DNA–nanoparticle micelles as supramolecular fluorogenic substrates enabling catalytic signal amplification and detection by DNAzyme probes†‡
Miao-Ping
Chien
,
Matthew P.
Thompson
and
Nathan C.
Gianneschi
*
Department of Chemistry & Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA. E-mail: ngianneschi@ucsd.edu
First published on 7th September 2010
Abstract
Catalytic DNA molecules have tremendous potential in propagating detection events vianucleic acid sequence selective signal amplification. However, they suffer from product inhibition limiting their widespread utility. Herein, this limitation is overcome utilizing a novel fluorogenic substrate design consisting of cooperatively assembled DNA–nanoparticle micelles.
To enzymatically amplify a signal one requires a triggering mechanism that connects the detection event to the catalytic signal transduction process. This is the cornerstone of biologically regulated catalytic systems for signal transduction in living systems. Several effective enzymatic systems have been reported that use various types of DNA sequence and structure selective processes for detecting DNAviacatalytic turnover for signal amplification.1–19 These have included several efforts to harness catalytic single-stranded DNA (ssDNA) sequences as phosphodiesterases with ribonuclease activity (DNAzymes20–23) in a variety of assay formats usually aimed at activating fluorogenic nucleic acid substrates via strand cleavage reactions (Fig. 1).24–32DNAzymes offer a unique opportunity for detection and signal amplification because unlike proteinaceous enzymatic endonucleases, DNAzymes are not restricted to a given sequence but rather are synthesized in the laboratory to desired specifications making them selective for any given sequence. In this respect, DNAzymes are unique and potentially powerful tools in biodiagnostics because they have tailorable sequence selectivity and are potentially catalytic. Herein, their utility as selective, triggerable catalysts for signal amplification by true turnover is demonstrated in the context of a DNA detection protocol enabled by supramolecular fluorogenic substrates.33 Given the numerous modes of selective recognition open to ssDNA (e.g. hybridization,34–36aptamer-based recognition,18,37 enzymatic substrates,38 thermal responsiveness39) it is expected that harnessing DNAzymes in catalytic detection protocols will have broad utility.
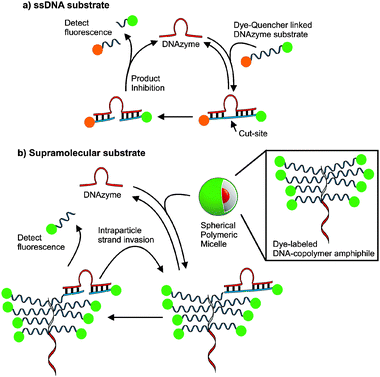 | ||
| Fig. 1 The design of supramolecular substrates capable of multiple turnovers compared to single-stranded DNA (ssDNA) substrates. Active DNAzymes recognize fluorogenic substrates and catalyze strand cleavage at RNA bases. (a) ssDNA substrates are typically modified with fluorophore and quencher pairs for activationvia sequence selective cleavage. (b) The DNA–nanoparticle micelles formed via the assembly of DNA–brush copolymer surfactants. Dye-labelled DNA strands within the particles are recognized and cleaved by a DNAzyme enabling detection of fluorescence. | ||
Currently, a major limitation of DNAzymes in signal amplification and detection via catalysis is that they are product inhibited and are therefore typically limited to a single turnover or less. This limitation is caused by substrate and product having the same DNA sequence and one must balance turnover of cleaved substrate with binding energy as with any system designed with substrate selectivity and turnover in mind. We reasoned that if one could generate a cooperatively assembled supramolecular fluorogenic substrate then DNAzyme catalytic activity would be enhanced by virtue of increasing effective DNA substrate concentration upon DNAzyme recognition of the probe.17,40–43 A DNA–brush copolymer capable of assembling into an aggregate structure was found to be capable of achieving this goal (Fig. 1). The key benefit to this assembly over material simply decorated with DNA is that in this system the substrate is within the internal structure increasing the density of the probe. We reasoned that a micellar aggregate consisting of a material formed from the DNA substrate itself would allow sequence selective turnover not accessible to single-stranded DNA fluorogenic substrates such as molecular beacons.34,36
The DNAzyme catalyst consists of a conserved DNA sequence forming a catalytic domain with 5′- and 3′-ends as recognition sequences.22 Therefore, a DNA substrate with a single RNA base (rA) as the site of cleavage was chosen for incorporation in the DNA–brush copolymer architecture used to form micellar aggregates (Fig. 2). The DNA substrate was designed to incorporate two sequences complementary to the recognition portions of the DNAzyme on either side of the RNA-base cut-site as well as incorporating a fluorescein moiety on the 3′-termini of the strand to allow for detection and analysis of the cleavage reaction by fluorescence spectroscopy. The polymers utilized in these systems were formed with low polydispersity and well-defined block structure amenable to post-polymerization modification with the 5′-amino modified oligonucleotide (see ESI‡). The resulting DNA–brush copolymer was dialyzed in buffered water (Tris, 20 mM, pH 7.4) to obtain spherical DNA nanoparticles approximately 20 nm in diameter as shown by transmission electron microscopy (TEM, Fig. 2), dynamic light scattering (DLS) and atomic force microscopy (AFM).33 Initially, particles were mixed with DNAzymes in 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 molecular weight cut off (MWCO) centrifuge tubes. Cleavage of the DNA shell at the RNA base (rA) cut-site releases a fluorescent 10 base ssDNA product into solution from the particle surface resulting in a shell containing a truncated 9-base long sequence. By centrifuging at 14000 × g for 30 seconds at given time points, the ssDNA product was separated from particles as it passed through the MWCO filter for analysis by fluorescence. The identity of the product was confirmed by MALDI mass spectrometry (see ESI‡). The DNAzyme catalytic efficiency in the supramolecular fluorogenic substrate particle is greatly enhanced compared to the single-stranded fluorescent DNA substrate (F-ssDNA) (Fig. 2). The multiple turnovers observed for the DNAzyme on the particle compared with the F-ssDNA is presumably due to the high effective concentration of substrate in the particle shell enabling enhanced, efficient shell strand truncation. Importantly, the ordinarily product inhibited DNAzyme is capable of multiple turnovers and complete shell degradation occurs within 15 minutes following addition to a solution of the particles (complete turnover was confirmed via calibration curve, see ESI‡). This is in comparison to the low turnover observed for F-ssDNA substrate over the same time scale.
000 g mol−1 molecular weight cut off (MWCO) centrifuge tubes. Cleavage of the DNA shell at the RNA base (rA) cut-site releases a fluorescent 10 base ssDNA product into solution from the particle surface resulting in a shell containing a truncated 9-base long sequence. By centrifuging at 14000 × g for 30 seconds at given time points, the ssDNA product was separated from particles as it passed through the MWCO filter for analysis by fluorescence. The identity of the product was confirmed by MALDI mass spectrometry (see ESI‡). The DNAzyme catalytic efficiency in the supramolecular fluorogenic substrate particle is greatly enhanced compared to the single-stranded fluorescent DNA substrate (F-ssDNA) (Fig. 2). The multiple turnovers observed for the DNAzyme on the particle compared with the F-ssDNA is presumably due to the high effective concentration of substrate in the particle shell enabling enhanced, efficient shell strand truncation. Importantly, the ordinarily product inhibited DNAzyme is capable of multiple turnovers and complete shell degradation occurs within 15 minutes following addition to a solution of the particles (complete turnover was confirmed via calibration curve, see ESI‡). This is in comparison to the low turnover observed for F-ssDNA substrate over the same time scale.
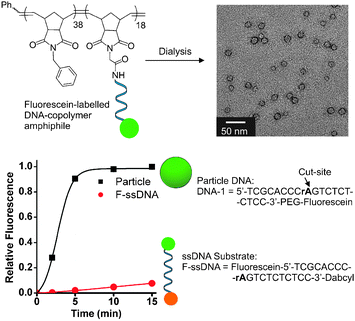 | ||
| Fig. 2 Structure of supramolecular substrate micelles and turnover of supramolecular fluorogenic substrate particle vs. single-stranded fluorescent substrate (F-ssDNA). TEM data indicate 20 nm particles assembled from the DNA–brush copolymer. PEG = polyethylene glycol incorporated viaphosphoramidite chemistry, see ESI‡ for structural details. Conditions: particle DNA (1 μM), F-ssDNA (1 μM), DNAzyme (5 nM). Buffer: Tris (20 mM, pH 7.4), MgCl2 (50 mM), room temp. DNAzyme utilized in these studies has sequence; DNAzyme-1: 5′-GGAGAGAGATCCGAGCCGGTCGAAGGGTGCGA-3′. | ||
To demonstrate the utility of this substrate design in signal amplification viaDNAzyme-mediated sensing,26,28 we designed an assay (Fig. 3) composed of a DNAzyme (DNAzyme-1), its inhibitor (DNA-Inh), a target sequence (DNA-T; sequence from HIV-1gag/polgene) and a supramolecular substrate particle, labelled P-1 in Fig. 3, encoded with single-stranded substrate (sequence = DNA-1). First, DNAzyme-1 was mixed for 30 min with DNA-Inh, designed to hybridize via the recognition sequences in the DNAzyme and block the active site. This inhibited DNAzyme complex was then mixed with varying concentrations of DNA-T and incubated for 30 min. Once added, DNA-T rapidly invades into the inhibited DNAzyme duplex (DNAzyme-1·DNA-Inh), releasing active DNAzyme-1 and forming the new, longer duplex DNA-T·DNA-Inh. Substrate particles (P-1) were added to the solution of activated DNAzyme-1 causing particle shell recognition and cleavage. Product strands were again separated by filtration from particles and analyzed by fluorescence. The observed pM sensitivities (Fig. 3a) for the detection and signal amplification of ssDNA confirm the turnover of substrate on the particle by DNAzyme-1.
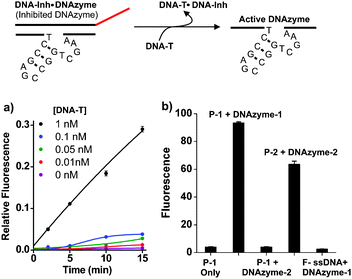 | ||
| Fig. 3 Selective and sensitive DNAzyme catalyzed DNA–shell truncation. Inhibited DNAzyme-1 (duplex: DNAzyme-1·DNA-Inh) is treated with target strand (DNA-T) over a range of concentrations causing displacement of DNAzyme-1 and formation of a new duplex, DNA-T·DNA-Inh. Activated DNAzyme-1 binds to and cleaves the particle shell releasing fluorescein-labelled ssDNA. (a) DNAzyme-1 triggering by target DNA (DNA-T). (b) Selectivity of DNAzyme catalyzed reactions; fluorescence measurements taken 5 minutes following addition of respective DNAzymes. Columns left to right: (1) signal from P-1 without DNAzyme. (2) P-1 mixed with DNAzyme-1. (3) P-1 mixed with DNAzyme-2. (4) P-2 (from copolymer with sequence, DNA-2) mixed with DNAzyme-2. (5) F-ssDNA mixed with DNAzyme-1. Conditions: particle DNA (1 μM), DNAzyme (5 nM). DNA-Inh (5 nM). Buffer: Tris (20 mM, pH 7.4), MgCl2 (50 mM), room temp. DNAzyme-2: 5′-AACACACACTCCGAGCCGGTCGAAAGCTTTCTGAT-3′. DNA-2: 5′-ATCAGAAAGCTrAGGTGTGTGTT-3′-PEG-Fluorescein. | ||
In addition, this process is sequence selective, as evidenced by the selective inhibition and triggering of DNAzyme-1 and the observation that the non-complementary DNAzyme-2 has no effect on P-1 (Fig. 3b). In turn, DNAzyme-2 catalyzes shell degradation when mixed with particles containing the complementary sequence, DNA-2 (P-2). The above studies illustrate two key features of this system: (1) particles are sensitive to low concentrations of catalytically active DNAzyme. (2) The particles are susceptible to degradation in a sequence selective and concentration dependent fashion.
In conclusion, the present study has introduced a novel approach to substrate design enabling a DNAzyme to be utilized in a truly catalysis-based amplification and detection assay. This affords a route toward DNAzyme detection assays of various types where the myriad ways of recognizing ssDNA sequences may be used as triggers.44
NCG thanks the Camille & Henry Dreyfus Foundation for support through a New Faculty Award and we acknowledge NSF for support through CHE-0741968.
Notes and references
- B. Schweitzer and S. Kingsmore, Curr. Opin. Biotechnol., 2001, 12, 21–27 CrossRef CAS.
- B. W. Kirk, M. Feinsod, R. Favis, R. M. Kliman and F. Barany, Nucleic Acids Res., 2002, 30, 3295–3311 CrossRef CAS.
- L. Zhu and E. V. Anslyn, Angew. Chem., Int. Ed., 2006, 45, 1190–1196 CrossRef CAS.
- A. J. H. Smith, Nucleic Acids Res., 1979, 6, 831–848 CrossRef CAS.
- L. H. Guo and R. Wu, Nucleic Acids Res., 1982, 10, 2065–2084 CrossRef CAS.
- R. K. Saiki, S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich and N. Arnheim, Science, 1985, 230, 1350–1354 CrossRef CAS.
- P. Duck, G. Alvarado-Urbina, B. Burdick and B. Collier, BioTechniques, 1990, 9, 142–148 CAS.
- K. Okano and H. Kambara, Anal. Biochem., 1995, 228, 101–108 CrossRef CAS.
- F. Bekkaoui, I. Poisson, W. Crosby, L. Cloney and P. Duck, BioTechniques, 1996, 20, 240–248 CAS.
- C. A. Mein, B. J. Barratt, M. G. Dunn, T. Siegmund, A. N. Smith, L. Esposito, S. Nutland, H. E. Stevens, A. J. Wilson, M. S. Phillips, N. Jarvis, S. Law, M. De Arruda and J. A. Todd, Genome Res., 2000, 10, 330–343 CrossRef CAS.
- T. M. Hsu, S. M. Law, S. Duan, B. P. Neri and P.-Y. Kwok, Clin. Chem., 2001, 47, 1373–1377 CAS.
- Z. Wang and J. Moult, Hum. Mutat., 2001, 17, 263–270 CrossRef CAS.
- M. Levy and A. D. Ellington, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6416–6421 CrossRef CAS.
- A. Saghatelian, K. M. Guckian, D. A. Thayer and M. R. Ghadiri, J. Am. Chem. Soc., 2003, 125, 344–345 CrossRef CAS.
- B. Ren, J.-M. Zhou and M. Komiyama, Nucleic Acids Res., 2004, 32, e42 CrossRef.
- K. Nakatani, ChemBioChem, 2004, 5, 1623–1633 CrossRef CAS.
- J. M. Gibbs, S.-J. Park, D. R. Anderson, K. J. Watson, C. A. Mirkin and S. T. Nguyen, J. Am. Chem. Soc., 2005, 127, 1170–1178 CrossRef CAS.
- V. Pavlov, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2005, 127, 6522–6523 CrossRef CAS.
- P. Simon, C. Dueymes, M. Fontecave and J.-L. Decout, Angew. Chem., Int. Ed., 2005, 44, 2764–2767 CrossRef CAS.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS.
- S. W. Santoro and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4262–4266 CrossRef CAS.
- J. Li, W. Zheng, A. H. Kwon and Y. Lu, Nucleic Acids Res., 2000, 28, 481–488 CrossRef CAS.
- M. N. Stojanovic and D. Stefanovic, Nat. Biotechnol., 2003, 21, 1069–1074 CrossRef CAS.
- M. N. Stojanovic, P. de Prada and D. W. Landry, Nucleic Acids Res., 2000, 28, 2915–2918 CrossRef CAS.
- A. V. Todd, C. J. Fuery, H. L. Impey, T. L. Applegate and M. A. Haughton, Clin. Chem. (Washington, DC), 2000, 46, 625–630 CAS.
- M. N. Stojanovic, P. d. Prada and D. W. Landry, ChemBioChem, 2001, 2, 411–415 CrossRef CAS.
- S. Schubert and J. Kurreck, Curr. Drug Targets, 2004, 5, 667–681 Search PubMed.
- Y. Tian and C. Mao, Talanta, 2005, 67, 532–537 CrossRef CAS.
- Y. Tian, Y. He and C. Mao, ChemBioChem, 2006, 7, 1862–1864 CrossRef CAS.
- J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2007, 46, 7587–7590 CrossRef CAS.
- D. P. Wernette, C. Mead, P. W. Bohn and Y. Lu, Langmuir, 2007, 23, 9513–9521 CrossRef CAS.
- A. K. Brown, J. Liu, Y. He and Y. Lu, ChemBioChem, 2009, 10, 486–492 CrossRef CAS.
- M. P. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076–5080 CrossRef CAS.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS.
- M. Singh-Zocchi, S. Dixit, V. Ivanov and G. Zocchi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7605–7610 CrossRef CAS.
- K. Wang, Z. Tang, C. J. Yang, Y. Kim, X. Fang, W. Li, Y. Wu, C. D. Medley, Z. Cao, J. Li, P. Colon, H. Lin and W. Tan, Angew. Chem., Int. Ed., 2009, 48, 856–870 CrossRef CAS.
- J.-L. He, Z.-S. Wu, H. Zhou, H.-Q. Wang, J.-H. Jiang, G.-L. Shen and R.-Q. Yu, Anal. Chem., 2010, 82, 1358–1364 CrossRef CAS.
- V. S. Trubetskoy, J. E. Hagstrom and V. G. Budker, Anal. Biochem., 2002, 300, 22–26 CrossRef CAS.
- H. S. Bisht, D. S. Manickam, Y. You and D. Oupicky, Biomacromolecules, 2006, 7, 1169–1178 CrossRef CAS.
- M. L. Collins, B. Irvine, D. Tyner, E. Fine, C. Zayati, C.-a. Chang, T. Horn, D. Ahle, J. Detmer, L.-P. Shen, J. Kolberg, S. Bushnell, M. S. Urdea and D. D. Ho, Nucleic Acids Res., 1997, 25, 2979–2984 CrossRef CAS.
- M. S. Shchepinov, I. A. Udalova, A. J. Bridgman and E. M. Southern, Nucleic Acids Res., 1997, 25, 4447–4454 CrossRef CAS.
- M. S. Shchepinov, K. U. Mir, J. K. Elder, M. D. Frank-Kamenetskii and E. M. Southern, Nucleic Acids Res., 1999, 27, 3035–3041 CrossRef CAS.
- A. K. R. Lytton-Jean and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 12754–12755 CrossRef CAS.
- S. K. Silverman, Chem. Commun., 2008, 3476–3485 Search PubMed.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental details including synthesis and characterization. See DOI: 10.1039/c0cc02291h |
| This journal is © The Royal Society of Chemistry 2011 |