Targeting glycolysis: a fragment based approach towards bifunctional inhibitors of hLDH-5†‡
Adam D.
Moorhouse
a,
Christian
Spiteri
a,
Pallavi
Sharma
a,
Mire
Zloh
b and
John E.
Moses
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: john.moses@nottingham.ac.uk; Fax: +44 (0)115 95 13564; Tel: +44 (0)115 951 3533
bThe School of Pharmacy, University of London, London, WC1N 1AX, UK
First published on 30th July 2010
Abstract
hLDH-5 has emerged as a promising target for anti-glycolytic cancer chemotherapy. Here we report a first generation of bifunctional inhibitors, which show promising activity against hLDH-5.
The lactate dehydrogenase (LDH) enzyme catalyses the interconversion of pyruvate and lactate with concomitant interconversion of NADH and NAD+. It converts pyruvate, the final product of glycolysis, to lactate when oxygen is absent or in short supply, and it performs the reverse reaction during the Cori cycle in the liver.1 At high concentrations of lactate, the enzyme exhibits feedback inhibition and the rate of conversion of pyruvate to lactate is decreased.
In humans, hLDH exists as five tetrameric isozymes, designated hLDH-1 through 5, each formed from a combination of two protein subunits M and H. hLDH-5 (M4) predominates in the liver and skeletal muscle while hLDH-1 (H4) prevails in the cardiac muscle, where it catalyses the reverse reaction using lactate as an aerobic source of energy.2
In tumour cells, an over-expression of oncogenes including Ras, Src, and HER-2/Neu through stabilisation of HIF-1α protein leads to an up-regulation of most proteins/enzymes involved in the glycolytic pathway, including hLDH-5 and the glucose transporters GLUT-1 and GLUT-3.3 This up-regulation is found in greater than 70% of human cancer cases, suggesting that tumour cells must acquire selective advantages by shifting from normal glucose metabolism (OXPHOS) to lactic acid production, even under normoxic conditions, the so called glycolytic switch4 or Warburg effect.5 Recent gene knock-out experiments demonstrated that reduction in the hLDH-5 activity of neu-initiated mammary tumor cells resulted in stimulation of mitochondrial respiration and a decrease of mitochondrial membrane potential.3b It also compromised the ability of these tumour cells to proliferate under hypoxia.
The diminished tumorigenicity of these hLDH-5 deficient cells could be reversed by complementation with the human ortholog hLDH-A (gene coding for the M unit) and hLDH-5 (enzyme), thus demonstrating that hLDH-5 plays a key role in tumour maintenance.3b Although the hierarchy of the possible genetic alterations, and/or adoption of optimum metabolic pathways, involved in the Warburg effect are still being uncovered,6hLDH-A and indeed the M4isozyme (hLDH-5) are primary targets for anti-glycolytic based therapeutic intervention.7
We report here a new class of bifunctional ligands which show selectivity towards hLDH-5. These novel compounds were prepared using a fragment based ‘click’ chemistry8 approach, taking advantage of the high fidelity of the CuAAC fusion reaction.9
The structure of hLDH-5 cocrystallised with the active site inhibitor oxamate and co-factor NADH has been solved,2 enabling the rational design of molecular inhibitors. Due to the close proximity between substrate and co-factor binding sites, we envisaged a fragment based approach could be used to unify, through a triazole linkage, the substrate and co-factor mimetics (Fig. 1).
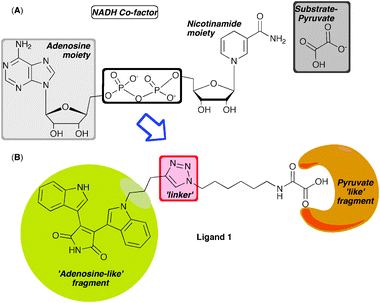 | ||
| Fig. 1 (A) Structure of the NADH cofactor and substrate, (B) designed bifunctional inhibitor using BIM as adenosine mimetic and oxamate-substrate mimetic tethered through a triazole linker. | ||
In the design of our inhibitors, we required synthetically viable ‘NADH-like’ fragments, and were drawn towards bis(indolyl)maleimides (BIM's) as potential candidates. BIM's are known adenosine mimetics and have been shown to be potent inhibitors of NAD+ dependent sirtuins.10 Considering their ready synthetic availability,11,12 opportunities for diversification and validated binding, BIM's were an excellent starting point for our ligand design. Oxamate on the other hand is a known inhibitor of LDH enzymes.13 Since oxamic acid derivatives have also proven to inhibit pfLDH,14 we hypothesised that N-alkylation to allow tethering would not adversely affect binding interactions. Furthermore, we envisaged replacing the oxamic acid functionality with a simple carboxylic acid group. The tether would comprise alkyl chains of varying length, fused through a triazole linker. Modelling studies using the known crystal structure of the human lactate dehydrogenase ternary complex with NADH and oxamate were performed with hypothetical ligand 1, which appeared to bind appropriately across both the substrate and co-factor binding site (see ESI†, Fig. S1).
Encouraged by these preliminary modelling studies, we began the syntheses of the ‘NADH-like’ and ‘substrate-like’ binding moieties, containing complementary azide and alkyne groups ready to be tethered using the CuAAC.8 Readily available azide functionalised fragments, aliphatic carboxylic acids 2–4,15 the aromatic azido carboxylic acids derivatives 5–8, and aromatic azido compounds 9–11 as potential substrate binding mimetics were prepared (Scheme 1).16
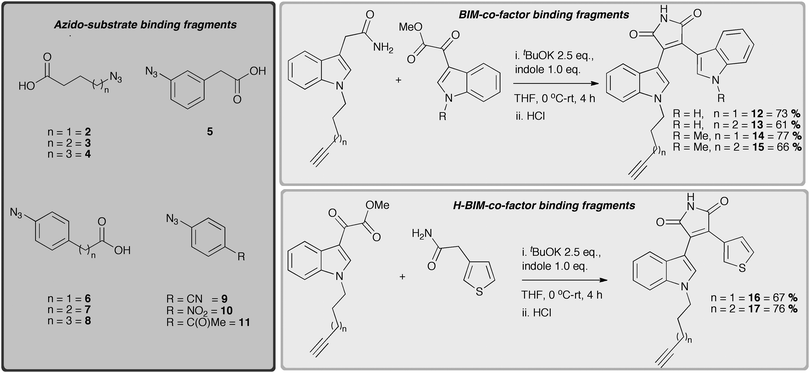
A selection of alkyne functionalised BIM's 12–15 were made by condensation of the corresponding indole-3-acetamides and indole-3-glyoxylates17 in the presence of tBuOK,12 as were the indoyl heteroaryl maleimide (H-BIM) substrates 16–17, using a similar condensation approach (Scheme 1). Uniting the corresponding azido-substrate and alkynyl-co-factor mimetics using microwave enhanced CuAAC16b gave a 31-member library of 1,4-triazole fused ligands (18–48, see ESI†, Table S1) of varied linker length/structure, in good yields (33–83%).18 Both the target ligands and fragments were screened for hLDH-5 activity at relatively high concentrations (50 and 100 µg mL−1) using a high-throughput 96-well plate UV assay, where decrease in the concentration of NADH was measured at 340 nm.19 The rate curves (ΔA/Δt) at these concentrations were compared with those of a blank, and against the curve of the known inhibitor sodium oxamate (at 50 and 100 µg mL−1) respectively. Compounds demonstrating inhibition more than/or similar to sodium oxamate were identified and their IC50 subsequently evaluated. Three compounds (∼10%), 24, 29 and 31, demonstrated promising activity at the concentration tested and were taken forward for further study (Fig. 2). Interestingly, the oxamate-like fragments did not show any significant activity, whereas the BIM fragments were insoluble in the assay medium and could not be evaluated.
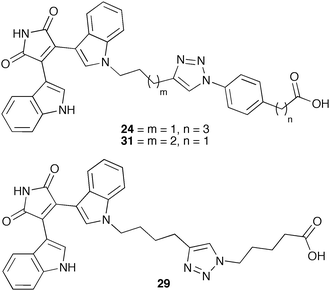 | ||
| Fig. 2 Lead hit hLDH-5 bifunctional ligands 24, 29 and 31. | ||
The activities against three enzymes were tested, human hLDH-5 (liver), rabbit rLDH-5 (muscle) and bovine bLDH-1 (heart). The IC50's of the three lead compounds 24, 29 and 31 were measured using the four-parameter logistic equation (GraphPad Prism for Windows V5.02). The IC50 of sodium oxamate was also recorded for comparative purposes (ESI†, Table S2). Interestingly, and unlike sodium oxamate, none of the bifunctional compounds (24, 29 and 31) demonstrated activity against the isozyme bLDH-1, which is comprised exclusively of the LDH-B gene product, the H-domain (H4). This unprecedented observation was very promising and a step closer towards achieving selective inhibition of hLDH-5, the major LDH in carcinoma. Such precise targeting of the LDH-A gene product will be essential for future development as an anti-cancer treatment.
The IC50 values revealed that compounds 24 and 29 were particularly promising lead structures, demonstrating notable selectivity for the hLDH-5 isozyme over other isozymes tested. Relatively low micromolar IC50's were exhibited for hLDH-5 (14.8 µM and 35.9 µM, respectively) (ESI†, Table S2), whereas neither compound displayed inhibition of the bLDH-1 even at high concentrations. The activity of 24 against hLDH-5 was 9-fold better than the standard sodium oxamate, making this candidate a promising target for further development. It is noteworthy that none of the bifunctional ligands bearing an N-methyl indole showed any kind of activity against LDH (ESI†, Table S1, 36, 37, 38, 39, 42, 43, 44, 45, 46). This may imply a possible H-bonding interaction of the indole group of 24, 29 and 31 in the enzyme active site. Unfortunately, bifunctional ligands bearing either a thiophene group (BIM fragment) or a non-carboxylic group (azido-fragment) were insoluble in the buffer medium (ESI†, Table S1, 25–27, 34, 35, 40–42, 47 and 48), and no conclusions could be drawn from these entries.
To help us understand the nature of ligand–target interaction, docking experiments on the lead compounds were performed.
Of the three compounds, ligand 24 was the best fit for the protein binding site, and overlayed well with the cofactor from the crystal structure. A favourable interaction between the protein and 24 was also apparent (binding energy = −10 kcal mol−1) with hydrogen bonds formed between the protein and carboxylic and BIM moiety (ESI†, Fig. S2). In line with the observed IC50's, ligands 29 and 31 were found to interact less favourably (cf.29 = −8.4 kcal mol−1 and 31 = −7.5 kcal mol−1).
Using a fragment based CC approach we have synthesised a small library of bifunctional compounds, specifically designed to target hLDH-5. We have identified three promising lead structures with impressive selectivity and activity. These ligands represent a new generation of bifunctional inhibitors specifically designed to target the hLDH-5 isozyme with a means to interfering with tumour metabolism. Molecular docking experiments corroborated our initial hypothesis and revealed favourable interactions with both the substrate and co-factor binding sites.
Detailed molecular modelling investigations, and further biological testing with a panel of cancer cell-lines are currently underway and will be reported in due course.
We thank EPSRC and The University of Nottingham for financial support. Thanks to Dr K. Barral and Sally Dempster for fruitful discussions.
Notes and references
- J. Everse and N. O. Kaplan, Adv. Enzymol. Relat. Areas Mol. Biol., 1973, 37, 61 Search PubMed.
- J. A. Read, V. J. Winter, C. M. Eszes, R. B. Sessions and R. L. Brady, Proteins: Struct., Funct., Genet., 2001, 43, 175 CrossRef CAS.
- (a) C. V. Dang and G. L. Semenza, Trends Biochem. Sci., 1999, 24, 68 CrossRef CAS; (b) V. R. Fantin, J. St-Pierre and P. Leder, Cancer Cell, 2006, 9, 425 CrossRef CAS.
- (a) S. Mazurek, C. B. Boschek and E. Eigenbrodt, J. Bioenerg. Biomembr., 1997, 29, 315 CrossRef CAS; (b) R. A. Gatenby and R. J. Gillies, Nat. Rev. Cancer, 2004, 4, 891 CrossRef CAS.
- (a) O. Warburg, The Metabolism of Tumors, Arnold Constable, London, 1930 Search PubMed; (b) O. Warburg, Science, 1956, 123, 309 CrossRef CAS.
- A. Le, C. R. Cooper, A. M. Gouw, R. Dinavahi, A. Maitra, L. M. Deck, R. E. Royer, D. L. V. Jagt, G. L. Semenza and C. V. Dang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2037 CrossRef CAS.
- (a) A. Giatromanolaki, E. Sivridis, K. C. Gatter, H. Turley, A. L. Harris and M. I. Koukourakis, Gynecol. Oncol., 2006, 103, 912 CrossRef CAS; (b) H. Xie, V. A. Valera, M. J. Merino, A. M. Amato, S. Signoretti, W. M. Linehan, V. P. Sukhatme and P. Seth, Mol. Cancer Ther., 2009, 8, 626 CrossRef CAS.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS; (c) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC; (d) C. J. Hawker, V. V. Fokin, M. G. Finn and K. B. Sharpless, Aust. J. Chem., 2007, 60, 381 CrossRef CAS; (e) A. D. Moorhouse and J. E. Moses, ChemMedChem, 2008, 3, 715 CrossRef CAS.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192 CrossRef CAS; (c) P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928 CrossRef CAS.
- J. Trapp, A. Jochum, R. Meier, L. Saunders, B. Marshall, C. Kunick, E. Verdin, P. Goekjian, W. Sippl and M. Jung, J. Med. Chem., 2006, 49, 7307 CrossRef CAS.
- (a) M. Brenner, H. Rexhausen, B. Steffan and W. Steglich, Tetrahedron, 1988, 44, 2887 CrossRef CAS; (b) P. D. Davis and R. A. Bit, Tetrahedron Lett., 1990, 31, 5201 CrossRef CAS.
- (a) M. M. Faul, L. L. Winneroski and C. A. Krumrich, J. Org. Chem., 1998, 63, 6053 CrossRef CAS; (b) S. Roy, S. Roy and G. W. Gribble, Org. Lett., 2006, 8, 4975 CrossRef CAS.
- For example see: J. H. Wilkinson and S. J. Walter, Enzyme, 1972, 13, 170 Search PubMed.
- S.-r. Choi, A. Pradhan, N. L. Hammond, A. G. Chittiboyina, B. L. Tekwani and M. A. Avery, J. Med. Chem., 2007, 50, 3841 CrossRef CAS.
- Z. B. Li, Z. Wu, K. Chen, F. T. Chin and X. Chen, Bioconjugate Chem., 2007, 18, 1987 CrossRef CAS.
- (a) K. Barral, A. D. Moorhouse and J. E. Moses, Org. Lett., 2007, 9, 1809 CrossRef CAS; (b) A. D. Moorhouse and J. E. Moses, Synlett, 2008, 2089 CAS.
- C. Menozzi, P. I. Dalko and J. Cossy, Chem. Commun., 2006, 4638 RSC.
- Note: the products of the 31-member library were all isolated, purified and characterised. Under the reaction conditions, some of the fragments decomposed or the products could not be isolated or adequately purified. In these cases, the samples were not taken forward for screening.
- M. Ryberg, D. Nielsen, K. Osterlind, T. Skovsgaard and P. Dombernowsky, Ann. Oncol., 2001, 12, 81 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Extended experimental data, experimental procedures and 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c0cc01166e |
| ‡ This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| This journal is © The Royal Society of Chemistry 2011 |