Construction of a functional IgG-binding luciferase fusion protein for the rapid detection of specific bacterial strains
Makiko
Nakamura
,
Masayasu
Mie
and
Eiry
Kobatake
*
Department of Biological Information, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, Nagatsuta, Midori-ku, Yokohama, 226-8501, Japan. E-mail: ekobatak@bio.titech.ac.jp; Fax: +81 45 924 5779
First published on 20th October 2010
Abstract
A fusion protein consisting of two IgG-binding domains of streptococcal protein G and firefly luciferase was constructed, and a simple and specific bioluminescent immunodetection system for bacterial strains was developed.
In recent years, antigen-specific immunosensors have played important roles in clinical immunoanalysis and detection of several pathogens.1,2 Among these, much attention has focused on the use of genetically fused proteins to produce enzyme-binding protein conjugates because of the controllable site-to-site binding of the constituting proteins. As the binding portion of a fusion protein, staphylococcal protein A and streptococcal protein G are ideal proteins because of their versatile capabilities of binding to the Fc region of immunoglobulin.3 In previous studies, we constructed a fusion protein consisting of protein A and firefly luciferase and reported that protein A-luciferase was a universal marker protein by the sandwich-ELISA method.4,5 The luciferin–luciferase bioluminescence reaction is known for its high quantum yield and its high sensitivity and low background have been demonstrated.6 However, protein A has limited binding to several species such as mouse, goat or sheep. In contrast, streptococcal protein G has three domains, denoted C1, C2 and C3, which bind to the Fc region of immunoglobulins of many species.7,8 Although these protein G domains have been widely applied to immunoassays, Maeda et al. reported that a fusion protein consisting of the C1 domain and Vargulaluciferase had no IgG-binding ability due to a conformational preference of the C1 domain.9 Therefore, some other strategy is required to engineer protein G–luciferase fusion proteins.
In this study, fusion of two repeats of the protein G C3 domain and firefly luciferase was investigated. Tandem repeats of the C3 domain have been reported to gain more IgG-binding versatility than a single domain,10 and we previously confirmed that a C3-repeated construct retained its activity when it was fused to designed multi-peptides.11 Therefore, a C3 dual repeat-luciferase fusion protein (C2-Luc) was constructed and the IgG-binding activity was determined in several mammalian species. Finally, application of C2-Luc to a bioluminescent immunodetection system was investigated. C2-Luc was immobilized to Escherichia coli (E. coli) cell surfacesvia a strain-specific antibody. Luciferase-labeled E. colicells were collected onto the surface of a 96-well plate through a 0.22 µm filter.
The C2-Luc gene fusion vector, pET-His-C2-Luc (Fig. 1A), was constructed, and the resulting fusion protein, consisting of two protein G C3 domains and firefly luciferase, is illustrated in Fig. 1B. Expression of C2-Luc was induced by IPTG and the expressed protein was purified by metal-ion affinity chromatography using His-tag.
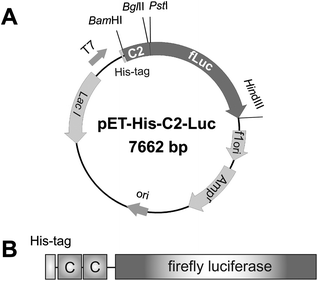 | ||
| Fig. 1 (A) The constructed plasmid, pET-His-C2-Luc. The fusion gene was under the control of T7 promoter. (B) Schematic illustration of the C2-Luc fusion protein. Two C3 domains (shown as C in this figure) were fused to the N-terminus of firefly luciferase. To aid purification of the recombinant protein, there is a His-tag at the N-terminus. | ||
IgG-binding capability of C2-Luc to several mammalian species was determined by measuring the luminescence relative to IgG absorbed onto a microplate surface. Aliquots (100 µl) of IgG (2 µg ml−1) were absorbed on a 96-well microplate (Costar 3912) for 1 h at 37 °C, then blocked with 0.25% Block Ace (Dainippon Seiyaku, Japan) in PBS. After washing, 100 µl of C2-Luc were added and incubated for 30 min at 37 °C, followed by the addition of 50 µl of substrate solution of luciferase. The luminescence of each well was determined for 10 s by a luminometer (Lmax II, Molecular Devices). Previously, streptococcal protein G was reported to bind rabbit, goat, mouse, and sheep IgG, but most efficiently to rabbit IgG. Chicken IgG was considered to be a rare example that showed little association to protein G7. Here, C2-Luc bound to rabbit IgG showed the highest luminescence signal in all samples (Fig. 2). Mouse, goat and sheep IgG-absorbed wells also showed high bioluminescent signals, but there was no detectable signal of C2-Luc bound to chicken IgG. Therefore, we confirmed that C2-Luc could bind IgG of several species.
 | ||
| Fig. 2 IgG-binding versatility of C2-Luc. IgGs of several mammalian species were absorbed to the surface of 96-well microplates and C2-Luc bound to IgGs was detected by the bioluminescent signal. Luminescent activity (relative light units (RLU)) was monitored for 10 seconds. NC, negative control wells where only PBS buffer was added. Each assay was performed in duplicate and repeated three times. Error bars represent the mean ± standard deviation (SD) values. | ||
Throughout the results, the tandem repeat design of the IgG-binding domain was revealed to be a successful strategy to produce a functional fusion protein for an immunodetector. The results shown in Fig. 2 indicate that the number of repetitive IgG-binding sequences could modulate the abilities of resultant fusion proteins. C2-Luc also retained the bioluminescent activity of luciferase in the fixed form, which indicated that C2-Luc had the potential for functioning as a sensor protein immobilized onto a specific antigen.
To investigate the application of C2-Luc fusion protein to an immunodetection system, we immobilized C2-Luc onto E. colicell surfacesvia an antibody. Since E. coli is a pathogen that causes diverse diseases,12 the detection of specific E. coli strains was studied in this paper. The microplate filtration system was introduced to our study to simplify experimental procedures. E. coli strains JM109 and BL21 (DE3) were pre-cultured in LB medium overnight at 37 °C. The cells were then diluted with PBS/1% BSA buffer and 1/1000 volume of rabbit anti-E. coliantibody was added. After incubation for 30 min at room temperature, 100 µl of the E. coli–antibody mixtures were applied to each well of a 96-well filter plate (Costar 3504, 0.22 µm-pored PVDF membrane). The filter plate was centrifuged at 1300g for 3 min and the fractions that flowed through the wells were discarded. C2-Luc (100 µl) diluted with PBS/1% BSA buffer was added to each well. After incubation for 30 min at room temperature, the filter plate was centrifuged again and washed three times with 100 µl of PBS. Finally, 50 µl of substrate solution were applied and the luminescence of each well was determined for 10 s. All buffers and C2-Luc were filtered with a 0.22 µm syringe filter before use.
By this method, E. colicells could be trapped onto the 0.22 µm filter pore by centrifugation or aspiration. In contrast, conventional immunodetection methods such as sandwich ELISA require overnight incubation with primary antibody and the plate.1 This target-immobilization step was not needed in our methodology. Therefore, the antigen-specific detecting ability of C2-Luc was determined by measuring luminescence relative to the concentration of E. coli. The results are shown in Fig. 3. In the present study, we applied an antibody that specifically recognized E. coli K or O strains. Bioluminescence of C2-Luc increased concomitantly with the number of E. coliJM109 (K-12 strain derivatives) cells, in contrast to bioluminescence of BL21 (DE3) (B strain derivatives) cells. The detecting limit was 5 × 106 colony forming units (cfu). From the results of Fig. 3, it was confirmed that C2-Luc could be immobilized onto a specific antigenvia an antibody, which suggests its potential as an immunodetector protein.
 | ||
| Fig. 3 Detection of specific E. coli strains by cell surface-immobilized C2-Luc. C2-Luc-labeled E. coli were trapped to each well surface of a filter plate by centrifugation and luminescence (RLU) was measured for 10 seconds after addition of substrate. The number of E. coli was measured by colony forming units (cfu). Each assay was performed in duplicate and repeated twice. Error bars represent the mean ± SD values. | ||
In conclusion, a simple immunodetection system with a C2-Luc fusion protein was developed. With our system, no more than 2 hours were required to determine a specific pathogen. Since C2-Luc binding abilities can be modulated by engineering of the IgG-binding domain, we consider it to be a powerful tool for immunosensing.
Acknowledgements
This work was in part supported by Grant-in-Aid for Japan Society for the Promotion of Science (JSPS) Fellows, and the Ministry of Education, Culture, Sports, Science & Technology (MEXT), Japan.Notes and references
- M. Magliulo, P. Simoni, M. Guardigli, E. Michelini, M. Luciani, R. Lelli and A. Roda, J. Agric. Food Chem., 2007, 55, 4933–4939 CrossRef CAS.
- C. Ricci, G. Volpe, L. Micheli and G. Palleschi, Anal. Chim. Acta, 2007, 605, 111–129 CrossRef CAS.
- M. Tashiro and G. T. Montelione, Curr. Opin. Struct. Biol., 1995, 5, 471–481 CrossRef CAS.
- T. Ebihara, H. Takayama, Y. Yanagida, E. Kobatake and M. Aizawa, Biotechnol. Lett., 2002, 24, 147–149 CrossRef CAS.
- E. Kobatake, T. Iwai, Y. Ikariyama and M. Aizawa, Anal. Biochem., 1993, 208, 300–305 CrossRef CAS.
- T. Minekawa, H. Ohkuma, K. Abe, H. Maekawa and H. Arakawa, Luminescence, 2009, 24, 394–399 CAS.
- B. Akerstrom, T. Brodin, K. Reis and L. Bjorck, J. Immunol., 1985, 135, 2589–2592 CAS.
- B. Guss, M. Eliasson, A. Olsson, M. Uhlen, A. K. Frej, H. Jornvall, J. I. Flock and M. Lindberg, EMBO J., 1986, 5, 1567–1575 CAS.
- Y. Maeda, H. Ueda, J. Kazami, G. Kawano, E. Suzuki and T. Nagamune, Anal. Biochem., 1997, 249, 147–152 CrossRef CAS.
- S. Y. Cai, Y. Y. Wang and Z. J. Yao, Sci. China B., 1994, 37, 454–461 CAS.
- G. Tanaka, H. Funabashi, M. Mie and E. Kobatake, Anal. Biochem., 2006, 350, 298–303 CrossRef CAS.
- J. B. Kaper, J. A. Nataro and H. L. Mobley, Nat. Rev. Microbiol., 2004, 2, 123–140 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |