Site-specific introduction of gold-carbenoids by intermolecular oxidation of ynamides or ynol ethers†‡
Paul W.
Davies
*,
Alex
Cremonesi
and
Nicolas
Martin
School of Chemistry, University of Birmingham, Edgbaston, B15 2TT, Birmingham, UK. E-mail: p.w.davies@bham.ac.uk; Tel: +44 (0)121 4144408
First published on 8th September 2010
Abstract
Ynamides and ynol ethers undergo intermolecular gold-catalysed reaction with a nucleophilic oxidant to access metal-carbenoid reactivity patterns. A site-specific oxidation/1,2-insertion cascade is used for a general access to functionalised α,β-unsaturated carboxylic acid derivatives and vinylogous carbimates.
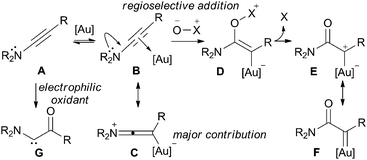
Metal carbenes, generated by the site-specific metal-promoted decomposition of a reactive functionality such as a diazo group [eqn (1), LHS], are employed extensively across an array of useful transformations.1,2 However, the need to introduce such potentially hazardous diazo functionality, often immediately prior to the carbene-based step, comes at the expense of synthetic efficiency and flexibility. In this paper we report a strategy that does not rely on the use of sacrificial functionality:3 we show that the ynamide, or ynol ether, functional group can be employed as a direct equivalent to an α,α-disubstituted-diazo imide, or ester, for regiospecific access to gold-carbenoid reactivity patterns.
 | (1) |
 | (2) |
To test our reactivity hypothesis, two ynamides were reacted in the presence of a gold catalyst and an oxidant (Scheme 1). Pyridine-N-oxide was selected as an O-nucleophilic, stable, crystalline and commercial oxidant, the by-product of which is readily removed. To our delight, products of oxidation processes were observed. For substrate 1a (R = Ph), the use of 2.2 eq. of pyridine-N-oxide led to the formation of oxoacetamide 2a in good yield. The use of stoichiometric pyridine-N-oxide with substrate 1b (R = nBu) resulted in isolation of the α,β-unsaturated carboxylic acid derivative 3b. Both transformations are consistent with the reactivity expected from the formation of a gold-carbenoid E/F, by subsequent oxidation11 or 1,2-insertion respectively.4,5b Significantly, this strategy provides the opposite regiochemistry to that previously established in oxidative ynamide chemistry (eqn (2), E/Fvs.G).12
 | ||
| Scheme 1 Gold-catalysed oxidation reactions of ynamides. Conditions: Ph3PAuCl/AgOTs (2a: 10 mol%; 3b: 5 mol%), pyridine N-oxide (2a: 2.2 eq.; 3b: 1.0 eq.), CH2Cl2, RT. | ||
While the formation of oxoacetamide 2a complements the known oxidation of ynamides with electrophilic reagents,13 the formation of α,β-unsaturated imide 3b provides a new method to access this important structural motif and building block for organic synthesis.14,15 The fact that ynamides 1 are now very readily prepared from accessible materials,16 a sulfonamide and a terminal alkyne,16c means that this approach constitutes an interesting alternative to classical disconnections for the α,β-unsaturated imides 3. We therefore decided to explore this transformation in greater detail.
Taking 1b as the test substrate, we varied reaction parameters, maintaining a just-over stoichiometric equivalence of N-oxide for efficiency (see ESI‡). An array of gold(I) and gold(III) species proved to be active for this process, whilst platinum(II) salts, Brønsted acids and an electrophilic bromine source gave no reaction and/or degradation. We chose to take forward two sets of conditions: system A uses air-stable dichloro(pyridine-2-carboxylato)gold(III) precatalyst Au-I17 in a chlorinated solvent at mildly-elevated temperature; system B avoids the use of chlorinated solvent, employing AuBr3 precatalyst at room-temperature in THF.
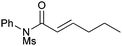
We subsequently prepared a range of ynamides to explore the scope of this transformation (Table 1). The reaction was successful using N-alkynylsulfonamides (entries 1–12) and N-alkynyloxazolidinones (entries 13–28). Both N-tosyl and N-mesityl substitution worked well, for instance 3b and 3e. Good to excellent yields were achieved across the substrates and in all cases the (E)-isomer was formed as the major, or sole product. The reaction tolerates a variety of functionality including alkyl chlorides (entries 3 and 4, 21 and 22), alkyl, benzyl and silyl ethers (entries 9 and 10, 17 and 18, 23 and 24), a phthalimido group (entries 19 and 20) and a thioester (entries 25 and 26). Synthetically valuable vinylogous carbimates 3d and 3h can also be prepared when employing substrates with an alkoxy group in the propargylic position (entries 5, 6 and 15). Similar yields were generally observed with both the catalysis systems. However, under conditions B more complex mixtures were obtained with silyl- and methyl-ether functionalised substrates 2h and 2i. In both cases, high yields of the desired product were obtained using system A (entries 15 and 17). E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶Z selectivity varied with conditions to a moderate or significant effect though system B generally afforded greater E∶Z selectivity. Trisubstituted olefins 5a and 5b can also be readily prepared in good yield applying either reaction system to both the N-alkynylsulfonamide 4a (entries 11 and 12) and the N-alkynyloxazolidinone 4b (entries 27 and 28).
∶Z selectivity varied with conditions to a moderate or significant effect though system B generally afforded greater E∶Z selectivity. Trisubstituted olefins 5a and 5b can also be readily prepared in good yield applying either reaction system to both the N-alkynylsulfonamide 4a (entries 11 and 12) and the N-alkynyloxazolidinone 4b (entries 27 and 28).

| Entry | Producta | Systemb | Yieldc [%] (E∶Z ratio) |
|
|---|---|---|---|---|
| a Mixtures of geometric isomers unless otherwise stated. b See ESI2 for reaction times. c Isolated after flash chromatography, ratio of isomers determined by 1H NMR spectroscopy. Isomers were separated for characterisation. d Only E-isomer was observed. e Major product is 3 in a complex mixture. f Starting material remaining. | ||||
| 1 |

|
3b | A | 71 (2.3∶1) |
| 2 | B | 70 (3.7∶1) |
||
| 3 |

|
3c | A | 73 (1.9∶1) |
| 4 | B | 70 (3.5∶1) |
||
| 5 |

|
3d | A | 70d |
| 6 | B | 65d | ||
| 7 |
|
3e | A | 75 (3.0∶1) |
| 8 | B | 71 (4.0∶1) |
||
| 9 |
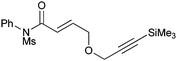
|
3f | A | 66 (2.8∶1) |
| 10 | B | 68 (3.2∶1) |
||
| 11 |

|
5a | A | 80 |
| 12 | B | 78 | ||
| 13 |

|
3g | A | 63 (3.2∶1) |
| 14 | B | 70 (5.0∶1) |
||
| 15 |
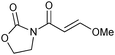
|
3h | A | 89d |
| 16 | B | —e | ||
| 17 |

|
3i | A | 75 (6.7∶1) |
| 18 | B | —e,f | ||
| 19 |
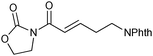
|
3j | A | 68 (5.6∶1) |
| 20 | B | 75 (6.7∶1) |
||
| 21 |
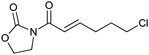
|
3k | A | 63 (2.9∶1) |
| 22 | B | 65 (4.0∶1) |
||
| 23 |
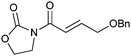
|
3l | A | 73 (2.8∶1) |
| 24 | B | 68 (3.1∶1) |
||
| 25 |

|
3m | A | 75 (2.6∶1) |
| 26 | B | 71 (7.7∶1) |
||
| 27 |

|
5b | A | 81 |
| 28 | B | 81 | ||
The reaction of 1f in particular demonstrates exquisite chemoselectivity between the ynamide and alkyne groups, whilst the retention of the silyl appendage demonstrates the mildness of both sets of conditions (entries 9 and 10).
The generality of this reactivity pattern was further explored using an ynol ether in place of the ynamides.18 Reaction of 6 afforded α,β,γ,δ-unsaturated carboxylic ester 7, predominantly as the (E,E)-isomer (Scheme 2).19
 | ||
| Scheme 2 Use of an ynol ether in the gold-catalysed oxidation reactions. | ||
The presence of a stereogenic centre in the α,β-unsaturated imide or vinylogous carbimate products of catalysis would further enhance their utility as building blocks for organic synthesis. Non-racemic chiral ynamides 8a and 8b were tested under both sets of reaction conditions. In all cases, the desired vinylogous amides 9a and 9b were formed in high yield (Scheme 3). Intriguingly, whilst simple N-alkynyloxazolidinone 3h had afforded only the (E)-isomer (Table 1, entry 13), reaction of the chiral analogue 8a saw (Z)-9a formed in appreciable quantities (Scheme 3). The reactions of the more highly substituted ynamide 8b, employed in the reaction as a 1∶1 mixture of diastereomers, diverged significantly with choice of reaction conditions.
 | ||
| Scheme 3 The oxidation/1,2-insertion reaction of chiral ynamides. | ||
Whilst the use of moderately elevated temperature and Au-I (system A) afforded (E)-9b as the major isomer, a result consistent with those observed in Table 1, the room temperature reaction using AuBr3 in THF (system B) afforded (Z)-9b as the major product. This stereoselective access to either geometric isomer of chiral β-methoxy unsaturated carboxylic imides from a mixture of diastereomers, could be of significant utility in synthesis.‡
In summary, we have demonstrated the gold-catalysed site-specific intermolecular oxidation of ynamides and ynol ethers. This method provides a means to access α-imido and α-ester metal carbenoid reactivity from these electron-rich π-systems. A general and efficient preparation of synthetically important α,β-unsaturated carboxylic ester and imide derivatives, as well as vinylogous carbimates is achieved from readily-accessible materials. The mild reaction conditions are tolerant of a wide range of functional groups, including other alkyne moieties. The general potential of this approach for diazo-free reaction development, as well as the specific synthetic utility of the reactions detailed herein, are under study in our lab.
We thank the University of Birmingham for financial support and Johnson Matthey plc for a generous loan of metal salts. The work was part supported through Science City Advanced Materials project 2, funded by AWM and the ERDF.
Notes and references
- (a) Carbene Chemistry, ed. G. Bertrand, Fontis Media, Lausanne, Switzerland, 2002 Search PubMed; (b) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley-Interscience, New York, 1998 Search PubMed.
- (a) G. Maas, Angew. Chem., Int. Ed., 2009, 48, 8186 CrossRef CAS; (b) M. Regitz and G. Maas, Diazo Compounds—Properties and Synthesis, Academic Press, Orlando, 1986 Search PubMed.
- P. W. Davies and S. J. C. Albrecht, Angew. Chem., Int. Ed., 2009, 48, 8372 CrossRef CAS.
- Reviews: (a) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef; (b) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef; (c) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS; (d) E. J. Núňez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef CAS; (e) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC.
- (a) N. D. Shapiro and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 4160 CrossRef CAS; (b) G. Li and L. Zhang, Angew. Chem., Int. Ed., 2007, 46, 5156 CrossRef CAS.
- (a) L. Cui, Y. Peng and L. Zhang, J. Am. Chem. Soc., 2009, 131, 8394 CrossRef CAS; (b) L. Cui, G. Zhang, Y. Pen and L. Zhang, Org. Lett., 2009, 11, 1225 CrossRef CAS.
- (a) H.-S. Yeom, J.-E. Lee and S. Shin, Angew. Chem., Int. Ed., 2008, 47, 7040 CrossRef CAS; (b) H.-S. Yeom, Y. Lee, J.-E. Lee and S. Shin, Org. Biomol. Chem., 2009, 7, 4744 RSC.
- (a) During the course of our study Zhang and co-workers reported the intermolecular oxidation of terminal alkynes to access and employ monosubstituted α-oxo gold carbenoids, L. Ye, L. Cui, G. Zhang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 3258 Search PubMed; also: (b) L. Ye, W. He and L. Zhang, J. Am. Chem. Soc., 2010, 132, 8550 CrossRef CAS; (c) For an intermolecular study of alkyne oxyarylation, see: A. B. Cuenca, S. Montserrat, K. M. Hossain, G. Mancha, A. Lledós, M. Medio-Simón, G. Ujaque and G. Asensio, Org. Lett., 2009, 11, 4906 Search PubMed.
- Recent reviews of ynamide reactivity: (a) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010 DOI:10.1021/cr100003s; (b) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840 CrossRef CAS.
- (a) F. Marion, J. Coulomb, C. Courillon, L. Fensterbank and M. Malacria, Org. Lett., 2004, 6, 1509 CrossRef CAS; (b) F. Marion, J. Coulomb, A. Servais, C. Courillon, L. Fensterbank and M. Malacria, Tetrahedron, 2006, 62, 3856 CrossRef CAS; (c) E. Soriano and J. Marco-Contelles, J. Org. Chem., 2005, 70, 9345 CrossRef CAS; (d) Y. Zhang, R. P. Hsung, X. Zhang, J. Huang, B. W. Slater and A. Davis, Org. Lett., 2005, 7, 1047 CrossRef CAS; (e) A. Buzas and F. Gagosz, Org. Lett., 2006, 8, 515 CrossRef CAS; (f) A. Buzas, F. Istrate and F. Gagosz, Tetrahedron, 2009, 65, 1889 CrossRef CAS; (g) A. S. K. Hashmi, R. Salathie and W. Frey, Synlett, 2007, 1763 CrossRef CAS; (h) F. M. Istrate, A. K. Buzas, I. D. Jurberg, Y. Odabachian and F. Gagosz, Org. Lett., 2008, 10, 925 CrossRef CAS; (i) G.-Y. Lin, C.-W. Li, S.-H. Hung and R.-S. Liu, Org. Lett., 2008, 10, 5059 CrossRef CAS; (j) S. Couty, C. Meyer and J. Cossy, Tetrahedron, 2009, 65, 1809 CrossRef CAS; (k) A. S. K. Hashmi, M. Rudolph, J. W. Bats, W. Frey, F. Rominger and T. Oeser, Chem.–Eur. J., 2008, 14, 6672 CrossRef CAS; (l) S. Couty, C. Meyer and J. Cossy, Angew. Chem., Int. Ed., 2006, 45, 6726 CrossRef CAS; (m) S. Kramer, K. Dooleweerdt, A. T. Lindhardt, M. Rottländer and T. Skrydstrup, Org. Lett., 2009, 11, 4208 CrossRef CAS.
- Capture of in situ generated gold carbenoids using sulfoxides: C. A. Witham, P. Mauleón, N. D. Shapiro, B. D. Sherry and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 5838 Search PubMed.
- (a) S. Couty, C. Meyer and J. Cossy, Synlett, 2007, 2819 CAS; (b) Z. F. Al-Rashid and R. P. Hsung, Org. Lett., 2008, 10, 661 CrossRef CAS.
- Z. F. Al-Rashid, W. L. Johnson, R. P. Hsung, Y. Wei, P.-Y. Yao, R. Liu and K. Zhao, J. Org. Chem., 2008, 73, 8780 CrossRef CAS.
- J. M. Concellón, H. Rodríguez-Solla and P. Díaz, J. Org. Chem., 2007, 72, 7974 CrossRef CAS and references therein.
- [2+2] cycloaddition/reversion with ynamides and carbonyl moieties to prepare α,β-unsaturated carboxylic imides: (a) R. P. Hsung, C. A. Zificsak, L.-L. Wei, C. J. Douglas, H. Xiong and J. A. Mulder, Org. Lett., 1999, 1, 1237 CrossRef CAS; (b) K. C. M. Kurtz, R. P. Hsung and Y. Zhang, Org. Lett., 2006, 8, 231 CrossRef CAS; (c) L. You, Z. F. Al-Rashid, R. Figueroa, S. K. Ghosh, G. Li, T. Lu and R. P. Hsung, Synlett, 2007, 1656 CAS; (d) N. Shindoh, Y. Takemoto and K. Takasu, Chem.–Eur. J., 2009, 15, 7026 CrossRef CAS.
- Recent advances in ynamide synthesis: see ref. 9 and (a) Y. Zhang, R. P. Hsung, M. R. Tracey, K. C. M. Kurtz and E. L. Vera, Org. Lett., 2004, 6, 1151 CrossRef CAS; (b) A. Coste, G. Karthikeyan, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2009, 48, 4381 CrossRef CAS; (c) T. Hamada, X. Ye and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 833 CrossRef CAS and references therein.
- A. S. K. Hashmi, J. P. Weyrauch, M. Rudolph and E. Kurpejovic, Angew. Chem., Int. Ed., 2004, 43, 6545 CrossRef CAS.
- Examples of ynol ethers in gold catalysis (a) L. Zhang and S. A. Kozmin, J. Am. Chem. Soc., 2004, 126, 11806 CrossRef CAS; (b) D. A. Engel and G. B. Dudley, Org. Lett., 2006, 8, 4027 CrossRef CAS; (c) A. S. K. Hashmi, M. Rudolph, J. Huck, W. Frey, J. W. Bats and M. Hamzić, Angew. Chem., Int. Ed., 2009, 48, 5848 CrossRef CAS.
- Rhodium-catalysed diazodecomposition: D. F. Taber, R. J. Herr, S. K. Pack and J. M. Geremia, J. Org. Chem., 1996, 61, 2908 Search PubMed.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Catalysis optimisation study; experimental details and analytical data for catalysis precursors and products; control reactions and a preliminary discussion of the interconversion of (E)/(Z)-9b. See DOI: 10.1039/c0cc02736g |
| This journal is © The Royal Society of Chemistry 2011 |