Engineering active sites for enhancing synergy in heterogeneous catalytic oxidations†‡
James
Paterson
a,
Matthew
Potter
a,
Enrica
Gianotti
b and
Robert
Raja
*a
aDepartment of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: R.Raja@soton.ac.uk; Fax: +44 (0)2380-593781; Tel: +44 (0)2380-592144
bDepartment of Chemistry IFM & NIS-Centre of Excellence, University of Turin, Via P. Giuria 7, 10125, Torino, Italy
First published on 1st November 2010
Abstract
The simultaneous isomorphous substitution of Al(III) and P(V) ions, in an aluminophosphate framework, with redox active Co(III) and Ti(IV) metal ions, generates highly active single-site heterogeneous catalysts that exhibit considerable synergy, compared to their corresponding monometallic analogues, in the catalytic epoxidation of olefins.
Engineering multifunctional active sites in microporous solids is a relatively new area of research that exploits the fundamental principles of nanoscience by taking cues from biological and chemical systems.1 Microporous solids, such as aluminophosphates2 (AlPO's), possess a wide-range of interconnecting pores and channels, where single-site catalytic entities can be judiciously designed for effecting a range of selective oxidation and acid-catalysed transformations.3–6
In this study, we have specifically focused our attention on designing bimetallic multifunctional active sites, by isomorphously replacing a few atom percent of the framework7Al(III) and P(V) cations with tetrahedrally-coordinated Co(III) and Ti(IV) ions respectively (Fig. 1). The rationale behind our design strategy was that, the simultaneous incorporation of a tetrahedral Ti(IV) centre, along with a tetrahedral redox site {Co(III)}, should facilitate enhancements in the observed synergy in catalytic transformations.
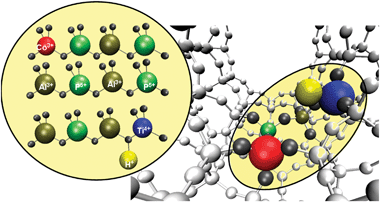 | ||
| Fig. 1 Graphical representation of an AlPO-framework; where some of the Al(III) and P(V) sites have been isomorphously substituted with Co(III) and Ti(IV) ions respectively. | ||
Titanium-based microporous (e.g. titanosilicates such as TS-1)8 and mesoporous solids (e.g.TiMCM-41)9 have proved particularly effective in epoxidation reactions. The high activity and selectivity of these catalysts has been attributed to the local structural environment and coordination geometry of the tetrahedral Ti(IV) centre. Furthermore, the oxophilic nature of the Ti(IV) ion facilitates the coordination of the peroxo species to the metal centre and its concomitant interaction with the olefinvia the Eley–Rideal mechanism,10 that leads to high catalytic turnovers. Whilst the activities and selectivities of these monometallic catalysts are noteworthy, we believed that, a combination of redox active metal centers that were located within close proximity to a oxophilic species,11 such as Ti(IV), should facilitate synergistic interactions, both from a structural and catalytic perspective.
The synthetic procedure that we have evolved for synthesising the CoIIITiIVAlPO-5 catalyst used the following gel composition: 0.96Al![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1.5P∶0.03Co∶0.03Ti∶0.8SDA∶50H2O. More details regarding the synthesis can be found in ESI.‡ Typical XRD patterns for the calcined monometallic (CoAlPO-5 and TiAlPO-5) and its corresponding bimetallic analogue (CoTiAlPO-5) are shown in Fig. 2. The indexed pattern for the latter is in good agreement with that reported in the literature for the corresponding monometallic analogues; displaying a high degree of crystallinity with no phase impurities12 (see inset and ESI‡ for refinement results). Inductively coupled plasma (ICP) results (Fig. 3A) show a good linear correlation between the individual metal loadings in the initial gel and in the final calcined catalyst; with more than 90% of the transition metal that was present in the initial gel being incorporated in the (final) calcined AlPO catalyst.
∶1.5P∶0.03Co∶0.03Ti∶0.8SDA∶50H2O. More details regarding the synthesis can be found in ESI.‡ Typical XRD patterns for the calcined monometallic (CoAlPO-5 and TiAlPO-5) and its corresponding bimetallic analogue (CoTiAlPO-5) are shown in Fig. 2. The indexed pattern for the latter is in good agreement with that reported in the literature for the corresponding monometallic analogues; displaying a high degree of crystallinity with no phase impurities12 (see inset and ESI‡ for refinement results). Inductively coupled plasma (ICP) results (Fig. 3A) show a good linear correlation between the individual metal loadings in the initial gel and in the final calcined catalyst; with more than 90% of the transition metal that was present in the initial gel being incorporated in the (final) calcined AlPO catalyst.
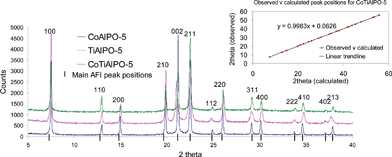 | ||
| Fig. 2 Powder X-ray diffraction pattern for CoIIIAlPO-5, TiIVAlPO-5 and CoIIITiIVAlPO-5. See ESI‡ (and inset) for refinement results. | ||
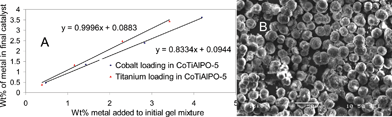 | ||
| Fig. 3 ICP results for metal loading correlations in the synthesis gel and final catalyst (A) and SEM image of the calcined CoIIITiIVAlPO-5 (B). | ||
Scanning electron microscopy (SEM, Fig. 3B) also showed a good agreement between the monometallic catalysts and their corresponding bimetallic analogues. In all cases, a spherical morphology was observed and the mean particle sizes that we obtained are in good agreement with previously reported13 literature values (10 to 30 μm). The metal loadings (Table 1) of the monometallic and bimetallic catalysts were ascertained using ICP and AAS (Atomic Absorption Spectroscopy) and BET measurements (see ESI‡ and Table 1) confirmed that all three samples had a comparable surface area, which is in good agreement with the AFI-type materials reported in literature.13
Diffuse reflectance (DR) UV-Vis (Fig. 4) measurements further substantiate the redox capabilities of the cobalt ions in the AlPO-5 architecture and provide further evidence for the presence of a greater fraction of tetrahedral Ti(IV) species in the CoIIITiIVAlPO-5 catalyst (compared to TiIVAlPO-5). The DR UV-Vis spectrum of calcined CoTiAlPO-5 (Fig. 4A, curve a) shows two strong absorptions in the 250–500 nm range due to LMCT transitions between the oxygen ligands and the tetrahedral CoIII sites. In the Vis region, the triplet bands that we observe at 530, 592 and 659 nm can be assigned to the d–d transitions of CoII ions in Td coordination.14 The presence of this triplet after calcination, suggests that not all of the CoII ions can be oxidized to the CoIII state, which is consistent with our earlier observations.15 We also observe a slight enhancement in the intensity of the triplet band after the reduction treatment (Fig. 4A, curve b). In order to simultaneously detect the cobalt and titanium sites in the CoTiAlPO-5 sample, it was necessary to completely reduce the catalyst, due to the overlap of the intense UV bands of CoIII with those of Ti(IV). Upon reduction in H2 at 400 °C (Fig. 4A, curve b), these strong absorptions associated with CoIII ions completely disappear, and a distinct band at 230 nm becomes apparent. This latter band can be assigned to isolated tetrahedral Ti(IV) LMCT transitions.16 It is also noteworthy that this absorption is shifted to a higher wavelength and becomes significantly broader in the monometallic TiIVAlPO-5 catalyst (Fig. 4B, curve b), when compared with the bimetallic CoIIITiIVAlPO-5 (Fig. 4B, curve a) analogue, thereby substantiating the presence of octahedral Ti(IV) sites as oligomeric species or TiO2-like clusters in the monometallic TiIVAlPO-5.17 These more coordinated Ti(IV) species are present in the bimetallic CoIIITiIVAlPO-5 (shoulder of the UV band extending up to 350 nm). However, the absorption band is much sharper when compared with the monometallic TiIVAlPO-5, suggesting the presence of a higher fraction of tetrahedral Ti(IV) sites, in the bimetallic CoIIITiIVAlPO-5 catalyst, when compared with that of its monometallic analogue (TiIVAlPO-5). For the sake of comparison the DR UV-Vis spectrum of TiIVMCM-41 (Fig. 4B, curve c) where, the presence of tetrahedral Ti(IV) species can be unambiguously established by deliberately grafting18Ti(IV) active centres to the inner walls of the mesoporous silica, is also shown.
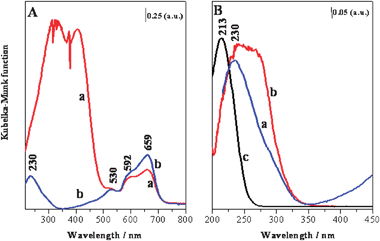 | ||
| Fig. 4 (A) DR UV-Vis spectra of the oxidized (a) and reduced (b) CoTiAlPO-5 catalyst. (B) Comparative DR UV-Vis spectra for CoTiAlPO-5 (a), TiAlPO-5 (b) and TiMCM-41 (c). All the spectra are recorded in vacuo at 298 K after the thermal treatments. | ||
The presence of redox (CoIII and FeIII) sites in AlPOs facilitates the in situ formation and generation of active oxygen species from acetylperoxyborate (APB),19 which have proved highly effective in the oxidation of hydrocarbons and in the production of fine-chemicals.4,5,20 Whilst the selectivities of these catalysed transformations has been high, the overall activities with the monometallic AlPO catalysts, has been, at best modest.20 With our bimetallic multifunctional analogues, that have been reported in this study for the first time, we hoped to achieve a synergistic enhancement in the overall catalytic potential, whilst at the same time, maintaining the high selectivities that have been previously associated with APB.
Further to the discovery of the Shell catalyst21 (TiIV–SiO2) for catalytic epoxidations, there have been numerous efforts devoted to the design of various Ti(IV)-framework-substituted molecular sieves for the epoxidation of olefins. In particular, the emergence of TS-1 was considered a major industrial break-through for selective oxidations with H2O28,22 and the use of TiIVMCM-41 with alkyl hydroperoxides for catalytic epoxidations has been highlighted in many reports.9,18 Hence, for the purpose of comparison, we have used these two catalysts to benchmark the performance of our monometallic (CoIII and TiIV) AlPO-catalysts. Furthermore, using the above approach, we could rationally demonstrate the influence of the synergy and the efficacy of our bimetallic catalyst (CoIIITiIVAlPO-5), when the two active sites were simultaneously substituted into the framework.
The catalytic epoxidation of cyclohexene, using APB as the oxidant, for the monometallic CoIIIAlPO-5 and TiIVAlPO-5 along with that of TS-1 and TiIVMCM-41 is summarised in Fig. 5. Whilst the redox sites in the monometallic CoIIIAlPO-5 are considerably inferior (12% conversion) for the epoxidation reaction, the Ti(IV) sites in TS-1, TiMCM-41 and TiAlPO-5 afford a modest level (30–35%) of conversion for the same reaction. The bimetallic catalyst (CoIIITiIVAlPO-5), on the other hand, displays a significantly higher level (82%) of activity when compared with its monometallic analogues or the conventionally used TS-1 {where the reaction could occur on the outer surface due to the smaller diameter of the channels (5.1 × 5.5 Å)} and Ti(IV)MCM-41 (30 Å mean pore-diameter), at comparable levels of metal loading (see turnover numbers (TON) in Fig. 5). In order to ascertain whether the higher activities observed with the CoIIITiIVAlPO-5 were not just due to the presence of the two metals (CoIII and TiIV), we prepared a physical mixture of the two individual catalysts (at similar levels of loading) and the activities observed with the physical mixture were only slightly higher (44%) than that of their corresponding monometallic analogues. The catalytic trend observed in Fig. 5 clearly demonstrates that the higher activities obtained with the CoIIITiIVAlPO-5 catalyst arise due to the simultaneous isomorphous incorporation of Co(III) and Ti(IV) ions within the AlPO-5 framework.
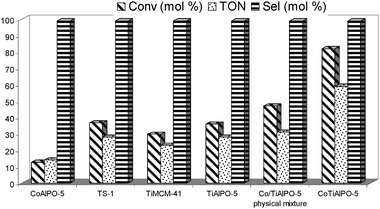 | ||
| Fig. 5 Synergistic effect of the bimetallic CoIIITiIVAlPO-5 catalyst in the epoxidation of olefins with APB (see ESI‡ for reaction conditions). | ||
Extensive catalyst-recycle studies and stringent tests (including hot-filtration experiments), aimed at analyzing the extent of leaching of the metal ions, have been carried out along the lines reported earlier.4,20 We did not observe any appreciable decrease in the catalysts' activity or selectivity even after four recycles and analysis of the reactions mixtures by ICP and atomic absorption spectroscopy (AAS)20 revealed only trace quantities (<3 ppb) of dissolved metal ions. Furthermore, preliminary computer modeling studies indicate that these two (CoIII and TiIV) metal ions in the CoIIITiIVAlPO-5 catalyst are present within close proximity to each other within the framework structure and, we believe, that it is this close proximity that leads to the enhanced synergy we observe in the catalytic epoxidation. Further X-ray absorption studies (EXAFS and XANES) are currently in progress to establish the exact location of the two metal ions; but the origin and establishment of the synergy is already evident in recently filed patent applications for other industrially significant catalytic oxidations.23 We believe, from the preliminary structural analysis, that the simultaneous isomorphous substitution of the Al(III) and P(V) sites in the AlPO-5 framework with Co(II) and Ti(IV) metal ions results in a higher fraction of Co(III) sites in our bimetallic catalysts when compared with their corresponding monometallic (CoIIIAlPO-5) analogues.24 These (redox) cobalt centres provide the loci for the initiation and generation of free-radical intermediates15 that are involved in the catalytic epoxidation. Our continued spectroscopic investigations indicate, that it is highly likely, the peroxo species associated with the oxidant preferentially coordinate to the oxophilic Ti(IV) centre in the CoIIITiIVAlPO-5 catalyst and one can envisage its concomitant interaction with an olefin during catalysis. The benefits of the design strategy in stabilizing the porous framework architecture, thereby facilitating alternate mechanistic pathways for enhancing catalytic reactions, will have huge implications for both fundamental and applied research in this area.
Notes and references
- J. K. Nørskov, T. Bligaard, B. Hvolbaek, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, Chem. Soc. Rev., 2008, 37, 2163–2171 RSC.
- S. T. Wilson, B. M. Lok, C. A. Messina, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1982, 104, 1146–1147 CrossRef CAS.
- J. M. Thomas and R. Raja, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13732–13736 CrossRef.
- R. Raja, J. M. Thomas, M. Greenhill-Hooper, S. V. Ley and F. A. A. Paz, Chem.–Eur. J., 2008, 14, 2340–2348 CrossRef CAS.
- R. Raja, Top. Catal., 2009, 52, 322–332 CrossRef CAS.
- M. Hartmann and L. Kevan, Chem. Rev., 1999, 99, 635–663 CrossRef CAS.
- D. Arieli, D. E. W. Vaughan, K. G. Strohmaier and D. Goldfarb, J. Am. Chem. Soc., 1999, 121, 6028–6032 CrossRef CAS.
- B. Notari, Adv. Catal., 1996, 41, 253–334 CAS.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321–323 CrossRef CAS.
- D. A. Ruddy and T. D. Tilley, J. Am. Chem. Soc., 2008, 130, 11088–11096 CrossRef CAS.
- J. C. van der Waal, P. Lin, M. S. Rigutto and H. van Bekkum, Stud. Surf. Sci. Catal., 1997, 105, 1093–1100.
- Collection of simulated XRD powder patterns for Zeolites, ed. M. M. J. Treacy and J. B. Higgins, Elsevier Science and Technology, 5th edn Search PubMed.
- J. Y. Wang, J. W. Song, C. Y. Yin, Y. Y. Ji, Y. C. Zou and F. S. Xiao, Microporous Mesoporous Mater., 2009, 117, 561–569 CrossRef CAS.
- R. A. Schoonheydt, R. de Vos, J. Pelgrims and H. Loeman, Stud. Surf. Sci. Catal., 1989, 49, 559–568.
- J. M. Thomas and R. Raja, Chem. Commun., 2001, 675–687 RSC.
- L. Lenoc, D. T. On, S. Solomykina, B. Echchaed, F. Beland, C. C. D. Moulin and L. Bonneviot, Stud. Surf. Sci. Catal., 1996, 101, 611–620 CAS.
- E. Gianotti, V. Dellarocca, L. Marchese, G. Martra, S. Coluccia and T. Maschmeyer, Phys. Chem. Chem. Phys., 2002, 4, 6109–6115 RSC.
- T. Maschmeyer, F. Rey, G. Sankar and J. M. Thomas, Nature, 1995, 378, 159–162 CrossRef CAS.
- Peroxid. Chemie GMBH, US 5462692, 1995 Search PubMed.
- R. Raja, J. M. Thomas, M. Xu, K. D. M. Harris, M. Greenhill-Hooper and K. Quill, Chem. Commun., 2006, 448 RSC.
- Shell Oil, GP 1249079, 1971 Search PubMed.
- G. Bellussi and M. S. Rigutto, Stud. Surf. Sci. Catal., 1994, 85, 177–213 CAS.
- University of Southampton, WO2009004342, 2009, EP2170812, 2009, GB2450711, 2007 Search PubMed.
- P. A. Barrett, G. Sankar, C. R. A. Catlow and J. M. Thomas, J. Phys. Chem., 1996, 100, 8977–8985 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, analytical protocols and refinement results are provided. See DOI: 10.10.39/c0cc02341h |
| This journal is © The Royal Society of Chemistry 2011 |