Probing for non-statistical effects in dissociation of the 1-methylallyl radical†‡
Michael
Gasser
,
Jann A.
Frey
,
Jonas M.
Hostettler
and
Andreas
Bach
*
Laboratorium für Organische Chemie, Wolfgang-Pauli Strasse 10, ETH Zürich, CH-8093, Switzerland. E-mail: bach@org.chem.ethz.ch; Fax: +41 44 632 1280; Tel: +41 44 633 4785
First published on 5th August 2010
Abstract
The 1-methylallyl radical loses a hydrogen atom following photoexcitation to its lowest valence electronically excited state and displays statistical behavior in decomposition, implying that the presence of methyl rotors cannot be depended upon as an indicator for non-statistical dissociation dynamics in hydrocarbon radicals.
In the last decades experimental and computational evidence has revealed that dynamical non-statistical effects can play an important role in both photo-induced and thermal chemical reactions involving several types of reactive intermediates, including carbocations and radicals.1 In those type of reactions, the widely used statistical rate theories that underlie the more general understanding of reaction dynamics in all branches of chemistry2 are often inadequate to describe the experimentally observed behavior.
Several alkyl radicals, including the ethyl radical—playing a central role as the principal precursor to ethylene through loss of a hydrogen atom in combustion3—displayed non-statistical effects in photo-induced hydrogen abstraction.4 In stark contrast, the rate of unimolecular decomposition of the allyl radical to allene and H was well modeled by Rice–Ramsperger–Kassel–Marcus (RRKM) theory in an analogous experiment5 where C–H bond fission occurs following nonradiative decay of photoexcited allyl.6 In the case of ethyl, classical trajectory calculations suggested that the unusually long-lived highly energized radicals far above the dissociation threshold arise from episodes of regular type intramolecular motions that result in non-RRKM dissociation dynamics.7 Intriguingly, the nuclear motions involved an internal rotation about the C–C bond, and an inversion motion at the radical center with some C–C bond stretch character. We seek to find a structural motif in hydrocarbon radicals or topological features of the potential energy surfaces that leads to non-statistical behavior in unimolecular dissociation. One of the obvious structural differences between alkyl radicals and the allyl radical is the methyl rotors and we recently probed for any putative non-statistical behavior in dissociation arising from the structural change induced by the methyl rotor8 in the 2-methylallyl radical,9 but the complex dynamics with many competing dissociation channels10 prevented detailed insight into the nature of the dissociation dynamics for highly energized radicals.
In this report, we probe for non-statistical effects in the photodissociation dynamics of the 1-methylallyl (C4H7) radical at the lowest possible excitation energy to sample only a small part of the complex C4H7 potential energy surface.10
The experimental apparatus is described in detail elsewhere.9,11 Briefly, we produce supersonically jet-cooled 1-methylallyl radicals in the gas-phase by photolysis of either pent-3-enyl nitrite or 1-iodobut-2-ene freshly prepared according to the procedure in the ESI† seeded in 3 bars of helium at 266 nm inside a quartz capillary tube attached to a pulsed valve. First, we recorded excitation spectra by 1+1′ resonance-enhanced multi-photon ionization (REMPI) to reveal the spectrum of the first electronically excited state of 1-methylallyl in the gas-phase for the first time. In a second pump–probe experiment, we use time-delayed photoionization of the H-atom photo-product via the Lyman-α transition after photo-excitation of 1-methylallyl using pulsed dye lasers to probe the dissociation dynamics of C–H bond fission.
There are only a few spectroscopic studies of the 1-methylallyl radical,12 including early experimental observations by electron spin resonance13 and by absorption spectroscopy,14 which revealed a series of broad and diffuse absorption bands in the ultraviolet between 226 and 238 nm, but to the best of our knowledge, no low lying excited states have been observed or predicted ab initio so far. To guide our experimental search for the optical transition to the Ã-state, we calculated the geometries and energetics of (E)- and (Z)-1-methylallyl radicals in their ground and first electronically excited states at the MCSCF(3,3)/DZP++ level of theory as described in the ESI†. Our ab initio calculations predict adiabatic excitation energies to the first non-planar valence excited state of 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 284 cm−1 for (E)-1-methylallyl and 24191 cm−1 for (Z)-1-methylallyl, respectively.
284 cm−1 for (E)-1-methylallyl and 24191 cm−1 for (Z)-1-methylallyl, respectively.
Fig. 1(a) shows the REMPI spectrum of the 1-methylallyl radical in the spectral range from 23900 to 24600 cm−1 obtained with a fixed ionization laser at 40000 cm−1. The first band visible in the spectrum appears at 23980 cm−1 that we tentatively assign to the electronic origin of the à ← ![[X with combining tilde]](https://www.rsc.org/images/entities/i_char_0058_0303.gif) transition of one of the stereoisomers of the 1-methyallyl radical, based on the excellent agreement with our ab initio predictions. A second prominent band appears at 24080 cm−1 followed by several weaker bands, which correspond to transitions to vibrational levels in the Ã-state. A more detailed discussion of the vibronic structure of the Ã-state will be presented elsewhere.
transition of one of the stereoisomers of the 1-methyallyl radical, based on the excellent agreement with our ab initio predictions. A second prominent band appears at 24080 cm−1 followed by several weaker bands, which correspond to transitions to vibrational levels in the Ã-state. A more detailed discussion of the vibronic structure of the Ã-state will be presented elsewhere.
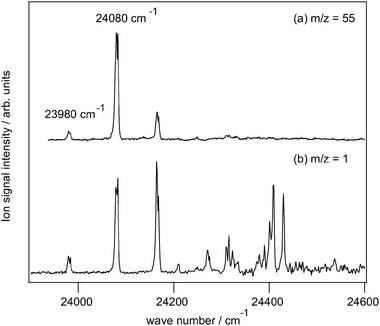 | ||
| Fig. 1 Excitation spectra of the 1-methylallyl radical (a) in the m/z = 55 and (b) in the m/z = 1 mass channel recorded 100 ns after photoexcitation. | ||
Following electronic excitation to its Ã-state, 1-methylallyl loses hydrogen as visible in the photofragment action spectrum, see Fig. 1(b). For each resonance in the REMPI spectrum we find a corresponding resonance in the action spectrum, confirming that hydrogen is lost from neutral 1-methylallyl. The band intensities, however, are different in the REMPI spectrum with rapidly decreasing band intensities to higher excitation energy. The REMPI ion signal intensity strongly depended on the temporal overlap of the excitation and ionization laser pulses and was sensitive to the nanosecond, which indicates that the decreasing band intensities in the REMPI spectrum most likely arise from short excited state lifetimes.
We also probed the dynamics of photo-induced hydrogen abstraction of the 1-methylallyl radical. Fig. 2 shows the appearance time of the hydrogen atom photoproduct following excitation of 1-methylallyl at 24080 cm−1. The signal intensity rises slowly and reaches a maximum at a time delay Δt ≈ 100 ns followed by a slow decay as the hydrogen atoms leave the detection volume in the source of our linear time-of-flight spectrometer. The solid line shown in Fig. 2 results from a fit of the experimental data to two exponentials, from which we obtain a rate constant k of 3.2(12) × 107 s−1 for the appearance of the H-atom photoproduct. The slow appearance rate of the photoproduct together with the presumably short excited state lifetimes inferred from the REMPI and action spectra suggests a nonradiative decay of 1-methylallyl radical after photoexcitation followed by dissociation in the ground electronic state, a mechanism similar to that observed for the allyl radical.5,6
 | ||
| Fig. 2 Appearance time of the H-atom signal following excitation of the 1-methylallyl radical at 24080 cm−1. The inset shows the Doppler profile recorded 70 ns after optical excitation of 1-methylallyl. | ||
To gain insight into the dissociation dynamics, we rely on the ab initio predictions by Miller10 of the complex ground state potential energy surface of C4H7. A scheme of the lowest energy pathways for the reaction of 1-methylallyl to C4H6 + H including low energy isomerization pathways appears in Fig. 3. In our photodissociation experiment we excite the 1-methylallyl radical at 24080 cm−1, which corresponds to 68.8 kcal mol−1, where the most favorable pathway for C–H bond fission according to extensive RRKM predictions (see ESI† for details) from 1-methylallyl is the formation of 1,3-butadiene and H over a barrier of only 45.3 kcal mol−1 for the E-isomer and 47.4 kcal mol−1 for the Z-isomer. The same photoproducts of C4H7 can also arise by isomerization to 3-buten-1-yl followed by C–H bond cleavage, which is energetically slightly less favorable than direct dissociation from 1-methylallyl.
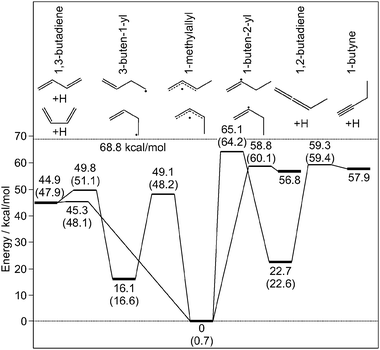 | ||
| Fig. 3 Schematic low energy reaction pathways for isomerization of the 1-methylallyl radical and C–H bond fission in C4H7 radicals using calculated energies from Miller for the E-isomers.10 The values in parentheses are the corresponding energies for the Z-isomers. See the ESI† for the structures of the transition states. | ||
Assuming 1,3-butadiene and H as the primary photofragments,15 we can predict the dissociation rate using RRKM theory. The calculated RRKM rate constant for direct dissociation of (Z)-1-methylallyl at an excitation energy of 68.8 kcal mol−1 is 5.80 × 107 s−1, while the corresponding rate for the E-isomer is slightly faster. The energized ground state 1-methylallyl radicals are expected to undergo fast E/Zisomerization16 with a RRKM rate of ∼3 × 1010 s−1 prior to dissociation and therefore from the point of view of the dissociation dynamics it does not matter which stereoisomer we selectively excited at 24080 cm−1.
We also measured Doppler profiles following excitation of 1-methylallyl at 24080 cm−1 by scanning the probe laser over the broadened Lyman-α transition. The FWHM for a Gaussian fit to the experimental Doppler profile shown in the inset in Fig. 2 is 3.2 cm−1, de-convoluted for the laser bandwidth. This corresponds9 to an average kinetic energy release of 8.6 kcal mol−1 or 36% of the available energy in the photoproducts using the calculated reaction energies10 for dissociation to (E)-1,3-butadiene and H or 40% for the Z-isomer, respectively. This is similar to the 42% that we found in the analogous photodissociation experiments of the allyl radical5 following excitation to its Ã-state where the nature of the dissociation dynamics agrees with a statistical mechanism.6
The measured dissociation rate constant of photoexcited 1-methylallyl agrees within a factor of two to the RRKM predictions for C–H bond fission to form 1,3-butadiene and H, the most favorable dissociation pathway in the electronic ground state following nonradiative decay,15 which supports a picture of statistical dissociation in the 1-methylallyl radical. We therefore consider it unlikely that methyl rotors as a structural element trigger the non-statistical effects observed in unimolecular decomposition of other hydrocarbon radicals.
We gratefully acknowledge the support of this work by the Schweiz. Nationalfonds Project No 200021-129478/1 and ETH Zürich.
Notes and references
- For selected reviews see: (a) I. Oref and B. S. Rabinovitch, Acc. Chem. Res., 1979, 12, 166 CrossRef CAS; (b) B. K. Carpenter, Angew. Chem., Int. Ed., 1998, 37, 3340 CrossRef; (c) B. K. Carpenter, Annu. Rev. Phys. Chem., 2005, 56, 57 CrossRef CAS; (d) U. Lourderaj, K. Park and W. L. Hase, Int. Rev. Phys. Chem., 2008, 27, 361 CrossRef CAS; (e) U. Lourderaj and W. L. Hase, J. Phys. Chem. A, 2009, 113, 2236 CrossRef CAS.
- T. Baer and W. L. Hase, Unimolecular Reaction Dynamics, Oxford University Press, New York, 1996 Search PubMed.
- J. Warnatz, Combustion Chemistry, Springer, New York, 1984 Search PubMed.
- (a) T. Gilbert, T. L. Grebner, I. Fischer and P. Chen, J. Chem. Phys., 1999, 110, 5485 CrossRef CAS; (b) M. Zierhut, W. Roth and I. Fischer, J. Phys. Chem. A, 2004, 108, 8125 CrossRef CAS; (c) B. Noller and I. Fischer, J. Chem. Phys., 2007, 126, 144302 CrossRef.
- L. Castiglioni, A. Bach and P. Chen, Phys. Chem. Chem. Phys., 2006, 8, 2591 RSC.
- J. Hostettler, L. Castiglioni, A. Bach and P. Chen, Phys. Chem. Chem. Phys., 2009, 11, 8262 RSC.
- A. Bach, J. Hostettler and P. Chen, J. Chem. Phys., 2006, 125, 024304 CrossRef.
- Substitution of functional groups has been shown to affect non-statistical dynamics in other chemical reactions, see e.g. (a) S. L. Craig, M. Zhong and J. I. Brauman, J. Am. Chem. Soc., 1999, 121, 11790 CrossRef CAS; (b) M. L. Chabinyc, S. L. Craig, C. K. Regan and J. I. Brauman, Science, 1998, 279, 1882 CrossRef CAS.
- M. Gasser, A. Bach and P. Chen, Phys. Chem. Chem. Phys., 2008, 10, 1133 RSC.
- J. L. Miller, J. Phys. Chem. A, 2004, 108, 2268 CrossRef CAS.
- M. Gasser, J. A. Frey, J. M. Hostettler, A. Bach and P. Chen, J. Phys. Chem. A, 2010, 114, 4704 CrossRef CAS.
- (a) J. C. Schultz, F. A. Houle and J. L. Beauchamp, J. Am. Chem. Soc., 1984, 106, 7336 CrossRef CAS; (b) D. J. Driscroll, W. Martir and J. H. Lunsford, J. Phys. Chem., 1987, 91, 3585 CrossRef CAS; (c) J. W. Hudgens, Advances in Multi-Photon Processes and Spectroscopy, World Scientific, Singapore, 1984 Search PubMed; (d) D. H. Tarrant, J. D. Getty, X. Liu and P. B. Kelly, J. Phys. Chem., 1996, 100, 7772 CrossRef CAS.
- J. K. Kochi and P. J. Krusic, J. Am. Chem. Soc., 1968, 90, 7157 CrossRef.
- A. B. Callear and H. K. Lee, Trans. Faraday Soc., 1968, 64, 308 RSC.
- The d4-1-methylallyl (CH3CDCDCD2) radical loses predominantly H-atoms, see Fig. S3 in the ESI†, which is only consistent with dissociation of 1-methylallyl to 1,3-butadiene + H as the dominant C–H bond fission process.
- P. J. Gorton and R. Walsh, J. Chem. Soc., Chem. Commun., 1972, 783 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Radical precursor synthesis, geometries, energies, vibrational frequencies of the ab initio calculations and RRKM rates. See DOI: 10.1039/c0cc01899f |
| ‡ This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| This journal is © The Royal Society of Chemistry 2011 |