In situ ion exchange synthesis of the novel Ag/AgBr/BiOBr hybrid with highly efficient decontamination of pollutants†
Hefeng
Cheng
a,
Baibiao
Huang
*a,
Peng
Wang
a,
Zeyan
Wang
a,
Zaizhu
Lou
a,
Junpeng
Wang
a,
Xiaoyan
Qin
a,
Xiaoyang
Zhang
a and
Ying
Dai
b
aState Key Lab of Crystal Materials, Shandong University, Jinan 250100, People's Republic of China. E-mail: bbhuang@sdu.edu.cn
bSchool of Physics, Shandong University, People's Republic of China
First published on 3rd May 2011
Abstract
A novel Ag/AgBr/BiOBr hybrid was prepared by a rational in situ ion exchange reaction between BiOBr hierarchical microspheres and AgNO3 in ethylene glycol followed by light reduction, which displayed superior visible light driven photocatalytic activities in sterilization of pathogenic organism and degradation of organic dye compared to N-doped P25.
For decades, the decontamination of noxious substances from wastewater, such as organic pollutants and pathogenic organisms, has always been a thorny issue people are concerned about. The conventional disinfection methods, involving autoclaving and chlorine dioxide treatments, usually consume a mass of energy or ineluctably give birth to harmful by-products. As an alternative, TiO2-based heterogeneous photocatalysis seems to be more attractive for its availability of abundant solar energy and benignity to environments.1–4 However, the wide band gap (WBG) of TiO2 renders it only responsive to ultraviolet (UV) light, which accounts for a small portion of sunlight (less than 5%). Moreover, the rapid recombination of the photogenerated carriers leads to low quantum efficiency.2 To take sufficient advantage of solar energy and indoor illumination predominately concentrating in the visible-infrared range, the development of efficient visible-light-driven photocatalysts is promising and indispensable.
Owing to the correlation between physical/chemical properties and the size, shape, and structure of materials, designed syntheses of novel nano- and/or microstructured semiconductor materials provide great versatility to tailor their photocatalytic activities.5 In particular, orderly deposited nanoparticles (NPs) or quantum dots (QDs) of narrow band gap (NBG) semiconductor such as CdS and CdSe on the WBG materials with suitable energetics, are capable of harvesting the visible light absorption and improving the interfacial charge transfer.6,7 Recently, supported-AgX (X = Cl, Br, I) on SiO2, TiO2, and other substrates4,8–11 have shown considerable visible-light activities in decomposition of organic pollutants. It is attractive that silver halides could maintain the stability with the assistance of the supports, which facilitates the transfer of the photoexcited carriers and avoid the photocorrosion of silver halides. Furthermore, upon light illumination, the surface of silver halides is partially reduced to Ag NPs, which exhibit strong visible light absorption due to their localized surface plasmon resonance (SPR).12 The resultant Ag/AgX plasmonic photocatalysts, which use plasmonic noble metal NPs to sensitize the WBG semiconductor and trap the photoexcited electrons, have been developed and shown efficient photocatalytic performances as well as good stability under visible light irradiation.13 However, direct precipitation of silver halides on the supports inevitably brings about severe aggregation, poor dispersity and loose connection between them, which consequently impedes the interfacial charge transfer. In contrast, nanomaterials could endow higher surface-to-volume ratio and more reactive sites to the photocatalysts.14 Therefore, designing a rational synthetic approach to highly dispersive silver halide NPs, which possess superior activity and repeatability, is a necessity for both researches and applications.
Herein, we report a novel Ag/AgBr/BiOBr hybrid synthesized via an in situ ion exchange reaction between BiOBr hierarchical architectures and AgNO3 solution followed by light-reduction at room temperature. The BiOBr nanosheets not only provide the reactive sites for in situ ion exchange with Ag+, but also perform as clapboards to spatially separate AgBr NPs, avoiding their aggregation. Evaluated by the sterilization of Escherichia coli (E. coli) and degradation of methyl orange (MO), Ag/AgBr/BiOBr hybrid shows superior photocatalytic activity under visible irradiation (λ ≥ 400 nm), which is supposed to be associated with the effective transfer of the photogenerated carriers during the photocatalytic reaction.
Fig. 1a illustrates the synthetic procedure of the novel Ag/AgBr/BiOBr hybrid. Firstly, uniform hierarchical BiOBr microspheres were prepared by an ion liquid-assisted solvothermal method (Fig. S1, ESI†). With an average diameter of 1–2 μm, the BiOBr microspheres are constructed by numerous interlaced two-dimensional nanosheets (Fig. 1b). Secondly, through the ion exchange reaction between BiOBr and Ag+ in ethylene glycol, AgBr NPs deposited on the surface of BiOBr nanosheets with high dispersity, while the produced BiO+ ions dissolved in the solvent (BiOBr + Ag+ → AgBr↓ + BiO+). As AgBr NPs were produced in situ on the surface of BiOBr nanosheets, an intimate connection between them was established. Finally, under visible irradiation AgBr NPs were partially reduced to Ag0 species and Ag/AgBr NPs with 20–50 nm in size are uniformly anchored on the BiOBr nanosheets (Fig. 1c). To demonstrate the role of BiOBr hierarchical microspheres in the modulation of AgBr NPs, AgBr samples were also prepared by direct precipitation and ion exchange with BiOBr nanoplates (see details in the ESI†). As shown in Fig. S2†, AgBr particles with poor dispersity and aggregation were observed. Consequently, the BiOBr hierarchical architecture offers a favorable scaffold to in situconstruct Ag/AgBr/BiOBr hybrid.
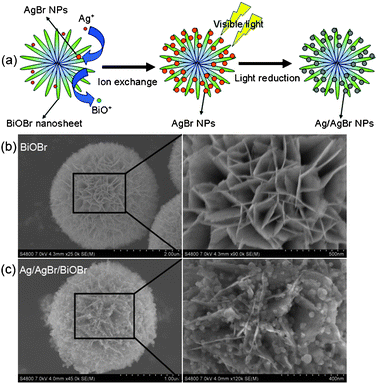 | ||
| Fig. 1 (a) The schematic synthetic route to Ag/AgBr/BiOBr hybrid. Typical SEM images of (b) BiOBr, and (c) Ag/AgBr/BiOBr hybrid. | ||
The structure of Ag/AgBr/BiOBr hybrid was investigated by X-ray diffraction (XRD, Fig. S3, ESI†), which reveals the coexistence of tetragonal BiOBr (JCPDS No. 73-2061) and cubic AgBr (JCPDS No. 6-0438). No diffraction peaks assigned to Ag0 are observed, probably due to its low content and high dispersity.10Fig. 2A shows the UV–Vis diffuse reflectance spectra of the samples. In contrast to BiOBr that has an absorption edge at ca. 460 nm, Ag/AgBr/BiOBr hybrid exhibits strong absorption in the visible range, even extending to near infrared (NIR) region. The remarkable photoabsorption enhancement of Ag/AgBr/BiOBr hybrid stems from the SPR of Ag NPs, further confirming the presence of Ag0 species.13 Moreover, X-ray photoelectron spectroscopy (XPS) results show that both Ag 3d5/2 and 3d3/2 peaks (Fig. 2B) can be divided into two separate peaks. Among them, the peaks at 367.6 and 373.7 eV are attributed to Ag+ in AgBr, and those at 368.1 and 374.1 eV are assigned to Ag0 species.9 The Br 3d XPS peaks (Fig. S4, ESI†) can also be resolved into two typical peaks, of which 68.3 and 69.3 eV are ascribed to BiOBr; whilst the peaks at 69.8 and 68.9 eV for AgBr.9 In addition, two peaks at 159.35 and 164.65 eV are separately attributed to Bi 4f7/2 and Bi 4f5/2 of Bi3+ in BiOBr (Fig. S5, ESI†). On the basis of XPS peak areas, the surface Ag0 and Ag+ contents are individually calculated to be 2.38 and 17.56 at %.
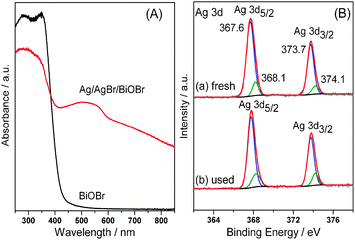 | ||
| Fig. 2 (A) UV–Vis diffuse reflectance spectra of BiOBr and Ag/AgBr/BiOBr hybrid. (B) Ag 3d XPS spectra of (a) fresh Ag/AgBr/BiOBr hybrid and (b) Ag/AgBr/BiOBr used after five cycling experiments. | ||
E. coli, which is typically gram-negative, was employed as a model bacterium to evaluate the bactericidal property of the samples. As shown in Fig. 3a, Ag/AgBr/BiOBr hybrid displays highly efficient anti-bacterial activity, for which about 75% percentage is inactivated under visible irradiation for only 3 min. and all are killed within 9 min. Neither photolysis (without photocatalyst) nor catalysis (without light) shows bactericidal effects on E. coli, suggesting that anti-bacterial activity is induced by photocatalysis. For comparison, Ag/AgBr sample, which was obtained by photoreduction of AgBr derived from direct precipitation (see the ESI†), could inactivate about 42% amount of E. coli. However, only about 15% percentage is inactivated over N-doped P25 after irradiation for 30 min. It is generally believed that the inactivation of E. coli is due to the oxidative species (e.g., O2−, hVB+) produced by photocatalyst under irradiation, which could oxidize the cell membranes and afterwards kill the cell.4 The photocatalytic property was also examined by MO degradation (Fig. 3b). In the absence of photocatalyst, MO self-degradation is almost negligible. It is worth noting that Ag/AgBr/BiOBr hybrid displays superior photocatalytic activity and the insert of Fig. 3b shows the corresponding color changes of MO solution, in which about 93% of MO dye molecules are bleached only in 4 min. While in the case of Ag/AgBr, about 65% of MO dye molecules could be bleached under visible irradiation for 60 min. Assuming that the photodegradation procedure belongs to pseudo-first-order reaction, MO degradation rate over Ag/AgBr/BiOBr (0.396 min−1) is sixteen times faster than Ag/AgBr (0.024 min−1), and faster than N-doped P25 (0.007 min−1) by a factor of fifty-six as well.
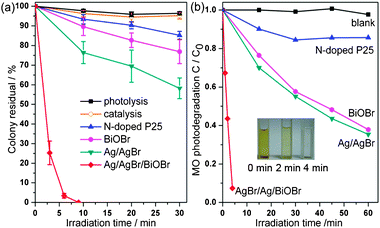 | ||
| Fig. 3 Photocatalytic (a) inactivation of E. coli and (b) degradation of MO under visible light irradiation (λ ≥ 400 nm). The insert demonstrates the color changes of MO over Ag/AgBr/BiOBr hybrid. | ||
In view of practical applications, besides efficiency, the stability and durability are also indispensable to photocatalysts. In our recycling experiments of MO photodegradation (Fig. S6, ESI†), Ag/AgBr/BiOBr hybrid exhibits an inconspicuous decrease after five cycles. Furthermore, the XRD patterns (Fig. S3, ESI†) and Ag 3d XPS spectra (Fig. 2B) of Ag/AgBr/BiOBr hybrid before and after recycling experiments are almost identical. The stability in structure and property ensures that Ag/AgBr/BiOBr hybrid can be used as an efficient and stable visible-light-driven photocatalyst.
It is fascinating to study the plausible reaction mechanism for the superior photocatalytic activity of Ag/AgBr/BiOBr hybrid. Band calculations (see the ESI†) elucidate that BiOBr and AgBr have matching band potentials, which facilitates the transfer of the photoexcited carriers between the two semiconductors. Given the aforementioned experimental and theoretical results, a plasmon-induced electron transfer process was proposed and illustrated in Scheme 1. Under visible light irradiation, photogenerated electron-hole pairs are formed on the surface of Ag-NPs owing to the SPRs. The electrons transfer from the photoexcited Ag-NPs (labeled as Ag-NPs*) to the conduction band (CB) of AgBr.10 Since the CB of AgBr is more negative than that of BiOBr, the electrons would sequentially migrate to the surface of BiOBr, which can be trapped by molecular oxygen in solution to form O2− and other oxidative species.13 Once releasing the photoexcited electrons, Ag-NPs* would shift to more positive potentials to generate positively charged Ag-NPsn+, which was considered to be one of the primary active species.10 By oxidation of the organic pollutants or accepting electrons from the valence band (VB) of AgBr, Ag-NPsn+ return to the primal Ag-NPs.15 Such plasmon-induced oscillation processes of Ag-NPs upon light irradiation endow the Ag/AgBr/BiOBr hybrid light harvesting in Vis-NIR region and efficient transfer of photoexcited electrons, which ensures the highly activity and well stability of the hybrid. On the other hand, both AgBr (2.6 eV) and BiOBr (2.7 eV) can be excited by visible light. The photoinduced electrons could migrate to the surface of BiOBr from AgBr and are further trapped by O2 to form O2−, preventing its combination with the interstitial Ag+. Meanwhile, the photoinduced holes in the VB of BiOBr and AgBr could directly oxide the organic pollutants.11 These oxidative species will result in the degradation of azo dyes and inactivation of pathogenic organisms.
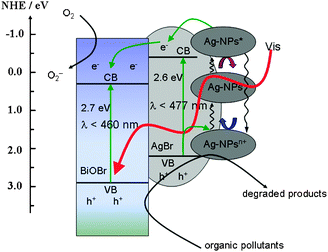 | ||
| Scheme 1 Schematic photocatalytic reaction process and charge transfer of the Ag/AgBr/BiOBr hybrid under visible light illumination. | ||
In summary, Ag/AgBr/BiOBr as a novel hybrid has been synthesized via an in situ ion exchange route. The Ag/AgBr/BiOBr hybrid, which integrates the synergetic effect of nanosized plasmonic Ag/AgBr and AgBr/BiOBr composite photocatalysts, is superior for the transfer of photogenerated electrons and holes, thereby improving the photocatalytic efficiency dramatically. Our work provides a highly efficient plasmon-induced Ag/AgBr/BiOBr hybrid photocatalyst, and elucidates a designed approach to rational semiconductor NPs/QDs-modified microstructures, which benefit the effective interfacial charge transfer and thus maximize the photocatalytic efficiency.
This work was financially supported by the National Basic Research Program of China (No. 2007CB613302), the National Natural Science Foundation of China (Nos. 20973102, 51021062, 51002091).
Notes and references
- T. Matsunaga, R. Tomoda, T. Nakajima and H. Wake, FEMS Microbiol. Lett., 1985, 29, 211 CrossRef CAS.
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- M. R. Elahifard, S. Rahimnejad, S. Haghighi and M. R. Gholami, J. Am. Chem. Soc., 2007, 129, 9552 CrossRef CAS.
- Y. Q. Lan, C. Hu, X. X. Hu and J. H. Qu, Appl. Catal., B, 2007, 73, 354 CrossRef CAS.
- (a) Y. W. C. Cao, R. C. Jin and C. A. Mirkin, Science, 2002, 297, 1536 CrossRef CAS; (b) H. C. Zeng, J. Mater. Chem., 2006, 16, 649 RSC; (c) H. F. Cheng, B. B. Huang, K. S. Yang, Z. Y. Wang, X. Y. Qin, X. Y. Zhang and Ying Dai, ChemPhysChem, 2010, 11, 2167 CrossRef CAS; (d) H. F. Cheng, B. B. Huang, J. B. Lu, Z. Y. Wang, B. Xu, X. Y. Qin, X. Y. Zhang and Y. Dai, Phys. Chem. Chem. Phys., 2010, 12, 15468 RSC; (e) J. Zhang, F. J. Shi, J. Lin, D. F. Chen, J. M. Gao, Z. X. Huang, X. X. Ding and C. C. Tang, Chem. Mater., 2008, 20, 2937 CrossRef CAS; (f) X. Zhang, Z. H. Ai, F. L. Jia and L. Z. Zhang, J. Phys. Chem. C, 2008, 112, 747 CrossRef CAS.
- A. Kongkanand, K. Tvrdy, K. Takechi, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2008, 130, 4007 CrossRef CAS.
- Y. Tak, H. Kim, D. Lee and K. Yong, Chem. Commun., 2008, 4585 RSC.
- J. G. Yu, G. P. Dai and B. B. Huang, J. Phys. Chem. C, 2009, 113, 16394 CrossRef CAS.
- P. Wang, B. B. Huang, X. Y. Qin, X. Y. Zhang, Y. Dai and M.-H. Whangbo, Inorg. Chem., 2009, 48, 10697 CrossRef CAS.
- C. Hu, T. W. Peng, X. X. Hu, Y. L. Nie, X. F. Zhou, J. H. Qu and H. He, J. Am. Chem. Soc., 2010, 132, 857 CrossRef CAS.
- H. F. Cheng, B. B. Huang, Y. Dai, X. Y. Qin and X. Y. Zhang, Langmuir, 2010, 26, 6618 CrossRef CAS.
- (a) G. Mie, Ann. Phys., 1908, 330, 377 CrossRef; (b) R. C. Jin, Y. W. Cao, C. A. Mirkin, K. L. Kelly, G. C. Schatz and J. G. Zheng, Science, 2001, 294, 1901 CrossRef CAS; (c) H. Y. Zhu, X. Chen, Z. F. Zheng, X. B. Ke, E. Jaatinen, J. C. Zhao, C. Guo, T. F. Xie and D. J. Wang, Chem. Commun., 2009, 7524 RSC.
- (a) P. Wang, B. B. Huang, X. Y. Qin, X. Y. Zhang, Y. Dai, J. Y. Wei and M.-H. Whangbo, Angew. Chem., Int. Ed., 2008, 47, 7931 CrossRef CAS; (b) P. Wang, B. B. Huang, X. Y. Zhang, X. Y. Qin, H. Jin, Y. Dai, Z. Y. Wang, J. Y. Wei, J. Zhan, S. Y. Wang, J. P. Wang and M.-H. Whangbo, Chem.–Eur. J., 2009, 15, 1821 CrossRef CAS; (c) Y. P. Bi and J. H. Ye, Chem. Commun., 2009, 6551 RSC; (d) Z. Z. Lou, B. B. Huang, X. Y. Qin, X. Y. Zhang, Z. Y. Wang, Z. K. Zheng, H. F. Cheng, P. Wang and Y. Dai, CrystEngComm, 2011, 13, 1789 RSC.
- (a) S. Ko and J. Jang, Angew. Chem., Int. Ed., 2006, 45, 7564 CrossRef CAS; (b) C. An, S. Peng and Y. Sun, Adv. Mater., 2010, 22, 2570 CrossRef CAS.
- P. Xu, J. Lu, T. Xu, S. M. Gao, B. B. Huang and Y. Dai, J. Phys. Chem. C, 2010, 114, 9510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Experimental details and calculations, Fig. S1–S6. See DOI: 10.1039/c1cc11525a |
| This journal is © The Royal Society of Chemistry 2011 |