Toward the synthesis of sequence-controlled vinyl copolymers†
Xinming
Tong
,
Bao-hua
Guo
and
Yanbin
Huang
*
Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: yanbin@tsinghua.edu.cn; Tel: +86-10-62797572
First published on 2nd December 2010
Abstract
An ATRA based strategy to synthesize vinyl copolymers with monomer-level sequence control is proposed. In each cycle, one allyl alcohol is added to the ATRP chain end, and then the hydroxymethyl residue is oxidized to carboxylic acid and a side group is introduced via esterification, making the new chain end active for the next cycle.
Sequence-controlled polymer synthesis is one of the ultimate goals of polymer chemistry.1–3 In analogy to proteins and nucleic acids, synthetic polymers consisting of multiple types of monomers with defined sequence may also be able to fold into regular three-dimensional structures and possess fascinating capabilities such as molecular recognition and enzymatic catalysis. However, even with the great progress of controlled polymerization chemistry in the past decades (i.e., ionic polymerization, ATRP, RAFT, etc.), we can only control the sequence of blocks of common monomers, especially vinyl monomers, but not the precise sequence at the monomer level. Recently, several studies were aimed at sequence-controlled vinyl copolymer synthesis,4–8 but a general strategy is not available.
In this communication, we propose and preliminarily tested the feasibility of such a strategy to synthesize vinyl copolymers with monomer-level sequence control. The idea is inspired by the solid-phase step-growth synthesis of proteins,9 where a monomer is added at each synthesis cycle, and the new chain end is reactivated for the next monomer addition (Scheme 1). In our approach (Scheme 2), at the beginning of each cycle, a low-activity vinyl monomer, allyl alcohol, is added at the vinyl polymer chain end via atom transfer radical addition (ATRA), as first demonstrated by Matyjaszewski and co-workers.10,11 Without stabilizing groups connected to the Br–C, the new chain end is practically inactive for further monomer addition viaATRA or ATRP (atom transfer radical polymerization). Then, the primary alcohol group is oxidized to carboxylic acid, followed by esterification to introduce specific side groups and to make the new chain end reactivated for the next ATRA reaction. By repeating this protocol with different side groups introduced at each esterification step, a vinyl copolymer with sequence of side groups controlled at the monomer level may be synthesized. It should be noted that Sawamoto and co-workers, independent from us, recently proposed a similar idea via living cationic polymerization to incorporate functional groups into a vinyl polymer chain at desired positions.12
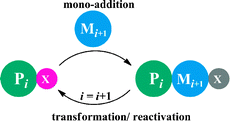 | ||
| Scheme 1 Step-growth polymerizationvia mono-additions of protected monomer. | ||
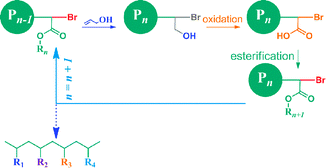 | ||
| Scheme 2 Sequence controlled polymerization utilizing allyl alcohol as the monomer precursor. | ||
In the following part, we report our proof-of-concept study to complete a full synthesis cycle (Scheme 3). Poly (methylacrylate) (PMA) was used as a polymer synthesis support, which was prepared through a typical ATRP procedure (details of all the experiments and characterizations are in ESI†). In the first step, The bromide-terminated PMA was reacted with an excess of allyl alcohol, using Cu(0)/CuBr2/Me6TREN (tris[2-(dimethylamino)ethyl]amine) as catalyst and MeOH as solvent. The ESI-MS results (Fig. 1) showed the major product (2) of this step was the expected mono-adduct of allyl alcohol and with bromide at the new chain end. The minor peak series in the ESI-MS spectrum corresponded to PMA with the bromide atom replaced by a hydrogen atom at the chain end, likely due to the known chain transfer side reaction of ATRA/ATRP after long reaction time.13–15 To quantify the yield of the targeted structure, we followed the approach of Matyjaszewski and co-workers,10,11 and modified the product mixture with trichloroacetyl isocyanate before 1H NMR analysis, and then used the NMR peaks (Fig. 1) at 4.5 ppm (d), 4.1 ppm (e) and 1.1 ppm (a) to determine that the synthesis efficiency of target structure was more than 80%.
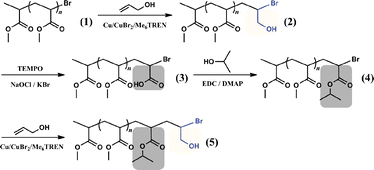 | ||
| Scheme 3 Proof-of-concept study of sequence controlled polymerization using PMA support. | ||
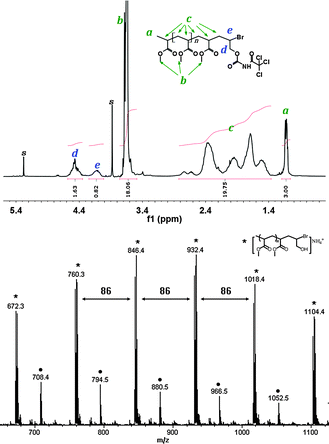 | ||
| Fig. 1 1H NMR and ESI-MS spectrum of product 2. 1H NMR analysis was carried out with addition of trichloroacetyl isocyanate to confirm the hydroxyl group, and the amount of hydroxyl functionality can be calculated from the integration ratio of peaks d and a, while the amount of Br functionality can be calculated from peaks e and a; Peak series with (*) in ESI-MS spectrum represents mono-adducts with Br end group and those with (●) represents PMA without Br end. The m/z values of ESI-MS peaks are consistent with the calculated values (difference less than 0.2 Da) and the isotope distribution patterns were confirmed. | ||
In the second step, the hydroxymethyl group at the chain end was oxidized to a carboxylic acid group, through a typical Anelli oxidation procedure, in which TEMPO was used as oxidant and NaOCl/KBr as secondary oxidant.16 To ensure the alcohol was oxidized to carboxylic acid instead of stopping at the aldehyde stage, acetonitrile–water mixture was used as solvent and excess NaOCl was used. The 1H NMR and ESI-MS results indicated that the oxidation was very effective, and the hydroxyl group was completely oxidized to carboxylic acid without affecting the bromide atom (Fig. 2).
 | ||
| Fig. 2 1H NMR and ESI-MS spectrum of product 3. (*) in the ESI-MS spectrum represents product 3 and (●) represents PMA without Br end from the previous step. The m/z values of ESI-MS peaks are consistent with the calculated values (with difference less than 0.2 Da) and the isotope distribution patterns were confirmed. | ||
Next, isopropanol was used as a model side group precursor to react with the newly-formed carboxylic acid group. The esterification was carried out using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) as condensing agent and catalyzed by dimethylaminopyridine (DMAP). The conversion efficiency was also close to complete without affecting the bromide at the chain end, as confirmed by 1H NMR and ESI-MS analysis (Fig. 3).
 | ||
| Fig. 3 1H NMR and ESI-MS spectrum of product 4. (*) in the ESI-MS spectrum represents product 4 and (●) represents PMA without Br end from the first step. The m/z values of ESI-MS peaks are consistent with the calculated values (difference less than 0.2 Da) and the isotope distribution patterns were confirmed. | ||
To complete the full cycle of synthesis, a new mono-addition of allyl alcohol was carried out at the new chain end. Loss of some Br functionality also occurred like in the first mono-addition step, but more than 80% of the product chains kept the Br atom through conversion from product 4 (calculated from the 1H NMR spectrum, shown in Fig. 4). This means there is no noticeable difference in reaction activity between two cycles of the synthesis process, and subsequent addition–transformation–functionalization cycles are feasible. The product was also confirmed by 1H NMR and ESI-MS analysis (Fig. 4).
 | ||
| Fig. 4 1H NMR and ESI-MS spectrum of product 5. 1H NMR analysis was carried out with addition of trichloroacetyl isocyanate, and the amount of hydroxyl functionality can be calculated from the integration ratio of peaks g and a, Br functionality can be calculated from d and a; (*) in the ESI-MS spectrum represents product 5 with Br end group and (■) represents the Br-loss subsidiary product of 4, while (●) represents PMA without Br end. The m/z values of ESI-MS peaks are consistent with the calculated values (difference less than 0.2 Da) and the isotope distribution patterns were confirmed. | ||
Among the three reaction steps in each synthesis cycle, the efficiency of the hydroxyl group oxidation and esterification was found close to 100% without affecting the bromide atom at the chain end. In this feasibility study, loss of bromide as a side reaction in the step of mono-addition of allyl alcohol occured and resulted in the total coversion efficiency for each synthesis cycle to be around 80%. While the efficiency of this specific synthesis route certainly needs to be improved to synthesize long polymer chains, we hope our experiment results shows the possibility to combine mono-addition and chemical transformation as a strategy for sequence-controlled vinyl copolymers.
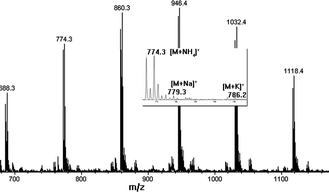
Further optimization of the reaction protocol is being made to improve the efficiency of that addition step. An alternative approach is to remove the unwanted byproduct by separation. Considering the carboxylic group of product 3 provided high polarity, it is possible to separate this main product from the less polar byproducts after the oxidation step in each cycle. This possibility was confirmed by doing a HPLC separation, and the purified product 3 was confirmed by ESI-MS analysis (Fig. 5).
In summary, one cycle of mono-addition–transformation–functionalization in the proposed step-by-step polymerization was carried out to show the possibility of such strategy to control the sequence of vinyl copolymers. It should be pointed out that allyl alcohol is just one of the possible low-activity monomers applicable to this synthesis strategy, and other routes are being investigated. After improving the mono-adduction to a higher yield by optimizing the reaction protocol, this synthesis strategy may also be realized in a solid-phase synthesis system and even possible for automation, and thus provide a true route to synthesize sequence controlled vinyl copolymers, similar as the chemical synthesis of polypeptides and nucleic acids.
This study is supported by National Natural Science Foundation of China (NSFC) through Project No. 21074064. We are grateful for the help from Dr Zhibo Li (ICCAS), Dr Chunbo Li and Dr Di Miao (Analysis Center, Tsinghua University) on mass spectroscopy characterizations, and Dr Minlian Zhang and Dr Diannan Lu for HPLC separation.
Notes and references
- N. Badi and J. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- J. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- K. Satoh, S. Ozawa, M. Mizutani, K. Nagai and M. Kamigaito, Nat. Commun., 2010, 1, 1–6 Search PubMed.
- S. Pfeifer and J. Lutz, J. Am. Chem. Soc., 2007, 129, 9542–9543 CrossRef CAS.
- S. Pfeifer, Z. Zarafshani, N. Badi and J. Lutz, J. Am. Chem. Soc., 2009, 131, 9195–9197 CrossRef CAS.
- K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120–5156 CrossRef CAS.
- S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 10808–10809 CrossRef CAS.
- K. Nakatani, T. Terashima and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 13600–13601 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- V. Coessens and K. Matyjaszewski, Macromol. Rapid Commun., 1999, 20, 127–134 CrossRef CAS.
- V. Coessens, J. Pyun, P. J. Miller, S. G. Gaynor and K. Matyjaszewski, Macromol. Rapid Commun., 2000, 21, 103–109 CrossRef CAS.
- S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3375–3381 CrossRef CAS.
- W. Lin, B. Huang, Q. Fu, G. Wang and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2991–2999 CrossRef CAS.
- S. Pioge, L. Fontaine, J. Soutif, E. Nicol and S. Pascual, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1526–1537 CrossRef CAS.
- M. Bednarek, T. Biedron and P. Kubisa, Macromol. Chem. Phys., 2000, 201, 58–66 CrossRef CAS.
- P. L. Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559–2562 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary spectra. See DOI: 10.1039/c0cc04807k |
| This journal is © The Royal Society of Chemistry 2011 |