FT-IR metrology aspects for on-line monitoring of CO2 and CH4 in underground laboratory conditions
Cristelle
Cailteau
*abc,
Jacques
Pironon
a,
Philippe
de Donato
b,
Agnés
Vinsot
d,
Thomas
Fierz
e,
Christophe
Garnier
f and
Odile
Barres
b
aNancy Université-CNRS-CREGU, G2R laboratory, BP 70239, 54506, Vandoeuvre-lès-Nancy, France
bNancy Université-CNRS, LEM laboratory, BP 40, F-54501, Vandoeuvre-lès-Nancy Cedex, France
cAndra, 1–7, rue Jean-Monnet, 92298, Châtenay-Malabry, France
dAndra, Centre de Meuse/Haute-Marne, Route Départementale 960, 55290, Bure, France
eSolexperts AG, Mettlenbachstrasse 25, Postfach 122, CH-8617, Mönchaltorf, Switzerland
fUniversity Paul Verlaine, LCME, EA 4164—rue Victor Demange B.P. 80105, 57503, Saint-Avold Cedex, France
First published on 11th March 2011
Abstract
The French National Radioactive Waste Management Agency (ANDRA) has recently developed a new experimental set-up which allows sampling water from marl rock formations, together with an in situ characterisation of the composition and migration mechanisms of the gases dissolved in the marl porewater. Gases and liquids are collected from vertical borehole drillings in underground laboratories. The analytical design, Fourier transformed infrared spectroscopy based, allows powerful and long term on-line monitoring of gases released by low-permeability media. The IR system is designed to cope with the unfavourable measurement conditions occurring in an experimental underground laboratory (moisture, dust, etc.). Because the working conditions in such an underground laboratory make complete purging of the IR spectrometer difficult, the IR spectra of geological gases are often perturbed by contributions from atmospheric CO2 and water vapour. The metrology aspect is based on an IR low resolution sensor equipped with two measurement compartments. In the internal compartment linked to the borehole layout, gases are monitored on-line through a cell with a variable optical path, whereas in the external compartment, atmospheric CO2 is measured through a short open path configuration. The experimental method and data processing procedure used to determine the real partial pressure of CO2 arising from the marl rock formation are described in this paper. Results of the on-line gas (CO2 and CH4) monitoring conducted in the Mont Terri underground laboratory are presented and compared with punctual gas chromatography analyses.
Introduction
Sedimentary rocks, and among them, argillaceous rocks, contain various gases (CO2, alkanes, H2, O2, N2, H2S, SO2, COS, NH3, noble gases) of organic and inorganic origin. They are present as vapour, or as supercritical phase and/or dissolved in porewater according to their concentrations and the conditions of total pressures and temperatures. Organic gases originate from the diagenesis of fossil organic matter trapped during sedimentation. Alkanes and CO2 are the most common gases found in sedimentary rocks and are sometimes associated with H2S and N2.1CO2 is in chemical equilibrium with carbonate minerals. They result from the biotic–abiotic decay of organic matter and can also be derived from bacterial fermentation, whereas CH4 mainly forms through the thermal or bacterial degradation of organic matter. CH4 thermogenesis generates large amounts of heavier gas molecules (C2, C3, C4, C5), whereas bacterial methanogenesis does not produce any additional alkanes.2,3 The presence of dissolved gases and their respective concentrations influence the geochemistry of porewater, and consequently alter the texture and mineralogy of the rock through chemical interactions. Carbonaceous clay formations are the main gas reservoir rocks in sedimentary basins.Clay-based rocks are also considered as potential host formations for deep geological disposal of high-level and long-lived intermediate-level radioactive wastes.4,5 Clay formations contain interstitial porewater but display a low permeability, thus limiting radionuclide mobility. However, the retention properties of clay formations can vary depending on the chemistry of interstitial water.6,7 Characterising porewater is intrinsically difficult because its extraction from clayey media is complex (low content and low permeability).8,9 Furthermore, the extraction techniques can alter the porewater chemistry through atmospheric exposure (i.e. pH variation and Eh increase). To avoid any atmospheric contamination of porewater during sampling, a new experimental concept of porewater extraction was implemented for the first time at the Mont Terri rock laboratory (Switzerland) through the PC-C (Porewater Chemistry-C) experiment.10 The experimental concept was duplicated in the underground research laboratory (URL) in Bure (North-East of France) built by the French National Radioactive Waste Management Agency (ANDRA). Early 2005, a borehole was drilled in the drift at a depth of 445 m (experiment PAC2002). At the end of 2005, another borehole was drilled at a depth of 490 m and was equipped (experiment PAC1002)11 with a specific device called a completion. In each experiment, a borehole interval was filled with argon at a pressure close to 1.3 bar in order to follow the evolution of the composition of the gas mixture by exchange with the host rock and to sample porewater under controlled conditions. All conditions were designed to avoid rock oxidation and microbiological perturbation in either the borehole or the gas circuit (dry-drilling with nitrogen, aseptic tool and total pressure >1 bar). A water-sampling module pumped the water flowing from the rock due to the hydraulic pressure difference between the rock and the interval (40 bar to 1.3 bar). This module allowed online analysis of water and guaranteed that the main part of the interval remained free of liquid water. Gas in contact with rock and seepage water circulated in an external gas module. The gas mixture was then sampled at various dates, thus providing the evolution of its composition upon exchange with the rock. The gas circulation module also allowed on-line non-destructive gas monitoring.12
The main objective of this paper is to introduce a system, which allows powerful and long term on-line monitoring of gases released by low-permeability media. Knowledge of the nature, partial pressure and transfer mechanisms of various gases is very valuable for assessing porewater chemistry and consequently, for predicting the behaviour of radionuclides in deep geological waste storages.13
Several in situ gas analysers have been developed to monitor atmospheric trace species (CO2, CH4, N2O, CO,…). The most frequently used ones for measuring CO2 are based on non-dispersive infrared spectroscopy (NDIR). In standard air composition the accuracy can reach 0.01 µmol mol−1 for punctual measurements and 0.003% for hourly measurements.14 Using Fourier transform spectroscopy with a resolution of 1 cm−1, various gases can be analyzed simultaneously with an accuracy of 0.04% for 360 ppmv CO2, 0.05% for 1700 ppmv CH4, 0.5% for 50 ppmv CO and 0.1% for 310 ppmv N2O.15 Such accuracy can be obtained by using an optical path length of 22 m and averaging 256 scan spectra in recording between 2300 and 3000 cm−1 during 8 min. Non-optical techniques such as mass spectroscopy or gas chromatography16 can also be used for measuring CO2, CH4, N2O, CO, etc. Both these methods require sample extraction and preconditioning, and the use of specific sensors. An accuracy of 0.02% for CO2 (360 µmol mol−1), 0.2% for CH4 (1700 nmol mol−1),17 0.1% for N2O (300 nmol mol−1)18 and 1% for CO (100 nmol mol−1) can be obtained from these experimental methods.19 Recent technological advances exhibit the potentialities of quantum cascade laser (QCL) for monitoring gases.20,21 The advance of QCL technology allows the use of very promising tuneable, miniature, non-cryogenic mid-IR sources for online, in situ and continued monitoring of gases,22–24 which cover down to 100 cm−1spectrum range, with high resolution (<1 cm−1) and high speed measurement (to 100 cm−1 per second). Coupled to photoacoustic spectroscopy, QCLs could lower the detection limit to ppb or sub-ppb.25
Choosing a sensor for on-line non-destructive and non-altering analysis of a quasi-unknown gas mixture in the context of an underground laboratory appears as a complex task that must take into account various key-parameters. The first stage is to choose the most appropriate technique for gas analysis, taking into account four critical constraints: (i) full spectrum analyses are needed because the gas composition is not sufficiently well-known at the beginning of the project, (ii) accurate analyses are needed because gas concentrations can be rather low, (iii) the technique must be insensitive to argon (carrier gas in the borehole), and (iv) the technique must be sensitive to CO2 and to the main organic gases (light alkanes) present in the porewater of the rock formation. Infrared spectroscopy is a vibrational spectroscopy frequently used for analyzing natural gases such as CO2 and light alkanes.15,26,27 In the mid-IR range (5000–600 cm−1), the fundamental vibration frequencies28 of C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC are all active and precisions down to the ppb can be reached under favourable conditions.25,29,30 The first constraint leads to the choice of a whole spectrum technique, like Fourier transform infrared spectroscopy (FT-IR) which can cover all the mid-IR range. QCL technology doesn't provide a full spectrum analysis (applicable only to ca. 100 cm−1) and the composition of the gas mixture must be previously known in order to choose the type of sensor and select the appropriate line for each compound. Furthermore, Fourier transformed infrared spectrometers can be equipped with a variable long path White cell,31,32 which presents two advantages: (i) it is versatile in terms of sensitivity since a variable optical path can be used, (ii) such a cell can be linked to a gas collection circuit and allows on-line and flux analyses.28,33,34 The use of a White cell allows the second constraint to be addressed.
O and CC are all active and precisions down to the ppb can be reached under favourable conditions.25,29,30 The first constraint leads to the choice of a whole spectrum technique, like Fourier transform infrared spectroscopy (FT-IR) which can cover all the mid-IR range. QCL technology doesn't provide a full spectrum analysis (applicable only to ca. 100 cm−1) and the composition of the gas mixture must be previously known in order to choose the type of sensor and select the appropriate line for each compound. Furthermore, Fourier transformed infrared spectrometers can be equipped with a variable long path White cell,31,32 which presents two advantages: (i) it is versatile in terms of sensitivity since a variable optical path can be used, (ii) such a cell can be linked to a gas collection circuit and allows on-line and flux analyses.28,33,34 The use of a White cell allows the second constraint to be addressed.
Working conditions in underground galleries require specific adaptations of the FT-IR spectrometer taking into account moisture, vibrations, dust, and the non-controlled atmosphere. Moreover, for security reasons, it was not allowed to continuously use pressurized gases in the URL. As a consequence, the FT-IR spectrometer must work without any atmospheric purge, in contrast to conventional laboratory equipment. In such conditions the infrared beam of the IR device crosses the gas inside the cell and the gas from the atmosphere, both containing CO2. Therefore, the IR signature due to atmospheric CO2 is collected together with the signature from the borehole gas mixture. Part of this paper will focus on the development of an original analytical procedure specific to the quantitative determination of CO2 in the presence of a large contribution from atmospheric CO2. An example of gas measurements in natural samples acquired in the Mont Terri rock laboratory will be presented and discussed. Because first results showed that CO2 and CH4 are the two major species identified by FT-IR, this paper focuses on their evolutions. The obtained spectra allow the determination of other gases, the study of which will be developed in future work.
Equipment and methods
IR system design: a double compartment system
One possible way to subtract the contribution of atmospheric CO2 from measurements is to record a reference spectrum before each measurement. However, recording reference spectra requires either emptying the gas cell, or filling it with a neutral gas. Evacuating the gas cell induces a loss of gas from the circuit. Filling it with a neutral gas requires, after recording the reference spectra, that the neutral gas must be transferred into the circuit or into the atmosphere. Both solutions were, however, rejected because (i) gas transfer in the circuit dilutes the original gas mixture and impacts the equilibrium of the gas mixture/borehole system, (ii) neutral gas transfer in the atmosphere requires vacuum pumping that could lead to an important head loss when the cell is returned on-line. Consequently, the FT-IR spectrometer must work without any atmospheric purge and without recording new reference spectra before each measurement.As the infrared beam crosses the gas cell and the atmosphere (internal compartment Fig. 1), IR signals due to atmospheric water vapour and carbon dioxide are collected together with the signals of water vapour and carbon dioxide from the borehole gas mixture. In order to decompose this superimposition of signals, a second compartment specifically dedicated to the monitoring of atmospheric gases must be added to the IR device. It uses the same source and interferometer (external compartment Fig. 1). In this second compartment, the IR beam crosses only the atmosphere from the source to the detector. Note that the geometry of this optical bench implies that the length of the optical path crossing the atmosphere is different for each compartment, and by consequence, must be determined for quantitative CO2 measurements. Such a configuration has already been used for other purposes.35
 | ||
| Fig. 1 IR spectrometer with a double compartment system. The modulated IR beam can be switched between the two compartments through the use of a dual-position rotational mirror. The internal compartment is in the left part and the external compartment in the right one. | ||
The chosen FT-IR device is a double compartment low resolution Fourier transform infrared spectrometer (TENSOR 27 from BRUKER), as presented in Fig. 2. To minimise the impact of an open system, it is equipped with a “water resisting” ZnSe beamsplitter and windows. The first compartment, called the internal compartment, is equipped with a multipass gas cell (variable long path gas cell A 136/2-L BRUKER) connected to the gas collection circuit. The optical path length of this gas cell can vary from 0.8 m to 8 m and its volume is 2 L. The gas cell is equipped with CaF2 windows. A drawback associated with such a choice is that the infrared range accessible to CaF2 is limited to wavenumbers at least higher than 1000 cm−1. The second compartment, called the external compartment, is dedicated to the monitoring of the atmosphere and acts as an open path system working in active mode. The modulated beam is switched between the two compartments through a software controlled rotational mirror. Each compartment is equipped with a DTGS sensor.
 | ||
| Fig. 2 View of the IR spectrometer with a double compartment system in use at the Mont Terri laboratory. | ||
 | ||
| Fig. 3 Layout of the experimental device used in the Mont Terri and the ANDRA experimental underground laboratories A: sectional drawing of the borehole and completion, B and C: layout of the device for sampling and online analyses. | ||
| f(A) = k1A + k2A2 + k3A3 = lpCO2 | (1) |
Our study36 confirmed that coefficients k1, k2, and k3 are dependent on the bulk pressure according to polynomial relations. Consequently, eqn (1) becomes:
| fPbulk(A) = k1(Pbulk)A + k2(Pbulk)A2 + k3(Pbulk)A3 = lpCO2 | (2) |
The same type of result was obtained for CH4 quantification.
Analytical development
This section describes how the spectra recorded in the two compartments are treated to acquire the CO2 partial pressures in the borehole. CH4 partial pressures are obtained with the calibration procedure developed in the companion paper of this work.36 | ||
| Fig. 4 Representation of the double compartment system. The full line represents the IR beam crossing the gas cell filled with borehole gas mixture. The dotted line represents the IR beam crossing the atmosphere. α, β and α′ refer to the corresponding simulated gas cells. | ||
The second compartment can be simulated as a simple gas cell noted α′ (Fig. 4, right), filled with atmospheric gases at atmospheric pressure (Patm). The optical path length lα′ of the α′ cell represents the optical path length of the external compartment which crosses the atmosphere from the IR source to the DTGS sensor of the external compartment (Fig. 1). The difference between the two virtual α and α′ gas cells is their optical path length lα and lα′, which is unknown and must be determined.
| pαCO2 = pα′CO2 | (3) |
 | (4) |
 | ||
| Fig. 5 Upper part: internal (a) and external (a′) compartment spectra with β gas cell emptied. Lower part: internal (b) and external (b′) compartment spectra with β gas cell filled with a standard gas mixture containing argon and 750 ppmv of CO2 (pCO2β = 0.825 mbar) with a bulk pressure Pcell of 1.1 bar. | ||
 | ||
| Fig. 6 Evolution of (a) bulk atmospheric pressure, (b) fPatm(Aα), (c) fPatm(Aα′) and (d) κ ratio between 28 November 2006 and 28 December 2006. Measurements were obtained in the ANDRA underground laboratory in Bure (−490 m). fPatm(Aα) and fPatm(Aα′) represent pCO2α and pCO2α′ values balanced by lα and lα′ optical path lengths, respectively. | ||
Experiments were first performed in a surface laboratory with the same equipment in experimental conditions replicating those encountered in the URL. The gas cell (β) can then be either empty or filled with standard gas mixtures (from Messer France SAS). Bulk pressures in the cell (β) and in the atmosphere are recorded (α and α′ gas cell). The equipment design is that of Fig. 4. The lower part of Fig. 5 shows an example of spectra obtained when the β gas cell is filled with a standard gas mixture containing argon and 750 ppmv of CO2 (pCO2β = 0.825 mbar) with a bulk pressure Pcell of 1.1 bar. The optical path length lβ in the cell is fixed at 1.6 m. The bulk pressure involved in α and α′ gas cells is the atmospheric bulk pressure Patm (983 mbar). Aαβ and Aα′ are the integrated areas of this band measured in the internal (both α and β gas cell) and external compartment (α′ gas cell), respectively.
According to the mathematical development of Plass,38,39 the IR beam crosses the cells in series, with independent bulk pressures, temperatures and concentrations of absorbing gas. Plass modelled a multi-cell system with jcells in series by a virtual homogeneous (h) one-layer system associated with a bulk pressure Ph, a temperature Th, an amount of absorbing gas uh and an optical path length lh. The signal received by the detector corresponds to the signal produced by a virtual homogeneous one-layer system (i.e. from all the jcells in series). In the case of the integration of a non-well defined and non-opaque band and if the temperature in the cells is constant, Plass gives the following simple relation linking the virtual homogeneous one-layer system with the model of jcells in series:
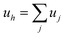 | (5a) |
| with uj = ρjlj | (5b) |
 | (6) |
As our internal compartment has been simulated as a two-cells in series system (α and β gas cell), uh becoming noted uαβ, eqn (5a) is as follows:
| uαβ = uα + uβ | (7) |
The combination of eqn (6) and (7) gives eqn (8):
| fPαβ(Aαβ) = fPatm(Aα) + fpcell(Aβ) | (8) |
For the rest of this work, it will be assumed that the bulk pressure Pαβ characteristic of the bulk signal received by a detector from the internal compartment is the average of Pcell and Patm:
 | (9) |
The final objective of the procedure is to determine pCO2β, the partial pressure of CO2 in the β gas cell representing the multipass gas cell of the internal compartment. Determination of pCO2β means determining fpcell(Aβ) (eqn (2)) or uβ (eqn (6)). Combining eqn (4) and (8) we arrive at:
| fpcell(Aβ) = fPαβ(Aαβ) − κfPatm(Aα′) | (10) |
The partial pressure of CO2 (pCO2β) in the cell is calculated by dividing fpcell(Aβ) values by the value of the optical path length in the cell (lβ):
 | (11) |
Results and discussion
Validation of the method for the absolute determination of CO2
Gas cells filled with standard gas mixtures, with known bulk pressure and the same optical path length as used in the URL experimentation (i.e. 0.80 m), were analyzed to validate both the experimental and data treatment aspects. Six standard gas mixtures were used; they were different to those used for the calibration step.36 They contained 152, 344, 490, 1480, 1970 and 2500 ppm of CO2, respectively, the rest being argon. The atmospheric bulk pressure during the experiments was around 1000 ± 5 mbar. Triplicate spectra for each sample were acquired with five different bulk pressures, between 900 and 1300 mbar. Gas mixture concentrations were certified with a relative uncertainty of 3% and bulk pressures were measured with an accuracy of ±4 mbar. In each measurement, the CO2 spectra were recorded from both the internal and external compartments, which allowed us to calculate Aαβ and Aα′, coupled with bulk pressure in the cell and atmospheric pressure (Pcell and Patm) measurements. Aαβ values higher than 100 U.A. were not taken into account because repeatability of the measurements is not sufficient.36Results are presented in Table 1: the mean obtained value for all gas cell bulk pressures is given with its relative standard deviation (RSD) and the relative root mean squared error of prediction (RMSEP) of the determination method of absolute CO2. Taking into account that the RMSEP of the calibration model is included between 1 and 3%, the relative RMSEP obtained for absolute CO2 determination is reasonable. The calculations are considered as truly valid for values higher than 0.3 mbar m, as the relative RMSEP in the calibration model increases for values lower than 0.3 mbar m.36,40 It can be concluded that the performance of the proposed calculation algorithm depends on the amount of CO2 in the cell. In the case of low CO2 amounts, close to 0.3 mbar m, it is proposed to increase the optical path length in the cell in order to increase accuracy.
| Declared (ppm) | Mean obtained value (ppm) | RSD (%) | RMSEP (%) |
|---|---|---|---|
| 152 | 141 | 19 | 18 |
| 344 | 339 | 5 | 5 |
| 490 | 480 | 2 | 3 |
| 1480 | 1484 | 2 | 2 |
| 1970 | 2016 | 2 | 3 |
| 2590 | 2623 | 1 | 1 |
Measurements of gases in natural samples
The PC-C experiment borehole located in the underground rock laboratory of Mont-Terri (St Ursanne, Switzerland) was studied for gas phase exchange with the rock from March to December 2004.10 During this period of 301 days, the test interval, which was filled with pure argon the first day, was purged twice with argon after 44 and 232 days for technical reasons. Before the second argon filling, experimental disturbances took place, impacting mainly on the homogeneity of the total pressure in the gas circuit. Only the last 70 days are considered to have yielded significant results. These results are presented in Fig. 7 for CO2 (a) and CH4 (b), respectively. During this period, 838 pairs of IR spectra were acquired (with an optical path length of 0.8 m) and 7 gas sampling cells were extracted from the circuit and analyzed by gas chromatography. For CO2 concentration evolution, measurements by the IR system were stopped on the 254th day while the quantitative measurement of CH4 continued until the end of the experimental period. After the 254th day, the intensity of CO2 band (sum of atmospheric and borehole CO2 signals, i.e.Aαβ) exceeded the limit of quantification (100 U.A.).36 The evolution of the CO2 concentration showed a sharp increase in the first 5 days of the experiment, followed by a slight regular increase during the rest of the time. These results are in good agreement with the 6 gas chromatography results acquired on sampling days 231, 253, 286, 294, 296, 299 and 301, except for the datum acquired on day 273 (Table 2), that is not in accordance with the other five gas sampling GC data.10CH4 concentration evolution shows a quasi-monotonic increase with time during the same period, that is confirmed by seven gas samples analysed by gas chromatography.10 The continuous measurements obtained through the IR system show a weak dispersion compared with those obtained through the gas sampling method. Dispersion of gas sampling data can be due to several reasons: (i) the gas cells are not located exactly at the same place in the gas circuit and data dispersion could represent heterogeneities in CO2 amount in the circuit, (ii) manipulation or storage of gas cells can induce slight leakages by transfer operations from one container to another, (iii) local adsorption of CO2 on the wall of containers, (iv) low reproducibility of GC data with time because analyses were not acquired together on the same day. The advantage of the IR technique is the low dispersion of data giving direct access to the gas transfer curves. Gas transfer models in porous media can be numerically resolved taking into account the experimental parameters. The curves resulting from this modelisation can then be adjusted with the experimental gas transfer curves in order to quantify the respective contributions of the diffusion, advection and/or adsorption involved in gas transfer in the porous media. Such curves can be used to determine the gas concentration in the source-rock responsible for gas migration towards the borehole. It can also be used to discuss the reliability of some geological data such as effective diffusion coefficient and petro-physical parameters (porosity, permeability, adsorption surfaces). | ||
| Fig. 7 Evolution of CO2 (a) and CH4 (b) gas concentration between day 230 and day 310 of the PC-C experiment in the Mont Terri laboratory obtained by gas chromatography on gas samplings (circles) and by IR device monitoring (dots). | ||
| Sampling days/d | CO2 (ppmv) | CH4 (ppmv) |
|---|---|---|
| 231 | 1800 ± 75 | 125 ± 3 |
| 253 | 3900 ± 162 | 1410 ± 33 |
| 273 | 3400 ± 141 | 2700 ± 64 |
| 286 | — | 2800 ± 66 |
| 294 | 4300 ± 178 | 3400 ± 81 |
| 296 | 4400 ± 183 | 3800 ± 90 |
| 299 | 4400 ± 183 | 3800 ± 90 |
| 301 | 4000 ± 166 | 4900 ± 116 |
The high sensitivity of this method allows us to detect slight variations in the partial pressure of gas. Fig. 8 represents a period of two days and a few hours of the PAC1002 experiment carried out in the Bure URL.11 This period occurred 573 days after drilling of the borehole and after argon refilling of the gas circulation circuit and the gas cell, while the borehole was isolated from the rest of the circuit. The gas mixture inside the borehole contained a given quantity of CO2, CH4 and other gases (mainly argon) resulting from the 573 days of gas equilibration with the rock formation (marl). The pressure in the gas circuit was slightly higher than in the borehole. When we reconnected the borehole with the gas circulation circuit filled with argon, we observed very specific behaviours for CO2 and CH4. The evolution of CH4 concentration during the days after the connection initially followed a sine cardinal (aka sampling function (sinc)) profile, and then stabilised. Whereas, the concentration of CO2 reached a maximum value after 2 hours, then decreased slightly to tend towards stabilization after 30 hours. The same behaviours could be observed for both gases after starting and stopping the gas circulation pump, but with lesser amplitudes.
 | ||
| Fig. 8 Evolution of CO2 (green circles) and CH4 (orange circles) concentrations between the day 573 and the day 576 of the PAC1002 experiment in the Bure laboratory monitored by IR device after a dilution of the borehole gas mixture by addition of argon. | ||
Concerning the behaviour of CH4, the duration of the sine cardinal period was 10 hours, which corresponds to the time necessary for the circulation pump to pump out the whole volume of the gas circuit (about 11.14 L at 20 mL min−1). The transfer mechanism of CH4 in clayey rock, probably diffusion–advection, is not rapid enough to compensate for the CH4 decrease induced by argon input. The behaviour of CH4 concentration seen by the IR device could thus be the result of three phenomena acting in concert: gas flow induced by the pump, diffusion of the mixture of the two gases (gas in the borehole and argon in the circulation circuit) and pressure re-equilibrium between the borehole and the circulation circuit.
Concerning the behaviour of CO2, its concentration reaches a maximum and decreases slightly to tend towards stabilization. The fast increase at the beginning of the cycle could have the same origin as for CH4: the action of the pump coupled to pressure re-equilibrium allows the CO2 present in the borehole to reach the infrared cell. In the borehole, the arrival of argon (i.e. decrease of CO2 concentration) is quickly compensated by CO2 production from the rock. The equilibrium of the gas mixture in the borehole with fluid in the rock system is governed here by the dissolution of the carbonate minerals within the porewater, with regard to the pH.
Thus, the difference of behaviour between CO2 and CH4 is attributed to the difference of chemical activity of the two components. The behaviour of CH4 is representative of a purely mechanical mixing of two gases in a circuit while the concentration in CO2 is influenced mainly by the chemical reaction between CO2, porewater and the rock. These details of the evolution of the two gases are only accessible with continuous, online, and non-destructive measurement.
Conclusions
A new FTIR procedure for on-line monitoring of molecular gases (mainly CO2 and CH4) released by low-permeability geological media has been presented. In an underground laboratory context, gas monitoring by FT-IR is specific. In contrast to conventional surface laboratory equipment, the FT-IR spectrometer must work without any atmospheric purge. Such conditions are highly problematic for CO2 monitoring, because this gas is present in the rock formation (e.g. the atmosphere of the URL) at high concentrations. To deal with such specific conditions, an original procedure based on FT-IR analysis using a spectrometer with two compartments sharing the same source and interferometer has been developed. The first internal compartment is connected to the borehole through a variable optical path gas cell connected to the gas circuit. The external compartment is open to the atmosphere. In the case of the internal compartment, the bulk signal recorded by the IR system comes from two contributions: atmospheric and borehole CO2. Deconvolution allows extraction of the borehole CO2 concentration expressed in terms of absolute partial pressure or concentration. The results of this procedure are limited by the accuracy of calibration and the quantification limit of the device: explored partial pressure ranges are between 0.3 and 4 mbar m for CO2 and 0.3 and 12 mbar m for CH4, and relative RMSEP of the models is included between 1 and 3% in the explored pressure ranges for each gas. Quantification limits can be lowered by using gas cells with higher path lengths, which increases the signal/background ratio.Automatic spectra recording, coupled with an appropriate calculation procedure, permits on-line measurements of partial pressures of CO2 and associated gases such as CH4 over short to long periods of time. The periodicity of measurement can be adapted or modified by the operator as needed. Quantitative calculations take into account bulk pressure variations in the gas circuit because the experiment is not carried out in isobaric conditions.36 Comparison of on-line measurements acquired by the IR device and measurements acquired by gas sampling coupled to GC analysis favour the FT-IR technique. It is not affected by gas manipulation and allows the determination of accurate physical laws to describe on-site gas transfer from porous geological media. Gas content determination is a very useful tool to complete porewater geochemistry as well as the measurement of accurate pH deduced from CO2 partial pressure at equilibrium with water. Such data are of great interest for the description of rock formations acting as host rocks for nuclear waste disposal. It is also useful to monitor CO2 or CH4 geological storage: survey during the injection or post-injection period requires comparison with the baseline acquired before injection.41 The development of the FT-IR analysis of other natural gases (alkanes, H2O) by statistical spectral analyses (classical least square method)33 and new advances in 13C isotopes determination of CO2 and CH4 offer new challenges for the immediate future. This novel approach used to quantify gas emissions in a rock formation over time is an important contribution to the knowledge of gas transfer in the lithosphere. It is also a comprehensive way to monitor gas exchanges between the geosphere and the atmosphere.
Nomenclature
| A | intensity of the CO2ν3 band integrated between 2220 and 2400 cm−1 |
| A α, Aα′, Aβ, Aαβ | intensity of the CO2ν3 band integrated between 2220 and 2400 cm−1 considering the α, α′, β or αβ gas cell |
| f | polynomial calibration function |
| f P | polynomial calibration function for a given constant pressure P |
| k 1, k2, k3 | polynomial coefficients of function f |
| k 1(P), k2(P), k3(P) | polynomial coefficients of the function fP |
| l | optical path length |
| l α, lα′, lβ, lαβ | optical path length in the α, α′, β or αβ cell |
| M m | molar mass of the considered gas |
| P α, Pα′, Pβ, Pαβ | bulk pressure of the sample in the α, α′, β or αβ cell |
| P atm | atmospheric pressure around the system |
| P bulk | bulk pressure of the considered sample |
| P cell | bulk pressure in the cell |
| p CO 2 α, pCO2α′, pCO2β | partial pressure of CO2 in the α, α′, or β cell |
| R | Rydberg constant |
| T | temperature of the gas sample (K) |
| u α, uβ, uαβ | mass absorbing gas per unit area, nomenclature derived from Plass' nomenclature, considering the α, β or αβ cell |
| κ | optical path ratio crossing the atmosphere between the two compartments of the IR system |
| α, β, αβ, α′ | indices referring to the virtual gas cell |
Plass' Nomenclature38
| h | index referring to a single-layer homogeneous system |
| j | index referring to a single cell in series |
| u h | mass absorbing gas per unit area for all the jcells in series |
| u j | mass absorbing gas per unit area for the jcell |
| r j | density of absorbing gas for the jcell |
| l j | optical path length of jth cell in series |
Acknowledgements
This work was financially supported by the French National Agency for Radioactive Waste Management (ANDRA) to whom we express our gratitude. Experiments in the surface laboratory were performed at Nancy University (LEM laboratory). We gratefully acknowledge Gordon Filby and Anne de Henning for improving the English.References
- J. M. Hunt, Petroleum Geochemistry and Geology, W.H. Freeman & Co, New York, 2nd edn, 1996 Search PubMed.
- A. Prinzhofer and A. Battani, Oil Gas Sci. Technol., 2003, 58, 299 CrossRef CAS.
- J.-P. Girard, C. Fléhoc and E. Gaucher, Selected Papers from the 5th International Symposium on Applied Isotope Geochemistry, Heron Island, Great Barrier Reef, Australia, 26–30 May 2003, 2005, 20, p. 713 Search PubMed.
- Andra, Dossier 2005 Argile: Evaluation of the Feasibility of a Geological Repository in an Argillaceous Formation, Dossier 2005 Argile, 2005 Search PubMed.
- M. Thury, C. R. Phys., 2002, 3, 923–933 Search PubMed.
- É. C. Gaucher, P. Blanc, F. Bardot, G. Braibant, S. Buschaert, C. Crouzet, A. Gautier, J.-P. Girard, E. Jacquot, A. Lassin, G. Negrel, C. Tournassat, A. Vinsot and S. Altmann, C. R. Geosci., 2006, 338, 917–930 CrossRef CAS.
- C. Degueldre, A. Scholtis, A. Laube, M. Turrero and B. Thomas, Appl. Geochem., 2003, 18, 55 CrossRef CAS.
- M. H. Bradbury and B. Baeyens, Geochim. Cosmochim. Acta, 1998, 62, 783–795 CrossRef CAS.
- P. Leroy, A. Revil, S. Altmann and C. Tournassat, Geochim. Cosmochim. Acta, 2007, 71, 1087–1097 CrossRef CAS.
- A. Vinsot, C. A. J. Appelo, C. Cailteau, S. Wechner, J. Pironon, P. De Donato, P. De Cannière, S. Mettler, P. Wersin and H. E. Gäbler, Phys. Chem. Earth, 2008, 33, S54–S60 Search PubMed.
- A. Vinsot, S. Mettler and S. Wechner, Phys. Chem. Earth, 2008, 33, S75–S86 Search PubMed.
- Solexperts, Rapport d'installation—Instrumentation hydrogéologique après creusement de la niche, rapport ANDRA D.RP.SOP.05.0007, 2005 Search PubMed.
- P. Marschall, S. Horseman and T. Gimmi, Oil Gas Sci. Technol., 2005, 60, 121–139 CrossRef CAS.
- W. D. Komhyr, T. B. Harris, L. S. Waterman, J. F. S. Chin and K. W. Thoning, J. Geophys. Res., 1989, 94, 8533–8547 CrossRef CAS.
- M. B. Esler, D. W. T. Griffith, S. R. Wilson and L. P. Steele, Anal. Chem., 2000, 72, 206–215 CrossRef CAS.
- D. Helmig, J. Chromatogr., A, 1999, 843, 129 CrossRef CAS.
- L. P. Steele, P. J. Fraser, R. A. Rasmussen, M. A. K. Khalil, T. J. Conway, A. J. Crawford, R. H. Gammon, K. A. Masarie and K. W. Thoning, J. Atmos. Chem., 1987, 5, 125 CrossRef CAS.
- R. Prinn, D. Cunnold, R. Rasmussen, P. Simmonds, F. Alyea, A. Crawford, P. Fraser and R. Rosen, J. Geophys. Res., 1990, 95, 18369–18385 CrossRef.
- P. C. Novelli, J. W. Elkins and L. P. Steele, J. Geophys. Res., 1991, 96, 13109–13121 CrossRef CAS.
- M. Pushkarsky, M. Weida, T. Day, D. Arnone, R. Pritchett, D. Caffey and S. Crivello, Proc. SPIE-Int. Soc. Opt. Eng., 2008, 6871, 68711X.
- S. Barbieri, J. P. Pellaux, E. Studemann and D. Rosset, Rev. Sci. Instrum., 2002, 73, 2458 CrossRef CAS.
- M. Taslakov, V. Simeonov and H. Van Den Bergh, Spectrochim. Acta, Part A, 2006, 63, 1002–1008 CrossRef CAS.
- E. Normand, I. Howieson, M. McCulloch and P. Black, Proc. SPIE-Int. Soc. Opt. Eng., 2006, 6402, 64020G.
- Z. A. Allah, D. Sawtell, V. L. Kasyutich and P. A. Martin, J. Phys.: Conf. Ser., 2009, 157, 012001 CrossRef.
- M. W. Sigrist, Rev. Sci. Instrum., 2003, 74, 486–490 CrossRef CAS.
- D. W. T. Griffith, R. Leuning, O. T. Denmead and I. M. Jamie, Atmos. Environ., 2002, 36, 1833 CrossRef CAS.
- M. Grutter, Atmosfera, 2003, 16, 1 Search PubMed.
- W. M. Doyle, Process Control Qual., 1992, 2, 11 CAS.
- C. P. Rinsland, A. Goldman, F. J. Murcray, T. M. Stephen, N. S. Pougatchev, J. Fishman, S. J. David, R. D. Blatherwick, P. C. Novelli, N. B. Jones and B. J. Connor, J. Geophys. Res., 1999, 104, 18667 CrossRef CAS.
- D. W. T. Griffith, Appl. Spectrosc., 1996, 50, 59–70 CAS.
- P. L. Hanst and S. T. Hanst, in Air Monitoring by Spectroscopic Techniques, ed. M. W. Sigrist, John Wiley & Sons Inc., New York, 1994, vol. 127, pp. 335–470 Search PubMed.
- J. U. White, J. Opt. Soc. Am., 1942, 32, 285 CrossRef.
- A. Hakuli, A. Kytokivi, E. L. Lakomaa and O. Krause, Anal. Chem., 1995, 67, 1881–1886 CrossRef CAS.
- E. Lopez-Anreus, S. Garrigues and M. De La Guardia, Anal. Chim. Acta, 1995, 308, 28 CrossRef CAS.
- D. W. T. Griffith and B. Galle, Atmos. Environ., 2000, 34, 1087 CrossRef CAS.
- C. Cailteau, J. Pironon, P. de Donato, A. Vinsot, C. Garnier and O. Barrès, Anal. Methods, 2011 10.1039/C0AY00622J.
- J. Bak and A. Larsen, Appl. Spectrosc., 1995, 49, 437–443 CAS.
- G. N. Plass, Appl. Opt., 1965, 4, 69–78.
- J. A. Jamieson, R. H. McFee and G. N. Plass, Infrared Physics and Engineering, McGraw Hill Publishing Company, New York, 1963 Search PubMed.
- C. Cailteau, PhD Thesis, Nancy Université, 2008.
- J. Pironon, P. de Donato, O. Barrès, C. Garnier, C. Cailteau, A. Vinsot and G. Radilla, Int. J. Greenhouse Gas Control, 2010, 4, 217–224 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |