimers and miktoarm polymers for therapeutics delivery has been due to their versatility in structure, multivalency, and low polydispersity.28–30 These features offer real life applications in enhancing drug loading efficiency, and in developing multi-tasking nanoparticle based technology for targeted delivery by conjugating drug, imaging and targeting moieties in the same scaffold.31,32DNA and siRNA can also be bound to terminal groups of dendrimers by electrostatic interactions resulting in better transfection efficiency.33–35 Similarly, hydrophobic drugs can be solubilised by physical incorporation in the cavities of dendrimers or inside self-assembled structures of miktoarm polymers.36–38 This article is not intended to be an exhaustive review of the literature on dendrimers and miktoarm polymers. Instead, it will focus on recent developments in their efficacy as targeted drug delivery vehicles.
2. Synthetic challenges in the construction of dendrimers and miktoarm polymers for biomedical applications
In the recent past, dendrimers and miktoarm star polymers have become prominent in the field of biomedical science due to their unique morphological and physical properties as compared to linear polymers. Synthetic challenges associated with dendrimers and miktoarm polymers have been widely addressed by the chemical community during the last 25 years. A brief summary of this account is provided below.2.1. Challenges in the synthesis of dendrimers
The most important aspect to synthesize a dendrimer for biological applications is to have proper control on the number and types of functional groups present at its periphery, as well as the type of backbone (i.e. if it is biodegradable/biocompatible).39 These groups are required to attach diverse functional moieties40 such as drug molecules, imaging41,42 and targeting agents43etc. Historically, dendrimer synthesis has been carried out by either convergent or divergent strategies, each with its own advantages and disadvantages. The dendrimer synthesis was initiated by Tomalia et al.,24 Newkome et al.44 and Vögtle et al.,23 using a divergent route, in which the construction starts from a core and progresses to the periphery with successive formation of new generations.45 The advantage of a divergent strategy is that one can synthesize higher generation dendrimers such as those reported for poly(amidoamine) or PAMAM and poly(propylene imine) or PPI dendrimers.46 But the drawback is that the dendrimers often have defects at higher generations due to incomplete reactions. To address this problem, often the monomers are added in excess which then require purification at every step. Accelerated synthetic strategies have been developed by Malkoch et al. to reduce the number of steps associated with the divergent synthesis. This methodology involves two different monomers having complementary functionalities, which eliminates the need for deprotection.47The convergent approach was introduced by Hawker and Fréchet,48 and it involves the prior construction of branches of a dendrimer, the so called dendrons, which are then attached to the central core. The convergent approach is synthetically more demanding but the dendrimers can be produced in much higher purity as the number of active sites present per reaction are limited, thus reducing structural defects in the dendrimer. The dendrons can also act as building blocks, and can be attached to the poly-functional cores to synthesize multimodal dendrimers. The disadvantage of the convergent approach is that it can be used only for lower generations as the attachment of larger dendrons on the core faces steric hindrances. However, lower generations may not be a limitation for dendrimers to be used for biological applications.38
The dendrimer synthesis has benefitted from the development of methodologies which involve reactions with fewer steps, high yields and tolerance to a variety of functional groups, and which could be carried out under mild conditions with minimum purification steps.47,49 Sharpless et al. described a set of reactions which meet above mentioned criteria, and coined the term “Click” chemistry.50 Although a number of reactions come under the category of “click” chemistry,25Cu(I) catalyzed Huisgen 1,3 dipolar cycloaddition reaction is the most popular one. Hawker et al. demonstrated the synthesis of dendrimers using this click methodology by reacting an azide terminated dendrimer with an alkyne monomer, followed by halogenation and azido substitution.51 This group used click reaction to synthesize dendrimers in a convergent manner in which a third generation dendron with an azide focal point was constructed and clicked onto the polyacetylene core to obtain a 4th generation dendrimer.52 Recently, Hawker et al. used thiol–ene click reaction to construct dendrimers of up to generation 4 using a divergent route. With this method, they constructed dendrimers by an orthogonal strategy that eliminated the use of any solvent and metal catalysts, and it reduced the structural defects related to typical divergent synthesis.53 For biomedical usage, there is still a need to construct dendrimers on a large scale in an efficient and reproducible manner, with minimum defects in the structure. These ‘fast’ reactions would provide a greater control over the synthesis and reduce the need for purification. The development of a wide variety of chemo-selective ligation reactions,54–56 like Staudinger ligation,54Cu(I) catalyzed alkyne–azide,55thiol–ene56 and Diels–Alder click57 reactions, has helped to overcome the synthetic challenges associated with dendrimer synthesis.
The introduction of multiple functionalities in the same dendrimer (Fig. 2) in a controlled manner is a synthetically challenging task, and only a few examples of dendrimers with more than two functionalities have been reported.16,26,31,32,58–61 Our group recently developed a versatile synthetic methodology to bi- and tri-functional dendrimers with orthogonal end groups, using sequential Cu(I) catalyzed alkyne–azide click reactions. Using this approach, we synthesized multifunctional dendrimers with the desired combination of a therapeutic agent (α-lipoic acid or niacin), an imaging agent (dipyrromethene boron difluoride) (BODIPY) and a solubilising agent (polyethylene glycol) in decent yields.16,31,32 An elaboration of this methodology allows covalent linking of any desired combination of functionalities at the periphery of dendrimers. The Cu(I) catalyzed alkyne–azide “click” reaction provides an efficient way to construct dendrimers in good yields with minimum purification. It has been reported that there is no detectable amount of the catalyst in the dendrimer products. In addition, these dendrimers could be further purified to remove any residual copper by incubation with ethylenediaminetetraacetic acid (EDTA), followed by dialysis, using oxide-capped metallic iron nanoparticles as Cu sequesters.62,63
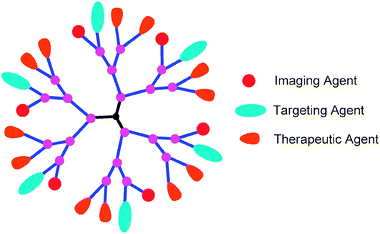 | ||
| Fig. 2 Multifunctional dendrimer. | ||
Until now, the most commonly used dendrimers for biomedical applications have been PAMAM based. Introduction of multiple functionalities to commercially available PAMAM dendrimers is achieved by a random statistical approach by partial functionalization of terminal amines.64,65 Kannan's group has reported nearly complete peripheral modification (87–93%) of G4 PAMAM dendrimers, in which they used one-step synthesis to convert 64 symmetrical end groups into two reactive, distinct, orthogonal and chemo-selective moieties.66 Ornelas and Weck have introduced a new strategy towards multifunctional dendrimers in which they attached two poly(amide) based dendrons bearing different functionalities and a fluorescent dye onto a tri-functional core.61
Functionalization of the dendrimers not only provides a route to introduce a variety of drugs, imaging and solubilising agents, but it can also fulfill the objectives of targeted drug delivery,67,68 increase in plasma residence time,69 and reduction in cytotoxicity.70 For example, cytotoxicity associated with amine terminated dendrimers has been overcome by PEGylation and acetylation.71–75 PEGylation makes the dendrimers biocompatible, as well as helps meet other objectives including improved biodistribution and pharmacokinetics, solubility, increase in drug loading, sustained and controlled drug release etc.76–78 Sometimes, surface modifications can lead to problems. For example, Roth and coworkers were able to decrease toxicity of PAMAM dendrimers to U87 cells, but higher degrees of amine neutralization reduced the gene silencing efficiency of PAMAM/siRNA delivery vectors.73,75 Thus, there are still opportunities for chemists to develop synthetic methodologies that could achieve surface modifications without compromising their overall properties, and achieve functionalization with chemically reactive groups suitable for attachment of desired moieties at a low cost and minimum purification steps.
2.2. Challenges in the synthesis of miktoarm polymers
Miktoarm stars continue to attract considerable attention because of their compact architecture.79–81 These were originally synthesized using living anionic polymerization methodologies.82–86 However, these often involve stringent polymerization conditions and are restricted to only a few monomers. Recent developments in controlled/living polymerizations such as controlled ring opening polymerization (CROP),87 atom transfer radical polymerization (ATRP),88–90nitroxide-mediated radical polymerization (NMP)91 and reversible addition–fragmentation chain transfer (RAFT) polymerization92 have provided efficient ways to design and synthesize well defined and complex miktoarm polymers. This is due to a wide variety of applicable monomers and mild polymerization conditions as compared to living anionic polymerization. Miktoarm stars are usually synthesized via one of the three common strategies: “core-first”, “arm-first” or “in-out”.80,93,94 The core-first method employs living polymerization from a heteromultifunctional core initiator with different initiating sites.95 With this method multiple arms can be grown simultaneously from a single core molecule. The major drawback of this method is that it involves complex multistep syntheses of heteromultifunctional initiators from small molecules, and often involves the use of protection/deprotection strategies which makes the chemistry complex.96–98 For instance, Webster and coworkers have reported the synthesis of miktoarm stars from propargyl diol which was used to initiate ROP of ε-caprolactone followed by alkyne/azide click reaction of a two arm ROP product with an azide functionalized poly(butyl acrylate) polymer.99 In the “arm-first” method, the chain ends of many linear macroinitiators formed from different controlled radical polymerization methods are used to polymerize a divinyl compound. The synthesis of miktoarm star polymers becomes problematic with this method when macroinitiators with different activities are used. The “in-out” method involves a living macroinitiator which initiates the polymerization of a cross linking agent to form a homoarm star polymer, with its initiating sites preserved within the core. It is used as a multifunctional initiator for the subsequent growth of second generation of arms. This is an important method for synthesizing miktoarm star copolymers with multiple arms. Its disadvantage is that all the initiating sites cannot participate in the reaction because of congestion around the core, thus decreasing the number of second generation arms as compared to first generation in miktoarm star copolymers.100 The grafting of second generation arms in the core of star polymers has faced several obstacles such as star–star and intrastar couplings which can lead to broad molecular weight distribution.101 In order to overcome this problem, Matyjaszewski and co-workers have proposed the synthesis of miktoarm core cross-linked star polymers by copolymerization of linear monomers with cross-linkers using a low molecular weight ATRP initiator.102 The number of initiating sites and arms are independently controlled, thus reducing the star–star coupling reactions leading to smaller molecular weight distribution.For applications in the field of biomedicine and nanotechnology, the miktoarm star polymers with novel architectures are required. The synthesis of AnBm multimiktoarm star polymers having more than 10 arms is very complex and these are rarely synthesized by simple controlled living polymerizations. Matyjaszewski and co-workers have reported a new method for the synthesis of multi-miktoarm star copolymersvia one-pot ATRP cross-linking reaction of several different linear macroinitiators and a divinyl cross-linker. Using this strategy, the average number of arms can be extended to 50.100,103,104 But this strategy could not lead to miktoarm polymers with a precise architecture. The alternate method to synthesize these multi-miktoarm stars with a precise arm number and arm length is by designing heteromultifunctional AnBm initiators, but the synthesis of these initiators with more than 10 functionalities is a tedious task. To resolve this issue, polymer chemists started using naturally occurring cyclodextrins having fixed numbers of primary and secondary hydroxyl groups with different reactivities.105–107 Shen and co-workers have successfully synthesized A14B7 multimiktoarm star copolymers composed of 14 PCL arms and 7 poly(acrylic acid) (PAA) arms with β-cyclodextrin as a core using a combination of CROP and ATRP.108
The synthetic methodologies discussed above have their own limitations, and efforts are continually being made to develop facile and efficient routes to the synthesis of miktoarm polymers. Recent addition of “click chemistry” which in combination with different living radical polymerization techniques has provided a useful tool in diversifying the synthetic strategy of these miktoarm polymers.50,93,94,109–113 A wide variety of miktoarm star copolymers have been synthesized using Cu(I) catalyzed azide–alkyne click reaction with living radical polymerization techniques.99,109,114–121 We have recently reported the synthesis of A2B and ABC type miktoarm stars containing a combination of polyethylene glycol, polycaprolactone and polystyrene arms, using a core with orthogonal functionalities on which Cu(I) catalyzed alkyne–azide click reactions and ring-opening polymerization are carried out in sequence.114,115
Another click methodology is the Diels–Alder reaction, which generally consists of coupling of a diene and a dienophile by intra- or intermolecular reaction. Recently, Tunca and coworkers have synthesized multi-miktoarm star block copolymers where they have employed sequential double click reactions involving azide–alkyne and Diels–Alder reactions.122 Deng and coworkers have synthesized miktoarm star copolymers by a combination of a RAFT arm first technique and aldehyde–aminooxy click reaction. The advantage of using this click reaction is that except the reacting reagents no other auxiliaries such as metallic catalysts are required.123 In summary, numerous methods have been developed for the synthesis of miktoarm polymers, each with its own advantages, and the judicious choice of a synthetic methodology clearly depends on the type of the desired miktoarm architecture.
3. Modalities of drug incorporation into dendrimers and miktoarm polymers
Active pharmaceutical agents can be loaded into the internal voids of dendrimers and into self-assembled structures of miktoarm polymers by physical incorporation, or by covalent conjugation into the nanocarrier architecture.3.1. Drug loading by physical incorporation
Due to their inherent architecture, drug molecules can be loaded into dendrimers using their well defined internal crevices by hydrophobic interactions or hydrogen bonding or by electrostatic interactions between drug ionic groups and oppositely charged dendrimer surfaces. The exact location of the solubilised drug and the loading capacity are controlled by several factors, such as dendrimer generation, architecture, characteristics of internal cavities, drug molecular weight and pKa of drug ionisable groups.37,124–127 A detailed computational and experimental study by Tomalia and coworkers has shown that low generation β-alanine dendrimers (G1–G3) exhibit an oblong open structure while higher generations (≥G4) have a densely packed surface that is necessary to produce enclosed internal spaces that can incorporate and solubilise hydrophobic drugs.128,129 Using poly(amidoamine) (PAMAM) dendrimers of different generations (G2–G6) and four different drugs, Cheng et al. compared the effect of internal hydrophobic interactions versus external electrostatic interactions on hydrophobic drug solubilisation.130 By combining solubility and NMR studies, they demonstrated that external electrostatic interactions between the dendrimer surface and the drug contributed more to drug solubility enhancement than hydrophobic interactions in the PAMAM cavities. The ability of dendrimers to solubilise a variety of drugs including anticancer, anti-HIV and anti-inflammatory agents has recently been reviewed.21,28,131,132Compared to chemical conjugation, physical incorporation of drugs has the advantage of straightforward, rapid preparation without adversely affecting drug pharmacological activity. Its disadvantages include low stability in terms of storage and premature drug release, variation of the concentration of the solubilised drug from batch to batch and low drug loading capacity. In vitro release of physically incorporated drugs from dendrimers is usually rapid, and depends on several factors such as the drug partition coefficient between hydrophobic and aqueous environments, strength of drug/dendrimer interactions, dendrimer generation and surface groups (Fig. 3).133–135
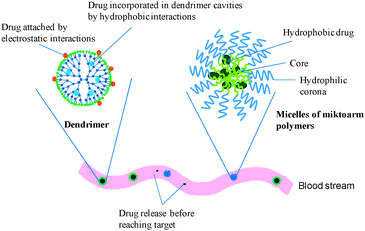 | ||
| Fig. 3 Schematic illustration of physical drug incorporation into dendrimers and micelles of miktoarm polymers with rapid drug release in the blood stream. | ||
It has been shown that drug release and loading efficiency can be enhanced by PEGylation of dendrimers. For instance, the anti-cancer drug 5-fluorouracil showed 6-fold lower release rate and 12-fold higher loading capacity in PEGylated G4 PAMAM dendrimers in which 25% of PAMAM surface groups were capped with PEG (5 kDa) compared to non-PEGylated dendrimers.136 In addition to PEGylation, entrapment of dendrimers in other nanocarriers, such as liposomes, has been shown to sustain drug release. Gardikis et al. showed that at 96 h cumulative doxorubicin release from pure liposomes was 74.6 ± 7.8% compared to 27.9 ± 2.8% when the G2 dendrimer was co-encapsulated with doxorubicin in liposomes.137 Despite this improvement, controlled drug release in physiological media is difficult to achieve for drugs incorporated into dendrimers.37 Covalently linking drugs to dendrimers may help overcome some of these disadvantages.
Similar to dendrimers, drugs are incorporated into miktoarm stars by hydrophobic interactions (Fig. 3). However, miktoarm polymers have to be self-assembled first into nanoparticles since their architecture is different from that of dendrimers. For successful self-assembly in aqueous solutions, miktoarm polymers should have at least one hydrophobic arm with a suitable balance between hydrophilic and hydrophobic arms. The unique self-assembly behaviour of miktoarm polymers has been demonstrated in bulk and in solution, and it is related to their architecture of three or more arms branching out from a central junction point.138 Based on their structure, chemical composition and length of the arms, miktoarm stars can form different nanoparticle morphologies, such as micelles, polymersomes, worm micelles or multicompartment micelles.115,139–141 This lends considerable advantages to miktoarm polymers as drug and gene delivery systems over analogue linear copolymers. For instance, Wang et al. reported that micelles obtained from PCL and PEG-based multiarm star block copolymers had a loading capacity of 22 weight% for etoposide, a hydrophobic anticancer drug.142,143 Yin et al. reported the preparation of a series of Y-shaped AB2 miktoarm polymers (A = polyethylene glycol (PEG); B = poly(L-lactide)) that mimic the natural structure of phospholipids.140 These polymers formed polymersomes (polymeric vesicles) in aqueous solutions with a size that ranged from 33–75 nm in radius, depending on polymer composition. The polymersomes had an incorporation efficiency of 72 weight% for doxorubicin HCl, a hydrophilic anticancer drug, and sustained its release for more than 48 h. Polymersome formation for the miktoarm polymers was observed at PEG volume fractions of 0.2–0.7 compared to PEG volume fractions of 0.2–0.4 for their linear diblock copolymer counterparts confirming the versatility of miktoarm polymers. We have recently reported the incorporation of nimodipine into polymeric micelles of A2B miktoarm polymers (A = PEG; B = polycaprolactone (PCL)).115Nimodipine is a poorly water soluble drug used primarily for the prevention and treatment of delayed ischemic neurological disorders. The micelles showed nimodipine incorporation efficiency up to 78 weight% and sustained drug release for more than 3 days. Enhanced micellar stability and smaller sizes were obtained for these A2B miktoarms compared to their linear counterparts of similar composition and molecular weight. Other reports have also suggested that miktoarm polymers self-assemble differently from their linear counterparts.144,145 Gou et al. have reported the synthesis and drug loading of ibuprofen-conjugated amphiphilic A14B7 miktoarm stars composed of 14 PCL arms and 7 PEG arms with β-cyclodextrin (β-CD) as a core moiety.146 These amphiphilic star polymers self-assembled into multimorphological aggregates in aqueous solution and showed higher drug loading capacity compared to the corresponding non-drug conjugated copolymers. Güç et al. showed that a fatty acid-based hyperbranched resin (HBR) formed nanoparticles with an average size ranging from 206–276 nm.147 The nanoparticles had loading efficiencies up to 74% for tamoxifen and idarubicin, both hydrophobic anticancer drugs. Compared to free tamoxifen, the nanoparticles-loaded drug exhibited up to 13-fold higher cytotoxicity against MCF-7 breast cancer cells, in vitro.
In order to widen the spectrum of drugs that can be incorporated into the same nanodelivery system, Radowski et al. prepared a liposome-like nanodelivery dendritic multishell system based on a hyperbranched polymeric core surrounded by a double layered shell.148 The core was hyperbranched poly(ethylene imine) (PEI) surrounded by a shell of amphiphilic alkyl dicarboxylic acids and a corona of PEG. Nimodipine and β-carotene were used as model hydrophobic guest molecules whereas congo red and vitamin B6 monohydrochloride were used as model polar guest molecules. The nanoparticles were able to increase the solubility of these guest molecules. While the incorporation efficiency of the guest molecules was slightly affected by the PEG molecular weight, it was strongly dependent on the size of the PEI polar core and the size of the hydrophobic inner shell. Another interesting amphiphilic hyperbranched polymeric system suitable for selective incorporation of guest molecules was recently reported.149 The system is based on a hydrophobic core of poly(β-cyclodextrin) (β-CD) and a hydrophilic corona of poly(N,N-dimethylaminoethyl methacrylate) (PDMA). The authors hypothesized that selective incorporation of two different guest molecules could be achieved due to the presence of two different cavities; the internal cavity of β-CD and the cavity created between the different polymeric arms (Fig. 4). Levofloxacin lactate (LL) and phenolphthalein (PP) showed different release profiles suggesting their incorporation into different molecular cavities with different microenvironments: PP being encapsulated in β-CD cavities and LL in the hyperbranched cavities. The release of LL dominates at an early stage in comparison with PP, subsequently the release rate of PP increases to play a determinate role in the release system.
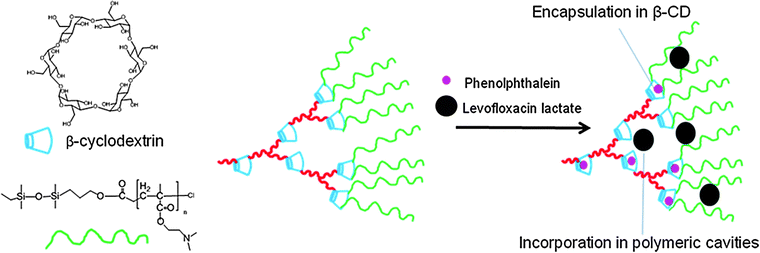 | ||
| Fig. 4 Selective incorporation based on drug molecular size into copolymers of poly(β-cyclodextrin) (β-CD)–poly(N,N-dimethylaminoethyl methacrylate) (PDMA). | ||
The above mentioned examples show a potential of dendrimers and miktoarm stars as nanodelivery vehicles for drugs by physical drug incorporation which is superior to other carriers. However, premature drug release from these systems is still a hurdle, and chemical conjugation of drugs to dendrimers and miktoarms is one of the promising strategies to overcome it.
3.2. Drug loading by chemical conjugation
Some of the drawbacks of physical drug incorporation can be reduced by covalent linking of a drug to the dendrimer or miktoarm polymer framework, using a chemical approach which will also bring about selective drug release in vivo. Such an approach can generate structures with a pre-measured drug content and enhanced stability. In addition, targeted drug delivery and controlled release can be achieved by attaching the drug to the polymeric scaffold through stimuli responsive bonds where drug release can be obtained by change in the biological microenvironment such as variation in pH, temperature or concentration of a specific enzyme.The type of bond linking dendrimer–drug conjugates not only influences the stability as well as release profile of drugs, but it also affects the efficacy of the drug. Methotrexate (MTX), an anticancer drug, has two possible sites for covalent attachment i.e. through its carboxylic acid group forming an ester linkage by reacting with a hydroxyl group of the carrier or through its amine group by reacting with carboxylic acid present on the carrier producing an amide linkage. MTX linked via an ester bond to an acetamide-functionalized G5 PAMAM dendrimer was found to be four times more active than free MTX, whereas MTX conjugated by amide bonds to the same dendrimer was less active than free MTX.158,159 Different studies have shown that the conformation of anticancer drugs attached on the dendrimers surface also affects their cytotoxic activity.150,158,160 Kannan and co-workers compared the anticancer activity of MTX when attached through its amine group to the carboxylic acid group of G2.5-COOH dendrimers forming amide linkages with MTX attached through its carboxylic acid group to the primary amines of G3-NH2 dendrimers. G2.5-MTX conjugates were found to be 3-fold more toxic as compared to free MTX toward lymphoblastic leukaemia cells, whereas G3-MTX conjugates were 10 fold less toxic than the free MTX.150Dendrimer conjugation of anticancer drugs, including methotrexate,150,151,161doxorubicin162–165 and camptothecin,166,167 has shown to provide a high payload, due to which the administered dose can be reduced to minimize side effects and maximize the therapeutic effect. Also, the retention of efficacy of the drug after conjugation to a certain scaffold is very important. There are few studies which show that the efficacy depends on the type of scaffold. For instance, some studies showed that there was a considerable reduction in efficacy of streptokinase (SK) upon conjugation to PEG and dextran due to considerable loss in enzymatic activity (67%, 50%) respectively. In contrast, Kannan and co-workers have reported that SK–PAMAM G3.5 conjugates with an equimolar ratio of dendrimer to SK retained highest enzymatic activity (80%).168 These examples show that a choice of the scaffold is really important in order to retain the activity of the drug after chemical conjugation.
As in the case of dendrimers, the covalent attachment of drugs to miktoarm polymers has also been reported.31,32,169–172 Recently, Shen and co-workers have reported that the miktoarm-drug conjugates not only affect the drug release behaviour, but the covalent attachment of the drug to these polymers can also change their drug loading and drug incorporation efficiencies.118,146 They have developed A2B2 miktoarm star copolymers having PEG and PCL chains with ibuprofen attached on PCL chains through ester linkage. The drug loading capacities of miktoarm polymer micelles were better in drug conjugated miktoarm star copolymers than their non-drug counterparts.118
4. Dendrimers and miktoarm polymers for targeted drug delivery
The ultimate goal of targeted drug delivery is to increase drug's therapeutic index through maximizing its concentration at its site of action while minimizing it in non-target tissues. This usually allows administration of lower doses and reduced side effects which is particularly important for anticancer drugs whose accumulation in healthy tissues results in serious side effects.180Dendrimers and miktoarm polymers are excellent vehicles for targeted drug delivery by virtue of their small size (5–100 nm) and availability of many surface groups to conjugate drugs, imaging tags and targeting moieties. Drug targeting using dendrimers and miktoarm polymers can be achieved through passive or active targeting.4.1. Passive targeting
Passive targeting of nanoparticles refers to their accumulation at a particular site in the body due to physicochemical and pathophysiological factors.181Nanoparticles made of dendrimers and miktoarm polymers can be passively targeted to solid tumors and inflamed tissue via the enhanced permeability and retention (EPR) effect.182 This effect takes advantage of both tissue pathophysiological properties as well as nanoparticles physicochemical properties. Tumors usually have a leaky vasculature and impaired lymphatic drainage, which allows 10–30 times higher drug concentration in tumors compared to the blood.183–186 The EPR effect was first described in the 1980s by Maeda who demonstrated that Evans blue dye bound to plasma albumin selectively accumulated in tumor tissue after intravenous administration.182 Moreover, radiolabeled transferrin (∼90 kDa) and immunoglobulin G (IgG) (∼160 kDa) exhibited similar behavior, whereas small proteins such as neocarzinostatin (∼12 kDa) and ovomucoid (∼29 kDa) did not accumulate in tumors. Based on this and on evidence accumulated by independent research during the last 25 years, it was concluded that the EPR effect takes place in most solid tumors for biocompatible macromolecules with molecular weight ≥40 kDa and for nanoparticles of size up to 1 μm.187 However, for successful EPR-mediated-targeting, nanoparticles should have long circulation time in the blood, ideally more than 6 h and their size should be less than 200 nm to avoid uptake by the reticuloendothelial system (RES) (Fig. 5).184,188,189 Long circulation properties of nanoparticles can be achieved by surface modification with hydrophilic polymers such as PEG or polyvinylpyrrolidone (PVP).190–193 These hydrophilic polymers can decrease the adsorption of opsonin proteins in the blood, help nanoparticles escape recognition by RES and circulate longer in the blood.Nanoparticles should not be filtered in the kidneys in order to attain longevity in the blood. The filtration size cut-off for the kidney is known to range from a hydrodynamic diameter of 3.7 to 6.0 nm.194 Therefore, water soluble nanoparticles and dendrimers with a hydrodynamic diameter less than ∼4 nm are expected to be rapidly filtered by the kidneys and excreted in the urine (Fig. 5). Indeed, Choi et al. have shown that quantum dots with hydrodynamic diameter <5.5 nm were rapidly and efficiently eliminated from the body via urinary excretion.195 Miktoarm polymers form nanoparticles with hydrodynamic diameters larger than the threshold of renal filtration suggesting their possible extended blood circulation; taking into account surface charge and molecular weight. PAMAM dendrimers, on the other hand have generation and surface group-dependent hydrodynamic diameter. Thus, PAMAM–OH dendrimers were shown to have smaller hydrodynamic size compared to their PAMAM–NH2 counterparts of the same generation due to a more extended structure of PAMAM–NH2.196,197 G0–G7 PAMAM dendrimers with an ethylenediamine core have a hydrodynamic diameter in the range of 1.4–8.8 nm whereas G1–G5 poly(propylene imine) (PPI) dendrimers have a hydrodynamic diameter in the range of 0.6–2.0 nm.198,199 In addition to the dendrimers hydrodynamic diameter, the nature of their surface groups also affects their residence time in the blood. Thus, PAMAM–NH2 dendrimers were rapidly eliminated from the blood and accumulated in the liver, lung and spleen possibly due to electrostatic interactions between the surface cationic groups and anionic sulfated proteoglycans on cell surfaces.200–203 Approaches such as acetylation, succinylation or PEGylation of dendrimers have been effective in prolonging their circulation times and decreasing their accumulation in the liver and kidneys.69,161,201,204 For instance, PEGylated polylysine dendrimers were concentrated ∼8-fold and 3-fold in Walker 256 tumors more than in muscle and heart, respectively.205 Moreover, PEGylated G4 PAMAM–doxorubicin conjugates showed time dependent tumor accumulation after intravenous injection in mice inoculated with SKOV-3 cells. Higher concentration of the conjugates in the tumor tissue was detected as early as 1 h after injection with a maximum at 24 h and small decrease within 48 h post injection. Conjugates with the highest PEG content (∼20 PEG molecules per PAMAM molecule) showed the highest accumulation in tumors compared to other conjugates.162
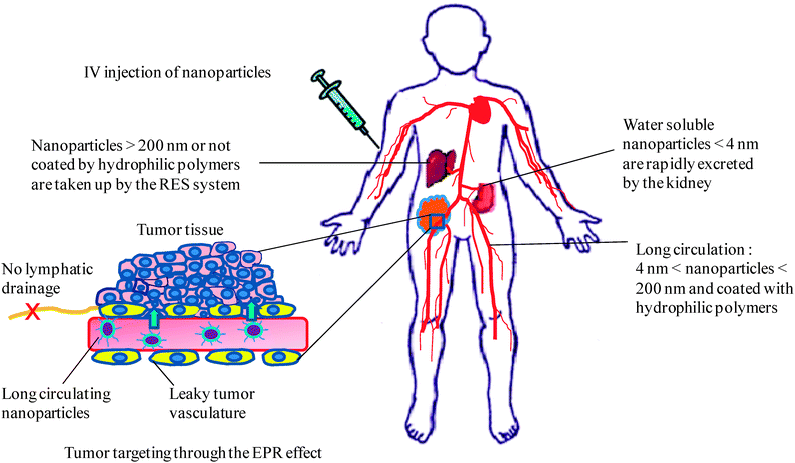 | ||
| Fig. 5 Schematic illustration of biodistribution of intravenously injected nanoparticles showing nanoparticles accumulation in tumors due to the EPR effect, nanoparticles uptake by the RES system, and nanoparticles excretion by the kidney. | ||
The EPR effect was more extensively studied with nanoparticulate formulations based on linear copolymers rather than those of branched and hyperbranched polymers.7 Recently, Sadekar et al. reported a comparative study on the biodistribution of the linear copolymer N-(2-hydroxylpropyl)methacrylamide (HPMA) and hydroxyl-terminated PAMAM dendrimers of generations 5, 6 and 7 in ovarian-tumor-bearing mice.196 They reported that the molecular weight, hydrodynamic size and polymer architecture were the major factors that influenced the biodistribution profiles of these polymers. G5 PAMAM–OH (hydrodynamic diameter of 4.6 nm) was retained in the kidney over 1 week, whereas a linear HPMA copolymer of a similar molecular weight (hydrodynamic diameter of 2.8 nm) was excreted through the kidney within 2 h. G6 PAMAM–OH with a hydrodynamic diameter of 6 nm was not filtered in the kidney; instead, it accumulated in the liver. The HPMA copolymer of comparable molecular weight had a three times higher plasma circulation time compared to G6 PAMAM–OH. Among the studied polymers, the G7 PAMAM–OH dendrimer showed extended plasma circulation, enhanced tumor accumulation, and prolonged retention with the highest tumor/blood ratio (T/B ratio). This dendrimer showed a time-dependent accumulation in the tumor with a maximum at 6 h and persisted for up to 1 week with a T/B ratio of ∼12.75. This was attributed to the rigid sphere-like conformation of this dendrimer and its hydrodynamic diameter (i.e., ∼8 nm) above the filtration size cut-off of the kidney (i.e., 3.7–6 nm).194,206 The size of nanoparticles also affects their biodistribution profiles after in vivo administration. Thus, paclitaxel-loaded nanoparticles of amphiphilic telodendrimers (PEG-b-dendritic oligocholic acid) of small size (17–60 nm) were accumulated in the tumors in xenograft models whereas larger paclitaxel-loaded micelles (150 nm) were mostly accumulated in the liver and lungs.207 The antitumor efficacy of these paclitaxel-loaded micelles was greater than that of Taxol® and Abraxane® in subcutaneous and intraperitoneal ovarian cancer mouse models.208 In order to overcome the disadvantages of dendrimers small size, several approaches have been reported to facilitate dendrimers self-assembly into larger multimolecular micelles. For instance, Zhang et al. have shown that modification of G1 PAMAM dendrimer by grafting cholic acid to its surface amino group endowed the dendrimer with self-assembly properties.209 The modified PAMAM dendrimer had 2 cholic acid molecules per PAMAM molecule, and self-assembled into micelles with an average size of 50–70 nm. Moreover, the micelles showed pH-dependent incorporation of camptothecin, a hydrophobic anticancer drug. The anticancer activity of micelle-incorporated camptothecin was higher than that of the free drug, in vitro. The in vivo biodistribution of these camptothecin-containing micelles remains to be determined.
In addition to tumors, dendrimers have also shown passive accumulation in inflamed tissues. Although arthritic joints do not lack lymphatic drainage, indomethacin incorporated in the G4 PAMAM dendrimer was 2.29 times more concentrated in inflamed joints compared to the free drug.210Flurbiprofen incorporated in the PAMAM dendrimer had 2–3 fold higher concentration in inflamed tissue of a carrageenan induced paw edema model in male albino rats compared to the free drug.211 Furthermore, Dai et al. have shown that G4 PAMAM–OH dendrimers with no targeting moieties exhibit intrinsic targeting properties to activated microglia and astrocytes following subarachnoid administration in newborn rabbits with maternal inflammation-induced cerebral palsy (CP).212 Using fluorescently labelled PAMAM dendrimers, it was shown that a 15-fold greater PAMAM concentration was observed in the CP rabbits, compared with control (Fig. 6). The increased dendrimer uptake in the CP rabbits was attributed to increased phagocytic ability of activated microglia and hypertrophic astrocytes compared to healthy cells. The usefulness of PAMAM dendrimers and other nanoparticles remains to be determined in different neurodegenerative disorders associated with inflammation.
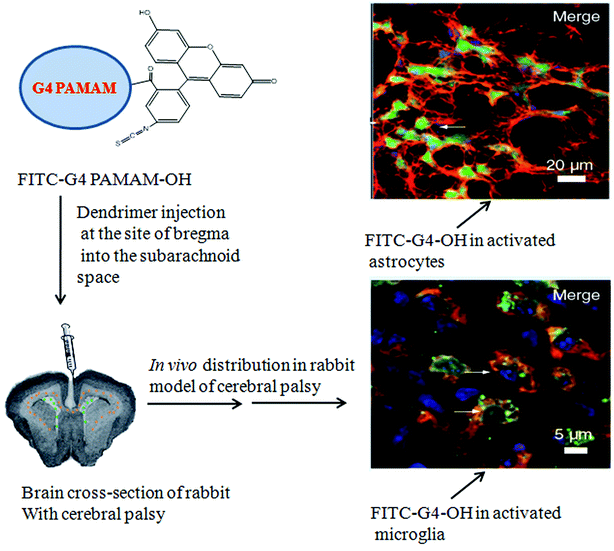 | ||
| Fig. 6 Selective uptake of G4 PAMAM–OH–FITC dendrimer in activated microglia and astrocytes following subarachnoid injection in the cerebral palsy of rabbit model. Adapted from ref. 212. | ||
4.2. Active targeting
Active targeting of nanoparticles refers to their accumulation at a particular site in the body using specific interactions between tissue or cell components and nanoparticles.181 Active targeting can be achieved by making stimuli-responsive nanoparticles, or by attaching specific targeting ligand molecules to the nanoparticles surface.3 Directing drug–dendrimer conjugates to tumor cells can be achieved by attaching a variety of targeting moieties e.g.folic acid (FA),72,158,159,204,213,214biotin,177,215 or macromolecules, for example, cell penetrating peptides,216–218 or antibodies.151,219,220FA has affinity towards receptors expressed on tumors. FA-bound dendrimer–drug conjugates largely accumulated in the tumor cells, as compared to the free drug,217,221 or the dendrimer–drug conjugates without a targeting agent.158,222 For example, Baker and co-workers have reported that FA-acetylated PAMAM–MTX conjugates showed accumulation in tumor and liver tissue over 4 days after intravenous administration into immunodeficient mice with human KB tumors. Treatment with conjugates led to 10 times higher suppression of tumor growth as compared to free MTX of equivalent dose.204 Wen and co-workers have reported partially acetylated G5 PAMAM dendrimers with biotin as a targeting moiety, and fluoresceinisothiocyanate (FITC) as an imaging agent. PAMAM–biotin–FITC conjugates exhibited much higher cellular uptake in HeLa cells as compared to non-targeted counterparts.215 More recently, Jia and co-workers reported the PEGylated PAMAM G4 dendrimer with a dual targeting drug carrier bearing transferrin (Tf) and wheat germ agglutinin (WGA) as brain-targeting ligands. Dox was loaded in the interior of this dendrimer. PAMAM–PEG–WGA–Tf showed an enhanced transport ratio (13.5%) of drug across the blood–brain barrier, as compared to a single targeting carrier, PAMAM–PEG–WGA (8%), PAMAM–PEG–Tf (7%), or free Dox (5%) within 2 h. The dual targeting conjugate significantly decreased cytotoxicity of Dox to healthy cells, but decreased the growth of C6 glioma cells.223Bornhop's group has designed a translocator protein (TSPO) targeted imaging agent based on the G4 PAMAM dendrimer using 1-(2-chlorophenyl)isoquinoline-3-carboxylic acid (CIPhIQ Acid) and lissamine dye. TSPO is a translocator protein expressed on the outer mitochondrial membrane in steroid producing cells. These conjugates colocalized with mitotracker green, demonstrating their ability to target mitochondria.224 More recently, our group has demonstrated targeting of cytoplasmic lipid droplets with dendrimers and miktoarm polymers covalently linked to a therapeutic agent (niacin or α-lipoic acid) and lipophilic fluorescent dye, BODIPY.16,31,32 These conjugates localized in the cytoplasmic lipid droplets in living hepatocytes and microglia cells (Fig. 7).31
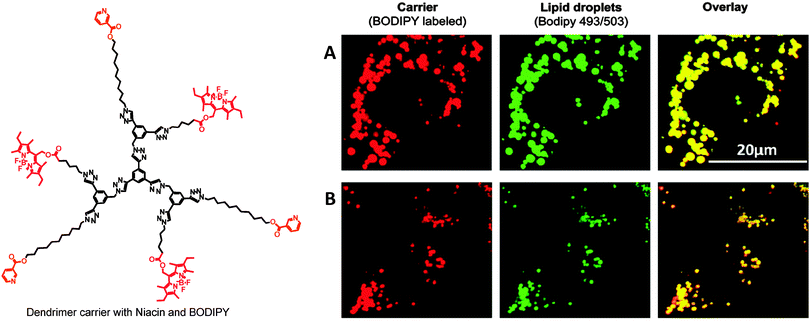 | ||
| Fig. 7 Colocalization of niacin- and BODIPY-conjugated dendrimer with lipid droplets in human hepatocytes (A) and murine microglia (B). | ||
Stimuli responsive nanoparticles have offered considerable potential for targeted drug delivery,3,225 since stimuli produced in a diseased tissue can trigger the release of drug only in the affected area, and thus reducing the side effects. The drug release can be triggered by various stimuli including temperature, light, pH, redox microenvironment and enzyme overexpression. Dendrimer–drug conjugates with pH sensitive covalent linkages have been discussed in the section on chemical conjugation. The difference in pH can allow the release of drug payload by breaking pH sensitive linkages or by protonation of the dendrimer structure to release the incorporated drug. Jain and co-workers reported the release of incorporated aceclofenac, an anti-inflammatory drug from PEGylated PPI dendrimers in a pH responsive manner.226 In another study, they demonstrated the release of incorporated histamine H2receptor agonist, famotidine, from the PEGylated PPI dendrimer under acidic pH.227 pH responsive polymers can also be attached to dendrimers to make pH sensitive drug delivery systems.228 Recently, Baker and co-workers used a strategy of photocaging, involving the inactivation of the drug using a photocleavable protecting group, which upon UV irradiation can irreversibly release the drug in its active form. They used G5 PAMAM dendrimers conjugated to folic acid as a targeting ligand and doxorubicin was attached through a photocleavable group. This doxorubicin-caged nanoconjugate showed toxicity to the KB cells only after exposure to UV light when the drug was released.229 A novel concept for the simultaneous release of all the peripheral groups on the surface of dendrimers in the presence of an enzymatic trigger has been reported by Shabat and co-workers.179,230,231 Different terms have been used for this type of mechanism, such as ‘dendrimers disassembly’, ‘cascade release’ or ‘self immolative dendrimers’.232–235 Thus, combining the advantages of dendrimers with stimuli responsive modalities can produce smart nanocarriers for site specific drug delivery with increased therapeutic efficacy and reduced side effects.
5. Dendrimers and miktoarm polymers cytotoxicity
Nanoparticles intended for biomedical applications and their degradation products should not be cytotoxic. Most miktoarm polymers intended for drug delivery applications are based on biocompatible and/or biodegradable polymers such as PEG, PCL and PLA.146,236,237 We recently reported the synthesis and characterization of a series of A2B miktoarm polymers (A = PEG and B = PCL) for the incorporation and delivery of hydrophobic drugs.115 Micelles of these miktoarm polymers did not induce cytotoxicity in N9 microglia cells at polymer concentrations high enough to produce clinically relevant drug concentrations in aqueous medium. Micelles of star shaped 6sPCL-S-S-PEG were not cytotoxic against MCF-7 cells at concentrations as high as 1000 mg L−1. In contrast to miktoarm polymers, a wide range of building blocks including biodegradable and non-biodegradable blocks is being used in dendrimers synthesis. Dendrimers based on biodegradable backbones such as polylysine, dimethylolpropionic acid (bis-MPA) or poly(glycerol succinic–adipic acid) (PGLSA–OH) are preferred for drug delivery applications over non-biodegradable ones.38 With repeated administration accumulation of non-biodegradable dendrimers and nanoparticles could result in serious side effects. The dendrimer cytotoxicity is dependent on the dendrimer generation, nature of building blocks, concentration and surface groups.201,238–241 Cationic dendrimers such as PAMAM, PLL and PPI have shown significant cytotoxicity both in cell cultures and in vivo models, which certainly limits their clinical utility.242–244 Naha et al. reported a generation dependent cytotoxicity of PAMAM dendrimers in mouse macrophage cells (J774A.1) where the degree of cytotoxicity followed the order G6 > G5 > G4. The degree of cytotoxicity was correlated with the number of PAMAM surface amino groups.240 Mechanistic studies suggest that PAMAM–NH2 induces cell death by stimulating an overproduction of reactive oxygen species (ROS) and proinflammatory cytokines (macrophage inflammatory protein-2 (MIP-2), tumour necrosis factor-α (TNF-α) and interleukin-6, (IL-6)). Cytotoxicity of G5 PAMAM–NH2 dendrimers was evaluated in mouse embryonic fibroblast cells (NIH 3T3 cells) by measuring the IC50 values (concentration at which 50% of mitochondrial dehydrogenase activity was inhibited).243 While the IC50 was 0.3 mg mL−1 for unmodified PAMAM–NH2, this value increased by 12–105 times by PEGylation of PAMAM amino groups pointing towards the importance of PAMAM surface groups in determining their toxicity. Indeed, cationic dendrimers cytotoxicity has been shown to be due to interactions between the cationic surface groups and the negatively charged cell surface. Hong et al. studied the interactions between PAMAM–NH2 and supported 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) lipid bilayers and KB and Rat2 cell membranes using atomic force microscopy (AFM), enzyme assays, flow cell cytometry, and fluorescence microscopy.245 G7 PAMAM–NH2 dendrimers (10–100 nM) formed holes of 15–40 nm in diameter in aqueous, supported lipid bilayers. In contrast, G5 PAMAM–NH2 and PAMAM–acetamide dendrimers did not form holes within a similar concentration range, yet G5 PAMAM–NH2 expanded already existing defects. G5 PAMAM–NH2 and PAMAM–acetamide dendrimers were not cytotoxic up to a 500 nM concentration in KB and Rat2 cells. However, G5 PAMAM–NH2 decreased the integrity of the cell membranes as evidenced by a dose dependent release of the cytoplasmic proteins, lactate dehydrogenase (LDH) and luciferase (Luc), in contrast to the PAMAM–acetamide dendrimer which had little effect on membrane integrity at a concentration up to 500 nM. PAMAM–NH2 induced permeability was not permanent however, since leaking of cytosolic enzymes returned to normal levels upon dendrimers removal.In contrast to the detailed and numerous in vitro cytotoxicity studies of dendrimers there are fewer studies dealing with side effects associated with their in vivo administration. Using zebrafish embryo, Heiden et al. evaluated the developmental toxicity of low generation (G3.5 and G4) PAMAM dendrimers, as well as Arg–Gly–Asp (RGD)-conjugated PAMAM. G4 PAMAM–NH2 was toxic and attenuated growth and development of zebrafish embryos. Mortality was observed at low dendrimer concentration (0.2 μM) and was dependent on both PAMAM dose and exposure time. In contrast, G3.5 PAMAM–COOH dendrimers were not toxic to zebrafish embryos. Furthermore, RGD-conjugated G4 dendrimers were less toxic than G4 PAMAM–NH2 dendrimers. RGD-conjugated G3.5 dendrimers did not show toxicity at the highest concentrations tested.242 Neerman et al. studied the acute toxicity of melamine dendrimers by administering 2.5, 10, 40 and 160 mg kg−1 of dendrimers to mice by i.p. injection. All mice died after 6–12 h when injected 160 mg kg−1. Mice treated at a dose of 40 mg kg−1 showed hepatotoxicity as indicated by increased liver enzyme activity. In subchronic toxicity studies (three i.p. injections of 2.5–40 mg kg−1 in 3-week intervals) did not cause renal damage or mortality, except for the extensive liver necrosis with the highest dose (40 mg kg−1) after 48 h. These studies clearly demonstrate the importance of detailed in vivo toxicity studies of dendrimers and miktoarm polymers before any clinical implementation.
In view of the great potential of dendrimers in nanomedicine, several strategies have been proposed to eliminate or to reduce their toxicity. The most common approaches include designing biodegradable dendrimers such as polyester-based dendrimers164 and surface engineering of cationic dendrimers to mask their surface cationic groups by strategies such as PEGylation,205acetylation, and conjugation with amino acids and peptides.246 A detailed review of dendrimers toxicity and strategies to minimize it has been recently published.244
Despite hurdles in the synthesis and biocompatibility of dendrimers several dendrimer-based formulations are in clinical trials or have already made it to the market. For instance, a polylysine dendrimer-based microbiocide topical gel formulation, VivaGel™, is being developed by Starpharma for the prevention of transmission of HIV and other sexually transmitted diseases. SuperFect® of Qiagen is an activated dendrimer used for gene transfection in many cell lines. Another application of dendrimers is in the use of multiple antigen peptides (MAPs).247–249 The first example of MAPs was based on a polylysine dendrimer core reported by Tam and coworkers. Since these first reports, several studies have been carried out where MAP structures have been used to produce peptide-specific antibodies. A MAP based malaria vaccine is in phase I human clinical trials.250 As diagnostic tools, several dendrimer-based systems have been developed. Thus, a polylysine dendrimer functionalized with gadolinium chelates, Gadomer-17, was developed by Schering AG as a magnetic resonance imaging (MRI) contrast agent. Stratus® CS, commercialized by Dade Behring, has been used as a rapid detection tool of suspected myocardial ischemia. The system is based on a G5 PAMAM dendrimer used as a linking agent of monoclonal antibodies to a glass fiber matrix.251 Moreover, the US army research laboratory has developed a dendrimer-based detection tool of anthrax called Alert Ticket™.252
6. Conclusions
Macromolecule based drug therapy has come a long way from the use of simple polymer matrices to nano-therapeutics. It has brought scientists with varied backgrounds to a common goal of achieving high specificity in directing pharmaceutical agents to the tissue, cell and cell-organelle. Considerable progress has been made in this direction, and it suggests that dendrimers and miktoarm stars, relatively new additions to the macromolecular field, are important and key players in this quest. Chemists have stood up to the challenge, and tremendous success has been achieved in streamlining their synthesis and characterization. Introduction of “click” chemistry to the repertoire of chemical reactions available for their construction has significantly contributed to the rapid pace with which these macromolecules and their conjugates could be synthesized. In this article, we have attempted to highlight some of the recent developments in chemistry and biology of dendrimers and miktoarm stars, and their efficacy as vehicles for different drugs and imaging agents. Biological studies indicate that both dendrimers and miktoarm polymers are promising nanomaterials for biomedical applications. However, stringent criteria and multiple biological assays have to be used to determine which physical and chemical properties should be considered to minimize/avoid adverse effects in experimental biological systems and eventually humans.Notes and references
- V. I. Shubayev, T. R. Pisanic Ii and S. Jin, Adv. Drug Delivery Rev., 2009, 61, 467–477 CrossRef CAS.
- S. M. Janib, A. S. Moses and J. A. MacKay, Adv. Drug Delivery Rev., 2010, 62, 1052–1063 CrossRef CAS.
- C. Kojima, Expert Opin. Drug Delivery, 2010, 7, 307–319 Search PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- I. K. Voets, A. de Keizer and M. A. C. Stuart, Adv. Colloid Interface Sci., 2009, 147–148, 300–318 CrossRef CAS.
- V. Torchilin, Eur. J. Pharm. Biopharm., 2009, 71, 431–444 CrossRef CAS.
- Y. Matsumura and K. Kataoka, Cancer Sci., 2009, 100, 572–579 CrossRef CAS.
- M. Wang and M. Thanou, Pharmacol. Res., 2010, 62, 90–99 CrossRef CAS.
- Z. L. Tyrrell, Y. Q. Shen and M. Radosz, Prog. Colloid Polym. Sci., 2010, 35, 1128–1143 Search PubMed.
- M. Talelli, C. J. F. Rijcken, C. F. van Nostrum, G. Storm and W. E. Hennink, Adv. Drug Delivery Rev., 2010, 62, 231–239 CrossRef CAS.
- Y. Matsumura, Jpn. J. Clin. Oncol., 2008, 38, 793–802 Search PubMed.
- E. Miele, G. P. Spinelli, F. Tomao and S. Tomao, Int. J. Nanomed., 2009, 4, 99–105 Search PubMed.
- A. Agarwal, Y. Lvov, R. Sawant and V. Torchilin, J. Controlled Release, 2008, 128, 255–260 CrossRef CAS.
- H. Chen, S. Kim, W. He, H. Wang, P. S. Low, K. Park and J.-X. Cheng, Langmuir, 2008, 24, 5213–5217 Search PubMed.
- R. Savić, T. Azzam, A. Eisenberg and D. Maysinger, Langmuir, 2006, 22, 3570–3578 CrossRef CAS.
- R. Hourani, M. Jain, D. Maysinger and A. Kakkar, Chem.–Eur. J., 2010, 16, 6164–6168 Search PubMed.
- D. A. Tomalia and J. M. J. Frechet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719–2728 CrossRef CAS.
- C. C. Lee, J. A. MacKay, J. M. J. Frechet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS.
- R. Hourani and A. Kakkar, Macromol. Rapid Commun., 2010, 31, 947–974 CrossRef CAS.
- S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2005, 57, 2106–2129 CrossRef CAS.
- S. Svenson, Eur. J. Pharm. Biopharm., 2009, 71, 445–462 CrossRef CAS.
- C. M. Paleos, D. Tsiourvas, Z. Sideratou and L.-A. Tziveleka, Expert Opin. Drug Delivery, 2010, 7, 1387–1398 Search PubMed.
- E. Buhleier, W. Wehner and F. Vögtle, Synthesis, 1978, 155–158 CrossRef CAS.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J. (Tokyo), 1985, 17, 117–132 Search PubMed.
- G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536–1544 RSC.
- G. Franc and A. Kakkar, Chem. Commun., 2008, 5267–5276 RSC.
- K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171–1185 RSC.
- B. K. Nanjwade, H. M. Bechra, G. K. Derkar, F. V. Manvi and V. K. Nanjwade, Eur. J. Pharm. Sci., 2009, 38, 185–196 CrossRef CAS.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef CAS.
- M. Irfan and M. Seiler, Ind. Eng. Chem. Res., 2010, 49, 1169–1196 Search PubMed.
- A. Sharma, A. Khatchadourian, K. Khanna, R. Sharma, A. Kakkar and D. Maysinger, Biomaterials, 2011, 32, 1419–1429 Search PubMed.
- A. Sharma, K. Neibert, R. Sharma, R. Hourani, D. Maysinger and A. Kakkar, Macromolecules, 2011, 44, 521–529 Search PubMed.
- P. Wang, X.-H. Zhao, Z.-Y. Wang, M. Meng, X. Li and Q. Ning, Cancer Lett., 2010, 298, 34–49 Search PubMed.
- J. Wang, Z. Lu, M. G. Wientjes and J. L. S. Au, AAPS J., 2010, 12, 492–503 Search PubMed.
- C. M. Paleos, L. A. Tziveleka, Z. Sideratou and D. Tsiourvas, Expert Opin. Drug Delivery, 2009, 6, 27–38 Search PubMed.
- J. R. Baker, Jr., Hematology, 2009, 2009, 708–719 Search PubMed.
- S. H. Medina and M. E. H. El-Sayed, Chem. Rev., 2009, 109, 3141–3157 CrossRef CAS.
- M. A. Mintzer and M. W. Grinstaff, Chem. Soc. Rev., 2011, 40, 173–190 RSC.
- L. Röglin, E. H. M. Lempens and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 102–112 CrossRef.
- N. Fischer-Durand, M. Salmain, B. Rudolf, L. Jugé, V. Guérineau, O. Laprévote, A. Vessières and G. Jaouen, Macromolecules, 2007, 40, 8568–8575 CrossRef CAS.
- H. Kobayashi, S. Kawamoto, S.-K. Jo, N. Sato, T. Saga, A. Hiraga, J. Konishi, S. Hu, K. Togashi, M. W. Brechbiel and R. A. Star, Kidney Int., 2002, 61, 1980–1985 Search PubMed.
- H. Kobayashi, T. Saga, S. Kawamoto, N. Sato, A. Hiraga, T. Ishimori, J. Konishi, K. Togashi and M. W. Brechbiel, Cancer Res., 2001, 61, 4966–4970 CAS.
- M. Saad, O. B. Garbuzenko, E. Ber, P. Chandna, J. J. Khandare, V. P. Pozharov and T. Minko, J. Controlled Release, 2008, 130, 107–114 Search PubMed.
- G. R. Newkome, Z. Yao, G. R. Baker and V. K. Gupta, J. Org. Chem., 1985, 50, 2003–2004 CrossRef CAS.
- M. Fischer and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 884–905 CrossRef.
- E. M. M. de Brabander-van den Berg and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1993, 32, 1308–1311 CrossRef.
- P. Antoni, D. Nystrom, C. J. Hawker, A. Hult and M. Malkoch, Chem. Commun., 2007, 2249–2251 RSC.
- C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef CAS.
- S. J. E. Mulders, A. J. Brouwer, P. G. J. van der Meer and R. M. J. Liskamp, Tetrahedron Lett., 1997, 38, 631–634 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. J. Joralemon, R. K. O'Reilly, J. B. Matson, A. K. Nugent, C. J. Hawker and K. L. Wooley, Macromolecules, 2005, 38, 5436–5443 CrossRef CAS.
- P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3932 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- M. Köhn and R. Breinbauer, Angew. Chem., Int. Ed., 2004, 43, 3106–3116 CrossRef.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS.
- G. Franc and A. K. Kakkar, Chem.–Eur. J., 2009, 15, 5630–5639 CrossRef CAS.
- P. Wu, M. Malkoch, J. N. Hunt, R. Vestberg, E. Kaltgrad, M. G. Finn, V. V. Fokin, K. B. Sharpless and C. J. Hawker, Chem. Commun., 2005, 5775–5777 RSC.
- A. P. Goodwin, S. S. Lam and J. M. J. Fréchet, J. Am. Chem. Soc., 2007, 129, 6994–6995 CrossRef CAS.
- P. Antoni, Y. Hed, A. Nordberg, D. Nyström, H. von Holst, A. Hult and M. Malkoch, Angew. Chem., Int. Ed., 2009, 48, 2126–2130 CrossRef CAS.
- C. Ornelas and M. Weck, Chem. Commun., 2009, 5710–5712 RSC.
- J. E. Macdonald, J. A. Kelly and J. G. C. Veinot, Langmuir, 2007, 23, 9543–9545 CrossRef CAS.
- C. D. Hein, X. M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS.
- R. K. Tekade, P. V. Kumar and N. K. Jain, Chem. Rev., 2008, 109, 49–87 Search PubMed.
- D. G. Mullen, E. L. Borgmeier, M. Fang, D. Q. McNerny, A. Desai, J. R. Baker, B. G. Orr and M. M. Banaszak Holl, Macromolecules, 2010, 43, 6577–6587 Search PubMed.
- R. S. Navath, A. R. Menjoge, B. Wang, R. Romero, S. Kannan and R. M. Kannan, Biomacromolecules, 2010, 11, 1544–1563 CrossRef CAS.
- T. Okuda, S. Kawakami, T. Maeie, T. Niidome, F. Yamashita and M. Hashida, J. Controlled Release, 2006, 114, 69–77 CrossRef CAS.
- M. L. Patil, M. Zhang, O. Taratula, O. B. Garbuzenko, H. He and T. Minko, Biomacromolecules, 2009, 10, 258–266 CrossRef CAS.
- L. M. Kaminskas, B. J. Boyd, P. Karellas, G. Y. Krippner, R. Lessene, B. Kelly and C. J. H. Porter, Mol. Pharmaceutics, 2008, 5, 449–463 CrossRef CAS.
- K. M. Kitchens, M. E. H. El-Sayed and H. Ghandehari, Adv. Drug Delivery Rev., 2005, 57, 2163–2176 CrossRef CAS.
- I. J. Majoros, B. Keszler, S. Woehler, T. Bull and J. R. Baker, Macromolecules, 2003, 36, 5526–5529 CrossRef CAS.
- I. J. Majoros, T. P. Thomas, C. B. Mehta and J. R. Baker, J. Med. Chem., 2005, 48, 5892–5899 CrossRef CAS.
- R. Qi, Y. Gao, Y. Tang, R.-R. He, T.-L. Liu, Y. He, S. Sun, B.-Y. Li, Y.-B. Li and G. Liu, AAPS J., 2009, 11, 395–405 Search PubMed.
- R. B. Kolhatkar, K. M. Kitchens, P. W. Swaan and H. Ghandehari, Bioconjugate Chem., 2007, 18, 2054–2060 CrossRef CAS.
- C. Waite, S. Sparks, K. Uhrich and C. Roth, BMC Biotechnol., 2009, 9, 38 CrossRef.
- V. Gajbhiye, P. Vijayaraj Kumar, R. K. Tekade and N. K. Jain, Eur. J. Med. Chem., 2009, 44, 1155–1166 Search PubMed.
- D. Bhadra, A. K. Yadav, S. Bhadra and N. K. Jain, Int. J. Pharm., 2005, 295, 221–233 Search PubMed.
- V. Gajbhiye, P. V. Kumar, R. K. Tekade and N. K. Jain, Curr. Pharm. Des., 2007, 13, 415–429 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS.
- A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5–32 CrossRef CAS.
- T. Higashihara, T. Sakurai and A. Hirao, Macromolecules, 2009, 42, 6006–6014 CrossRef CAS.
- A. Hirao, M. Hayashi, S. Loykulnant, K. Sugiyama, S. W. Ryu, N. Haraguchi, A. Matsuo and T. Higashihara, Prog. Polym. Sci., 2005, 30, 111–182 CrossRef CAS.
- N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 857–871 CrossRef CAS.
- H. Iatrou and N. Hadjichristidis, Macromolecules, 1993, 26, 2479–2484 CrossRef CAS.
- A. Mavroudis and N. Hadjichristidis, Macromolecules, 2005, 39, 535–540 Search PubMed.
- S. Christodoulou, P. Driva, H. Iatrou and N. Hadjichristidis, Macromolecules, 2008, 41, 2607–2615 CrossRef CAS.
- S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Slomkowski, Prog. Polym. Sci., 2007, 32, 247–282 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- K. Ohno, B. Wong and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2206–2214 CrossRef CAS.
- D.-H. Han and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 789–799 CrossRef CAS.
- W. Yuan, J. Yuan, M. Zhou and C. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2788–2798 CrossRef CAS.
- M. Le Hellaye, C. Lefay, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3058–3067 CrossRef CAS.
- A. Vora, K. Singh and D. C. Webster, Polymer, 2009, 50, 2768–2774 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 11828–11834 CrossRef CAS.
- J. Du and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2263–2271 CrossRef CAS.
- H. Gao, S. Ohno and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 15111–15113 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 7216–7223 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2008, 41, 4250–4257 CrossRef CAS.
- R. Hoogenboom, B. C. Moore and U. S. Schubert, Chem. Commun., 2006, 4010–4012 RSC.
- P.-F. Gou, W.-P. Zhu, N. Xu and Z.-Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6455–6465 CrossRef CAS.
- Z. Ge, J. Xu, J. Hu, Y. Zhang and S. Liu, Soft Matter, 2009, 5, 3932–3939 RSC.
- P.-F. Gou, W.-P. Zhu, N. Xu and Z.-Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2961–2974 Search PubMed.
- C. Li, Z. Ge, H. Liu and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4001–4013 CrossRef CAS.
- A. Gozgen, A. Dag, H. Durmaz, O. Sirkecioglu, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 497–504 CrossRef CAS.
- Y. Zhang, H. Liu, H. Dong, C. Li and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1636–1650 CrossRef CAS.
- Q. Fu, G. Wang, W. Lin and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 986–990 CrossRef CAS.
- H. Durmaz, A. Dag, E. Erdogan, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 99–108 CrossRef CAS.
- K. Khanna, S. Varshney and A. Kakkar, Macromolecules, 2010, 43, 5688–5698 CrossRef CAS.
- G. M. Soliman, R. Sharma, A. O. Choi, S. K. Varshney, F. M. Winnik, A. K. Kakkar and D. Maysinger, Biomaterials, 2010, 31, 8382–8392 Search PubMed.
- G. Deng, D. Ma and Z. Xu, Eur. Polym. J., 2007, 43, 1179–1187 CrossRef CAS.
- M. R. Whittaker, C. N. Urbani and M. J. Monteiro, J. Am. Chem. Soc., 2006, 128, 11360–11361 CrossRef CAS.
- P.-F. Gou, W.-P. Zhu, N. Xu and Z.-Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6962–6976 CrossRef CAS.
- P.-F. Gou, W.-P. Zhu, N. Zhu and Z.-Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2905–2916 CrossRef CAS.
- Y. Zhang, C. Li and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3066–3077 CrossRef CAS.
- A. Dag, H. Durmaz, O. Sirkecioglu, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2344–2351 Search PubMed.
- A. Dag, H. Durmaz, V. Kirmizi, G. Hizal and U. Tunca, Polym. Chem., 2010, 1, 621–623 RSC.
- Z.-M. Wu, H. Liang, J. Lu and W.-L. Deng, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3323–3330 Search PubMed.
- J. Hu, Y. Cheng, Y. Ma, Q. Wu and T. Xu, J. Phys. Chem. B, 2008, 113, 64–74 Search PubMed.
- J. Hu, Y. Cheng, Q. Wu, L. Zhao and T. Xu, J. Phys. Chem. B, 2009, 113, 10650–10659 CrossRef CAS.
- L. Zhao, Y. Cheng, J. Hu, Q. Wu and T. Xu, J. Phys. Chem. B, 2009, 113, 14172–14179 CrossRef CAS.
- X. Feng, Y. Cheng, K. Yang, J. Zhang, Q. Wu and T. Xu, J. Phys. Chem. B, 2010, 114, 11017–11026 Search PubMed.
- A. M. Naylor, W. A. Goddard, G. E. Keifer and D. A. Tomalia, J. Am. Chem. Soc., 1989, 111, 2339 CrossRef.
- M. T. Morgan, M. A. Carnahan, C. E. Immoos, A. A. Ribeiro, S. Finkelstein, S. J. Lee and M. W. Grinstaff, J. Am. Chem. Soc., 2003, 125, 15485 CrossRef CAS.
- Y. Cheng, Q. Wu, Y. Li and T. Xu, J. Phys. Chem. B, 2008, 112, 8884–8890 CrossRef CAS.
- S. Svenson and A. S. Chauhan, Nanomedicine, 2008, 3, 679–702 CrossRef CAS.
- P. Sharma and S. Garg, Adv. Drug Delivery Rev., 2010, 62, 491–502 CrossRef CAS.
- C. Kojima, K. Kono, K. Maruyama and T. Takagishi, Bioconjugate Chem., 2000, 11, 910 CrossRef CAS.
- A. K. Patri, J. F. Kukowska-Latallo and J. R. Baker, Adv. Drug Delivery Rev., 2005, 57, 2203 CrossRef CAS.
- M. T. Morgan, Y. Nakanishi, D. J. Kroll, A. P. Griset, M. A. Carnahan, M. Wathier, N. H. Oberlies, G. Manikumar, M. C. Wani and M. W. Grinstaff, Cancer Res., 2006, 66, 11913 CrossRef CAS.
- D. Bhadra, S. Bhadra, S. Jain and N. K. Jain, Int. J. Pharm., 2003, 257, 111–124 CrossRef CAS.
- K. Gardikis, S. Hatziantoniou, M. Bucos, D. Fessas, M. Signorelli, T. Felekis, M. Zervou, C. G. Screttas, B. R. Steele, M. Ionov, M. Micha-Screttas, B. Klajnert, M. Bryszewska and C. Demetzos, J. Pharm. Sci., 2010, 99, 3561–3571 CrossRef CAS.
- J. Babin, D. Taton, M. Brinkmann and S. Lecommandoux, Macromolecules, 2008, 41, 1384–1392 CrossRef CAS.
- F. Nederberg, E. Appel, J. P. K. Tan, S. H. Kim, K. Fukushima, J. Sly, R. D. Miller, R. M. Waymouth, Y. Y. Yang and J. L. Hedrick, Biomacromolecules, 2009, 10, 1460–1468 CrossRef CAS.
- H. Yin, S.-W. Kang and Y. H. Bae, Macromolecules, 2009, 42, 7456–7464 CrossRef CAS.
- T. P. Lodge, A. Rasdal, Z. B. Li and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 17608–17609 CrossRef CAS.
- F. Wang, T. K. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2005, 16, 397–405 CrossRef CAS.
- F. Wang, T. K. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2008, 19, 1423–1429 CrossRef CAS.
- J. Yun, R. Faust, L. S. Szilágyi, S. Kéki and M. Zsuga, Macromolecules, 2003, 36, 1717–1723 CrossRef CAS.
- S. Pispas, N. Hadjichristidis, I. Potemkin and A. Khokhlov, Macromolecules, 2000, 33, 1741–1746 CrossRef CAS.
- P.-F. Gou, W.-P. Zhu and Z.-Q. Shen, Biomacromolecules, 2010, 11, 934–943 CrossRef CAS.
- E. Güç, G. Gündüz and U. Gündüz, Drug Dev. Ind. Pharm., 2010, 36, 1139–1148 Search PubMed.
- M. R. Radowski, A. Shukla, H. von Berlepsch, C. Böttcher, G. Pickaert, H. Rehage and R. Haag, Angew. Chem., Int. Ed., 2007, 46, 1265–1269 CrossRef CAS.
- W. Tian, X. Fan, J. Kong, Y. Liu, T. Liu and Y. Huang, Polymer, 2010, 51, 2556–2564 CrossRef CAS.
- S. Gurdag, J. Khandare, S. Stapels, L. H. Matherly and R. M. Kannan, Bioconjugate Chem., 2006, 17, 275–283 CrossRef CAS.
- R. Shukla, et al. , Nanotechnology, 2008, 19, 295102 CrossRef.
- M. Najlah, S. Freeman, D. Attwood and A. D'Emanuele, Int. J. Pharm., 2006, 308, 175–182 CrossRef CAS.
- Y. E. Kurtoglu, M. K. Mishra, S. Kannan and R. M. Kannan, Int. J. Pharm., 2010, 384, 189–194 CrossRef CAS.
- S.-T. Lo, S. Stern, J. D. Clogston, J. Zheng, P. P. Adiseshaiah, M. Dobrovolskaia, J. Lim, A. K. Patri, X. Sun and E. E. Simanek, Mol. Pharmaceutics, 2010, 7, 993–1006 Search PubMed.
- Y. E. Kurtoglu, R. S. Navath, B. Wang, S. Kannan, R. Romero and R. M. Kannan, Biomaterials, 2009, 30, 2112–2121 Search PubMed.
- M. Najlah, S. Freeman, D. Attwood and A. D'Emanuele, Int. J. Pharm., 2007, 336, 183–190 CrossRef CAS.
- X. Bi, X. Shi, I. J. Majoros, R. Shukla and J. R. Baker, J. Comput. Theor. Nanosci., 2007, 4, 1179–1187 CAS.
- A. Quintana, E. Raczka, L. Piehler, I. Lee, A. Myc, I. Majoros, A. Patri, T. Thomas, J. Mulé and J. Baker, Pharm. Res., 2002, 19, 1310–1316 CrossRef CAS.
- T. P. Thomas, I. J. Majoros, A. Kotlyar, J. F. Kukowska-Latallo, A. Bielinska, A. Myc and J. R. Baker, J. Med. Chem., 2005, 48, 3729–3735 CrossRef CAS.
- J. J. Khandare, S. Jayant, A. Singh, P. Chandna, Y. Wang, N. Vorsa and T. Minko, Bioconjugate Chem., 2006, 17, 1464–1472 CrossRef CAS.
- L. M. Kaminskas, B. D. Kelly, V. M. McLeod, B. J. Boyd, G. Y. Krippner, E. D. Williams and C. J. H. Porter, Mol. Pharmaceutics, 2009, 6, 1190–1204 CAS.
- S. Zhu, M. Hong, L. Zhang, G. Tang, Y. Jiang and Y. Pei, Pharm. Res., 2010, 27, 161–174 CrossRef CAS.
- C. C. Lee, E. R. Gillies, M. E. Fox, S. J. Guillaudeu, J. M. J. Fréchet, E. E. Dy and F. C. Szoka, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16649–16654 CrossRef CAS.
- D. G. van der Poll, H. M. Kieler-Ferguson, W. C. Floyd, S. J. Guillaudeu, K. Jerger, F. C. Szoka and J. M. Fréchet, Bioconjugate Chem., 2010, 21, 764–773 CrossRef CAS.
- S. Zhu, M. Hong, G. Tang, L. Qian, J. Lin, Y. Jiang and Y. Pei, Biomaterials, 2010, 31, 1360–1371 CrossRef CAS.
- M. E. Fox, S. Guillaudeu, J. M. J. Fréchet, K. Jerger, N. Macaraeg and F. C. Szoka, Mol. Pharmaceutics, 2009, 6, 1562–1572 CAS.
- G. Thiagarajan, A. Ray, A. Malugin and H. Ghandehari, Pharm. Res., 2010, 27, 2307–2316 Search PubMed.
- X. Wang, R. Inapagolla, S. Kannan, M. Lieh-Lai and R. M. Kannan, Bioconjugate Chem., 2007, 18, 791–799 CrossRef CAS.
- O. Perumal, J. Khandare, P. Kolhe, S. Kannan, M. Lieh-Lai and R. M. Kannan, Bioconjugate Chem., 2009, 20, 842–846 CrossRef CAS.
- X. Li, M. Lu, Q. Wu, D.-s. Lv and X.-F. Lin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 117–126 Search PubMed.
- Z. Xie, X. Hu, X. Chen, J. Sun, Q. Shi and X. Jing, Biomacromolecules, 2007, 9, 376–380 Search PubMed.
- J. Liu, P. Zahedi, F. Zeng and C. Allen, J. Pharm. Sci., 2008, 97, 3274–3290 CrossRef CAS.
- W.-C. Shen and H. J. P. Ryser, Biochem. Biophys. Res. Commun., 1981, 102, 1048–1054 CrossRef CAS.
- S. J. Guillaudeu, M. E. Fox, Y. M. Haidar, E. E. Dy, F. C. Szoka and J. M. J. Fréchet, Bioconjugate Chem., 2008, 19, 461–469 CrossRef CAS.
- K. Kono, C. Kojima, N. Hayashi, E. Nishisaka, K. Kiura, S. Watarai and A. Harada, Biomaterials, 2008, 29, 1664–1675 CrossRef CAS.
- H. R. Ihre, O. L. Padilla De Jesús, F. C. Szoka and J. M. J. Fréchet, Bioconjugate Chem., 2002, 13, 443–452 CrossRef CAS.
- H. Yuan, K. Luo, Y. Lai, Y. Pu, B. He, G. Wang, Y. Wu and Z. Gu, Mol. Pharmaceutics, 2010, 7, 953–962 CAS.
- O. L. Padilla De Jesús, H. R. Ihre, L. Gagne, J. M. J. Fréchet and F. C. Szoka, Bioconjugate Chem., 2002, 13, 453–461 CrossRef CAS.
- A. Gopin, S. Ebner, B. Attali and D. Shabat, Bioconjugate Chem., 2006, 17, 1432–1440 CrossRef CAS.
- S. R. MacEwan, D. J. Callahan and A. Chilkoti, Nanomedicine, 2010, 5, 793–806 CrossRef CAS.
- M. C. Garnett, Adv. Drug Delivery Rev., 2001, 53, 171–216 Search PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- S. A. Skinner, P. J. M. Tutton and P. E. O'Brien, Cancer Res., 1990, 50, 2411–2417 Search PubMed.
- K. Greish, J. Fang, T. Inutsuka, A. Nagamitsu and H. Maeda, Clin. Pharmacokinet., 2003, 42, 1089–1105 CrossRef CAS.
- H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CrossRef CAS.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS.
- S. Stolnik, L. Illum and S. S. Davis, Adv. Drug Delivery Rev., 1995, 16, 195–214 CrossRef CAS.
- V. C. F. Mosqueira, P. Legrand, A. Gulik, O. Bourdon, R. Gref, D. Labarre and G. Barratt, Biomaterials, 2001, 22, 2967–2979 CrossRef CAS.
- F. Ravenelle, S. Gori, D. Le Garrec, D. Lessard, L. Luo, D. Palusova, J. Sneyd and D. Smith, Pharm. Res., 2008, 25, 313–319 Search PubMed.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403–419 CrossRef CAS.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS.
- L. M. Kaminskas, Z. Wu, N. Barlow, G. Y. Krippner, B. J. Boyd and C. J. H. Porter, J. Pharm. Sci., 2009, 98, 3871–3875 CrossRef CAS.
- C. Rippe, D. Asgeirsson, D. Venturoli, A. Rippe and B. Rippe, Kidney Int., 2006, 69, 1326–1332 Search PubMed.
- H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS.
- S. Sadekar, A. Ray, M. Janàt-Amsbury, C. M. Peterson and H. Ghandehari, Biomacromolecules, 2010, 12, 88–96 Search PubMed.
- S. Uppuluri, S. E. Keinath, D. A. Tomalia and P. R. Dvornic, Macromolecules, 1998, 31, 4498–4510 CrossRef CAS.
- D. A. Tomalia, A. M. Naylor and W. A. Goddard, Angew. Chem., Int. Ed. Engl., 1990, 29, 138–175 CrossRef.
- R. Scherrenberg, B. Coussens, P. van Vliet, G. Edouard, J. Brackman, E. de Brabander and K. Mortensen, Macromolecules, 1998, 31, 456–461 CrossRef CAS.
- S. Nigavekar, L. Sung, M. Llanes, A. El-Jawahri, T. Lawrence, C. Becker, L. Balogh and M. Khan, Pharm. Res., 2004, 21, 476–483 Search PubMed.
- N. Malik, R. Wiwattanapatapee, R. Klopsch, K. Lorenz, H. Frey, J. W. Weener, E. W. Meijer, W. Paulus and R. Duncan, J. Controlled Release, 2000, 65, 133–148 CrossRef CAS.
- M. Bernfield, M. Gotte, P. W. Park, O. Reizes, M. L. Fitzgerald, J. Lincecum and M. Zako, Annu. Rev. Biochem., 1999, 68, 729–777 CrossRef CAS.
- B. J. Boyd, L. M. Kaminskas, P. Karellas, G. Krippner, R. Lessene and C. J. H. Porter, Mol. Pharmaceutics, 2006, 3, 614–627 Search PubMed.
- J. F. Kukowska-Latallo, K. A. Candido, Z. Cao, S. S. Nigavekar, I. J. Majoros, T. P. Thomas, L. P. Balogh, M. K. Khan and J. R. Baker, Cancer Res., 2005, 65, 5317–5324 CrossRef CAS.
- L. M. Kaminskas, B. D. Kelly, V. M. McLeod, G. Sberna, D. J. Owen, B. J. Boyd and C. J. H. Porter, J. Controlled Release, 2011 DOI:10.1016/j.jconrel.2011.02.005.
- D. A. Tomalia, L. A. Reyna and S. Svenson, Biochem. Soc. Trans., 2007, 35, 61–67 CrossRef CAS.
- J. Luo, K. Xiao, Y. Li, J. S. Lee, L. Shi, Y.-H. Tan, L. Xing, R. Holland Cheng, G.-Y. Liu and K. S. Lam, Bioconjugate Chem., 2010, 21, 1216–1224 CrossRef CAS.
- K. Xiao, J. Luo, W. L. Fowler, Y. Li, J. S. Lee, L. Xing, R. H. Cheng, L. Wang and K. S. Lam, Biomaterials, 2009, 30, 6006–6016 CrossRef CAS.
- K. Zhang, Y. Wang, A. Yu, Y. Zhang, H. Tang and X. X. Zhu, Bioconjugate Chem., 2010, 21, 1596–1601 CrossRef CAS.
- A. S. Chauhan, N. K. Jain, P. V. Diwan and A. J. Khopade, J. Drug Targeting, 2004, 12, 575–583 Search PubMed.
- A. Asthana, A. S. Chauhan, P. V. Diwan and N. K. Jain, AAPS PharmSciTech, 2005, 6, E536–E542 Search PubMed.
- H. Dai, R. S. Navath, B. Balakrishnan, B. R. Guru, M. K. Mishra, R. Romero, R. M. Kannan and S. Kannan, Nanomedicine, 2010, 5, 1317–1329 Search PubMed.
- Y. Zhang, T. P. Thomas, A. Desai, H. Zong, P. R. Leroueil, I. J. Majoros and J. R. Baker, Bioconjugate Chem., 2010, 21, 489–495 CrossRef CAS.
- Z. Sideratou, C. Kontoyianni, G. I. Drossopoulou and C. M. Paleos, Bioorg. Med. Chem. Lett., 2010, 20, 6513–6517 Search PubMed.
- W. Yang, Y. Cheng, T. Xu, X. Wang and L.-p. Wen, Eur. J. Med. Chem., 2009, 44, 862–868 CrossRef CAS.
- H. Yang and W. J. Kao, Int. J. Nanomed., 2007, 2, 89–99 Search PubMed.
- A. Myc, A. K. Patri and J. R. Baker, Biomacromolecules, 2007, 8, 2986–2989 CrossRef CAS.
- L. Han, R. Huang, S. Liu, S. Huang and C. Jiang, Mol. Pharmaceutics, 2010, 7, 2156–2165 Search PubMed.
- C. Wängler, G. Moldenhauer, M. Eisenhut, U. Haberkorn and W. Mier, Bioconjugate Chem., 2008, 19, 813–820 Search PubMed.
- T. Miyano, W. Wijagkanalan, S. Kawakami, F. Yamashita and M. Hashida, Mol. Pharmaceutics, 2010, 7, 1318–1327 Search PubMed.
- I. J. Majoros, A. Myc, T. Thomas, C. B. Mehta and J. R. Baker, Biomacromolecules, 2006, 7, 572–579 CrossRef CAS.
- A. K. Patri, J. F. Kukowska-Latallo and J. J. R. Baker, Adv. Drug Delivery Rev., 2005, 57, 2203–2214 CrossRef CAS.
- H. He, Y. Li, X.-R. Jia, J. Du, X. Ying, W.-L. Lu, J.-N. Lou and Y. Wei, Biomaterials, 2011, 32, 478–487 Search PubMed.
- L. E. Samuelson, M. J. Dukes, C. R. Hunt, J. D. Casey and D. J. Bornhop, Bioconjugate Chem., 2009, 20, 2082–2089 Search PubMed.
- M. Calderón, M. A. Quadir, M. Strumia and R. Haag, Biochimie, 2010, 92, 1242–1251 CrossRef CAS.
- V. Gajbhiye, P. V. Kumar, A. Sharma and N. K. Jain, Curr. Nanosci., 2008, 4, 267–277 Search PubMed.
- V. Gajbhiye, P. V. Kumar, R. K. Tekade and N. K. Jain, Eur. J. Med. Chem., 2009, 44, 1155–1166 Search PubMed.
- W. Yuan, J. Yuan, S. Zheng and X. Hong, Polymer, 2007, 48, 2585–2594 CrossRef CAS.
- S. Ki Choi, T. Thomas, M.-H. Li, A. Kotlyar, A. Desai and J. J. R. Baker, Chem. Commun., 2010, 46, 2632–2634 RSC.
- K. Haba, M. Popkov, M. Shamis, R. A. Lerner, C. F. Barbas and D. Shabat, Angew. Chem., Int. Ed., 2005, 44, 716–720 CrossRef CAS.
- M. Avital-Shmilovici and D. Shabat, Soft Matter, 2010, 6, 1073–1080 RSC.
- D. V. McGrath, Mol. Pharmaceutics, 2005, 2, 253–263 CrossRef CAS.
- F. M. H. de Groot, C. Albrecht, R. Koekkoek, P. H. Beusker and H. W. Scheeren, Angew. Chem., Int. Ed., 2003, 42, 4490–4494 CrossRef CAS.
- R. J. Amir, N. Pessah, M. Shamis and D. Shabat, Angew. Chem., Int. Ed., 2003, 42, 4494–4499 CrossRef CAS.
- S. Li, M. L. Szalai, R. M. Kevwitch and D. V. McGrath, J. Am. Chem. Soc., 2003, 125, 10516–10517 CrossRef CAS.
- X. Zhang, J. Cheng, Q. Wang, Z. Zhong and R. Zhuo, Macromolecules, 2010, 43, 6671–6677 Search PubMed.
- T.-B. Ren, Y. Feng, Z.-H. Zhang, L. Li and Y.-Y. Li, Soft Matter, 2011, 7, 2329–2331 RSC.
- D. Q. McNerny, P. R. Leroueil and J. R. Baker, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 249–259 Search PubMed.
- J. C. Roberts, M. K. Bhalgat and R. T. Zera, J. Biomed. Mater. Res., 1996, 30, 53–65 CrossRef CAS.
- P. C. Naha, M. Davoren, F. M. Lyng and H. J. Byrne, Toxicol. Appl. Pharmacol., 2010, 246, 91–99 Search PubMed.
- H. B. Agashe, T. Dutta, M. Garg and N. K. Jain, J. Pharm. Pharmacol., 2006, 58, 1491–1498 Search PubMed.
- T. C. King Heiden, E. Dengler, W. J. Kao, W. Heideman and R. E. Peterson, Toxicol. Appl. Pharmacol., 2007, 225, 70–79 CrossRef.
- W. Wang, W. Xiong, J. L. Wan, X. H. Sun, H. B. Xu and X. L. Yang, Nanotechnology, 2009, 20, 7.
- K. Jain, P. Kesharwani, U. Gupta and N. K. Jain, Int. J. Pharm., 2010, 394, 122–142 CrossRef CAS.
- S. Hong, A. U. Bielinska, A. Mecke, B. Keszler, J. L. Beals, X. Shi, L. Balogh, B. G. Orr, J. R. Baker and M. M. Banaszak Holl, Bioconjugate Chem., 2004, 15, 774–782 CrossRef CAS.
- Y. Cheng, L. Zhao, Y. Li and T. Xu, Chem. Soc. Rev., 2011, 40, 2673–2703 RSC.
- C. Rao and J. P. Tam, J. Am. Chem. Soc., 1994, 116, 6975–6976 CrossRef CAS.
- K. Sadler and J. P. Tam, Rev. Mol. Biotechnol., 2002, 90, 195–229 CrossRef CAS.
- J. Ziegler, R. T. Chang and D. W. Wright, J. Am. Chem. Soc., 1999, 121, 2395–2400 CrossRef CAS.
- E. H. Nardin, J. M. Calvo-Calle, G. A. Oliveira, R. S. Nussenzweig, M. Schneider, J.-M. Tiercy, L. Loutan, D. Hochstrasser and K. Rose, J. Immunol., 2001, 166, 481–489 Search PubMed.
- P. Singh, F. Moll, 3rd, S. Lin, C. Ferzli, K. Yu, R. Koski, R. Saul and P. Cronin, Clin. Chem. (Washington, D. C.), 1994, 40, 1845–1849 CAS.
- R. Yin, M. S. Bratcher, A. L. Jenkins, R. P. Hydutsky, R. Cheng, H. D. Durst, P. A. Emanuel and G. L. Hagnauer, Abstracts of Papers, 221st ACS National Meeting, San Diego, CA, United States, April 1–5, 2001, PMSE-474.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2011 |