Rationally controlled helical organization of a multiple-hydrogen-bonding oligothiophene: guest-induced transition of helical-to-twisted ribbons†‡
Shiki
Yagai
*a,
Marina
Gushiken
a,
Takashi
Karatsu
a,
Akihide
Kitamura
a and
Yoshihiro
Kikkawa
b
aDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan. E-mail: yagai@faculty.chiba-u.jp; Fax: +81-(0)43-290-3039; Tel: +81-(0)43-290-3368
bPhotonics Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8562, Japan
First published on 28th September 2010
Abstract
A cyanuric acid-functionalized quaterthiophene self-aggregates to form helical ribbons which are transformed into twisted ribbons upon complexing with a complementary bismelamine receptor.
Nanostructures formed by the self-assembly of π-conjugated oligomers through noncovalent interactions are attracting enormous attention as novel materials being developed in the nanoscale.1 The further development of this research field requires a precise control of self-assembled nanostructures by rational molecular designs based on specific noncovalent interactions.2 Among various self-assembled nanostructures, helical architectures have attracted special attention in view of their chirooptical properties, supramolecular chiral chemistry and supramolecular electronics.1,3 Such helical architectures can be roughly classified into two different morphologies, i.e., helical and twisted ribbons.4 Numerous helical π-nanostructures have been constructed from specifically-designed π-conjugated oligomers such as oligo(p-phenylenevinylene)s (OPVs),5 oligo(p-phenyleneethynylene)s,6 oligo(p-phenylene)s7 and oligothiophenes (OTs).8 Ajayaghosh et al. have reported the formation of helical and twisted ribbons from OPVs that are mono- or di-substituted with cholesterol.9 We herein report a rational supramolecular strategy to obtain the two helical nanostructures of functional π-systems by using multiple hydrogen-bonding interactions.
Quaterthiophene 1, functionalized on one end by a cyanuric acid (CA) and on the other end by a tridodecyloxybenzyl (TDB) tail, was synthesized according to Scheme S1 (ESI‡). Since monosubstituted CA could form hydrogen-bonded tapes,10hydrogen bond-directed self-aggregation of 1 may lead to the formation of flat ribbons. Upon capping the CA site of 1 with a xylylene-linked bismelamine receptor BMx,11 the resulting complex may undergo one-dimensional aggregation directed by π-stacking (Scheme 1).
![Schematic representation of the self-assemblies of 1 and 1·BMx with pictures of their cyclohexane solutions ([1] = 5 × 10−4 M) under UV-light.](/image/article/2011/CC/c0cc02225j/c0cc02225j-s1.gif) | ||
| Scheme 1 Schematic representation of the self-assemblies of 1 and 1·BMx with pictures of their cyclohexane solutions ([1] = 5 × 10−4 M) under UV-light. | ||
UV/Vis and fluorescence spectra of chloroform solutions of 1 (Fig. 1a) show absorption and emission maxima at 414 and 489/513 nm, which is characteristic of the π–π* transition of the molecularly dissolved quaterthiophenes.8 Upon dissolving 1 in nonpolar solvents such as methylcyclohexane (MCH), the absorption spectrum undergoes a significant blue-shift to 378 nm whereas the emission band undergoes a red shift to 547 nm. These spectral changes are characteristic of the formation of π-stacked OT aggregates with a face-to-face (H-type) arrangement.8 Temperature-dependent UV/Vis measurement shows the thermoreversibility of the aggregates (ESI‡).
![(a) UV/Vis (left axis) and fluorescence (right axis) spectra of 1 (c = 1 × 10−5 M) in chloroform (red curve) and in MCH (blue curve). (b) UV/Vis spectral change of 1 (c = 1 × 10−5 M) upon addition of BMx (c = 0 to 1 × 10−5 M). Blue and red curves are the spectra recorded at 1 : BMx = 1 : 0 and 1 : 1, respectively. Arrows indicate the changes upon adding BMx. Inset: plot of ε at 465 nm versus [BMx]/[1]. Red dashed curve shows the fluorescence spectrum recorded at 1 : BMx = 1 : 1.](/image/article/2011/CC/c0cc02225j/c0cc02225j-f1.gif) | ||
Fig. 1 (a) UV/Vis (left axis) and fluorescence (right axis) spectra of 1 (c = 1 × 10−5 M) in chloroform (red curve) and in MCH (blue curve). (b) UV/Vis spectral change of 1 (c = 1 × 10−5 M) upon addition of BMx (c = 0 to 1 × 10−5 M). Blue and red curves are the spectra recorded at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BMx = 1:0 and 1:1, respectively. Arrows indicate the changes upon adding BMx. Inset: plot of ε at 465 nm versus [BMx]/[1]. Red dashed curve shows the fluorescence spectrum recorded at 1:BMx = 1:1. :BMx = 1:0 and 1:1, respectively. Arrows indicate the changes upon adding BMx. Inset: plot of ε at 465 nm versus [BMx]/[1]. Red dashed curve shows the fluorescence spectrum recorded at 1:BMx = 1:1. | ||
Self-assembled nanostructures of 1 were investigated by atomic force microscopy (AFM) and transmission electron microscopy (TEM). Elongated fibrous nanostructures were observed by AFM and TEM with lengths over several hundreds of nanometres (ESI‡). Remarkably, high-resolution AFM imaging revealed the formation of helical structures existing as isolated (i) or bundled (ii) states (Fig. 2a). The bundled helices exhibit more pronounced helical pitches compared to isolated ones, which might be attributed to the flattening of the latter by the adsorption to the surface. Cross-sectional analysis along the bundled helices reveals the helical pitch of 16 nm (inset in Fig. 2a), which is significantly longer than the molecular length of 1 with extended alkyl chains (ca. 4 nm, ESI‡). The average height and width of the helical structures are 3.4 nm and 16 nm, respectively (Fig. 2b), after reducing the tip-broadening factor.12 The significant difference between the two dimensions could be attributed to the formation of a hollow helical ribbon that is susceptible to deformation through an evaporation process.
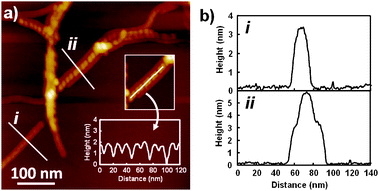 | ||
| Fig. 2 (a) AFM height images of nanostructures formed by 1 (c = 1 × 10−4 M) in MCH. For the sample preparation, see ESI. Inset is a height analysis along the helical ribbon (ii). (b) Cross-sectional analysis across the fibers (i) and (ii) in (a). | ||
In order to obtain insight into the aggregation structure, 1 in bulk state was investigated by X-ray diffraction (XRD). The XRD pattern obtained at 150 °C exhibited diffraction peaks at 6.35, 3.18 and 2.13 nm (ESI‡), which are ascribable to a lamellar structure. The layer spacing of 6.35 nm is significantly longer than the molecular length of 1 (ca. 4 nm), but shorter than the width of a linear tapelike aggregate (ca. 8 nm) formed by double hydrogen-bonding between CA moieties (Scheme 1a).10 Thus, it is suggested that a tilted packing of such hydrogen-bonded tapes results in the formation of sheets, which further stack into a lamellar structure. Although such a densely-packed lamellar structure could not be formed in diluted solutions, the hydrogen-bonded tapes would be stabilized by lateral coupling through multipoint π-stacking interactions of OT segments as shown by the pronounced hypsochromic shift of the π–π* transition, and the resulting bilayer tape is further stabilized by taking helical morphology (Scheme 1a).
Complexation of 1 with BMx was studied by using 1H NMR spectroscopy in CDCl3, a good solvent for the OT segment. CDCl3 solution of 1 (c = 5 × 10−3 M) showed well-resolved peaks reflecting the molecularly dissolved state (ESI‡). Upon adding BMx, the signals of all of the thiophene protons and one of the benzyl protons of the TDB moiety shifted upfield, indicating the formation of a hydrogen-bonded complex with suppressed conformational flexibility. The resonances of the CA imide protons are not observed. Instead, NH protons of added BMx become sharpest at 1:BMx = 1:1, after which they become too broad to be observed due to fast equilibrium between bound and free BMx.13 The binding constant was estimated to be 25100 M−1 by fitting the shift of the above protons to the 1:1 binding isotherm. The relatively high binding constant in CDCl3 ensures the stoichiometric complexation in more nonpolar solvents.
A pronounced UV/Vis spectral change is thus observed when varying amounts of BMx are mixed with 1 in MCH (Fig. 1b). The absorption band of the π-stacked OT undergoes a gradual red-shift to 412 nm, and the spectral change levels off when 1 equiv. of BMx is added. This observation indicates that BMx quantitatively disrupts the self-aggregates of 1 by rupturing hydrogen-bonded chains of CA moieties. The resulting 1:1 complex showed fluorescence maxima at 477 and 506 nm. Thus, the UV/Vis and fluorescence spectra of the 1:1 complex 1·BMx are similar to those of the molecularly dissolved 1 in chloroform (Fig. 1a). However, the former is characterized by the presence of an absorption shoulder at around 460 nm which disappears upon increasing the temperature (ESI‡). It is suggested that the 1:1 complexes exist as a stacked state in solution, where the OT moieties are longitudinally displaced by a slipping angle close to the magic angle 54.7°. This is further supported by the observation that UV/Vis spectra of 1·BMx in MCH do not show significant changes upon increasing the concentration to 5 × 10−4 M (gel state, vide infra).
High aspect ratio rodlike nanostructures were observed for 1·BMx and their lengths easily exceed 1 μm (Fig. 3). The width of the smallest resolved fibers is ca. 20 nm by TEM, and the height of the nanorods is 8 nm by AFM. This morphological feature is in great contrast to the deformed helical ribbons formed by 1, and suggests the formation of twisted ribbons with a solid interior (Scheme 1b). The nanorods show a periodic fine structure with pitches of ca. 80 nm, which can be attributed to periodic bending of twisted ribbons.14 The twisted ribbons of 1·BMx show a remarkable gelation ability while helical ribbons by 1 afford only viscous liquid at around 5 × 10−3 M in cyclohexane. The minimum gelation concentrations of 1·BMx in cyclohexane are 0.16 wt% (5 × 10−4 M), classified as super-gelator.15
 | ||
| Fig. 3 (a) TEM and (b) AFM height images of nanostructures formed by 1·BMx (c = 1 × 10−4 M) in MCH. Inset in (a) is a magnified image and arrows indicate the smallest-width fibers. (c) Magnified AFM height and (d) phase images of a smallest-width fiber. Inset in (c) shows cross-sectional analysis along the white line in the simultaneously obtained height images. | ||
We further investigated the twisted organization of 1·BMx by using a chiral receptor (S)-BMx and circular dichroism (CD) spectroscopy. Fig. 4a shows the CD spectral change of 1·(S)-BMx (c = 2 × 10−5 M) in MCH upon cooling from 90 to 20 °C. A bisignated CD signal with a negative maximum at 364 nm and a positive one at 393 nm gradually grows from 70 to 30 °C. The zero-crossing point is observed at 380 nm, which is close to the absorption maximum of this chiral complex (389 nm, ESI‡). Notably, the chiral complex shows a largely blue-shifted absorption maximum compared to the achiral complex (412 nm, Fig. 1b), indicating more H-type π-stacking of OT moieties taking place for the former. Below 25 °C, a strong negative CD signal emerges abruptly, which might be due to the artifact arising from macroscopic alignment of large aggregates by convective flow.16,17 Temperature-dependent linear dichroism (LD) measurements confirm the presence of a LD effect below 25 °C (Fig. 4b). The absence of the LD effect above 30 °C clearly demonstrates that the observed bisignate CD signal can be attributed to the formation of chirally twisted stacks of 1·(S)-BMx.8
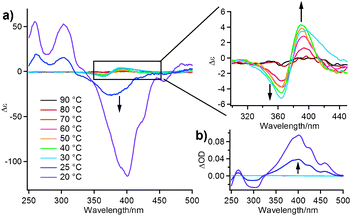 | ||
| Fig. 4 (a) CD and (b) LD spectral change of 1·(S)-BMx (c = 2 × 10−5 M) in MCH upon cooling from 90 to 20 °C using a 1-cm cuvette. Arrows indicate change upon cooling. | ||
In conclusion, two different types of self-assembled helical π-nanostructures, i.e., helical and twisted ribbons, have been rationally constructed upon changing hydrogen-bonding modes of the building block by the addition of guest. This study clearly demonstrates the power of complementary hydrogen-bonding interactions to organize functional molecules into desired nanostructures.
This work was partially supported by KAKENHI (20350061).
Notes and references
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS.
- For examples of our recent work on π-electronic nanostructures using multiple H-bonding interactions, see: (a) S. Yagai, S. Mahesh, Y. Kikkawa, K. Unoike, T. Karatsu, A. Kitamura and A. Ajayaghosh, Angew. Chem., Int. Ed., 2008, 47, 4691 CrossRef CAS; (b) S. Yagai, S. Kubota, H. Saito, K. Unoike, T. Karatsu, A. Kitamura, A. Ajayaghosh, M. Kanesato and Y. Kikkawa, J. Am. Chem. Soc., 2009, 131, 5408 CrossRef CAS; (c) S. Yagai, T. Kinoshita, Y. Kikkawa, T. Karatsu, A. Kitamura, Y. Honsho and S. Seki, Chem.–Eur. J., 2009, 15, 9320 CrossRef CAS.
- (a) T. Verbiest, S. Van Elshocht, M. Karuanen, L. Heliemans, J. Snauwaert, C. Nuckolls, T. J. Katz and A. Persoons, Science, 1998, 282, 913 CrossRef CAS; (b) H. Engelkamp, S. Middelbeek and R. J. M. Nolte, Science, 1999, 284, 785 CrossRef CAS; (c) T. Kato, T. Matsuoka, M. Nishii, Y. Kamikawa, K. Kanie, T. Nishimura, E. Yashima and S. Ujiie, Angew. Chem., Int. Ed., 2004, 43, 1969 CrossRef CAS; (d) J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481 CrossRef CAS; (e) V. K. Praveen, S. S. Babu, C. Vijayakumar, R. Varghese and A. Ajayaghosh, Bull. Chem. Soc. Jpn., 2008, 81, 1196 CrossRef CAS; (f) B. W. Messmore, P. A. Sukerkar and S. I. Stupp, J. Am. Chem. Soc., 2005, 127, 7992 CrossRef CAS; (g) A. Lohr, M. Lysetska and F. Würthner, Angew. Chem., Int. Ed., 2005, 44, 5071 CrossRef CAS; (h) C. C. Lee, C. Grenier, E. W. Meijer and A. P. H. J. Schenning, Chem. Soc. Rev., 2009, 38, 671 RSC.
- (a) J. V. Selinger, M. S. Spector and J. M. Schnur, J. Phys. Chem. B, 2001, 105, 7157 CrossRef CAS; (b) A. Brizard, R. Oda and I. Huc, Top. Curr. Chem., 2005, 256, 167 CAS; (c) T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401 CrossRef CAS.
- (a) P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80 CrossRef CAS; (b) A. Ajayaghosh, R. Varghese, S. J. George and C. Vijayakumar, Angew. Chem., Int. Ed., 2006, 45, 1141 CrossRef CAS.
- A. Ajayaghosh, R. Varghese, S. Mahesh and V. K. Praveen, Angew. Chem., Int. Ed., 2006, 45, 7729 CrossRef CAS.
- J. Bae, J.-H. Choi, Y.-S. Yoo, N.-K. Oh, B.-S. Kim and M. Lee, J. Am. Chem. Soc., 2005, 127, 9668 CrossRef CAS.
- (a) A. P. H. J. Schenning, A. F. M. Kilbinger, F. Biscarini, M. Cavallini, H. J. Cooper, P. J. Derrick, W. J. Feast, R. Lazzaroni, P. Leclere, L. A. McDonell, E. W. Meijer and S. C. J. Meskers, J. Am. Chem. Soc., 2002, 124, 1269 CrossRef CAS; (b) S.-i. Kawano, N. Fujita and S. Shinkai, Chem.–Eur. J., 2005, 11, 4735 CrossRef CAS; (c) E.-K. Schillinger, E. Mena-Osteritz, J. Hentschel, H. G. Börner and P. Bäuerle, Adv. Mater., 2009, 21, 1562–1567 CrossRef CAS.
- A. Ajayaghosh, C. Vijayakumar, R. Varghese and S. J. George, Angew. Chem., Int. Ed., 2006, 45, 456 CrossRef CAS.
- G. J. Sanjayan, V. R. Pedireddi and K. N. Ganesh, Org. Lett., 2000, 2, 2825 CrossRef CAS.
- A. G. Bielejewska, C. E. Marjo, L. J. Prins, P. Timmerman, F. de Jong and D. N. Reinhoudt, J. Am. Chem. Soc., 2001, 123, 7518–7533 CrossRef CAS.
- P. Samori, V. Francke, T. Mangel, K. Müllen and J. P. Rabe, Opt. Mater., 1998, 9, 390 CrossRef CAS.
- F. Wessendorf, J.-F. Gnichwitz, G. H. Sarova, K. Hager, U. Hartnagel, D. M. Guldi and A. Hirsch, J. Am. Chem. Soc., 2007, 129, 16057 CrossRef CAS.
- E. Jahnke, N. Severin, P. Kreutzkamp, J. P. Rabe and H. Frauenrath, Adv. Mater., 2008, 20, 409 CrossRef CAS.
- M. Zinic, F. Vogtle and F. Fages, Top. Curr. Chem., 2005, 256, 39 CAS.
- (a) A. Tsuda, M. A. Alam, T. Harada, T. Yamaguchi, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 8198 CrossRef CAS; (b) M. Wolffs, S. J. George, Z. Tomovic, S. C. J. Meskers, A. P. H. J. Schenning and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8203 CrossRef CAS.
- SEM visualized elongated fibrous aggregates with width of 200 nm (see ESI‡).
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures and characterization data of 1. UV/vis, AFM, 1H NMR, IR and SEM data of 1 and 1·BMx and 1·(S)-BMx. See DOI: 10.1039/c0cc02225j |
| This journal is © The Royal Society of Chemistry 2011 |