
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry†‡
Carlo
Sambiagio
 a,
David
Schönbauer
b,
Remi
Blieck
c,
Toan
Dao-Huy
b,
Gerit
Pototschnig
b,
Patricia
Schaaf
b,
Thomas
Wiesinger
b,
Muhammad Farooq
Zia
b,
Joanna
Wencel-Delord
d,
Tatiana
Besset
c,
Bert U. W.
Maes
a and
Michael
Schnürch
*b
a,
David
Schönbauer
b,
Remi
Blieck
c,
Toan
Dao-Huy
b,
Gerit
Pototschnig
b,
Patricia
Schaaf
b,
Thomas
Wiesinger
b,
Muhammad Farooq
Zia
b,
Joanna
Wencel-Delord
d,
Tatiana
Besset
c,
Bert U. W.
Maes
a and
Michael
Schnürch
*b
aOrganic Synthesis (ORSY), Department of Chemistry, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
bInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, A-1060 Vienna, Austria. E-mail: michael.schnuerch@tuwien.ac.at
cNormandie Univ, INSA Rouen, UNIROUEN, CNRS, COBRA (UMR 6014), 76000 Rouen, France
dLaboratoire de Chimie Moléculaire (UMR CNRS 7509), Université de Strasbourg, ECPM 25 Rue Becquerel, 67087 Strasbourg, France
First published on 23rd July 2018
Abstract
The present review is devoted to summarizing the recent advances (2015–2017) in the field of metal-catalysed group-directed C–H functionalisation. In order to clearly showcase the molecular diversity that can now be accessed by means of directed C–H functionalisation, the whole is organized following the directing groups installed on a substrate. Its aim is to be a comprehensive reference work, where a specific directing group can be easily found, together with the transformations which have been carried out with it. Hence, the primary format of this review is schemes accompanied with a concise explanatory text, in which the directing groups are ordered in sections according to their chemical structure. The schemes feature typical substrates used, the products obtained as well as the required reaction conditions. Importantly, each example is commented on with respect to the most important positive features and drawbacks, on aspects such as selectivity, substrate scope, reaction conditions, directing group removal, and greenness. The targeted readership are both experts in the field of C–H functionalisation chemistry (to provide a comprehensive overview of the progress made in the last years) and, even more so, all organic chemists who want to introduce the C–H functionalisation way of thinking for a design of straightforward, efficient and step-economic synthetic routes towards molecules of interest to them. Accordingly, this review should be of particular interest also for scientists from industrial R&D sector. Hence, the overall goal of this review is to promote the application of C–H functionalisation reactions outside the research groups dedicated to method development and establishing it as a valuable reaction archetype in contemporary R&D, comparable to the role cross-coupling reactions play to date.
 Carlo Sambiagio | Carlo Sambiagio studied chemistry at the University of Cagliari (IT), acquiring his Master's degree in 2011. He then moved to the University of Leeds (UK), completing his PhD on Cu-catalysed cross-couplings in 2015, under the supervision of Prof. Patrick McGowan. After that he spent two year as a postdoc in the Maes group in Antwerp (BE), mostly exploring aerobic oxidation chemistry. Carlo has recently moved to the Eindhoven University of Technology (NL) as a EU Marie Curie fellow to work on flow chemistry with Prof. Timothy Noël. His interests generally include metal-catalysed transformations and the development of new methodologies for organic synthesis. |
 David Schönbauer | David Schönbauer was born 1989 and is currently working towards his PhD at Technische Universität Vienna in the Institute of Applied Synthetic Chemistry, where he also finished his Master studies 2017. His current research in the field of C–H functionalisation is supervised by Professor Michael Schnürch and focuses on the activation of C(sp3)–H bonds. |
 Joanna Wencel-Delord | Joanna Wencel-Delord was educated in chemistry at the Ecole Nationale Supérieure de Chimie de Rennes, France and she received her PhD in 2010 from the University of Rennes 1, France (Dr C. Crévisy and Dr M. Mauduit). After postdoctoral studies with Prof. F. Glorius at the Westfälische Wilhelms-Universität Münster (Germany) and temporary assistant professor position (ATER) at the University of Strasbourg (Prof. P. Compain), she joined CNRS in 2013 as an associate researcher in the group of Prof. F. Colobert (University of Strasbourg, France). Her research focuses on the transition metal-catalysed asymmetric C–H activation. |
 Tatiana Besset | Dr Tatiana Besset obtained her doctoral degree in organic chemistry (2009) at Grenoble University (Dr Greene's group, France). She then moved to Münster WWU (Germany) as a postdoctoral fellow in the group of Prof. Glorius. In 2011, she joined the group of Prof. Reek (Amsterdam University, Netherlands), as a postdoctoral fellow in collaboration with Eastman company. Since 2012, she is working as a CNRS Associate Researcher in the team headed by Prof. Pannecoucke, UMR 6014-CNRS, Rouen University, INSA (France). Her research program is focused on transition metal-catalysed C–H activation and the development of new strategies to access fluorinated molecules. |
 Bert U. W. Maes | Bert Maes obtained his PhD from UAntwerpen and subsequently received a prestigious post doc position of the National Science Foundation in Belgium (FWO-Flanders). He performed postdoctoral work in France at the École Normale Supérieure in Paris with Prof. Anny Jutand, studying fundamental mechanisms in catalysis. In 2003 he was appointed assistant professor (docent) in the Department of Chemistry at UAntwerpen. He currently is a full professor of Organic Chemistry and spokesman of the Organic Synthesis division (ORSY). His research interests include heterocyclic chemistry, organometallic chemistry, homogeneous catalysis and sustainable chemistry. |
 Michael Schnürch | MS received his PhD in 2005 from TU Wien (supervisor Prof. Peter Stanetty). During his PhD-studies, he was on a 4 month sabbatical in the group of Prof. Victor Snieckus at Queens University (Kingston, Ontario). He was then post doc with Prof. Dalibor Sames at Columbia University as Erwin Schrödinger fellow. Then, he became Assistant Professor at TU Wien and completed his habilitation in 2013. Subsequently, he was promoted to Associate Professor for Organometallic Chemistry in 2016, a position he still holds. Additionally, he is currently the Chair of the COST Action CHAOS (C–H Activation in Organic Synthesis). |
1. Introduction
Metal catalysed C–H functionalisation chemistry is a rapidly expanding field with research going into many different directions.1–21 Due to the ubiquity of C–H bonds in organic molecules, selective functionalisation of a specific C–H bond is highly challenging. In aromatic heterocycles, the heteroatoms usually guide the regioselectivity of C–H functionalisation reactions due to their influence on the electron density of the different C–H positions.22–26 On arenes or aliphatic compounds, such intrinsic reactivity differences are significantly less pronounced, and selective cleavage of a specific C–H bond is more difficult, even though substituents in arenes can obviously have a significant effect. One way to overcome this problem is the assistance of directing groups (DGs), consisting of a coordinating moiety (an “internal ligand”), which directs a metal catalyst into the proximity of a certain C–H bond in the molecule, leading to its selective cleavage and subsequent functionalisation.17,27–33 This strategy allows to overrule innate reactivity in (hetero)arenes. A schematic example of this is shown in Fig. 1 for a number of common unsaturated heterocycles, where a comparison with classical electrophilic substitution is given.34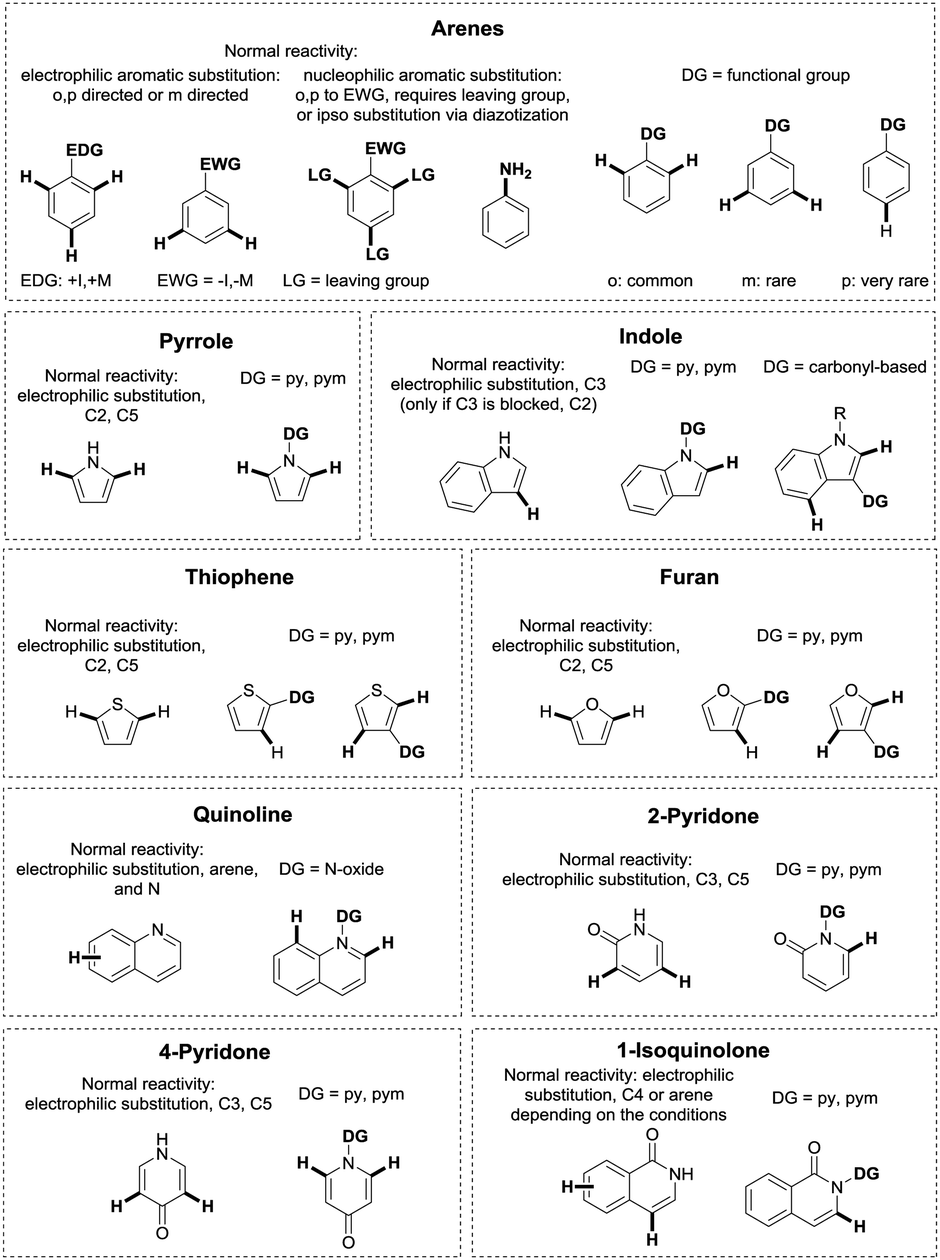 | ||
| Fig. 1 Examples of classical reactivity versus DG approach. | ||
DG-assisted C–H functionalisation started to gain momentum in the mid-1990s after a landmark contribution by the group of Murai.35 In this paper, a ketone DG enabled the Ru(II)-catalysed ortho-alkylation of arenes with alkenes, clearly illustrating that the regioselectivity issued might be selectively tackled if formation of a metallacyclic intermediate occurs. Since then, a huge number of papers on the topic have been published, expanding the scope of potential DGs and of chemical transformations which can be carried out.
1.1. Limitations to the use of C–H functionalisation in molecules for R&D or in total synthesis
Most of the published contributions can be classified as methodological papers. Typically, substrates are designed which align themselves nicely in order to allow a selective C–H cleavage. The complexity of these substrates is typically limited, since they are usually from the pool of readily available small molecules featuring a standard functional group (FG) compatibility. Applications in the field of R&D36–38 and total synthesis of complex molecules39–45 have been reported, however those remain the exception. For the former it is of course difficult to get an objective view on the situation as most results obtained in fine chemicals industry are never published. However, when comparing the number of cross coupling reactions and direct C–H functionalisations disclosed in the scientific literature by industry, one can safely conclude that the area is still in its infancy. This is somewhat surprising, considering the amount of transformations, which have been reported so far from academia and the wide variety of DGs which can potentially promote them. You might think that in a retrosynthesis, you frequently run into situations, in which transition metal-catalysed C–H functionalisation would be a potential and even the preferred option. So what are the reasons why C–H functionalisation has not yet reached the level of general applicability which related transformations, e.g. cross-coupling reactions, have achieved already quite some time ago? Considering cross-coupling reactions, it can be noticed that the majority of coupling partners organometals and (pseudo)halides (Suzuki–Miyaura, Stille, Negishi, Hiyama), or (pseudo)halides and olefins (Heck) or alkynes (Sonogashira) in order to create new C–C bonds without regioselectivity issues. Hence, in retrosynthetic analysis, cross-coupling reactions are quickly identified and nowadays these are standard transformations frequently applied in all contexts of synthesis, also including the most complex area of total synthesis of natural products. Additionally, the metals applied are also only a few with Pd being the single most important one dominating the field, followed by Cu and Ni (recently). All other metals play only relative minor roles at the moment, although research in this field is also increasing.In case of C–H functionalisation, the situation is much more complex due to the higher diversity in coupling partners which often react via distinctively different reaction mechanisms, and a large variety of specific additives which these reactions require (often complex mixtures of reagents and additives are required for each transformation). To facilitate the understanding and the choice of a specific procedure, below are listed the different types of additives and their roles in C–H functionalisation reactions. It is important to stress that, although the main classes of additives and roles are relatively clear, many compounds can have more than one distinct role in the reaction, therefore their choice and combinations is not straightforward.
• Oxidants: most often Cu salts (commonly Cu(OAc)2), and Ag salts (sometimes Mn salts) are used in stoichiometric or superstoichiometric amounts in oxidative reactions. Other oxidants, used alone or in combination with Cu or Ag are benzoquinones, peroxides, O2/air, K2S2O8 or hypervalent iodine compounds.
• Catalytic Ag salts: catalytic amounts of Ag salts are often used in combination with groups 8 or 9 metal halide dimers, commonly used as catalysts (e.g. [RhCp*Cl2]2). In these cases the Ag acts as a halide scavenger, and the counteranion (usually OTf, NTf2, or SbF6) promotes the in situ formation of cationic metal catalysts in solution. When a suitable preformed cationic metal catalyst is used, the addition of such additives is generally not required.
• Carboxylates: carboxylates (acetates, benzoates, pivalates, adamantanecarboxylates, trifluoroacetates and others) are very common additives in many C–H functionalisation protocols. Their role is mostly related to their ability to deprotonate the desired C–H bond, often via a concerted metallation–deprotonation (CMD) pathway (also termed ambiphilic metal ligand activation (AMLA)).2,46,47 These additives are generally added as acids in stoichiometric or catalytic amounts, often in combination with bases, or can be added as their Cu, Ag, Zn, Na salts.
• Ligands: although typically no external ligands are required in C–H functionalisation reactions (the DG acts as an “internal ligand”), in some cases an external auxiliary (catalytic amounts) is required to facilitate the reaction. These can be phosphines, carbenes, mono-protected aminoacids (MPAA) or other bidentate ligands. Noteworthy, the choice of the ligand will strongly depend on the mechanism of a targeted transformation (for instance Pd(II)/Pd(0) catalysis vs. Pd(II)/Pd(IV) catalytic cycle).
• Lewis acids: in this category many compounds can fall, including some of the above. Although most often simple compounds are used, such as Zn salts, the applied Lewis acids span over a wide range of reagents also including more exotic In/Gd salts or BPh3, as can be seen in the examples in this review. These can display different roles, including the activation of coupling partners (e.g. activation of ketones/aldehydes in alkylation reactions), and can be added in stoichiometric or catalytic amounts.
• Bases: a number of different bases, usually inorganic ones, e.g. carbonates are used to neutralize acids formed during the reaction, or to facilitate deprotonation of the starting materials/reactants/additives used. It is worth noting that common bases used for cross coupling reactions, such as Cs2CO3, are instead not often encountered in C–H functionalisation, while Na, K, or Li counterparts are generally more effective (Ag2CO3 is often used as both oxidant and base).
The variety of additives and reaction conditions encountered in this field is also reflected by the range of metals which are applied as catalysts. There is no such thing as a single dominant metal species, but Pd,48,49 Rh,50,51 and Ru52–54 share together the top spot. Moreover, increasing examples using Ni,55–58 Co,55,59–64 Ir,64–66 Cu,67–69 Fe,3,55,63,70,71 and Mn72–74 are appearing in the literature.
Naturally, such a manifold of metals with often distinctively different properties adds a lot of complexity to the field. This is of course an advantage for method development, since it is only logical that a larger pool of catalyst candidates allows also for a larger variety of possible transformations. On the other hand, the complexity has reached a level where it is very difficult to keep track of the developments and to get an overview of what is actually possible in C–H functionalisations.
What does this mean for a synthetic chemist planning a multistep synthesis of a molecule? The retrosynthetic disconnections might frequently lead to situations in which C–H functionalisation chemistry could be applied. However, because of the diversity of the field and the lack of a general and easy to consult overview, its application is unfortunately not even considered in many cases. This is the shortcoming which the authors attempted to address by this review. So overall, this review aims at giving a comprehensive overview on the types of FGs available for transition metal-catalysed C–H cleavage reactions and the types of transformation they allow, and the associated required “reaction cocktail”. This should be very useful for taking this research field to the next step, the more frequent application in syntheses, also at late stages of prolonged synthetic sequences.
1.2. Structure and scope of the review
Our aim in this review was to present the DGs and possible transformations in a concise way so that all readers can easily find the specific transformation looked for. Hence, the review is divided in sections dedicated to specific types of DGs: (i) monodentate carbonyl-containing DGs, (ii) bidentate DGs, (iii) heterocyclic DGs, and (iv) heteroatom-based DGs (N, S, P, Si…). These main sections are further subdivided into specific functional groups (e.g. ketones, amides, etc.), and then into alkylations, alkenylations, arylations, etc. In turns, for each of these subsections, the different procedures reported in the literature have been summarised in schemes, making it easier to compare them with each other. To facilitate this task, a list of advantages/disadvantages has been added for each procedure, concerning issues such as selectivity, substrate scope, reaction conditions, DG removal, and overall greenness. We realise that said advantages/disadvantages can be somewhat subjective, and what is good for one purpose may not be so good for another. For example, the use of halogenated solvents has been considered as a drawback in this review. This has not the aim of criticising the research methodology, neither reducing the value of the results reported, but the use of such solvents is a no-go in chemical manufacturing, and therefore is something that necessarily has disadvantages in terms of that procedure being used for such a purpose. The same reasoning applies to the other advantages/disadvantages considered in the review. In particular, among the drawbacks, the following have been specifically listed: toxic solvents; reaction temperatures > 120 °C; reaction time > 24 h; (most) yields < 70%; superstoichiometric amounts of (metal) oxidants in oxidative couplings; regio- or stereoisomeric mixtures; specific limitations in scope or FG tolerance (e.g. sensitivity to steric hindrance); use of additional ligands, unless in stereoselective reactions (the DG is already a ligand, and most non-stereoselective procedures do not require additional ligands). As positive aspects, the following are instead considered: green(er) solvents; reaction temperature < 50 °C; reaction times < 10 h; (most) yields > 80–90%; particularly good FG tolerance (halide, methoxy, alkyl, ester and cyanide are considered generally standard substituents, if something more advanced, such as alkene, alkyne, boryl, silyl, or free NH/OH groups are also tolerated, this has been mentioned as an advantage); other substrates and other DGs available for the reaction; DG removal (when specifically mentioned); in situ, one-pot, or cascade processes; scalability. Other aspects have not been mentioned as specific advantages/disadvantages, mostly because of the lack of a real distinction between the two; a catalyst loading up to 10 mol% metal has been considered standard, as most protocols use these amounts, though it does not mean this is what is acceptable for chemical development purposes (higher than 10 mol% metal for expensive catalyst has been considered specifically as a disadvantage); characteristic of reactants have also not been generally considered (e.g. availability or cost), as a large variety exist and price is heavily depending on the scale required.Another important aspect of the review is the stress that has been placed on the DG removal. The removal of DGs is an important drawback in C–H functionalisation chemistry, as the DG might not be required after the directed step, and its removal is therefore necessary (which is an extra synthetic step). Several DGs are actually not removable (depending on the substrate as well) which is no problem when they anyway constitute a (precursor) part of the target molecule, while others are easily removed or modified. We tried to incorporate all the possible protocols for the removal or modification of the DGs in each section, so that the reader will be immediately aware of what can currently be done with the DG in use. This information is often not reported in reviews dealing with C–H functionalisation. The removal of the DG has also been listed as a specific advantage in each procedure, in case this is specifically investigated by the authors. So not being mentioned does not mean it could not be done, it is just not reported in the original publication.
It has to be mentioned that sometimes different publications use different terms for the very same transformation. For the sake of simplicity, in this review each transformation has the same name in all entries. For example, the coupling of olefins with arenes to give alkylated arenes is either classified as alkylation or hydroarylation in literature. It was decided for this review to look at the reaction product and see what happened to the part of the molecule which carried the DG. Hence, in case the arene carried the DG, the reaction is always classified as an alkylation reaction.
This review aims at being comprehensive in the regard that all DGs and potential transformations with the DG are listed, however, reporting every single variant of a given transformation would just be beyond the scope of this review. In such cases a selection had to be made by critically looking at the reported protocols and choosing representative examples to give a clear flavour on chemical diversity searched for by the synthetic chemist. It is clear that such a selection will always be biased and readers might find that one or the other example should have been selected differently.
The speed in which new contributions are brought forward in the field is amazing. This shows the high relevance of this area of research and the potential of the field. For writers of a review it brings certain problems as well, most importantly which time frame to cover and which examples to include? As a starting point, we used the very comprehensive review by Zhanxiang Liu and Yuhong Zhang published in early 2015, which covers literature until the end of 2014.17 Hence, our review starts at the beginning of 2015 until the end of 2017.
2. Ketone DGs
The carbonyl group in ketones was historically amongst the first DGs to be used in C–H functionalisation chemistry. Already in 1993, a pioneering work for ketone-directed alkylations was published by Murai et al.35 In this landmark contribution, the first highly efficient and selective C–H cleavage with a simultaneous C–C bond formation mediated by a Ru complex on different aromatic ketones (e.g. naphtyl, furan, thiophene) with mono- and di-substituted olefins was shown. The mechanism proposed in this paper served as blueprint for many more contributions to come, and can be considered as one of the most important starting points for the field of C–H functionalisation chemistry, as we experience it today. It was proposed that the carbonyl function first pre-coordinates the metal catalyst, which brings it into a position in close proximity to the C–H bond ortho to the ketone function (Fig. 2). This allows C–H insertion of Ru into the C–H bond. Then, one PPh3 ligand is replaced by the olefin, first via π- then via σ-bonding. Finally, reductive elimination forms the new C–C bond and the catalyst dissociates again.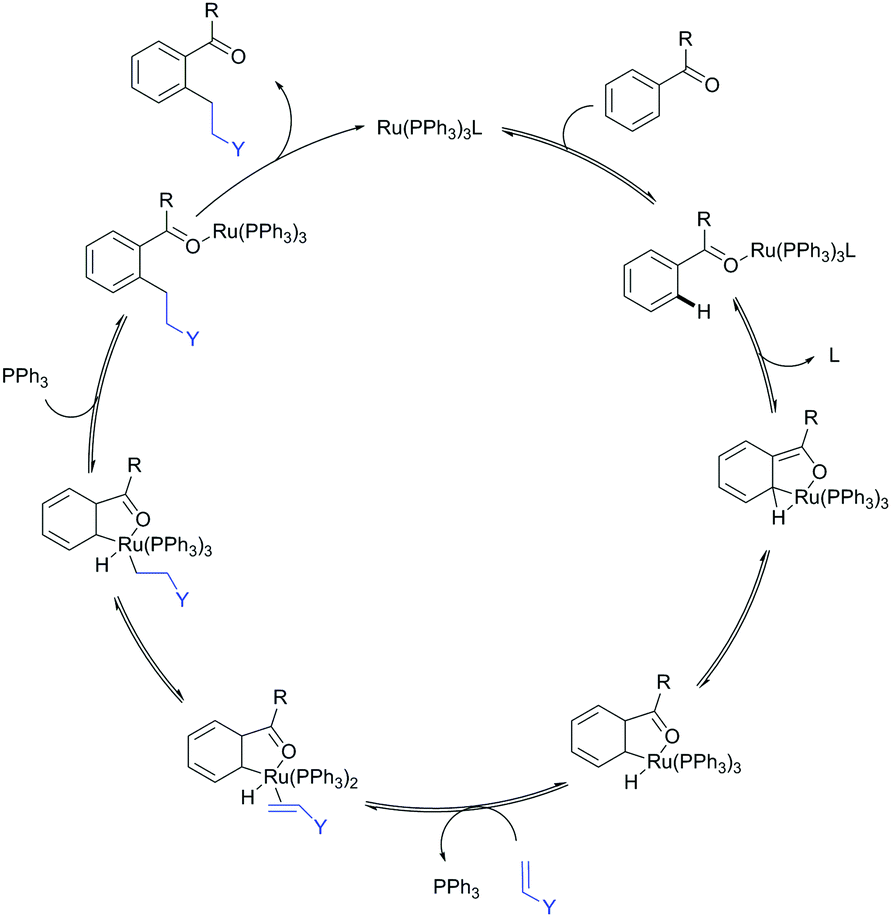 | ||
| Fig. 2 Murai's ketone-directed alkylation. | ||
25 years later, the field is still very active and new contributions are brought forward continuously and even though the field has been recently reviewed,75 a significant number of new contributions has been published, which will be treated in this section. This is certainly due to the manifold of opportunities of further manipulation a ketone allows, and also the ease of introduction via various methods, e.g. Friedel–Crafts acylation. From the very beginning, Ru proved to be especially well suited for ketone-directed C–H activation reactions followed by Rh, with other metals playing only a minor role.
2.1. Arylation
Kakiuchi and co-workers tackled the problem of arylating sterically demanding diaryl and aryl–alkyl ketones by designing Ru catalysts of the general structures [RuHCl(CO)(PAr3)3] and [RuH2(CO)(PAr3)3].76 3,3′,5,5′-Tetramethylbenzophenone served as model substrate and a boronic acid ester as the aryl source. For this substrate, [RuHCl(CO)(P(4-MeO-3,5-Me2C6H2)3)3] turned out to be the best performing catalyst, whereas for the pivalophenone derivative 1-(3,5-dimethylphenyl)-2,2-dimethylpropan-1-one [RuH2(CO)(P(3-MeC6H4)3)3] performed best (Scheme 1A).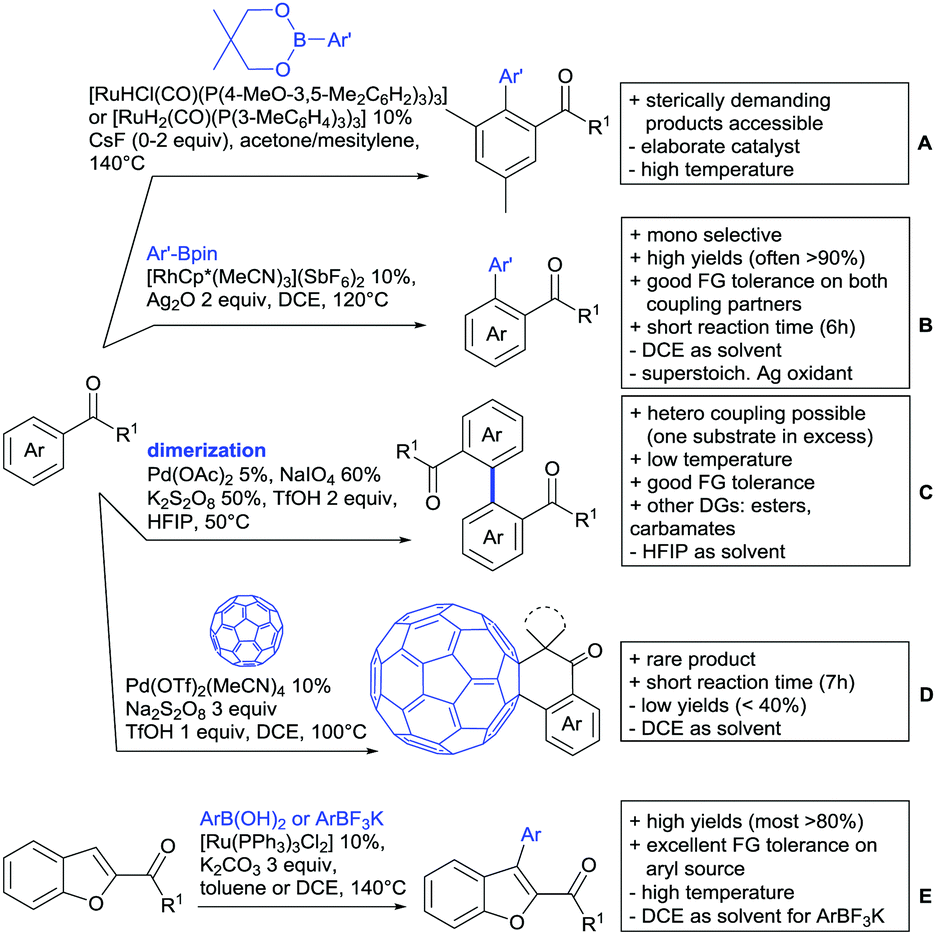 | ||
| Scheme 1 Ketone-directed arylations. | ||
Again using boronic esters as aryl sources, Lu and Sun developed a Rh catalysed protocol for arylating alkyl–aryl ketones exclusively at the C(sp2)–H bond.77 Di-arylated products were not observed, even in the absence of steric bulk. Additionally, yields were generally excellent and frequently >90% (Scheme 1B).
Rao reported on a dimerization of aromatic ketones leading to 2,2′-dicarbonylated biaryls via a Pd(II)–Pd(IV) catalytic cycle (Scheme 1C).78 Substituted benzophenone derivatives and also alkyl–aryl ketones worked well towards the homodimers. More importantly, the authors showed that also two different ketone substrates can be applied leading to heterocoupling products in yields between 61–82%. In these cases, the more electron-rich ketone had to be used in excess. This is a significant improvement to traditional routes towards the same type of products.79,80 Furthermore, the method was not limited to ketones as DG but also esters and carbamates were applied.
An example which can be counted to arylation reactions as well is the synthesis of [60]fullerene-fused tetralones via a Pd catalysed protocol.81 [60]Fullerene reacted with a series of alkyl–aryl ketones. Even though the yields were low (10–31%), this is an interesting example showing the diversity of possible coupling partners (Scheme 1D).
Ramana investigated the C3-arylation of 2-arylbenzofurans using either arylboronic acids or the corresponding potassium trifluoroborates as the aryl source.82 Almost the same conditions as for their alkylation protocol could be used (see Section 2.2) but interestingly AgOAc had a detrimental effect and only in its absence, high yields were obtained and also excellent FG tolerance was observed (Scheme 1E).
2.2. Alkylation
In 2015, the group of Prabhu reported on the addition of maleimides mainly to acetophenones, which is formally an alkylation reaction (Scheme 2A).83 Also 2-acetylthiophene was a suitable starting material, however with a decreased yield of 54%, whereas two benzophenone derivatives were as efficient as the acetophenones. Noteworthy are the DFT studies in this contribution, which strongly favour a CMD mechanism over an electrophilic aromatic substitution or a mechanism involving an oxidative addition. One year later, Kim and Kim reported a similar protocol applied to chromones and xanthones.84 By switching to a Rh catalyst, they could reduce the catalyst loading significantly (Scheme 2B). Acetyl-directed alkylation (as well as amidation and alkenylation) was also reported by Zhang (Scheme 2C).85 Relatively high catalyst and ligand loadings were required but on the upside, an uncommon substrate, tetraphenylene, was used in this transformation. | ||
| Scheme 2 Ketone-directed alkylations. | ||
Recently, an Fe catalysed alkylation protocol was published by Kakiuchi and co-workers (Scheme 2D).86 Using simple Fe(PMe3)4 as catalyst without any additional ligand or other additives, alkyl–aryl ketones were alkylated with substituted olefins (mainly alkenyl silanes) in excellent yields under neat conditions. Also cyclic alkyl–aryl ketones (e.g. 2,2-dimethyltetralone) could be used as substrates. Diaryl ketones did not react as efficiently and required higher catalyst loading and trialkylation was observed as the major product. The proposed mechanism followed the initial suggestion by Murai.35
One rare example of ortho methylation was recently disclosed by Ilies and Nakamura (Scheme 2E).87 AlMe3 served as alkyl source and simple Fe(acac)3 as catalyst. However, the addition of a less common ligand, Me2N-TP (see Scheme 2E) was necessary. As usual in most of such alkylation reactions, mixtures of mono- and di-alkylated products were obtained if permitted by the substrate structure. In the same paper also carboxylic acids were used as DGs (see Section 4).
All examples so far gave exclusively the synthesis of linear products. However, Ramana and co-workers showed that tuning the catalytic system allowed to switch between linear and branched products in the C3-alkylation of 2-aroylbenzofurans with acrylates (Scheme 3A).88 Whereas the selectivity for branched products was generally in the 9/1 (branched/linear) range, for linear products it was often significantly lower. Recently, the group published a follow-up paper trying to elucidate via DFT calculations how the linear/branched selectivity was controlled.89 They calculated transition states leading either to the linear or to the branched products, which are both feasible pathways. The transition state towards the branched product turned out to be significantly lower in energy, which goes in line with the experimental observations. Furthermore, the method was extended to the C2-alkylation of 3-aryloxybenzofurans.89 On this second type of substrates, the linear product was preferred with high selectivity (20/1) and no protocol for the branched products was disclosed (Scheme 3B).
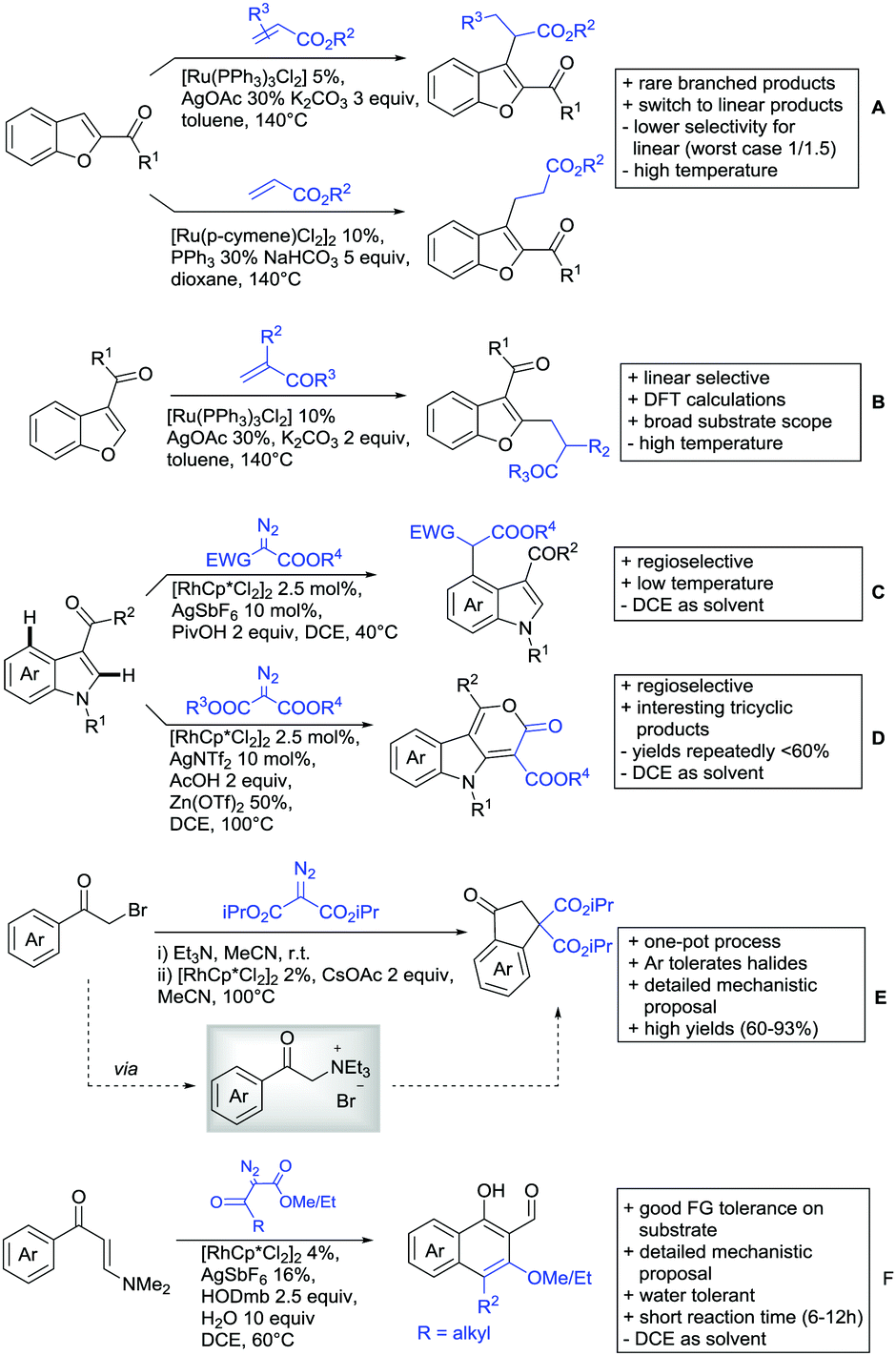 | ||
| Scheme 3 Ketone-directed alkylation on carbo- and heterocycles. | ||
The very same reagent was also used for another functionalisation/cyclisation sequence using 3-ketoindoles as substrates (Scheme 3C and D).90 Initially, the authors found two products, the one alkylated in C4, and one derived from alkylation and subsequent cyclisation in C2. At the beginning, they could successfully optimise the reaction towards selective formation of the C4 alkylated products with good to excellent yields (36–98%). Subsequently, also the second type of products could be obtained selectively, however, generally in lower yields (∼10% lower) for similar examples. In both cases, [RhCp*Cl2]2 was used as the catalyst, but the additives differed, as can be seen in Scheme 3C and D.
An interesting example has been reported by Wan and Li.91 Using phenylacyl ammonium salts as substrates (which can also be produced in situ) in combination with α-diazo esters led to benzocyclanone products (Scheme 3E). Key was the unprecedented presence of an oxidizing C–N bond in the substrates which allowed this overall redox-neutral process. Detailed mechanistic studies were carried out, including DFT calculations.
The group of Zhu used enaminone starting materials for the synthesis of naphthalene derivatives using either α-diazo-β-ketoesters (Scheme 3F) or alkynes (Scheme 4H) as coupling partners.92 In case of α-diazo-β-ketoesters as coupling partners the initial step is an alkylation and good FG tolerance on the substrate was observed. Synthetically useful yields between 50–85% were reported and various alkyl residues including cyclopropyl can be in position R2.
 | ||
| Scheme 4 Ketone-directed olefinations on various substrates. | ||
2.3. Alkenylation
Yu explored the application of distal weakly coordinating DGs, including ketones (Scheme 4A).93 In this report, the ketone functionality is not directly attached to the aromatic ring but separated by a –CH2–, or in one case a –CH2CH2– spacer. Yields for the reaction with acrylates are moderate and selectivity between mono- and di-alkenylation is an issue.Bakthadoss et al. showed that basically the same reaction conditions could be applied for the alkenylation of chromanones as well, in this case in dioxane as solvent (Scheme 4B).94 Yields were commonly good (60–81%) and the typical range of FGs was tolerated.
Again on tetraphenylenes, Zhang also reported alkenylation reactions (Scheme 4C).85 In this case, the catalyst loading could be significantly reduced and no ligand was required, although 2 equiv. of Cu had to be used. Besides acrylates, also alkenyl sulfoxides and -phosphates as well as styrene derivatives could be used.
Szostak's group showed the ketone-directed hydroarylation of internal alkynes leading to alkenylated arenes (Scheme 4D).95 A low Ru loading could be applied; however the E/Z ratio left room for improvement. In this contribution, the DG showed significant variation as opposed to many contributions which relied primarily on the acetyl group. Further elaboration of the obtained alkenylated products to a series of useful compounds was also demonstrated.
Prabhu and co-workers showed that a ketone DG in position 3 of indoles leads to alkenylated products either at the C2- or C4-position.96 The source of this regioselectivity is primarily the electronic nature of the DG. For R = Me/Et, alkenylation took place in C2-position under Ru catalysis (Scheme 4E).96 Also an amide DG gave the same regioselectivity. In case of R = t-butyl, di-alkenylation in C2- and C4-position was observed and for R = phenyl di-alkenylation in C2-position and on the DG occurred. For R = CF3 and using a Rh catalyst, exclusive C4 alkenylation was observed (Scheme 4F).96 Trying to elucidate the origin of this regioselectivity, the authors have reason to believe that COCF3 is the weaker coordinating DG, which eventually does not direct C–H activation by precoordination but rather stabilizes a metal–arene σ complex that forms due to electrophilic metallation. An ester or an aldehyde as DG also gave C4 alkenylation. Such examples, giving guidelines for achieving different regioselectivity, are of significant importance for further promoting the application of C–H functionalisation in complex syntheses.
Another peculiar ketone DG was developed by Lan and Li, namely phenylacyl ammonium salts (Scheme 4G).91 Using a Rh catalyst, ortho alkenylation was obtained in good yields with styrene derivatives. However, also alkyl substituted olefins could be applied with the drawback that sometimes, mixtures of E and Z isomers were obtained. It is noteworthy that the ammonium group was cleaved during the reaction, overall leading to ortho alkenylated acetophenone derivatives. Noteworthy, the reaction does not require an external oxidant since the ammonium salt of the DG takes over this role.
The group of Zhu used enaminone starting materials for the synthesis of naphthalene derivatives using either alkynes (Scheme 4H) or α-diazo-β-ketoesters (Scheme 3F) as coupling partners.92 In the case of alkynes, only 1,2-diphenylacetylene was used as reaction partner but good FG tolerance on the substrate was observed and synthetically useful yields between 50–85% were reported. According to the proposed mechanism, the initial step is a Rh-catalysed alkenylation.
2.4. Alkynylation and acylation
One example of an alkynylation reaction was reported by Jiang and Huo (Scheme 5A).97 TIPS protected bromo acetylene was used as alkyne source under Ir(III) catalysis. Aromatic and heteroaromatic ketones worked equally well and a range of substituents were tolerated with good yields (generally ∼70%). On the downside was the large amount of a mixture of three different Ag salts and the requirement for halogenated solvents. Esters can be used as alternative DGs, although the ketone showed a better directing effect, whereas an amide or pyrazole DG were superior to the ketone. Such studies, allowing the ranking of directing ability of different DGs, are unfortunately rare but extremely important for the application of C–H functionalisation to the synthesis/modification of complex molecules, carrying many FGs.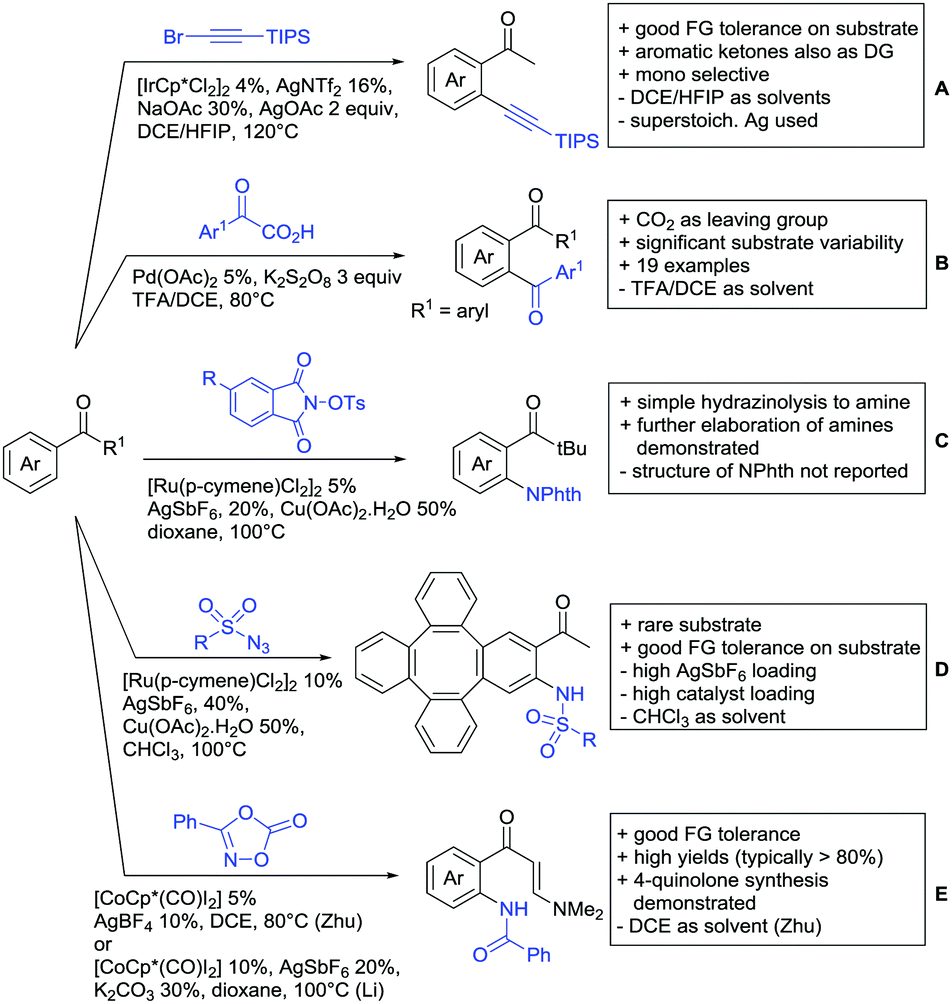 | ||
| Scheme 5 Ketone-directed alkynylation, acylation and C–N bond formation. | ||
Decarboxylative acylation was reported by Yu and co-workers (Scheme 5B).98 As acyl source, α-oxocarboxylic acids were used under Pd catalysis. The authors suggested an acyl radical as important intermediate, which was supported by a radical trapping experiment with TEMPO.
2.5. C–N bond formation
Ackermann disclosed Ru catalysed imidation of aryl–tbutyl ketones using N-tosyloxyphthalimides (Scheme 5C).99 The obtained products were easily transformed to the corresponding amines using standard conditions (N2H4), which gave valuable 2-amino-phenones. Those can be converted to a series of nitrogen containing heterocycles.Using aromatic or aliphatic sulfonyl azides, it was again Zhang who showed amidation of terpenylenes with high FG tolerance (Scheme 5D).85
Dioxazolones became popular amidating agents in recent years in the field of C–H activation. Interestingly, two groups almost simultaneously exploited this reagent on enaminones. Zhu and co-workers developed a Co(III)-catalysed protocol,100 whereas Li developed Co(III)-and Rh(III)-catalysed reactions.101 The two Co(III)-catalysed protocols used both [Cp*Co(CO)I2] as catalyst (Scheme 5E). The substrate scope and yield (typically >80%) were comparable and all common FGs seemed to be tolerated. The amidated product was then transformed to 4-quinolones under acidic conditions (not shown).
3. Aldehydes as DGs
Aldehydes are frequently applied in C–H functionalisation chemistry in case of the formation of so-called “transient” DGs, where a DG is formed and cleaved in situ. A prominent example is the formation of imines from aldehydes (or ketones) and amines. Since this subject has been reviewed very recently,102,103 this type of chemistry is not included in this chapter and only examples where the aldehyde function is the actual DG are discussed. Hence, in the timeframe covered by this review, only three examples have been reported and only in one example the aldehyde function remains in the final products, whereas in the other two examples it acts as traceless DG.The first example used the aldehyde function as a traceless DG in a Rh catalysed reaction with alkynes (Scheme 6A).104 2-Aryl-3-formylindoles were used as starting materials. In the first step (according to the proposed mechanism), the aldehyde-directed an alkenylation at C2-position of the indole core. Rh was then suggested to be involved in the decarbonylation process and further activation of a C–H bond of the N-aryl group as well leading overall to indolo[1,2-a]quinolones in high yields (up to 94%, frequently >80%), which is overall an elegant access to this system.
 | ||
| Scheme 6 Aldehyde-directed C–H functionalisation. | ||
Also in the second example, the aldehyde DG was removed under the applied reaction conditions, this time in an arylation protocol (Scheme 6B).105 Initially, salicylaldehyde was used as the starting material and aryl iodides as coupling partner. Arylation took place ortho to the formyl group, which was cleaved under the applied reaction conditions leading overall to meta substituted phenols in yields around 50%. Regarding the mechanism, experiments actually pointed towards a reaction sequence in which the first step is the oxidation of the salicylaldehyde to salicyclic acid (which would mean that the actual DG is the acid function), which is then removed via decarboxylation. The advantage of starting from the aldehyde function is that salicylic aldehyde derivatives could be prepared much more easily from the corresponding phenols than salicylic acids.
The one example keeping the aldehyde function reported sulfamidation of indoles in position 4 (Scheme 6C).106 This Ir catalysed method could be run at room temperature and delivered typically high yields of the desired products. Noteworthy, the application of the method to a gram scale synthesis was reported showing the scalability of this transformation.
4. Carboxylic acids as DG
Even though carboxylic acids are a frequently occurring FG, it has been added rather late to the repertoire of DGs. The majority of contributions regarding carboxylic acid-directed ortho C–H functionalisation were only made since the late 2000s and the field has been recently reviewed.107,108The benefits of carboxylic acids as DGs are quite striking as they are naturally abundant and relatively easy to introduce. They can either be cleaved or further transformed via decarboxylative coupling reactions, often possible to perform in situ or in one-pot fashion.109,110 Furthermore, the possibility to remove the group catalytically led to the development of its use as a traceless DG.111 However, carboxylates are usually weakly coordinating to the catalyst and are often disfavoured in comparison to stronger DGs (e.g. pyridines, bidentate amides). They can also undergo undesired side reactions such as hydrodecarboxylation as these are also catalysed by transition metals,112 which is an additional difficulty in developing carboxylic acid-directed C–H functionalisation processes.
4.1. C(sp2)–H activation
For example, the group of Gooßen developed a protocol using aryldiazonium salts as aryl sources, which was catalysed by Ir and proceeded at a mild temperature of 60 °C (Scheme 7A).113 Additionally, only a sub-stoichiometric amount of base and Ag salt was needed and acetone was used as solvent. Notably the weakly coordinating carboxylic acid controlled the regioselectivity even if “stronger” DGs are present (e.g. o-acetamide). The regioselectivity was greatly influenced by steric factors. If meta substituted acids were the substrates, the easier accessible position was arylated exclusively and if both meta positions were occupied, no conversion took place. The parent benzoic acid gave a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of mono- and di-arylated product. The transformation converted a wide range of different diazonium salts. However, heterocyclic salts did not react.113
:1 mixture of mono- and di-arylated product. The transformation converted a wide range of different diazonium salts. However, heterocyclic salts did not react.113
 | ||
| Scheme 7 Arylation with carboxylic acids as DG. | ||
Coupling with aryl halides was achieved with both Pd (Scheme 7B and C) and Ru (Scheme 7D–G). Ren (Scheme 7B) reported a selective mono-arylation with aryl iodides in water (with the use of TWEEN 20 as a surfactant).114 The yields were generally good to excellent. However, some sensitivity to steric hindrance in ortho substituted benzoic acids was observed. For instance, in comparison to 4-chlorobenzoic acid (88% yield), the 2-chloro acid reacted more slowly, and only 73% yield was reported.
Weiping Su reported another Pd catalysed arylation115 (Scheme 7C). The reaction used a mono-protected amino acid (MPAA) as a ligand, proceeded at room temperature and tolerated a broad range of substrates. However, this reaction was more sensitive to substitution in the ortho position to the acid moiety. Even the relative small methyl group reduced the yield by 20% compared to its para and meta regioisomers. The authors stated that the formation of the palladacycle might be prevented due to steric reasons, which also explains the exclusive monoarylation of this transformation.
Ru-Catalysed approaches were investigated by several groups (Scheme 7D–F). For example, Larrosa reported an arylation using aryl iodides in t-BuCN as a solvent (Scheme 7D).116 Several different aryl iodides were used as reactants in this protocol. Generally, para and meta substituted iodides gave better yields and electronic properties seemed to have a minor influence on the reactivity. Acids substituted in ortho position gave usually excellent yields. Remarkably, 3,6-disubstituted benzoic acids provided the arylated products as well, despite the considerable steric hindrance. However, if meta substituted acids were used 3.0 equiv. of water had to be added and di-arylation became an issue. Its extent depended mainly on steric congestion of the substituents. para substituted acids gave the di-arylated products exclusively. The protocol was also applicable to indole carboxylic acids in various positions and even in certain cases without any protection of the indole nitrogen.
With the help of Ru catalysis, significant improvement was made in making aryl bromides and chlorides applicable for this kind of chemistry: Gooßen developed a catalytic system with [Ru(p-cymene)Cl2]2 and PEt3·HBF4, which gave good yields with aryl bromides as substrates (Scheme 7E).117 The corresponding chlorides could be transformed as well, when pipecolinic acid was used as ligand instead of triethyl phosphine. Both protocols converted a broad range of different substrates. Synthetically useful was as well the possibility to protodecarboxylate the product in situ using a Cu catalyst. A very similar system by Weix (Scheme 7F) used aryl iodides (and bromides) as substrates.118 Even a broad scope of heteroaryl halides was reported for this transformation. Furthermore, when 2-iodophenols were used as substrates, an additional lactone formation offered an access to benzochromenones (see also Scheme 8A). Interestingly in both protocols, ketones or amides did not act as competing DGs, which opened possibilities for consecutive C–H functionalisation.
 | ||
| Scheme 8 Annulation reactions. | ||
Ackermann reported a protocol with PCy3 as ligand and ([Ru(MesCO2)2(p-cymene)]) as precatalyst (Scheme 7G).119 Arylation with the corresponding aryl bromides proceeded smoothly. Additionally, alkenes and alkynes were suitable substrates to achieve alkenylations/alkynylations similar to the transformations shown in the previous section. Both methods used NMP as solvent and K2CO3 as base.
All mentioned Ru catalysed protocols did not need any halogen scavenger (e.g. Ag salt) to facilitate the reaction (Scheme 7D–G). However, di-arylation could not be suppressed by neither of the protocols, which made the use of ortho substituted acids mandatory. This is in contrast to the Pd catalysed reactions (Scheme 7B and C). In such cases if a meta substituent at the carboxylic acid was used, the reactions proceeded regioselectively due to steric reasons.
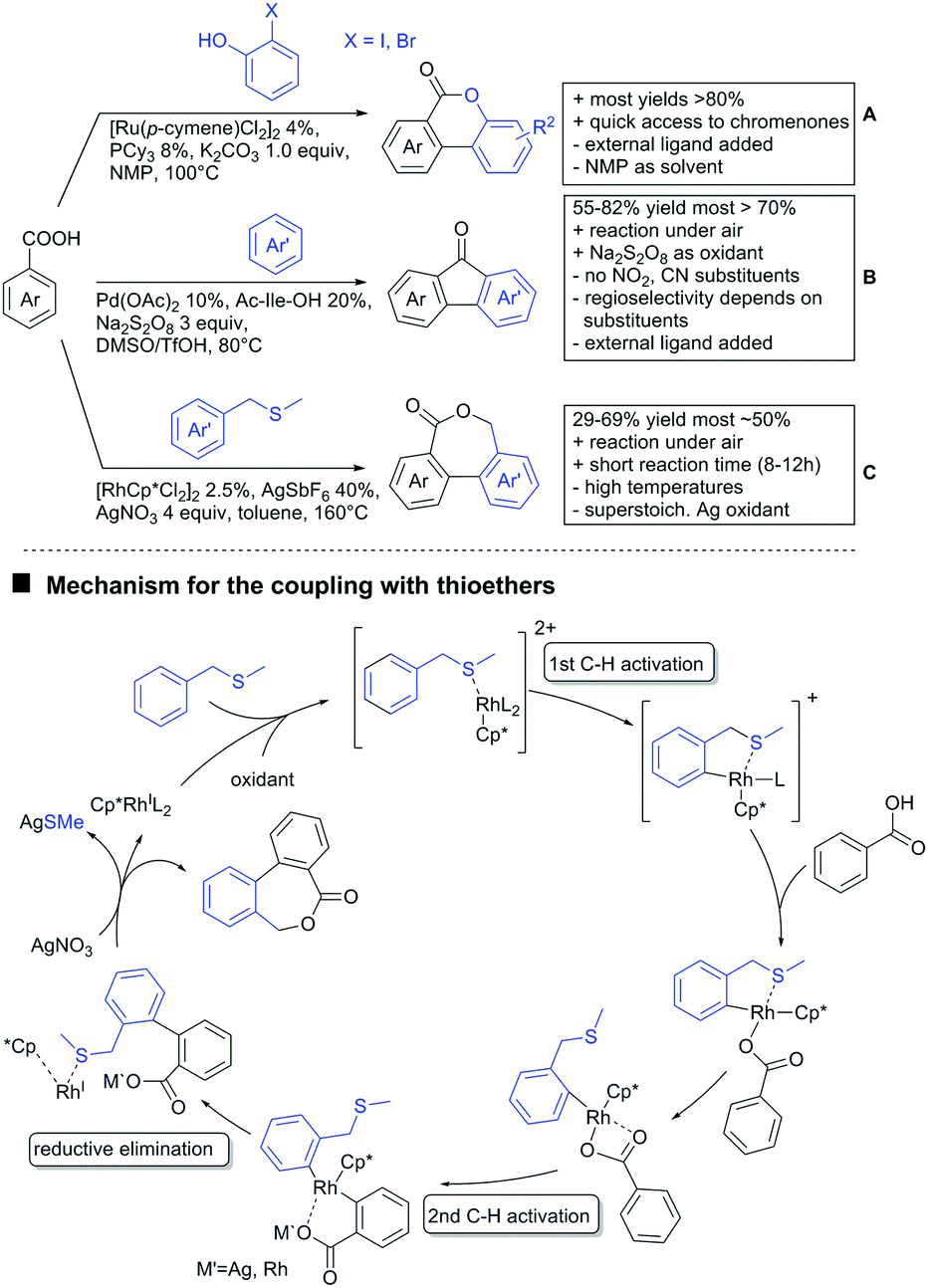
In the field of oxidative C–H activations, transformations with the retention of the DG and also annulations were reported (Schemes 7H, I and 8A–C). Chao-Jun Li reported an cross-dehydrogenative Rh catalysed homocoupling of benzoic acid derivatives.120 The protocol was later improved to achieve heterocoupling between electron-rich and electron-poor acids (Scheme 7H).121 Another cross-dehydrogenative C–H activation developed by You122 used thiophene and furan derivatives as coupling partners (see Scheme 7I). The reaction was again catalysed by a Rh species and showed good FG tolerance. Notably, a subsequent intramolecular Pd catalysed oxidative coupling between the acid and the C3 position of the heterocycle gave the corresponding lactones.
Arylation could also be achieved using phenylacetic acid derivatives under Pd catalysis via 6-membered metallacyles. One contribution used –OH protected mandelic acid to functionalise in ortho position to the residue (Scheme 7J).123 As expected under Pd catalysis, mono-arylation was observed and substrates with ortho substituents to the DG failed to react. It is noteworthy that the stereochemistry in case of a chiral substrate is completely retained.
Regarding arylations that are followed by a ring closing reaction, the already mentioned protocol developed in the group of Weix (see Scheme 7F) yielded chromenones if a hydroxy group was present ortho to the halide (Scheme 8A).124 In the group of Jingsong You an oxidative Pd catalysed arylation, followed by in situ ring closure via acylation (Scheme 8B) was developed.125
A rather unusual double C–H activation reaction also formed polycycles upon lactonization (Scheme 8C).126 Quite interestingly, the first C–H activation is actually directed by the sulfur atom of a thioether and only at a later stage of the catalytic cycle coordination to the carboxylic acid occurred.
As previously mentioned, carboxylic acids could also be applied as traceless DGs. The first (although in a separate reaction after workup) protocol using this idea was developed by Daugulis in 2007.127 The development towards a one-pot protocol in 2011 could be seen as the start of the quest towards the application of carboxylic acids as traceless DGs.128 This approach is very convenient and enhances the utility of directed C–H functionalisation, since the removal of DGs can be difficult. Recent progress in the traceless carboxylic acid-directed arylations over the last years are summarized in Scheme 9A–D.
 | ||
| Scheme 9 Arylation with carboxylic acids as traceless DG. | ||
The coupling of aryl iodides with salicylic acid derivatives was achieved by the use of PdPEPPSI-IPr as catalyst (Scheme 9A).129 Both electron-poor and -rich aryl iodides were good coupling partners in this regioselective protocol, although meta substituted iodides were not well tolerated, possibly due to steric reasons. Different salicylic acid derivatives could be converted with good yields. However, very electron-rich substrates gave lower yield, due to protodecarboxylation. Industrially appealing is the option to substitute the sub-stoichiometric amount of Ag salt with environmentally more benign tetramethylammonium chloride. In a previous study the same group showed that the typically required Ag salts, which are regenerating the catalyst after the catalytic cycle, can be replaced with a quaternary ammonium salt.130
Notably, pyridine carboxylic acids could also be functionalised (Scheme 9B), which is remarkable since C–H functionalisation on electron-poor heterocycles is rarely reported.131 The protocol was applicable with retention of the carboxylic acid. However, a decarboxylative protocol (Cu catalysis) was also reported in the same contribution. A competing experiment with 6-phenylpyridine-3-carboxylic acid showed that the carboxylic acid overrules the DG ability of pyridine. The compound is arylated in 4-position (43%) and only small amounts (4%) of arylation at the phenyl moiety were observed.
Oxidative heteroarylation (mainly with thiophenes) was established with [RhCp*Cl2]2 as catalyst and Ag2CO3 as oxidant (Scheme 9C and D).132,133 Both methods showed a good substrate scope, including nitriles, acetamides or halides, but differed in their use of additives and solvent.
 | ||
| Scheme 10 Alkylation and allylation reactions directed by carboxylic acids. | ||
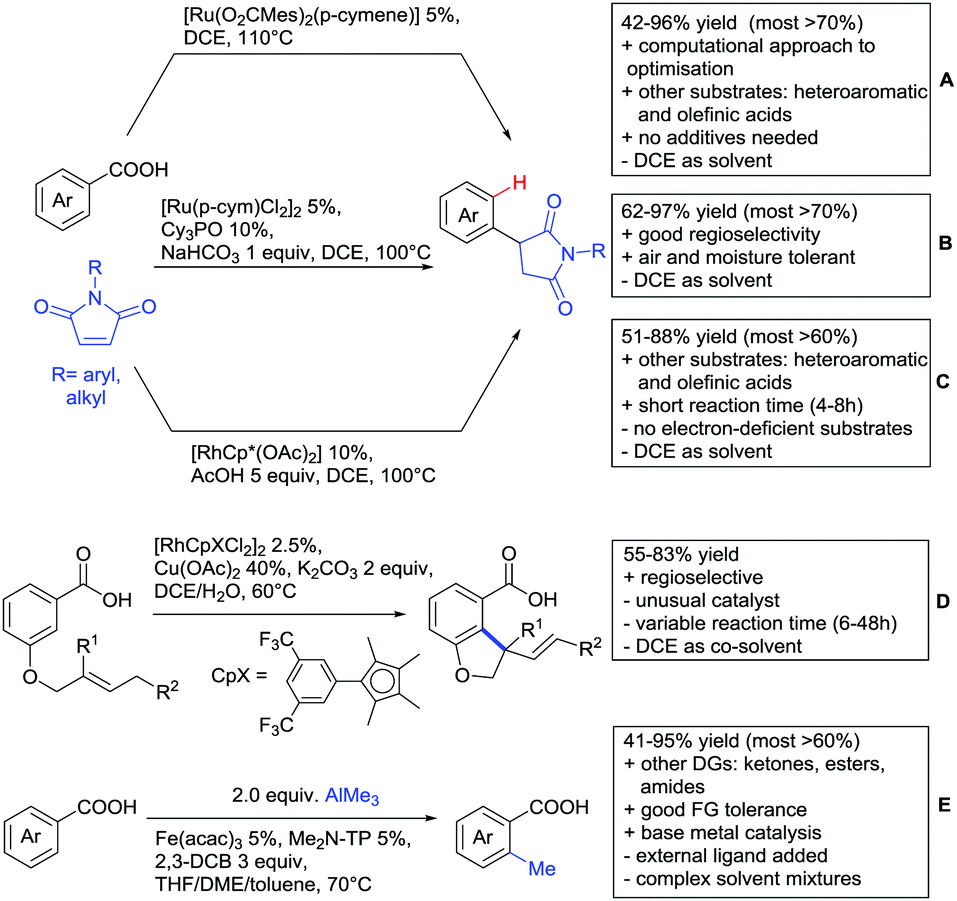
Quite interesting was an intramolecular allylation (Fujiwara–Moritani reaction) catalysed by an electron poor Rh complex (Scheme 10D).139 Presumably, the carboxylate-directed the ortho C–H functionalisation.
The ketone-directed methylation reported in Scheme 2 was also effective using carboxylic acids (Scheme 10E).140 This rare example of Fe catalysis proceeded under mild conditions with good FG tolerance. Furthermore, this transformation was also applicable with esters and amides.
 | ||
| Scheme 11 Alkenylations directed by carboxylic acids to access lactones. | ||
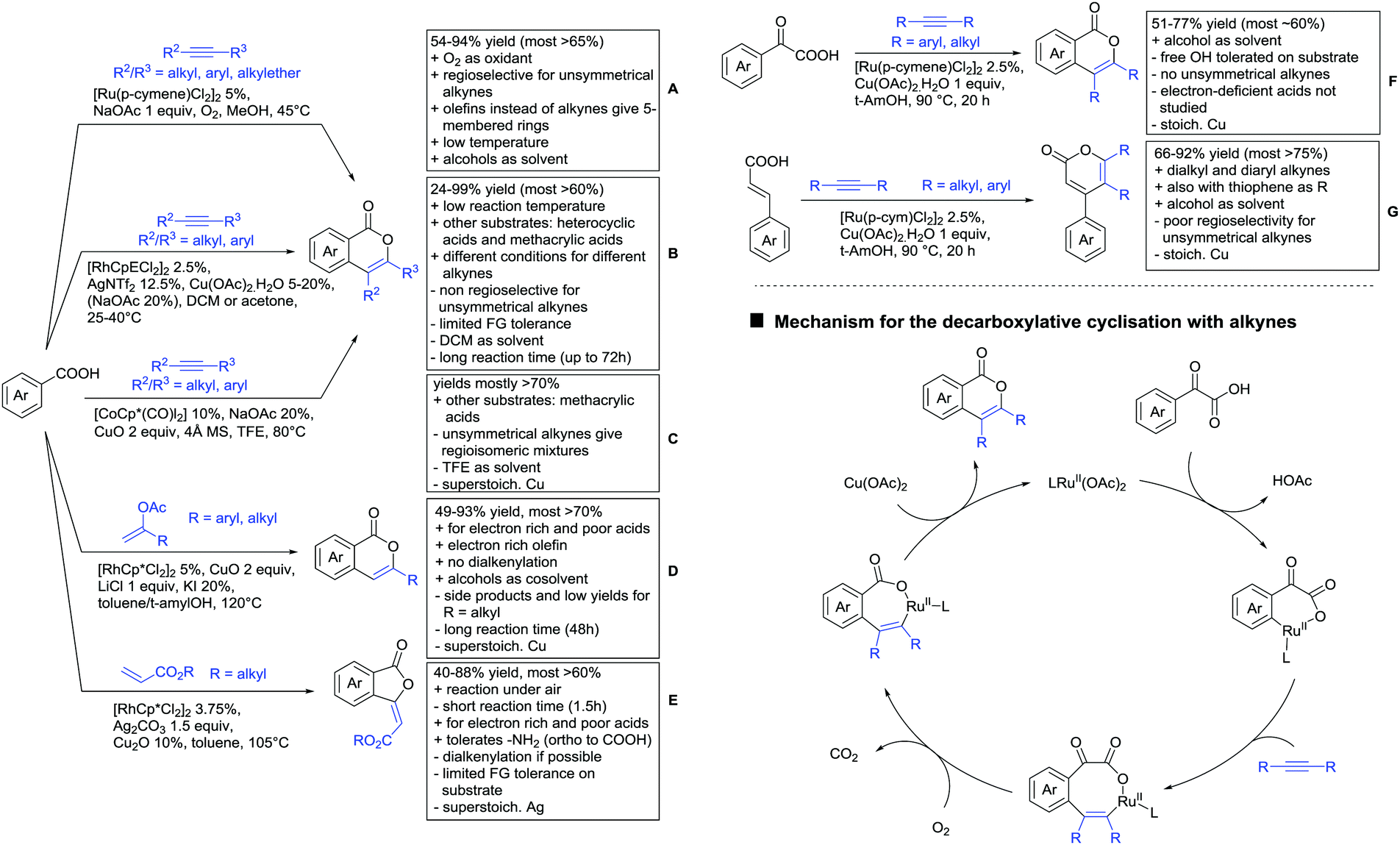
Major advances were made in the use of Ru catalysis and also a reaction with Co was published. Ackermann and co-workers reported a quite interesting example of a mild reaction, that used only oxygen as oxidant (Scheme 11A).143 The transformation gave good to excellent yields with good FG tolerance at 45 °C in methanol. Importantly, unsymmetrical alkyl–aryl alkynes gave a single regioisomer placing the alkyl group in the C4 position of the final product.
A Rh catalysed annulation developed by Tanaka also proceeded under very mild conditions (Scheme 11B).144 However, the use of DCM made this protocol environmentally less attractive. The authors classified the used alkynes into three groups depending on their coordination ability to the catalyst and developed three different protocols accordingly. They differ in the amount of Cu salt and the use of different additives and solvent. These variations enlarge the scope of applicable alkynes and might make the protocol applicable in the synthesis of more complex molecules and can be used as a guideline for a development of specific reaction conditions. Despite the low temperature, the reaction showed limited FG tolerance on the substrates and gave regioisomeric mixtures if unsymmetrical alkynes were used. Notably, though, the reaction also worked with heteroaromatic acids and methacrylic acids as substrates.144
The first carboxylic acid-directed C–H functionalisation with (mainly) diaryl alkynes catalysed by Co was reported by Sundararaju (Scheme 11C).145 Different acids could be converted with good to excellent yields (61–92%). Unsymmetrical alkyl–aryl alkynes gave regioisomeric mixtures with a preference for the isomer with the alkyl residue in C4-position and the aryl moiety in C3-position of the products. Quite interestingly, if a strong chelating group such as an azaindoyl moiety was installed at the para position to the carboxylic acid, the directing effect was still ensured by the acid. Similar to the Rh catalysed annulation by Tanaka (Scheme 11B), the protocol also converted methacrylic acids with good yields. Notably, unsubstituted acrylic acid could also be converted, albeit with low yield (33%).
One example for the formation of isocoumarines without the use of alkynes, was reported by Ting-Bin Wen 2016 (Scheme 11D).146 This protocol utilises vinyl acetates and is a rare example of a C–H functionalisation with electron-rich olefins. Both electron-rich and -poor carboxylic acids were tolerated and no di-alkenylated product was observed. The reaction proceeded smoothly with arylvinyl acetates as reactants. However, if alkylvinyl acetates were used, a five membered lactone with an exocyclic double bond (Z selective) was observed as a side product in considerable amounts (not shown).
The same Rh species gave the aforementioned 5-membered lactones as the main products, if electron-deficient olefins were used instead. Acrylic esters were transformed regioselectively with moderate to good yields into 5 membered lactones with exocyclic olefins with E selectivity (Scheme 11E).147 Although dialkenylation did not occur, ortho unsubstituted acids gave usually lower conversions.
Both mentioned protocols used stoichiometric amounts of Cu or Ag salts as oxidants, which is clearly a disadvantage regarding environmental sustainability. An interesting alternative to several transformations regarding the oxidant was reported by the group of Wang.148 N–O containing molecules were used as an external oxidant and might be an alternative approach to certain transformations.
A special decarboxylative annulation to six-membered lactones of α-ketoacids with Cu(OAc)2 as oxidant was also reported under Ru catalysis (Scheme 11F).149 The reported substrate scope included chloro and free hydroxy substituents on the acid moiety. However, only internal symmetric alkynes were reported (alkyl as well as aryl residues).
This reaction proceeds via the formation of an initial eight-membered ruthenacycle. After uptake of oxygen, incorporated in the carbonyl group of the product, carbon dioxide is released, and the catalytic Rh(II) species is reformed by oxidation (Scheme 11, bottom right).
As an example of an activation at a non-aromatic C(sp2)–H bond, the formation of α-pyranones was reported by Gogoi and co-workers (Scheme 11G).150 The reaction proceeded under Ru catalysis and the β carbon of a cinnamic acid derivative was activated. Subsequent insertion of an alkyne and reductive elimination furnished the six-membered ring. This transformation converted heteroaromatic cinnamic acids as well with good yields and accepted both diaryl as well as dialkyl alkynes. However, the use of unsymmetrical alkynes gave mixtures.
Under different reaction conditions, saturated lactones can be formed instead of unsaturated ones, depending on the use of oxidant and/or the used coupling partners (alkynes, olefins) of the protocols. As can be seen in Scheme 12, ortho-directed alkenylations and subsequent nucleophilic attack of the carboxylate group, followed by protonation, resulted in saturated 5-membered lactones. Shishido reported a transformation with unsymmetrical internal alkynes under ZrO2 supported Ru catalysis (Scheme 12A).151 Notably, the catalyst could be recycled for at least five runs and the reaction required only a catalytic amount of base. The regioselectivity was generally good. However, not a broad FG tolerance was reported.
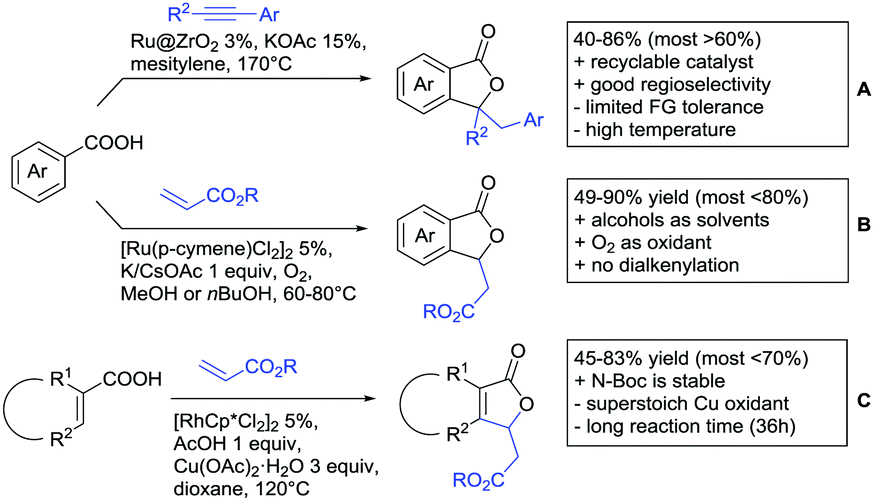 | ||
| Scheme 12 Alkenylation directed by carboxylic acids and subsequent annulation to alkylated lactones. | ||
Substantially milder reaction conditions were sufficient for the oxidative coupling with acrylates (Scheme 12B).152 Under O2 atmosphere and a reaction temperature of 60–80 °C, bromo and iodo substituents were tolerated and no di-alkenylated product was observed. A similar cyclisation was also reported by Zhu for cyclic acrylic acids (Scheme 12C).153 The reaction was catalysed by a Rh complex and Cu(OAc)2 was used as oxidant. Quite interestingly, under the reaction conditions (acidic, metal salts and 120 °C) N-Boc protected substrates were converted with moderate yields without any cleavage of the carbamate.
Alkenylation without annulation and subsequent decarboxylation could be achieved with alkenes under oxidative conditions or with alkynes under redox-neutral conditions. These transformations make meta substituted aryls more easily accessible if an ortho substituted acid is used as substrate and in situ protodecarboxylated. In recent literature, alkenylation protocols by the Ackermann154 (Scheme 13B), Hartwig/Zhao155 (Scheme 13C) and Gooßen156,157 groups (Scheme 13D) have appeared.
 | ||
| Scheme 13 Alkenylation directed by carboxylic acids. | ||
All three protocols used Ru complexes that catalysed the alkenylation as well as the in situ decarboxylation. Ackermann also included a report of an oxidative coupling between salicylic acids and electron-poor olefins, using V2O5 as oxidant (Scheme 13A).154 For this transformation, the FG tolerance regarding the olefin was quite good. However, the alkenylation with internal alkynes (Scheme 13B) converted also benzoic acid derivatives and was not restricted to salicylic acids.154 Furthermore, examples with aryl and alkyl (also unsymmetrical) alkynes were provided. Interestingly, nitro-, fluoro- and bromo groups ortho to the acid were tolerated and gave moderate to good yields. Both para and ortho residues to the acid naturally gave the corresponding meta products. However no example of meta substituted acids (to potentially generate the para alkenylated product) was reported.
A very similar protocol by Hartwig and Zhao (Scheme 13C) demonstrated a broad substrate scope.155 The same substitution pattern regarding the acid was reported with comparable yields (ortho and para substituents to COOH). As expected, meta substituted substrates delivered mixtures of regioisomers. However, if sterically more demanding substituents (e.g. –NMe2, –iPr, –tBu) were used, the para substituted product (after decarboxylation) was formed regioselectively.
Interestingly, in comparison to Ackermann's protocol, nitro groups on the acid were not tolerated, but the unsubstituted benzoic acid gave a much higher yield (80% vs. 43% in Ackermann's protocol). Upon the addition of 20 mol% Cu(OAc)2, good regioselectivity of aryl–alkyl alkynes was achieved (placing the aryl residue on the sterically less demanding position of the olefin product).
Gooßen developed a similar protocol for methyl aryl alkynes (not shown)157 that was tailored towards a larger substrate scope (see Scheme 13D).156 Additionally, a considerably faster version under microwave irradiation was also reported. The scope for both protocols was broad and no additional additives were necessary to convert asymmetric alkynes.
Regarding the mechanism of the reported Ru catalysed alkenylation reactions, a computational study by Hong gave possible insights to the selectivity of these transformations.135 It was shown, that the chemoselectivity (i.e. consecutive annulation versus decarboxylation) was strongly depending on the size of the ruthenacylce prior to the decarboxylation and the polarity of the solvent used.
The alkenylation of indoles was achieved via Rh catalysis under relatively harsh conditions, since DCM had to be heated to 100 °C (Scheme 13E).158 This transformation made fluorescence-active indoles easily accessible as it gave the di-alkenylated compounds exclusively.
One example of a decarboxylative annulation was the formation of polyarenes by C–H functionalisation of diphenyl acetic acids (Scheme 13F).159 The transformation was catalysed by Pd(OAc)2 with a MPAA as ligand. First the substrate was alkenylated via typical carboxyl-directed C–H activation. Subsequently, the ortho C–H bond of the second ring was activated and an intramolecular annulation occurred. The oxidation was mediated by benzoquinone under O2 atmosphere. Notably, this method constructed different (hetero)anthracene derivatives in a direct way.
The previously described mono-arylation of OH protected mandelic acids (see Scheme 7J) was not only accepting aryl halides but also electron poor olefins as substrates (among them one example with phosphonates) (see Scheme 13G).123 More importantly, the reaction proceeded also with N-protected α-phenylglycine instead of mandelic acid, which enabled a fast route to unnatural amino acids. The yield of the corresponding alkenylated amino acids was generally good to excellent (62–88%).
 | ||
| Scheme 14 Alkynylation directed by carboxylic acids. | ||
Protocol B, the alkynylation catalysed by [RuCl2(p-cymene)]2 showed a similar substrate scope (see Scheme 14B).161 The conversion for electron-poor substrates was also lower than for more activated compounds.
A similar procedure was reported by Dorel (Scheme 14C).162 This time DCE instead of an alcohol was used as solvent, a disadvantage regarding green chemistry efforts. As the protocol was also applied in a phenol-directed peri alkynylation, free phenols were also accepted as substrates for this protocol. Additionally, also nitro groups and unprotected aldehydes were tolerated FGs for this transformation.
Ackermann reported a transformation with a Ru carboxylate as the catalytic species (Scheme 14D).163 In comparison to the transformations above, this protocol showed slightly better yields for electron-poor substrates. However, the reaction conditions were not as mild and di-alkynylation still remained a problem. A decarboxylative protocol was also reported. Under elevated temperatures and addition of acetic acid after the product formation, the carboxyl group was removed without the typically employed Ag or Cu salts.
One example of alkynylation/lactonisation was reported making use of terminal alkynes and Ag2O as oxidant (Scheme 14E).164 This very fast reaction gave moderate to good yields with a relatively broad substrate scope. Notably, only stoichiometric amounts of alkyne were used and the reaction gave the Z isomer exclusively. However, both positions ortho to the acid were alkenylated and the alkylated lactone was formed as a side product.
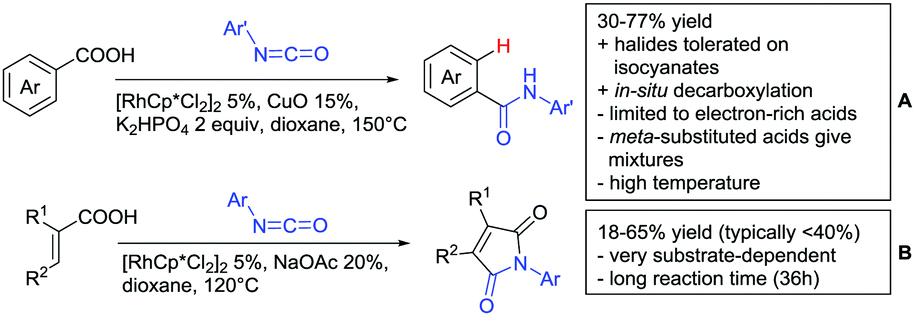 | ||
| Scheme 15 Aminocarbonylation directed by carboxylic acids. | ||
Arylisocyanates could also be applied in a synthesis of cyclic N-aryl imides (Scheme 15B).166 In this case, α substituted acrylic acids reacted with the isocyanates under Rh catalysis via a β vinyl C–H activation.
 | ||
| Scheme 16 C–Heteroatom bond formation directed by carboxylic acids. | ||
Selenation and thiolation could be achieved by reaction with diselenides or disulfides under Ru catalysis (Scheme 16B).168 The process proceeded generally to mono-chalcogenation for meta substituted arenes. However, apart from MOM-protected 4-hydroxybenzoic acid, para substituted acids were functionalised in both ortho positions to the acid. Different substituted diselenides could be transformed with good yields and the protocol also tolerated diphenyl disulfides. More importantly, this method provides the possibility to synthesise chalcogenxanthones via an intramolecular Friedel–Craft acylation by addition of triflic acid to the crude mixture after the reaction. For this annulation benzoic acids carrying halides, ethers, alkyl, and aryl groups were tolerated and also thiophene carboxylic acid was used as substrate giving good yields (58–86%) over two steps.
4.2. C(sp3)–H activation
Advances have been made in the β-C(sp3)–H activation and arylation of propionic (also α-amino) acids. Zhao,169 Yu170 and van Gemmeren171 all reported Pd catalysed protocols for this transformation (Scheme 17A–C).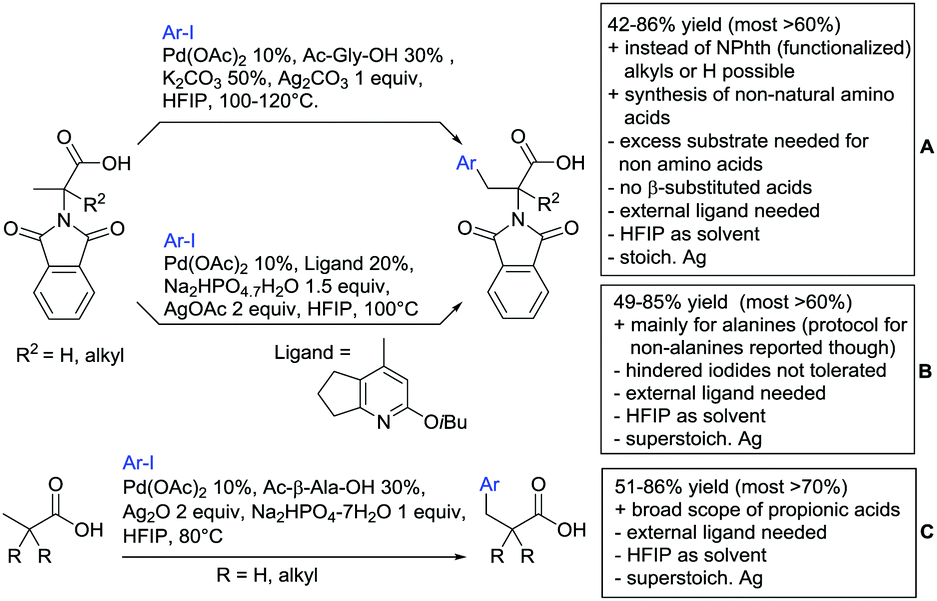 | ||
| Scheme 17 Functionalisation of C(sp3)–H bonds directed by carboxylic acids. | ||
Zhao (Scheme 17A) focused on the arylation of N-phtaloyl alanines to synthesise non natural amino acids in a one-step procedure.169 Generally, the scope of the transformation regarding aryl iodides was good, and the yields reported are in the 62–90% range. The reactions covered a wide range of para- and meta-substituted aryl iodides, one example of ortho substitution was also reported (1-fluoro-2-iodobenzene). Notably, different substituents in the α position of the acids were also accepted, and gave good yields if the aryl iodide was the limiting substrate (45–75%). Interestingly, it was even possible to arylate substrates with a quaternary α carbon. Limitations were shown with acids substituted in β position as they gave only traces of the product.169
The protocol developed by Yu (Scheme 17B) focused again on N-phtaloyl alanine as substrate.170 Here instead of a MPAA a fused pyridine ligand was used. This protocol also accepted acids with a quaternary carbon adjacent to the site of activation.
An improvement regarding the generality of the method of Zhoa has been achieved with the use of another MPAA as ligand and by using a different base and Ag salt (Scheme 17C).171 This protocol showed a broad substrate scope regarding the propionic acid derivatives and also tolerated a quaternary centre at the α position to the acid. In contrast to the above method, the acid derivatives were used as limiting substrate here.
5. Esters as DGs
The ester group is omnipresent in natural as well as synthetic organic molecules. Furthermore, esters can be introduced or interconverted by a variety of methods and the need for the development of reliable ester-directed C–H functionalisation procedures is therefore obvious. Pioneering work was performed in the 90s by Trost172 and Murai.173 However, esters coordinate weakly to metal catalysts, which make the development of successful protocols difficult. Therefore, not many contributions have yet been reported in the field. Compared to the free acids much fewer examples have been disclosed in the last three years, which will be discussed in the following section.5.1. C(sp2)–H activation
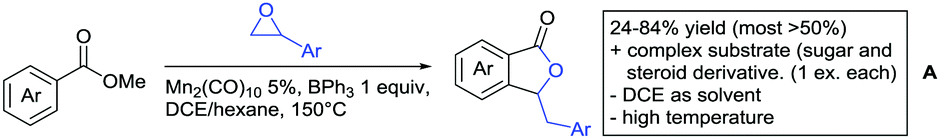 | ||
| Scheme 18 Mn-Catalysed lactonization directed by esters. | ||
Also 2,2-diphenyloxiran was converted, albeit with low yield (23%). If the aryl residue was replaced with a t-butyl group the unsubstituted furanone was formed. Presumably a Meinwald rearrangement leads to the formation of formaldehyde that is incorporated into the five membered ring.
:1 for ester DG in comparison to 1:2 for Weinreb amide DG on phenylacetate). The authors argued that the products were easier released by the metal species and hence di-alkenylation did not preferentially occur.
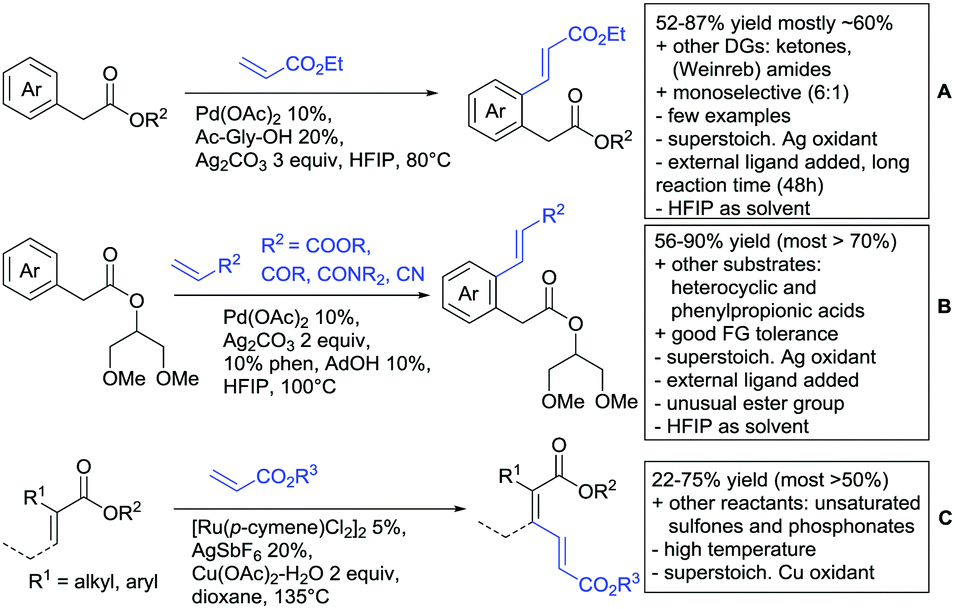 | ||
| Scheme 19 Ester-directed oxidative alkenylation reactions. | ||
Also under Pd catalysis, Zhao and co-workers reported a similar transformation, with quite an broad substrate scope (Scheme 19B).176 Ac-Gly-OH was replaced with 1,10-phenantroline as bidentate ligand, and instead of alkyl esters, they employed a 2-(1,3-dimethoxy)propyloxy handle. The authors speculated that this residue enhanced the coordination ability of the substrate (compared to methyl, ethyl or 2-methoxyethyl residues) to prevent an unselective Fujiwara–Moritani reaction and mono selectivity of the reaction (33:1) was achieved.
Notably, with this method also synthetically useful yields were reported for starting materials carrying electron withdrawing groups (60–82%). Heterocyclic esters also proved to be good substrates. Additionally different esters were converted with good yields: instead of phenyl acetates, also phenylpropionic acid esters and α substituted phenylacetates were possible substrates. Additionally, the method was not limited to acrylates but also α,β unsaturated nitriles, amides, and ketones were applicable as reaction partners.
Loh reported alkenylations of α-substituted acrylates with olefins (mostly acrylates) (Scheme 19C).177 The vinylic C–H activation was catalysed by Ru and used Cu(OAc)2 as oxidant. Different alkyl and aryl substituents were reported as possible substrates for the transformation. To achieve full conversion of the α-substituted acrylate, 2 equiv. of the coupling partner were necessary, as homodimerization of the unsubstituted acrylate was also observed. The stereoselectivity (Z/E) was usually about 9:1 and increased if a bulkier α substituent (R1) was used as coupling partner. Regarding olefin coupling partners, α,β-unsaturated sulfones (56%) and phosphonates (22%) could be also converted.
Alkenylation reactions with consecutive annulation were also reported with ester DGs as depicted in Scheme 20.
 | ||
| Scheme 20 Ester-directed alkenylation and consecutive annulation. | ||
The synthesis of indenones under Co catalysis was reported by Li (Scheme 20A)178 and Zhang (Scheme 20B).179 Both protocols employ [CoCp*(CO)I2] as precatalyst and a Ag salt as additive. The method developed by Li et al. used additionally Zn(OAc)2 to enhance reactivity by coordination of the metal to the ester. If the substrate carried a meta substituent, the regioselectivity depended on the size and ligating ability of the residue. A meta-methoxy ester gave a regioisomeric mixture of 1.2/1, whereas the smaller fluorine atom also coordinated to the catalyst, directing the alkyne into the more hindered position (15/1 regioisomeric ratio).179
The second Co catalysed annulation (Scheme 20B) seemed to be more sensitive to the electronic effects of the aryl substituents. Activating (OMe, Me) residues led to good yields, whereas low yields were observed if esters, –CF3 or other electron-withdrawing substituents were present in the substrate. Different symmetrical aryl alkynes were converted in moderate to good yields (44–72%). Additionally, the authors provided one example of an unsymmetrical alkyne (methyl, phenyl). In this case the regioisomer with the phenyl group α to the carbonyl was preferred (2:1).
Lactones can also be used as DGs. Patel et al. reported on an oxidative annulation of alkynes to 2-arylbenzoxazinones (Scheme 20C).180 The authors suggest the opening of the oxazinone to N-benzoylanthranilic acid under AgOAc/AcOH mediation. The free acid is then coordinating to the Rh catalyst to give a five membered ruthenacycle, which directs the alkyne to be incorporated into the desired position. The coordinating ability of the nitrogen in the substrates did not hinder the transformation in any way and the use of different internal alkynes (alkyl, aryl and alkoxy substituted) were demonstrated. Furthermore, it was also shown in one example that an unsymmetrical alkyne gives only one regioisomer. In this case the smaller residue was situated para to the carbonyl group. Different FGs on the lactone were tolerated such as halides and SMe. Annulation with 2-phenylquinolin-4(1H)-one as substrate was also demonstrated.
Cui reported a Rh catalysed alkenylation/cyclisation directed by peroxyesters, serving as internal oxidant (Scheme 20D).181 Several different peroxyesters were used in the reaction. However, only limited FG tolerance was observed. Different internal alkynes (alkyl and aryl) were also used as substrates. Additionally, also unsymmetrical alkynes were converted with very good regioselectivity (again the smaller residue is situated adjacent to the aromatic ring).
Li reported on a N,B ligand that enables ortho-directed C–H borylations with esters as DGs (Scheme 21A).182 In combination with [IrCl(cod)]2 as precatalyst, the authors reported high yields for the C–H borylation with B2pin2. Di-borylation could be excluded if large alkoxy groups (tBu) were used at the ester.
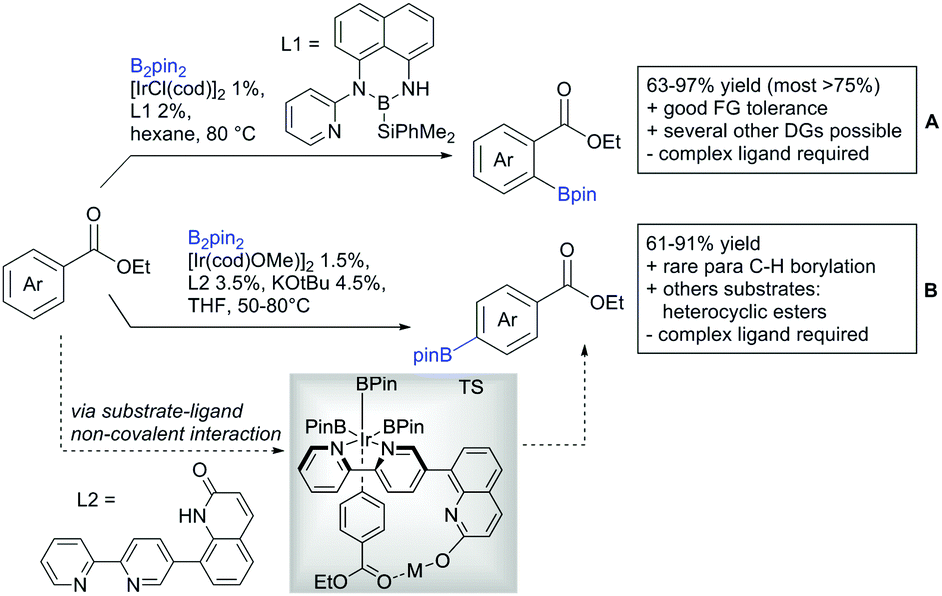 | ||
| Scheme 21 Ligand-assisted ortho and para borylation. | ||
Although not strictly a group-directed reaction (the ester moiety does not coordinate to the catalyst itself but to its ligand), para selective borylation was achieved by the group of Chattopadhyay (Scheme 21B).183 This reaction was also catalysed by Ir. The excellent para selectivity was achieved by non-covalent interaction between the ester moiety of the substrate and the ligand (see TS in Scheme 29B). Different aryl esters were borylated with good to excellent yields. The transformation was also applicable to electron poor and rich heteroaryls. The authors showed, that esters were necessary to direct the C–H activation, since ketones, aldehydes, and carboxylic acids were all reduced under the reaction conditions, and did not yield the desired product.
6. Monodentate amides as DGs in C–H functionalisation reactions
Substituted as well as unsubstituted amides proved to be extremely valuable as DGs in C–H activation. The first example utilizing a primary amide (CONH2) in C–H activation was presented by Li et al. in 2012.184 By now, amides are amongst the most frequently applied DGs in C–H functionalisation chemistry. The largest part of these contributions takes advantage of bidentate DGs including an amide moiety. However, these groups are treated in a separate chapter (see Section 14). Within this chapter, solely amide DGs are treated which use a mono-dentate binding mode.Due to the plethora of examples reported since 2015 contributions with great variations regarding the introduced residue have been disclosed, and also the substrate variety is significant including the activation of C(sp2)–H and C(sp3)–H bonds alike. Most contributions still take advantage of precious metals, but it can be seen that base metal catalysis is moving forward and becomes more and more important.
6.1. C(sp2)–H functionalisation
 | ||
| Scheme 22 Amide-directed arylation reactions. | ||
The group of Kianmehr introduced arylsulfonyl chlorides as aryl source in amide-directed ortho arylation (Scheme 22B).187 The reaction system was rather simple comprising of PdCl2 and Li2CO3 but required 10 mol% catalyst loading. Still, yields were high (71–96%) and the reaction gave only mono-arylated products. In fact, the presence of an ortho methyl group shut down the reaction completely.
A quite peculiar transformation was reported by Park and Hong. They developed an efficient strategy to build up triphenylene frameworks by using a cyclic diaryliodonium salt as aryl source, which basically resulted in ortho and meta arylation of mainly N-phenyl pivalamide substrates (Scheme 22C).188 The reaction required the presence of a substituent in one of the ortho position for obtaining high yields (71–95%), since otherwise only a maximum of 30% was obtained. Switching to N-aryl-2-pyrrolidinone as substrate allowed a two-fold reaction with the iodonium salt species leading to heptacyclic products in moderate yields.
Dimerisation of benzamides towards 5H-dibenzo[c,e]azepine-5,7(6H)-diones was reported by the group of Bao (Scheme 22D).189 Since the reaction required a relatively strong oxidant to proceed, the authors favoured a Pd(II)/Pd(IV) catalytic cycle, however, they did not carry out mechanistic investigations. Regarding the benzamide substrate, meta and para substitutents were tolerated (Me, OMe, tBu, F, Br, Cl) but ortho substitution shut down the reaction.
Kumar and co-workers used primary phenylacetamides as substrates, which suggested that in this case not a 5-membered, but a 6-membered palladacycle was involved in the catalytic cycle.190 In this case, aryl iodides were the coupling partners, which allowed the presence of bromide in the substrate (Scheme 22E). Otherwise, mainly “inert” FGs were used such as Me, OMe, Cl, F and NO2. Noteworthy was the good selectivity for mono-arylation and a gram scale experiment which showed the potential for scale up.
Tan and co-workers focused on the arylation of 3-carboxamido thiophene and aryl bromides (Scheme 22F).191 The amide nitrogen contains a pentafluorphenyl. Arylation took initially part in position two and in some cases 2,4-diarylated compounds were observed (maximum 15%). Also heterocyclic bromides could be used as coupling partner and it has to be mentioned that 2 equiv. of halide were applied in order to get good yields (51–87%, 15 examples). Besides the thiophene substrate, in one case also a 3-furancarboxamide was arylated using phenyl iodide (59% mono, 17% diarylation). Additionally, gram scale experiments were carried out as well and starting from 2-aryl-3-thiophencarboxamide, bromination conditions for selectively brominating either in position 4 or 5 were disclosed.
A cascade of C(sp3)–H and C(sp2)–H activation has been reported by Stamos and Yu (Scheme 23 see also Section 15.5).192 According to the proposed catalytic cycle, the first step was a pyrazole-directed arylation of a t-butyl group (Scheme 23, blue bond formed), whereas the second step was an oxidative coupling between position 4 of that same pyrazole and the introduced aryl ring (Scheme 23, red bond formed). This C–H activation step of the pyrazole was indeed amide-directed. Both steps were Pd catalysed and could be run under air, not uncommon for an oxidative coupling step. The range of FGs on the aryl iodide was remarkable and yields between 45–78% were obtained. The t-butyl group could be substituted for other quaternary centres, whereas the arylation always took place at a methyl group.
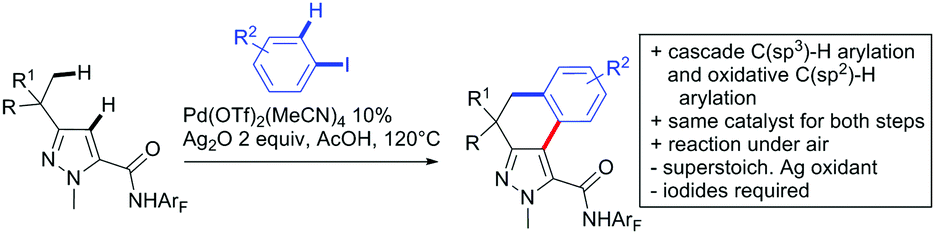 | ||
| Scheme 23 Tandem amide-directed arylation-oxidative coupling. | ||
The group of Glorius reported Co catalysed allylation using methyl allyl carbonate as the allyl source (Scheme 24A).193 Besides using pyrimidine as DG, N-alkylated amides were applied as well and it was shown that mono- and di-alkylated amides worked as well and that steric bulk on the amide was well tolerated, including a t-butyl group. Besides substituted benzamides, also acrylamides and 3-furan- and 3-thiophencarboxamides could be allylated as well. In most cases yields were in the 60–80% range.
 | ||
| Scheme 24 Amide-directed alkylation and allylation reactions. | ||
Another allylation procedure was reported by Yan, this time using allyl bromides and a Rh catalyst (Scheme 24B), which made the procedure less attractive but the advantage was the commercial availability of the Rh catalyst, which is unfortunately not yet the case for many Co species.194 Additionally, the reaction was carried out at room temperature. Yields were in a preparatively useful range (34–93%) but in this case also di-allylated products were formed. Further cyclisation of the allylated products to tetrahydroisoquinolin-1-ones was reported as well.
A significant challenge are typically methylation reactions since the often applied alkyl sources, namely olefines, are not an option and alternative methyl sources need to be found. Ilies and Nakamura have published a Mn catalysed protocol using MeMgBr as alkyl source, which worked with remarkable low catalyst loading (0.01–1 mol%) (Scheme 24C).195 Amongst the advantages of this protocol was the simplicity of the catalyst, MnCl2·LiCl without any ligand, the mild conditions, and high TONs. The use of Grignard species as methyl source naturally raised the question of FG tolerance. Remarkably, even bromide was tolerated. A disadvantage was again the formation of mono- and di-methylated products in case the substrate allowed a second methylation.
The group of Ackermann reported alkylation using methyl vinyl ketone as alkyl source, introducing a butanone side chain on an aryl ring (Scheme 24D).196 The reaction was Ru catalysed and required MesCOOH (1 equiv.) and MesCOOK (0.3 equiv.) as additives but water could be used as solvent. Yields were preparatively useful (45–81%) and electron donating and electron withdrawing substituents on the aryl ring are tolerated as well as bromine. Further cyclisation of the initial products to 2-methylquinolines was reported as well.
An Ir-catalysed reaction of aromatic amides with diazomalonates has been reported by the group of Wang (Scheme 24E).197 Using dimethyl diazomalonate led to the formation of products with both ester moieties still in place. Switching to methyl t-butyl malonates led to methyl esters of arylacetic acids, since the t-butyl group was cleaved under the reaction conditions with subsequent decarboxylation. The reaction worked also on electron rich heterocycles and tolerated the presence of halides (F, Cl, Br, I) and series of other FGs without significant influence on the yields (typically >80%!).
Very recently, they expanded this method, most importantly to methylation reactions as well.198 In this case the alkyl source was a sulfonium salt and stoichiometric amounts of Cu(OAc)2 were required. Yields were mediocre in most cases, but still it was one rare example for a methylation reaction (not shown).
N-Substituted succinimides have been used as alkylation source in the broader sense as well. Naturally, this allowed only the introduction of a very specific group. Prabhu and co-workers showed that this transformation works under Ru catalysis and several additives (Scheme 24F).199 The FG tolerance was not that broadly investigated, but iodine was tolerated which was important for further elaboration of the scaffold. In competition experiments it was shown that in presence of other potential DGs (ketone, aldehyde, acid, ester) it was still the amide which exhibited the directing effect.
The group of Novak disclosed a Pd catalysed protocol for the trifluoroethylation of N-aryl-anilides with an iodonium salt (Scheme 24G).200 The introduction of fluorine moieties onto molecules is of course highly interesting in context of medicinal chemistry projects and drug development. The protocol showed high yields (typically >70% and up to 95%) and remarkable FG tolerance. Most remarkable, esters, boronic acid esters and NO2 were tolerated. In the presence of an acetyl group it was still the amide which directed the trifluoroethylation.
A special case was the enamide directed α-alkylation with unactivated alkenes reported by Dong (Scheme 25A).201 As mentioned, the starting material was a cyclic enamide, which was first alkenylated. Adding afterwards hydrochloric acid to the reaction mixture cleaved the DG in situ and delivered α-alkylated ketones as products. With unactivated olefins, the new C–C bond is formed towards the C2 position of the olefin substrate, and not to C1 as it is the case with often applied acrylate species. Only few FGs were present in the starting materials and yields ranged from 20–73%.
 | ||
| Scheme 25 Amide-directed enamide and aldehyde alkylation. | ||
The group of Willis reported that also a formyl C–H bond can be alkylated and alkenylated.202 The carbonyl oxygen of the amide was in this case involved in the formation of an intermediate rhodacycle using β-amido aldehydes as substrates (Scheme 25B). However, the amide DG can be replaced by a keto- or ester group as well. FG tolerance on the alkene was quite good (Br, OH, acetal, ester, carbamate, aryl) and frequently yields >90% were observed.
A special case are alkylation reactions in which the alkylated product immediately cyclised under the applied reaction conditions to heterocyclic ring systems. Several such examples have been reported in the last three years. Patel and Borah reported the synthesis of oxindoles from acetanilides und Ir(III) catalysis.203 The diazo derivative of Meldrum's acid was the required reaction partner (Scheme 26A). Initially, the acid was attached with its C2 carbon to the ortho position of the amide function. Subsequent proton induced (formed from NaOAc) intramolecular cyclisation with the amide group delivered the target compounds.
 | ||
| Scheme 26 Amide-directed alkylation/allylation–cyclisation sequences. | ||
Huang and co-workers reported an allylation–cyclisation sequence to tetrahydroquinoline derivatives.204 Overall, they carried out a complex four component reaction using aniline derivatives, an anhydride (in most cases acetic anhydride), allylic alcohol, and 2,2,2-trifluoroethanol (Scheme 26B). The anhydride and the aniline derivative form in situ the corresponding amide, which directed the allylation with the allylic alcohol leading to a 3-arylpropanal derivative. Next, this intermediate cyclised to a 2-hydroxy-N-acetyl tetrahydroquinoline derivative and after a final etherification with trifluoroethanol (or other alcohols) the desired products were obtained. Due to this complex sequence there was a larger variation in yields (41–91%) but excellent functional group tolerance was observed.
Kapur and co-workers reported a similar sequence, but in absence of a second alcohol.205 Hence, the etherification did not occur but elimination of water and further aromatisation to quinoline derivatives (Scheme 26C). More than 30 examples have been reported in this contribution.
The group of Wei Zeng reported an alkylation/annulation sequence towards pyrimido[1,6-a]indole-1(2H)-ones under [Cp*RhCl2]2 catalysis with α-diazocarbonyl reagents (Scheme 26D).206 The method gave good access to this scaffold with the majority of examples in yields >70%. In the indole substrate Br and COOMe was tolerated amongst other less reactive FGs. The scope in R1 was mainly limited to carboxylic acid esters whereas R2 seemed to tolerate a wide range of alkyls and aryls.
Alkenylation using vinyl acetate was reported by Zhang and Wen under Rh catalysis.207 This Fujiwara–Moritani reaction required only 2.5 mol% [Cp*RhCl2]2 as catalyst and was carried out under air (Scheme 27A). Hence, 0.3 equiv. of Cu(OAc)2 were sufficient to get to good yields. Many FGs were tolerated including bromine. ortho and meta substituted N-arylacetanilides gave only mono-alkenylated products, however, the unsubstituted or para-substituted derivatives gave mixtures of mono- and di-alkenylated products. The reaction was also carried out on a 7 mmol scale and it was shown that the products can be further cyclised to N-acetylindoles by adding 6 M HCl.
 | ||
| Scheme 27 Amide-directed alkenylation reactions. | ||
Huang and Zhao showed that acrylates could be used to alkenylate N-acylindolines and tetrahydroquinolines in positions 7 and 8, respectively, under Ru catalysis (Scheme 27B).208ortho alkenylation of N-arylpivalamides was also demonstrated under the same conditions. Besides acrylates, many other olefinic substrates carrying an electron withdrawing group could be used as olefin source as well. Generally, good to excellent yields (typically 70–95%) were obtained.
Shortly thereafter, Xu and Pan showed another remote alkenylation, this time of indazoles and under Rh catalysis in which they also demonstrated the cleavage of the DG (Scheme 27C).209
An interesting example was reported by Yoshino and Matsunaga. In this example, the DG actually migrated from the indole nitrogen to the C2 carbon of the introduced olefin group (Scheme 27D).210 Usually, proto demetallation would deliver the N-acylated 2-alkenylated indoles using alkynes as olefin source. Slowing down this proto demetallation step was key in order to allow DG migration. [Cp*CoIII(CH3CN)3](SbF6)2 was identified as best performing catalyst and regarding the DG, morpholine amide performed significantly better as compared to other N-disubstituted amides. Good FG tolerance was observed and yields were frequently above 70% (up to 99%).
A second Co catalysed protocol was reported by Ellman (Scheme 27E).211 The first step was again an alkenylation reaction with a terminal alkyne. However, in the presence of NXS (mainly NIS), the olefine moiety was immediately halogenated. Mainly thiophene substrates were used (see Scheme 27E), but furan and pyrrole were also tested in single examples. The reaction performed at low temperature (40 °C) and the halide was incorporated on the C2 of the former alkyne with high regioselectivity.
If N-arylbenzamides were the substrates, the question arises whether the N-aryl or the C-aryl ring gets functionalised. Wang et al. found that in a [Cp*RhCl2]2 catalysed alkenylation reaction, the key to control the alkenylation side was the presence of a non-coordinating anion, here introduced by AgOTf (Scheme 28).212 Initially, N-tolylferrocencarboxamide was used in a test reaction and in the absence of AgOTf the ferrocene moiety was alkenylated exclusively with methyl acrylate. When adding 20 mol% AgOTf the regioselectivity switched completely and only the tolyl ring was alkenylated. This arylation of the N-aryl ring was then further explored with N-aryl–C-ferrocenyl substrates as well as with N-aryl–C-aryl substrates. C-Ring alkenylation with methyl acrylates led to isindolinones due to intramolecular cyclisation.
 | ||
| Scheme 28 Amide-directed alkenylation with acrylates. | ||
So far, only alkenylation of aromatic systems has been discussed. However, also olefinic systems have been alkenylated via amide-directed C–H activation. Zhang and Zhong reported two protocols for α,β-unsaturated amides, naturally in the β-position giving rise to products of 2Z configuration.213,214 In their first contribution (Scheme 29A), allyl acetate was the reaction partner, leading to (2Z,4E)-dienamide products. As could be expected, the allylated byproduct was formed as well but under the optimised conditions a selectivity of 95:5 or better for the desired product was reached. Also for the 2Z,4E configuration the reaction was not perfect, giving Z/E selectivities between 72:28 and 92:8 with typical yields in the 60–70% range. The second protocol used internal alkynes as alkenylation agent (Scheme 29B), which naturally eliminated the possibility for allylated byproducts giving 2Z,4Z configured products preferentially (frequently with >99:1 selectivity, depending on the nature of R3).
 | ||
| Scheme 29 Amide-directed alkenylation of non-aromatic C(sp2) centres. | ||
Also Bouillon and Poisson contributed to this field (Scheme 29C). In their case, 2-bromo-3,3,3-trifluoropropene was the coupling partner and gave the 2Z configuration.215 This is a rare case in which a bromide is the leaving group since typically oxidative couplings (e.g. with acrylates) are predominant. Position 2 of the substrate was mainly substituted by aryl groups and substituents on the aryl moiety had only little impact on the yield (typically ∼65%). Interestingly, when this position was not substituted, i.e. N,N-diisopropylacrylamide was used as the substrate, the configuration was E and not Z. The authors suggested that a Pd catalysed isomerisation might take place rapidly in this case.
The group of Willis disclosed an impressive amount of work on the C–H activation of aldehydes, mainly directed by amides but also examples for keto- and acid ester direction were reported.202 They showed that switching between two Rh catalysts they could either get the linear (Scheme 30A) or the branched olefin (Scheme 30B) when terminal alkynes were the coupling partner. Linear products were obtained in excellent yields, frequently >90%. However, relatively few FGs were present. For the branched products, yields were a bit lower and the branched: linear ratio varied between 2:1 and 17:1. Using olefins as coupling partner alkylated products were obtained (not shown).
 | ||
| Scheme 30 Amide-directed aldehyde olefination. | ||
Several protocols have been reported in which the final product is a quinoline. In all these cases, the amide was attached via its nitrogen to the aryl moiety and the amide nitrogen ends up as the heterocyclic nitrogen. Within a few weeks, the groups of Li (Scheme 31A)216 and Glorius (Scheme 31B)217 reported Co catalysed protocols for this type of quinoline synthesis. The conditions are quite similar, Li used [Cp*CoCl2]2 as catalyst whereas Glorius used [Cp*CoI2]2 both in DCE as solvent and a Ag salt as additive. Li's protocol required 1 equiv. AgNTf2 whereas Glorius used 20 mol% AgSbF6 combined with 10 mol% Fe(OAc)2 at slightly higher catalyst loadings. The yields and the substrate scope in both protocols were comparable as both reported that halides (amongst other FGs) were well tolerated. Typically, symmetrically substituted alkynes were used as coupling partners and in the few cases where unsymmetrically substituted alkynes were used, mixtures of regioisomers were obtained. In most examples acetamide was the DG, but also examples with other alkyls were reported to work, unless steric bulk was too large. In two examples, Glorius showed that addition of 1 equiv. Ag2O gave N-acylated indole products (Scheme 31C) but better yields for this product class were obtained with urea derived DGs (see Section 9).
 | ||
| Scheme 31 Amide-directed alkenylation–cyclisation sequences. | ||
If the amide DG was attached to the aryl system the other way round, i.e. via the carbonyl carbon, reactions with alkynes led upon cyclisation to isoquinolones (or a related system, depending on the specific substrate). Chuang and Chen reported such a protocol under quiet environmentally benign conditions, since the reaction could be carried out in water under air with a Rh catalyst (Scheme 31D).218 Again, symmetric aryl–aryl alkynes were the coupling partners in most cases but in three examples aryl–alkyl alkynes were used. The regioselectivity was actually quite good (87:13 up to 98:2) and the aryl ring always ended up in position 3 in the main product. One exception was the reaction of N-methyl-4-methoxybenzamide and N,N-dimethyl-3-phenyl-prop-2-yn-1-amine, where the regiochemistry was reversed and the aryl moiety was exclusively in position 4. A related transformation on thiophenecarboxamide substrates was reported by Miura and coworkers (not shown).219
Similar, but Ru catalysed cyclisations have been disclosed by the groups of Swamy220 and Urriolabeitia (not shown).221 The former one used chromene-3-carboxamides as substrates, the latter one various heterocyclic amides. Hence, in both cases a pyridinone ring was annulated to another heterocyclic ring, giving access to interesting scaffolds.
Tian and Van der Eycken published an intramolecular variant of this type of transformation (Scheme 31E).222 This allowed them the synthesis of polycyclic systems which occurred in natural products as well. In their paper, they applied this method for building up a variety of polycycles in good to excellent yields, and also demonstrated the synthesis of two natural products, rosettacin and oxypalmatine.
When using acrylates instead of alkynes as the olefination reagent, isoindolin-1-ones are the resulting products as demonstrated by Lu and Yu (Scheme 31F).223 The olefinated product could be isolated only in a few cases (e.g. with N-C6F5-2-furancarboxamide as substrate or when styrenes were used as olefinic coupling partner instead of acrylates) and mainly spontaneous cyclisation to the isoindolin-1-ones via base mediated 1,4-conjugate addition was observed. Typical yields were in the range of 80–93%.
ortho acylation of N-aryl acetamides with aldehydes was reported by Novak and co-workers (Scheme 32A).224 They found that the presence of a catalytic amount of B(C6F5)3 had a beneficial effect on catalysis and a series of aryl–aryl ketones were synthesized. Using aliphatic aldehydes, the yields were generally lower but overall there was a high variance of yields. Alternatively to aldehydes, α-oxocarboxylic acids can be used as acyl source in a decarboxylative reaction. Again Pd(OAc)2 is used as catalyst in presence of an oxidant, in this case K2S2O8 (not shown).225
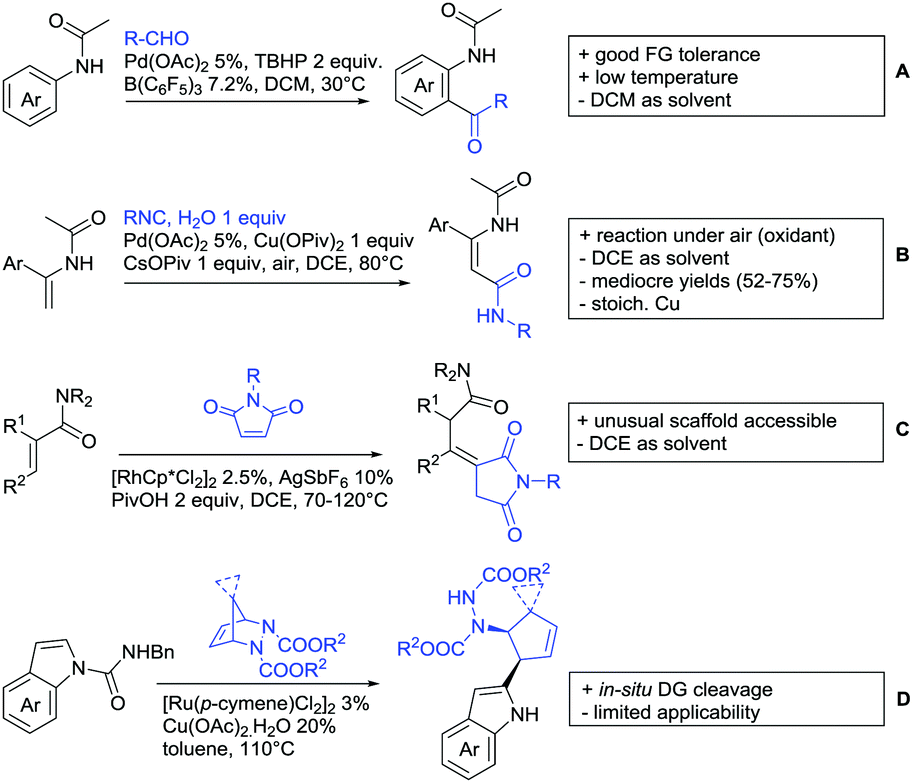 | ||
| Scheme 32 Miscellaneous amide-directed reactions. | ||
Carboxamidation of enamides by isocyanides was reported by Liang and Luo (Scheme 32B).226 So the amide-directed the introduction of another amide group at a terminal double bond. Fair to good yields were obtained and also good FG tolerance in this Pd(OAc)2 catalysed protocol was observed.
The protocol reported by In Su Kim is according to the proposed mechanism a C–H alkylation in the first step.227 However, this product was not isolated but a double bond migration occurred and hence this example was displayed in this special section (Scheme 32C). Acrylamides were the substrates and maleimides the coupling partners, which led to quite unusual but interesting product structures. With the optimized protocol ([Cp*RhCl2]2, AgSbF6, PivOH) almost two dozen examples were reported in low to excellent yield range. The high yields (also >90%) were obtained with acrylamide derived substrates but other types of substrates performed significantly worse.
Finally, a peculiar cyclopentenylation reaction has been reported by Radhakrishnan and co-workers (Scheme 32D).228 The reaction required a specific type of bicyclic diaza olefin and hence its utility was limited to very specific examples.
Halogenation reactions via directed C–H-activation are of particular interest since they can be used to override the directing effects of substituents present in the substrate. Mück-Lichtenfeld and Glorius developed an ortho chlorination protocol of benzamides using a Rh catalyst (Scheme 33A).229 Interestingly, often applied NCS did not work but trichloroisocyanuric acid (TCC) or 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) in combination with 2-trifluoromethylpyridine as activator. The most interesting examples were of course in which the chlorination was directed at the meta position compared to a usually ortho/para DG (and of course ortho to the carboxamide group) such as methoxy or methyl.
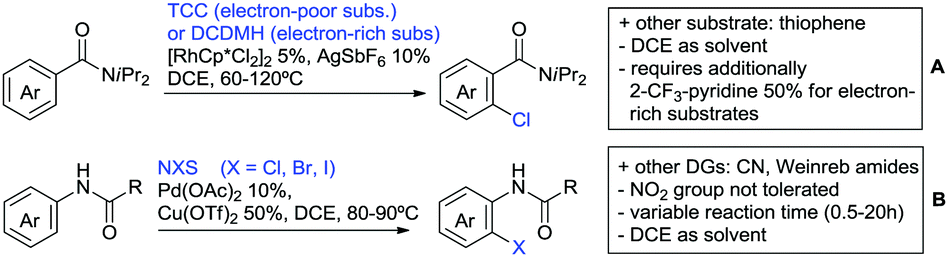 | ||
| Scheme 33 Amide-directed halogenation reactions. | ||
Kapur and Das showed that under Pd catalysis, Weinreb amides and N-acylanilines could direct NXS promoted halogenation in their ortho positions (Scheme 33B).230
A similar transformation but with a Pd@MOF nanocomposite catalyst was reported by Martin-Matute and co-workers. The work was mainly dedicated to pyridine-directed halogenation, but also amides worked and recyclability of the catalyst was demonstrated (not shown).231
 | ||
| Scheme 34 Amide-directed C–O bond forming reactions. | ||
C8-Triflation of naphthalenes was developed by Xiao and Fu (Scheme 34B).233 The optimised reaction conditions turned out to be quite complex since besides 10 mol% Pd(OAc)2 also 0.5 equiv. benzophenone (BP), 1.2 equiv. TfOH, 0.8 equiv. TMSOTf and 2 equiv. N-fluoro-1,3,5-trimethylpyridinium triflate reagent were required giving moderate to good yields.
Rh catalysed acyloxylation was reported by Xu and Li (Scheme 34C).234 The publication focused mainly on heterocyclic DGs but included also a table of amide-directed examples. Yields around 60% were typical.
Additionally, Chakraborti and co-workers developed a Pd catalysed hydroxylation protocol which they mainly applied to heterocyclic DGs (see Section 15.9).235 However, five examples of amide direction were reported but only three of them gave good yield (not shown).
 | ||
| Scheme 35 Amide-directed C–N bond forming reactions. | ||
The introduction of amine groups is a particular challenge in C–H activation chemistry. One example was disclosed by Sun and Yu using N-C6F5-benzamides and secondary dialkylamines as coupling partners (Scheme 35B).237 Hence, only tertiary amines were obtained. Switching to anilines as coupling partners, secondary N,N-diarylamines were the products. Yields are generally good in this [Cp*RhCl2]2 catalysed method and FGs are tolerated to a relatively good extend.
6.2. C(sp3)–H activation
The activation of C(sp3)–H bonds is typically more challenging than activation of their C(sp2)–H counterparts. Not surprisingly, much less work has been published on this topic in recent years. In fact only three amide-directed examples (i.e. mono-dentate amides!) have been reported all coming from the lab of Jin-Quan Yu.The first one dealt with arylation of methylene groups in β-position to the CONH(4-CF3-C6F4) DG (Scheme 36A). In this case, even an enantioselective method was developed using a chiral N-acetyl protected 2-aminoethyl quinoline (APAQ) as ligand.238 They obtained e.r. of typically 95:5 using Pd(OAc)2 as catalyst and aryl iodides as aryl source. Good FG tolerance and yields between 45–89% were reported. In a second contribution enantioselective alkenylation and alkynylation reactions were also reported (not shown).239
 | ||
| Scheme 36 Amide-directed C(sp3)–H functionalisations. | ||
The second contribution reported a Pd catalysed borylation with B2pin2 as coupling partner (Scheme 36B).240 Methyl or methylene groups in β-position were borylated whereas the less sterically demanding was favoured (CH3 over CH2) in cases where there was competition. Noteworthy, also saturated carbocyclic systems (C3–C7) were successfully borylated.
7. N-Alkoxy- and Weinreb amides
Besides CONH2, CONHR and CONR2 amides, also N-alkoxy amides i.e. Weinreb amides have been used as DGs, however less frequently. Not surprisingly, relatively similar catalytic systems as compared to ordinary amides are applied also in this case and also the types of reactions reported show a large overlap with the examples in Section 6.7.1. Arylation
Only two examples for arylation reactions of N-alkoxybenzamides have been reported in recent years. Bhanage and co-workers used anilines as aryl source, which were in situ converted to the corresponding diazonium salts (Scheme 37A).241 After the initial C–H activation and arylation, the alkoxyamide actually directs a second C–H activation on the previously introduced aryl ring and delivers phenantridinones as the final products after reductive elimination. Mild reaction conditions and good to excellent yields (57–91%) are the main advantages of this protocol. Additionally, halogens (F, Cl, Br) are tolerated on the N-alkoxybenzamide substrate. | ||
| Scheme 37 Arylation reactions directed by N-alkoxy and N-aryloxyamides. | ||
Land and You reported another arylation protocol, this time of N-aryloxyacetamides, under Rh catalysis. In this case, oxidative coupling between the position 2 of azoles and benzoazoles was realized with typical yields >70% (Scheme 37B).242 Mainly oxazoles and benzoxazoles were applied but one example with benzothiazole and one with N-methylbenzimidazole was reported as well. Under the applied reaction conditions, the N-aryloxy amide was N–O cleaved and overall ortho arylated phenols were obtained as the products.
Jin-Quan Yu and co-workers reported a C(sp3)–H arylation catalysed by simple Pd(OAc)2 and 2-picoline as ligand, which led to mono-arylation products (Scheme 37C).243 These mono-arylated products could be subjected to a second arylation when a different ligand, namely 2,6-lutidine was used (Scheme 37D). The substrates applied were typically enantiopure and it was demonstrated that the stereocenter was not compromised by the reaction conditions. After the second arylation step, the products were obtained in d.r. >20:1. Additionally, also the conversion of the N-methoxyalkanamide to a methyl ester was demonstrated and could be carried out in a single operation, again without compromising the stereocenter. Aryl- and heteroaryl iodides were the applied coupling partners and the yield range was relatively large spanning from 32–91%. Excellent FG tolerance was reported.
7.2. Alkylation and allylation
The group of Huang reported on the alkylation of N-methoxybenzamides using alkyl iodides as coupling partners (Scheme 38A).244 The reaction was mono-selective for substrates carrying a substituent already in ortho- or meta-position. In case of para substituted compounds or the parent N-methoxybenzamide, mixtures of mono- and di-alkylated products were obtained. On average, the mono-alkylated product was favoured in a 6:1 ratio, which is remarkable considering the large excess of iodide (6 equiv.). A weakly coordinating ligand was important for achieving this degree of mono-selectivity. Amongst others, bromine as FG was well tolerated in this Pd(OAc)2-catalysed protocol. Notably, N-methylbenzamide gave the alkylated product only in 17% yield, whereas the N-methoxybenzamides gave products in the 60–90% range.
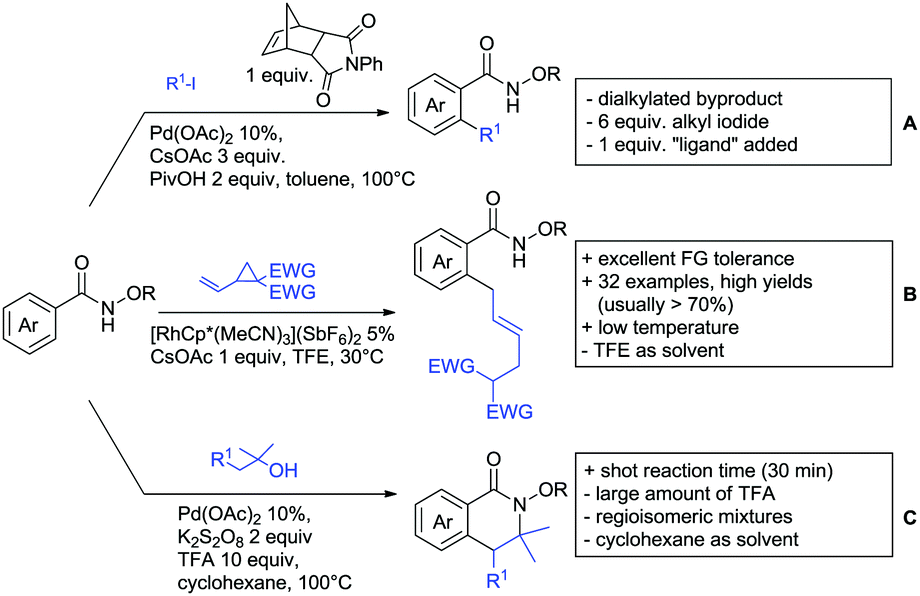 | ||
| Scheme 38 N-Alkoxyamide-directed alkylation/allylation reactions. | ||
Huang, Li, and Wang disclosed a Rh(III)-catalysed allylation protocol using 2-vinylcyclopropane-1,1-dicarboxylate (or related compounds) as allyl source (Scheme 38B).245 The reaction showed excellent FG tolerance (including Br, I, NO2, CN, COOMe) and yields around 80%. A large set of examples was reported and the products showed good E/Z selectivity (5:1 in the worst case, often ∼20:1).
The group of Huang reported the synthesis of dihydro-isoquinolinones via a Pd(OAc)2 catalysed alkylation–cyclisation sequence (Scheme 38C).246 Initially, di-tbutyl peroxide was the alkyl source and the reaction proceeded smoothly in cyclohexane, unfortunately requiring 25 equiv. of TFA. Notably, if N-alkyl- or N-arylbenzamides were used, the reaction was inefficient, whereas most examples for N-alkoxybenzamides gave a yield ∼80%. By adding 2 equiv. of K2S2O8 to the reaction mixture, the peroxide could be replaced with a tertiary alcohol. The reaction with t-butanol gave essentially the same yield as with the corresponding peroxide. In cases were one methyl group was replaced by a longer alkyl chain, cyclisation occurred either to the methyl, or the other alkyl group leading to mixtures of products.
7.3. Alkenylation
Moving to alkenylation reactions, several more examples as compared to alkylation reactions were reported. They can be clustered into reactions in which the alkenylated product is isolated as the target molecule, and in examples in which the alkenylation is followed by a cyclisation step to an N-containing heterocycle, without isolation of the intermediate product.Kapur reported pure alkenylation using cyclic aromatic Weinreb amides as substrates.247 The reaction was Ru-catalysed and acrylate or styrene derivatives were used as alkene sources (Scheme 39A). A wide range of electron withdrawing groups on the olefin were accepted, but yields were typically only in the 41–73% range. Cu(OAc)2·H2O was used as oxidant and AgSbF6 as additive. In the end, the authors demonstrated typically further transformations of Weinreb amides and converted them into an aldehyde or ketone moiety.
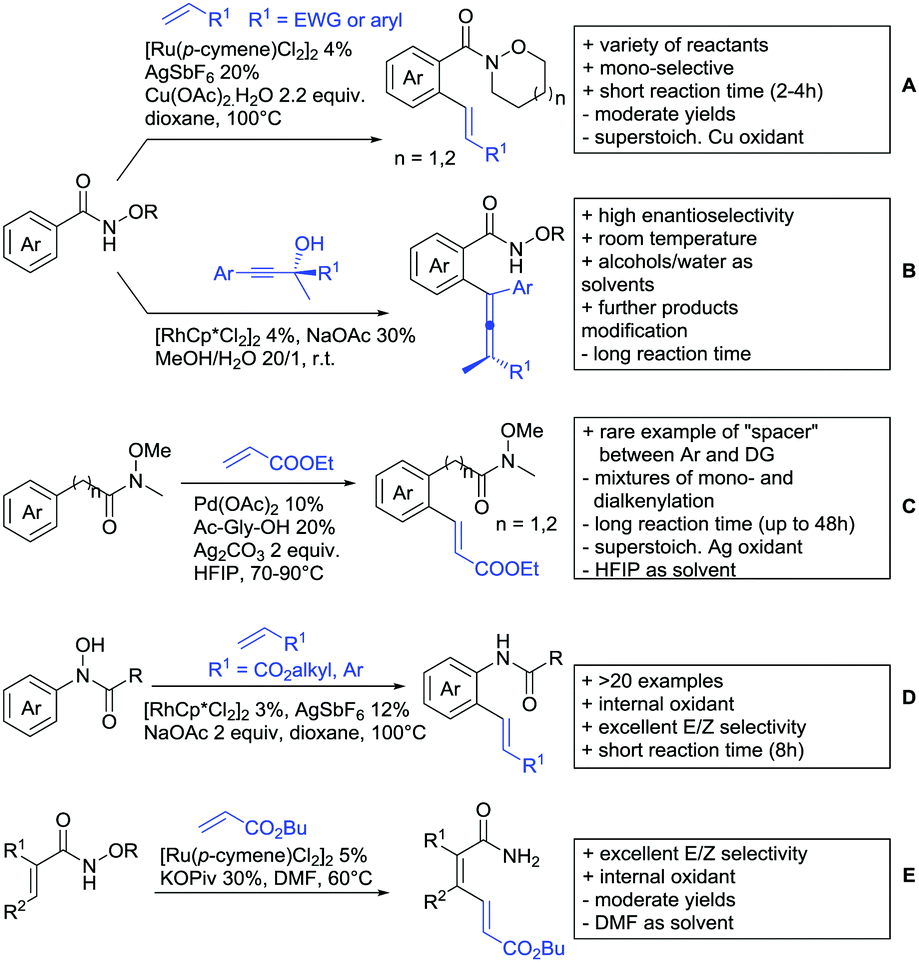 | ||
| Scheme 39 N-Alkoxy- and N-hydroxyamide-directed alkenylation reactions. | ||
A special case is the reaction with propargylic alcohols as coupling partners. Ma and co-workers showed that in this case an allene moiety was attached at the ortho position to the N-methoxybenzamide DG (Scheme 39B).63 The most remarkable feature of this transformation was the fact that in case of chiral propargylic alcohols, the allene products are formed in ees between 94–98%. It has to be mentioned that the reaction required long reaction times, typically 72 hours but delivered typical yields ∼70%. Furthermore, the intramolecular cyclisation of the products to several types of heterocycles was demonstrated as well, showing the synthetic potential of the disclosed method.
Alkenylation and also acetoxylation (not shown) of Weinreb amides was reported by Yu and co-workers (Scheme 39C).248 This protocol was also applied for other DGs such as ketones (see Section 2.3) and esters (see Section 5.1.2). In case of the Weinreb amides the substrate scope included only FGs which can be considered as innocent (F, Cl, CF3, OMe), since they do not undergo any transformations under a wide range of C–H functionalisation conditions. Yields for the alkenylation were high (70–92%) for examples in which no di-alkenylation was possible. In the other cases, mixtures of mono- and di-alkenylated products were obtained. For acetoxylation, the yields were generally ∼10% lower and the same issue with a two-fold functionalisation occurred.
In one case also an N-hydroxyanilide has been used as substrate.249 Acrylates were used as coupling partners in this Rh catalysed protocol (Scheme 39D). The resulting products were almost exclusively of E configuration but in two examples isomerisation occurred giving then E/Z mixtures. These were the t-butyl and ethyl acrylates, however why exactly these two examples showed isomerisation was not explained. Also in this case the final products were amides since the N-hydroxy group was cleaved in course of the reaction (internal oxidant).
Zhang and Zhong reported an alkenylation protocol of N-methoxy amides, in which the methoxy group is cleaved under the applied Ru catalysed conditions (internal oxidant), leading to alkenylated amides as final products (Scheme 39E).250 In this case not aromatic but α,β-unsaturated N-methoxyalkancarboxamides were used as substrate leading to the corresponding 2,4-diolefins with excellent Z/E selectivity often of 99/1. Yields however were low to good (22–70%).
7.4. Alkenylation with concomitant cyclisation
Several papers were published in which the alkenylation step is immediately followed by a cyclisation to a nitrogen containing heterocycle.Li, Yang, and Zhou showed that indolines carrying an N-methoxycarbamoyl in position 1 can be alkenylated with alkynes in position 7 and then further cyclised to 6- or 7-membered rings, depending on the nature of the alkyne (Scheme 40A).251 Dialkyl alkynes gave the 6-membered cyclisation product, whereas diaryl alkynes gave the 7-membered derivative. Otherwise, the reaction conditions in this Rh catalysed protocol were identical. Also alkylation with olefins and further cyclisation was reported. For this transformation the additive and solvent had to be changed (from NaOAc and MeOH to CsOAc and DMF) and acrylates were the reaction partners.
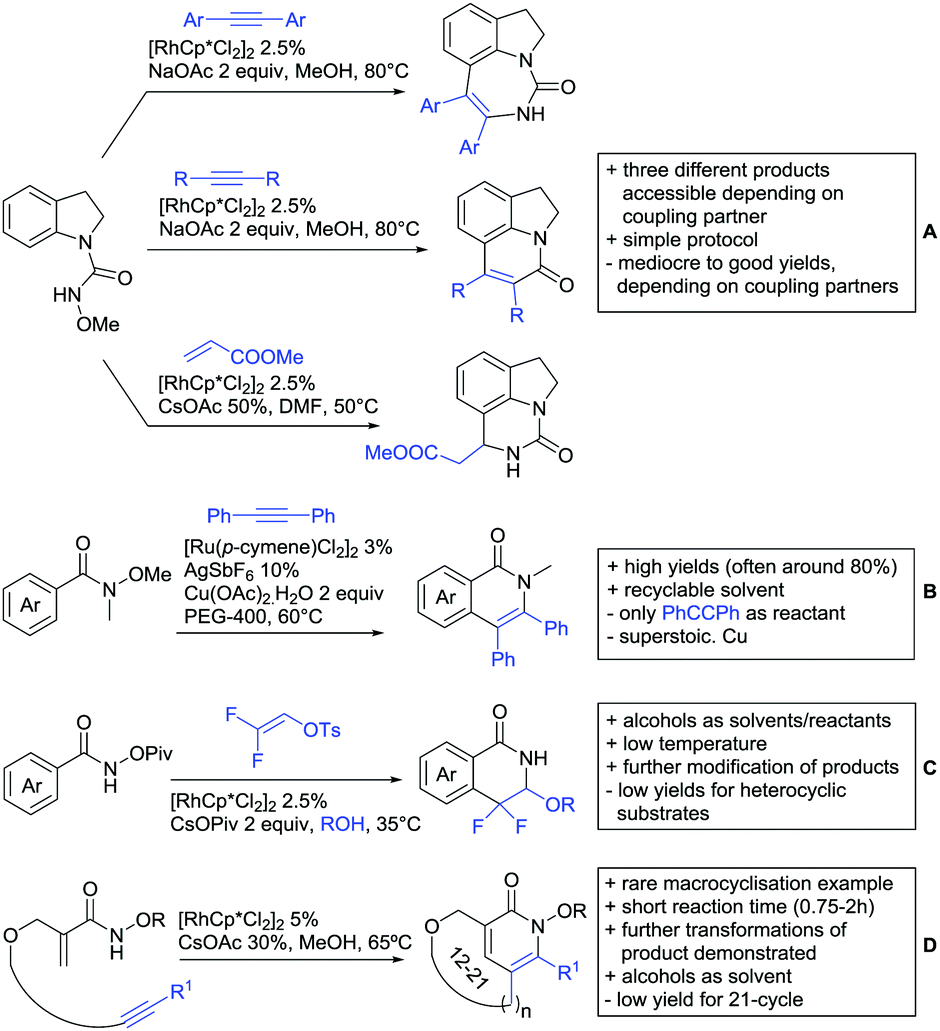 | ||
| Scheme 40 Alkenylation–cyclisation sequences directed by N-alkoxyamide/ureas. | ||
A Ru catalysed alkenylation–cyclisation reaction was reported by Bhanage and co-workers in PEG-400 as recyclable solvent (Scheme 40B).252N-Alkoxy or Weinreb amides were the best substrates and diphenylacetylene was the sole coupling partner and after cyclisation, in which the N–OMe bond is broken, isoquinolinones were the obtained products. Also carboxylic acids were applicable DGs for this reaction sequence giving isocoumarins. If styrenes were the olefin source, no subsequent cyclisation occurred (not shown). For all transformations yields were typically >80%.
Li and Wang investigated the reaction of 2,2-difluorovinyl tosylate with N-acyloxybenzamides under various Rh(III)-catalysed conditions, which gave access to different types of product.253N-(Pivaloyloxy)benzamides led to 4,4-difluoro-3-alkoxy-3,4-dihydroisoquinolin-1(2H)-ones (Scheme 40C). If the pivaloyloxy group was exchanged for a methoxy group, no cyclisation took place but β-fluoride elimination delivered the alkenylated products (not shown). In addition, detailed DFT calculations were carried out in order to elucidate the reaction mechanism.
Finally, Meyer and Cossy reported an N-alkoxycarboxamide-directed macrocyclisation with an intramolecularly accessible alkyne (Scheme 40D).254 Macrocyclisation reactions are important transformations for natural product synthesis and new methods therefore highly welcome. The formation of 12-, 14, and even a 21-membered macrocyclic pyridinone were demonstrated. For such a challenging transformation, yields were actually quite good, getting as high as 82% in one example. The 21-membered ring was still formed in 32% yield.
7.5. Other transformations
The group of Chien-Hong Chen disclosed a Ru-catalysed alkynylation of aryls and heteroaryls using hypervalent iodine–alkyne reagents (Scheme 41A).255 FG tolerance was excellent (including bromine) and yields were typically around 70%. | ||
| Scheme 41 Various transformations directed by N-alkoxy- and N-aryloxy- and Weinreb amide. | ||
Ketenimines were used for the annulation of N-methoxybenzamides under Rh catalysis (Scheme 41B).256 Initially, 3-amino-1,1-diarylmethylisoindolin-3-ones were isolated as products, which can be further converted to 3-(diarylmethylene)isoindolin-1-ones under Lewis acidic conditions via elimination of the amine. Using N-methoxy-1-naphthamides as substrates, this elimination occurred even spontaneously without any additional Lewis acid.
The halogenation of Weinreb amides (besides nitriles and amides, see Sections 10 and 6.1.6) was reported by Kapur (Scheme 41C).230 NXS was used as halide source and 22 examples with yields between 49–81% were reported in this Pd(OAc)2 catalysed protocol. The main focus was on bromination, but also iodination and chlorination was demonstrated. The presence of a nitro group shut down the reaction completely.
A quite peculiar transformation was reported by the group of Glorius (Scheme 41D).257 They used N-aryloxyacetamides as starting materials which were converted to N-(2-hydroxyaryl)acetamides. So overall, an intramolecular amidation took place via migration of the amide group. The reaction required a bicyclic olefin as co-catalyst which enabled the amide migration. DFT-calculations and experimental mechanistic studies allowed them to propose a mechanism for this transformation. Electron withdrawing groups (CF3, COOEt) directly on the aryl ring were not well tolerated, however, if they were separated by an aliphatic spacer yields were again high (typically 70–90%).
8. Carbamates
Only a few examples of carbamate-directed C–H activation reactions have been disclosed in the past three years. Hence, it is avoided to divide this part into further subsections. All reported examples deal with activation of C(sp2)–H bonds, either of aromatic or olefinic systems.Kim and co-workers developed an allylation protocol for vinylic C(sp2)–H bonds using cyclic or acyclic allylic carbonates as the reaction partner (Scheme 42A).258 The reaction was catalysed by [RhCp*Cl2]2 and performed at room temperature but consequently also took 36 h of reaction time. The yields in this transformation were low to good (28–82%) and in cases were Z or E products can be obtained, the selectivity is never better than 5:1. The same group also reported “alkylation” with maleimides under almost identical conditions.259
 | ||
| Scheme 42 Carbamate-directed transformations. | ||
Boc-protected 2,3-dehydropiperidines can be arylated with arylboronic acids in position 2 using [RuCl2(p-cymene)]2 as catalyst (Scheme 42B).260 The method tolerated also heterocyclic boronic acids and delivered good yields, typically >70%. Formyl protection of the dehydropiperidine worked as well.
Halogenation protocols using NXS under Pd catalysis have been reported with a number of different DGs. Also carbamates promote this chemistry as disclosed by Moghaddam and Jafari (Scheme 42C).261 The halogenation is mono-selective and the second ortho position stayed untouched. In fact, if one ortho position carried a substituent already, no reaction occurred. Chlorine, bromine and iodine could be introduced equally well and yields were generally >80%.
Finally, Satoh and Miura disclosed a carbamate directed alkenylation protocol via Rh catalysis. The transformation required 2 equiv. of Cu(OAc)2 as oxidant, and could be carried out in an environmentally benign alcoholic solvent, namely tBuOH (Scheme 42D). It was shown, that after Boc-cleavage and imine formation, a thermal cyclisation to a pyridine ring could be carried out, leading to polyannelated ring systems.262
9. Urea derivatives as DGs
Urea derivatives have not been used as frequently as DGs as compared to amides or esters, but some progress has been made in the last three years. In several cases, urea derivatives are often tested concomitantly with related DGs such as amides or N-alkoxyamides, often in these contributions only for showing broader applicability of a method. In this section, examples will be presented in which urea based DGs make up for a large part of the corresponding contribution and are not only treated as a side note.The group of Novak reported the alkylation of amides and urea derivatives using alkyl sulfonium salts as alkylating agents (Scheme 43A).198 The most interesting application is in methylation reactions, since methods for introducing the CH3 group via C–H activation are actually quite rare. The reaction conditions in this Pd(OAc)2 catalysed reaction were quite mild (50 °C) but 5 equiv. of TFA were required. It has to be mentioned that FG tolerance was not really investigated in this report and yields were between 48–89%. The same group reported also an ortho-trifluoroethylation reaction of N-arylureas, where they basically applied a protocol they had previously used for the same transformation with amide substrates (Scheme 24G) and obtained excellent yields (Scheme 43B).263
 | ||
| Scheme 43 Urea-directed alkylation reactions. | ||
A third alkylation protocol was reported by Nishimura and co-workers.264 Mainly starting materials of the type N,N′-diaryl-N-methylurea were used. Alkylation took place at the N-methyl and not on the arene system (!), which was an unusual finding (Scheme 43C). Actually, the reaction worked with excellent yields, regularly >90%. More than 30 examples were reported which showed excellent FG tolerance. This Ir-catalysed method used terminal olefins as the starting material and required one free NH group of the urea in order to allow coordination of the Ir species. Then, a 5-membered metallacycle was formed with the metal group before alkene insertion and reductive elimination delivered the final products.
The group of Lipshutz reported Pd catalysed arylation of N-aryl-N′,N′-dimethylureas on the aryl systems.265 Depending on the choice of catalyst, they could use either aryl iodides (Scheme 44A, Pd(OAc)2) or arylboronic acids (Scheme 44B, [Pd(MeCN)4](BF4)2) as aryl source. Both arylation protocols gave good to excellent yields, whereas the boronic acid protocol gave higher yields in those examples where the same product was formed via both methods. Additionally, oxidative alkenylation, i.e. Fujiwara–Moritani reaction, was also reported, again with remarkably high yields in most cases. This transformation was particularly interesting since it could be run in water with low amounts of a surfactant (wt 2% PTS) at room temperature as well (Scheme 44C).
 | ||
| Scheme 44 Urea-directed arylation and olefination. | ||
Later, a Rh catalysed variant was reported by Yi and co-workers (Scheme 44D).266 Also in this case N-aryl-N′,N′-dimethylureas were the starting materials to be alkenylated on the aryl systems. However in this case, the alkenylated urea products were not isolated but further converted by ammonolysis and esterification (MeOH is the solvent in this reaction) to the corresponding O-methyl carbamates. Quite good yields (frequently >90%) and substrate scope as well as good E-selectivity was observed.
The group of Jana reported reaction conditions, which gave two different products, just depending on the electronic nature of the applied olefine coupling partner.267 Again the same substrate class as in the previous examples was used in combination with styrene derivatives. In case of styrenes with electron withdrawing or only slightly donating (e.g. CH3) substituents, 2-arylated indoles were the product stemming from ortho alkenylation and subsequent aza-Wacker cyclisation as suggested by the authors (Scheme 45A). In case of 4-methoxystyrene or vinyl-naphthalenes, the corresponding 2-arylindolines were the products (Scheme 45B). The authors proposed that a π-benzyl Pd species was formed which was better stabilised by methoxy-arenes or naphthalenes, hence avoiding β-hydride elimination and allowed the immediate attack of this species with the free NH group.
 | ||
| Scheme 45 Urea-directed alkenylation–cyclisation sequences. | ||
A Pd catalysed alternative indoline synthesis was reported by the group of Han (Scheme 45C).268 In fact, the authors disclosed even an asymmetric variant of this transformation using Pd(OTs)2(MeCN)2 as precatalyst and a chiral sulfoxide-oxazoline (SOX) ligand. 1,3-Butadiene derivatives were the coupling partners of choice. Yields were mainly in the 50% range and the butadiene needed to carry an electron withdrawing group. However, e.r. of up to 95:5 were obtained.
A quite special case is the meta selective borylation of benzamides as reported by Kuninobu and Kanai.269 Of course the question arises why this example is presented in the urea section, and arguments could be found for both cases (and even for putting this example in the amine section). Actually, the amide group does not coordinate the metal, but the two NH groups of a urea derivative, and it is this urea derivative, which has included a bidentate pyridine group which coordinates the Ir catalyst (Scheme 46 bottom). So a secondary weak interaction via hydrogen bonding delivers the metal species via the ligand in proximity of the C–H bond to be broken. A wide variety of substituted benzamides and also carboxylic acid esters, and phosphoric acid esters (and derivatives thereof) have been used as starting materials demonstrating that the oxygen acting as hydrogen bond acceptor did not need to come from an amide. Interestingly, yields (only determined as NMR yields from meta/para mixtures) were either ∼50% or >90% whereas electron withdrawing groups lead to the high yields. The meta selectivity showed quite some variation ranging from 3.3:1 up to >30:1.
 | ||
| Scheme 46 Ligand-assisted meta borylation. | ||
10. Nitriles as DGs
In recent years, the application of nitriles as DGs has been dominated by directing functionalisation of an aromatic system meta to the DG.270–273The nitrile function typically sits at the end of a spacer and points towards the C–H bond to be activated. Due to its linearity, the nitrile function is perfectly suited for this kind of activation mode. Only few ortho directing transformations have been reported. These will be treated here at the beginning of this chapter.
10.1. ortho directing
1,3-Dicyanobenzene has been arylated in position 2 under Pd(OAc)2 catalysis using aryl bromides as the aryl source (Scheme 47A).274 Arylation occurred in position 2 in yields ranging from 45–81%. Interestingly, 2-bromo pyridine did not react whereas 3-bromo- and 4-bromopyridine did. One cyano group could be replaced by another electron withdrawing group such as Cl, F, or NO2. 1,2-Dicyanobenzene reacted as well. Mechanistic studies of a similar arylation protocol were reported by the group of Ren.275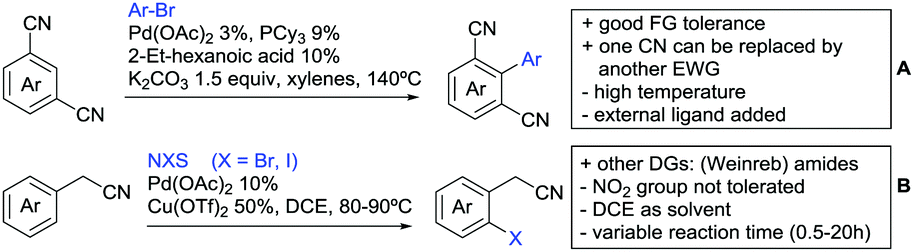 | ||
| Scheme 47 Cyano-directed ortho functionalisation. | ||
Finally, Kapur reported on ortho halogenation using several DGs, including benzyl nitriles (Scheme 47B).230 The reaction conditions were identical with those reported in previous sections (see Sections 6.1.6 and 7.5) and bromination and iodination with NBS and NIS were reported. As compared to the other DGs, namely Weinreb amides and anilides, yields were pretty much the same for comparable substrates.
10.2. meta and para directing
Further examples directed by the cyano function were mainly meta directing. As already mentioned, the cyano group has to be attached to a linker, in order to be able to bring the ligated metal in proximity to the C–H bond in meta position. What all examples have in common is the ortho substituted benzonitrile marked in blue in Fig. 3. The nature of the linker chain in between can vary. Interestingly, the ortho position of the benzonitrile group does not get functionalised and the functionalisation of the more distant ring is favoured! It can be suspected that the metal catalyst gets coordinated by the nitrile nitrogen, which of course disfavours ortho functionalisation. The reported protocols differ in the length of the linker chain. However, most often a 3 or 4 atom linker was used whereas typically one or two of them are heteroatoms. Regarding the transformations reported in recent years, there was a strong focus on alkenylation reactions as can be seen in the subsequent section. | ||
| Fig. 3 General strategy for designing meta-directing moieties. | ||
All examples in Scheme 48 used a four atom linker to connect the benzonitrile group, however, different solutions for this bridge were reported. Shangda Li and co-workers used N-nosylated amides (Scheme 48A),276 the group of Maiti relied on a sulphonic- or carboxylic acid ester (Scheme 48B and C),277 and Gang Li and co-workers applied a carbamate linker (Scheme 48D).278 All three protocols used Pd(OAc)2 as precatalyst and a mono-protected amino acid (MPAA) mainly Ac-Gly-OH as ligand and olefins carrying electron withdrawing groups as coupling partners in HFIP as solvent. The unique features of this solvent have been highlighted recently.279,280 Differences can be found in the choice of oxidant. Also the yields of these three protocols were in a similar range. Li's work had the largest substrate scope regarding the aryl ring,276 whereas Maiti showed that a wide range of electron withdrawing groups is tolerated in his protocol.277 However, the reaction was not exclusively meta selective and also other regioisomers are obtained in ratios between 6:1–20:1. Additionally, Shangda Li and Gang Li showed in their reports also a few examples of meta acetoxylation.276,278
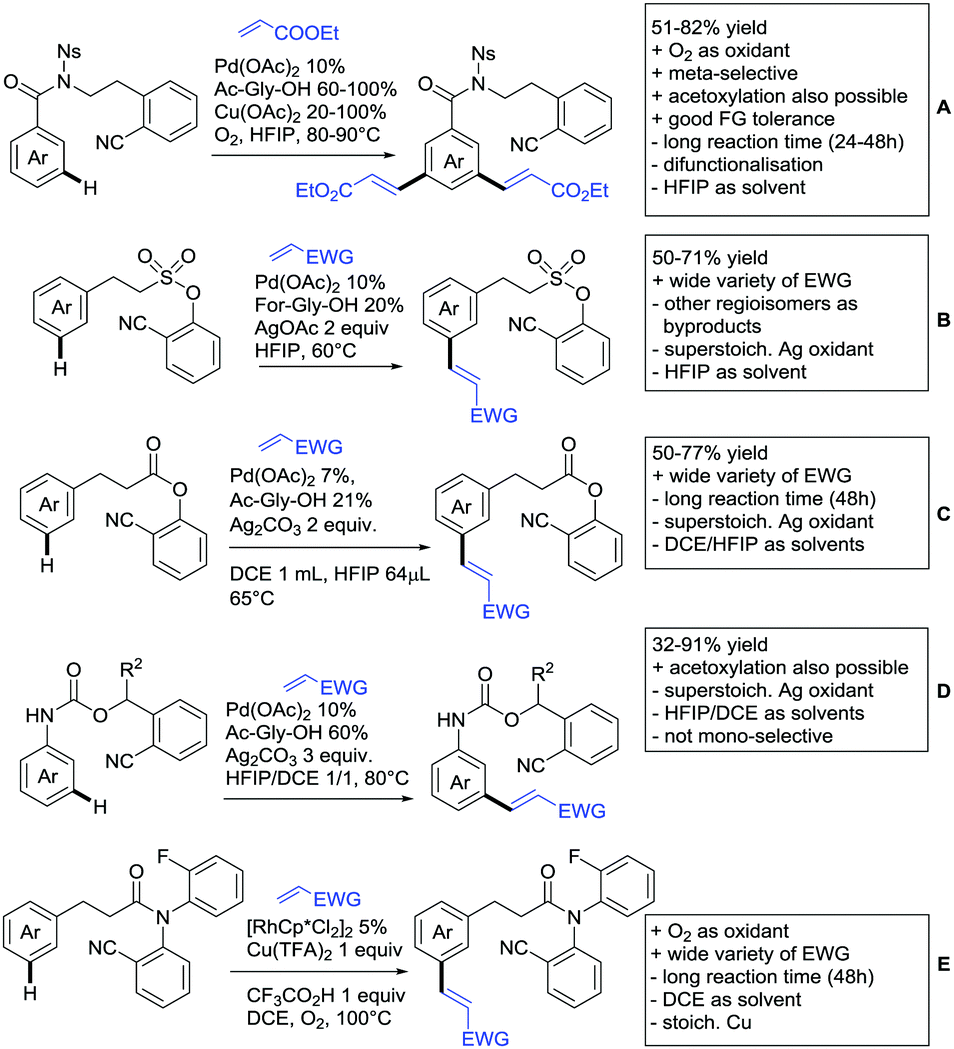 | ||
| Scheme 48 Cyano-directed meta-alkenylation. | ||
Yu and Sun also used an amide linker, however his protocol is Rh catalysed and did not require Ac-Gly-OH or HFIP (Scheme 48E). In his catalytic system, [RhCp*Cl2]2 was the catalyst and Cu(CO2CF3)2 the oxidant with additional amounts of CF3COOH in oxygen atmosphere and DCE as solvent. Also in this protocol, the reaction was not perfectly meta selective and ratios of meta:other isomers between 10:1–20:1 were observed. The yields are in the same range as for the previously discussed examples (49–85%).
The same group also showed that the number of linker atoms could be reduced to three as well and still the protocol using Pd(OAc)2/MPAA in HFIP as solvent could be applied.281 Instead of Ac-Gly-OH, formyl-Gly-OH was the ligand in this case. Again, meta selectivity was 20:1 at best, and no significant difference between the scope and yield of the above mentioned examples was observed (Scheme 49A).
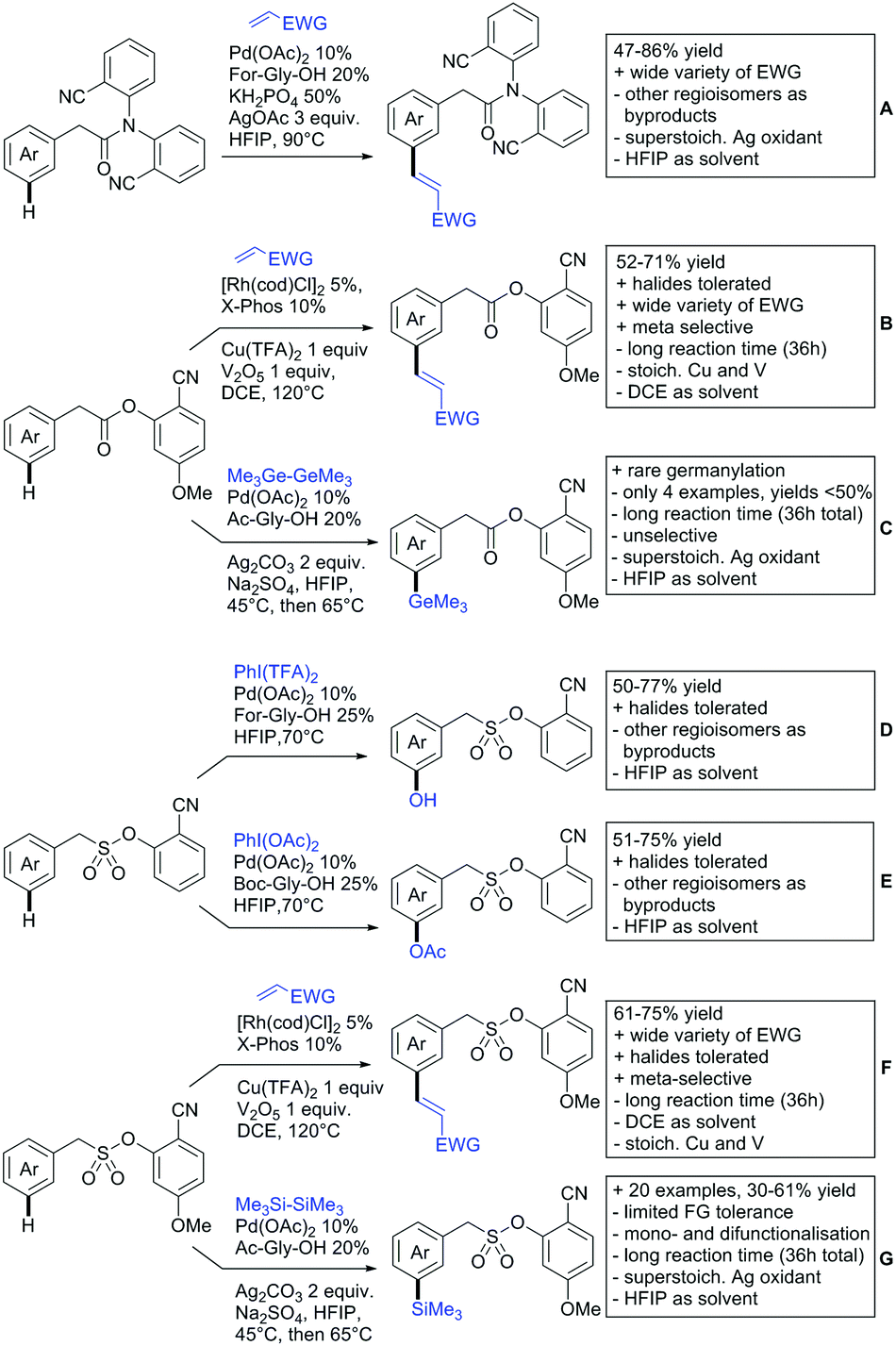 | ||
| Scheme 49 Cyano-directed meta-functionalisation. | ||
The group of Maiti also used “shortened linker” DGs for meta functionalisation of arenes. In a first publication they demonstrated hydroxylation (Scheme 49D) and acetoxylation (Scheme 49E)282 in a second one alkenylation (Scheme 49B and F)283 and finally also silylation (Scheme 49G) and germanylation reactions (Scheme 49C).284 For the hydroxylation, acetoxylation, silylation, and germanylation protocol the linker was sulphonic acid ester based. For the alkenylation, both sulphonic acid ester and carboxylic acid ester linkers were applied. The olefination reactions showed the same features as for the previously discussed 4-atom linker system. Hydroxylation reactions worked in yields between 50–77% and good to excellent meta-selectivity. Acetoxylations worked in the same yield range but with lower selectivity. In silylation and germanylation reactions only between 40–60% yield was obtained.
Again the group of Yu showed alkenylation reactions using a nitrile DG with a 5 atom linker (not shown). The method showed the same features discussed previously: Pd(OAc)2, Ac-Gly-OH as ligand and HFIP as solvent. However, Ac-Gly-OH seemed not to be ideal for starting materials already carrying a substituent on the aromatic ring. Hence, Ac-Val-OH was identified as superior ligand in a second screening campaign. The results regarding meta selectivity were comparable to those in the examples discussed previously. The same was true for the yields and substrate scope. As an add-on in this publication, significant effort has been put into elucidation of the reaction mechanism via experimental and theoretical methods.
The group of Maiti investigated biphenyl-templates as para-DGs in olefination reactions (Scheme 50A and B).285,286 Therein, the directing nitrile moiety was attached to a biphenyl system, which was linked to the arene system to be alkenylated via a silyl ether. It was shown that it did not matter which of the aromatic systems was directly linked to the oxygen. Crucial for success was that the silyl moiety in the linker chain was bulky, since this forced the arene and biphenyl system to face into the same direction and hence enabled the nitrile to bring the metal catalyst in proximity of the para C–H bond. Yields for both types of substrate were in the synthetically useful range of >50%, in a good number of cases around 70%. Besides the para-alkenylated products also other regioisomers were formed favouring the para product in ratios between 5:1 and 20:1. A few examples of acetoxylation were reported as well.285
 | ||
| Scheme 50 Cyano-directed para-(and meta-)alkenylation. | ||
By “inverting” the substrate, meaning that now the nitrile DG is placed on the arene part, meta alkenylation of biphenyls was achieved (Scheme 50C).287
11. Imines as DGs
Imines have been prominently used as DGs in recent years. One large application field was their use as so called “transient” DGs, in which the imine is formed in situ, enables a C–H activation reaction, and then gets cleaved again. Reactions such as hydroacylation, hydroarylation, hydroalkylation, arylation, alkenylation, amination, and halogenation have been realised with this approach. Since this field has been reviewed just now,102,103 these examples are excluded from this chapter and only examples are presented in which the imine is present from the beginning (and eventually cleaved after directing the metal catalyst), or in situ formed but then remains in the final product.11.1. Arylation
Imine-directed arylations have not been the centre point of recent research in the field as can be seen by the small amount of examples. Szostak and co-workers reported a Ru(0)-catalysed protocol which works under neutral conditions (Scheme 51A),288 which have been applied previously for C(sp3)–H arylation of saturated heterocycles.289 Arylboronic acid esters were the aryl source. The protocol gave good to excellent yields (up to 96%) and tolerated the most common FGs. In one example it was shown that the imine could be formed in situ as well, which would then fall into the category of transient DGs.102,103 | ||
| Scheme 51 Imine-directed arylation reactions. | ||
A second example was disclosed by Yoshikai using Co(acac)3 as catalyst and aryl chlorides as aryl source (Scheme 51B).290 Both of these features make the protocol highly attractive, with the drawback of the requirement of 2.5 equiv. of a Grignard species and the resulting limited FG tolerance. Still, the protocol worked well on suitable examples with typical yields around 80% and the DG could be converted to a nitrile function.
11.2. Alkylations and allylations
Alkylation reactions represent a very special case in imine directed C–H functionalisation. Interestingly, non-precious metals play a dominant role in this field and examples for Fe, Co, and Mn catalysis have been reported.55,61,70,72Yoshikai, one of the pioneers in non-precious metal catalysed C–H activation reported on the C2 alkylation of indoles carrying an imine group in position three (Scheme 52A).291 In the same contribution also alkenylation was disclosed, which will be discussed in the subsequent section. As catalyst simple Fe(acac)3 could be applied, however an NHC ligand had to be added. One drawback in this type of chemistry was the necessity for a Grignard species, which naturally limited the FG tolerance. Still, in the presented examples good to excellent yields (up to 93%) were reported. Notably, the reaction worked well on indole but was inefficient on simple arene systems, however, a protocol for this type of substrates was reported previously.292
 | ||
| Scheme 52 Imine-directed alkylation/allylation reactions. | ||
This aforementioned protocol also had the limitation of the requirement for Grignard reagents. In subsequent work, they tried to address this issue and it was found that addition of 50 mol% magnesium served the same purpose (Scheme 52B).293 However, still only examples with FGs not sensitive to Grignard species were included. Noteworthy, with styrenes as the alkyl source the branched products were obtained, whereas with vinyl-trimethylsilane the linear products were formed, potentially due to the steric demand of the SiMe3 group.
The two previous reports always used PMP protected imines as DG. However, the same group also demonstrated that unprotected imines can be used as well if ketimines are used (Scheme 52C).294 In fact, in competition experiments between PMP-protected imines and free imines, it was found that the latter reacted preferentially. In the present example vinyl silanes and terminal aliphatic olefins were the coupling partners always giving the linear products in good to excellent yields (up to 99%). In this contribution, the alkylated imines were also isolated in a number of cases. In case of pivalophenone imine, excellent selectivity for mono-alkylation products was observed due to the steric bulk introduced by the t-butyl group. In a later contribution, the alkylated imines and their further photochemical transformation to nitriles was the focal point (Scheme 52D).290 This time, alkyl bromides were used as the alkyl source and it can be speculated that the alkyl bromides might eliminate to the corresponding olefins in situ.295 Again, the reactions worked typically with high yields, often in the 70–90% range. In both contributions the presence of a Grignard reagent was required again, leading to the same issues with FG tolerance.
Ackermann and co-workers showed N-PMP protected ketamine-directed introduction of a substituted allyl-group stemming from dioxolanone (Scheme 52E).296 The reaction was catalysed by MnBr(CO)5 and gave the final products in decent yields with an E/Z selectivity of at least 7.3/1, often >20/1.
11.3. Alkenylation
C2-Alkenylation of indoles was reported by Yoshikai and co-workers under Fe catalysis (Scheme 53A).291 Reaction conditions were similar to the ones for the alkylation disclosed in the same contribution. It has to be mentioned that E/Z selectivity was not good in 3 out of 8 examples and also yields varied between 18–84%, the lowest one being obtained by a dialkyl alkyne. | ||
| Scheme 53 Imine-directed alkenylation and alkenylation–cyclisation reactions. | ||
As we have seen in previous chapters, alkenylation reactions are often followed by a cyclisation reaction with a heteroatom nucleophile leading to heterocyclic systems. Imine DGs showed the same behaviour, since the lone pair of nitrogen could attack suitable groups which were introduced via the C–H activation step.
Several isoquinoline syntheses have been reported. Li and Wang developed a reaction between aromatic imines and alkynes using Cp*Co(CO)I2 as catalyst (Scheme 53B).297 In most cases the reaction was quite efficient giving yields >70%. Most examples used diaryl alkynes, but also dialkyl or terminal alkynes were applied successfully. Additionally, the transformation showed good FG tolerance. Later, a slight variation of that protocol was reported as well.148
Another base metal catalysed isoquinoline synthesis was reported by Glorius and co-workers, this time using MnBr(CO)5 under redox neutral conditions (Scheme 53C).298 As coupling partners, tertiary propargylic carbonates were applied. The authors proposed that after C–H activation and olefination a carbonate elimination took place to deliver a reactive allene species, which was intramoleculary attacked by the imine to deliver the isoquinoline products. Yields were good to excellent, however not much FG tolerance was explored.
Using diazomalonates as reaction partners, isoquinolin-3-ones were the obtained products, again under Co catalysis (Scheme 53D).299 The reaction performed in TFE and unfortunately, transesterification of the product took place leading to mixtures of compounds in an otherwise high yielding protocol.
Almost simultaneously, Zhu300 and Jiao301 reported two different protocols for the synthesis of quinazolines starting from carboximidates (Scheme 54A and B). Both groups relied on the same catalyst, namely [Cp*RhCl2]2 but used different sources for the nitrogen ending up in position 1 of the quinazoline. Zhu used dioxazolones whereas Jiao preferred benzylic azides in most cases. Comparing both protocols it can be seen that the use of dioxazolones gives a bit better yield, but also with azides >70% are achieved in almost every example. FG tolerance was basically identically good. Overall, Zhu's protocol was operationally simpler as could be seen by comparing the reaction conditions in Scheme 54A and B.
 | ||
| Scheme 54 Imine-directed functionalisation–cyclisation sequences. | ||
A rare case of rhenium catalysis was reported by Wang and colleagues (Scheme 54C).302 In this contribution, an imine starting material was again reacted with an internal alkyne, as seen previously in isoquinoline syntheses. However, under ReBr(CO)8 catalysis a [3+2] cycloaddition took place and the imine nitrogen did not end up in the ring but in position 1 of an indenamine product. The reaction worked well with a number of alkynes (diaryl, dialkyl, and aryl–alkyl). Good to excellent yields were obtained (up to 98%) in this operationally simple protocol.
A second protocol in which the imine nitrogen ended up as amine function in an exocyclic position was disclosed by Zhang and co-workers.303 They reported that carboximidates could be alkenylated in ortho position using acrylates and [Cp*RhCl2]2 as catalyst (Scheme 54D). Until this point, this would be a quite standard transformation, however the consecutive sequence to the indenone products was quite interesting. It was proposed that an initial intramolecular cyclisation led to an 1H-isoindole intermediate which was oxidised by Mn(OAc)2via a single electron transfer forming in presence of O2 a peroxide intermediate (detected by LC-HRMS). This intermediate eventually ring opened and cyclised again, this time to the final product (Scheme 54, right). For a detailed discussion of this pathway we refer to the original paper.
The group of Jun reported a similar cyclisation, however without the subsequent oxidation-ring opening-cylisation sequence and hence delivering 1H-isoindoles as the products (not shown).304
Also iminoquinones have been used as DG, which have certainly different properties than ordinary imines, still this example is presented in this section (Scheme 54E and F).305 The reaction allowed fast access to a tricyclic system involving a number of olefins carrying different electron withdrawing groups. Depending on the catalytic system, different products were obtained. If only Pd(OAc)2 was the catalyst, the saturated products of Scheme 54E were isolated, whereas if additionally 0.5 equiv. Cu(OAc)2 oxidant was present, the products with an exocyclic double bond were formed (Scheme 54F), both in high yields.
In all previously discussed examples the imine group was already present in the starting material. There were also examples reported in which the imine was only formed in situ and then cyclisation took place to pyridium derived heterocycles. Besides the starting materials in Scheme 55, the imine intermediates are shown as well.
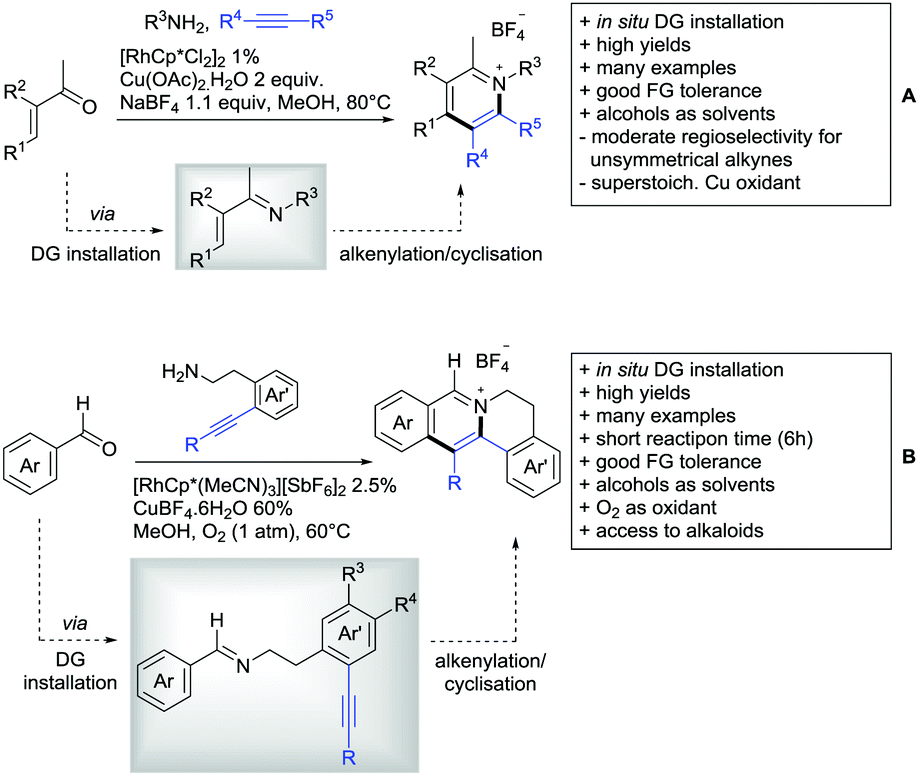 | ||
| Scheme 55 In situ formed imine DGs and subsequent cyclisation. | ||
Both contributions in this field stem from the group of Cheng and run under Rh catalysis.306,307 In the first report, a three component reaction was carried out between a ketone or an aldehyde, an amine, and an internal alkyne. Via the imine intermediate, the desired pyridinium salts were formed quite efficiently with typical yields of >80%.306 The second protocol was only a two component reaction, because the amine carried already an alkyne function for a subsequent intramolecular cyclisation.307 Also for this much more complex ring systems, good to excellent yields were reported surpassing 80% in almost every example. Via this route, facilitated access to protoberine alkaloids was granted.
12. Oximes and oxime derivatives
Oxime containing DGs are quite frequently applied and used in several variants, namely ketoximes, aldoximes and ethers and esters thereof. In 2016 the subject has been comprehensively reviewed,308 but since then again a good number of examples have been reported, which are now covered in this section. In several cases, besides the oximes also N-heterocyclic DGs have been tested in the same transformation. However, this chapter focuses solely on the oxime examples.12.1. Arylation
Arylation reactions are usually amongst the most detailed studied transformation with any DG. For oximes, only a few examples have been reported in the last three years.The group of Xu developed a site-selective biaryl synthesis via dehydrogenative C–H/C–H arylation under ball milling conditions (Scheme 56A).309 Mechanochemistry is an interesting, yet underrepresented method to introduce energy into a reaction system. In a dehydrogenative approach, the question of regioselectivity is a major issue in cases were substituted arenes are the coupling partners. In this protocol, arenes bearing electron donating groups showed excellent para selectivity, whereas arenes with electron withdrawing groups such as acetophenone or methyl benzoate gave good meta selectivity, presumably because the meta position is more reactive towards SEAr reactions. Overall, the transformation showed excellent FG tolerance and gave yields in the 50–80% range.
 | ||
| Scheme 56 O-Methyl oxime-directed arylation reactions. | ||
Pd-Catalysed ortho-C–H arylation of acetophenone oxime ethers with aryl boronic acid esters was reported by Lee and co-workers (Scheme 56B).310 The corresponding biaryl products were obtained in yields between 42–80%. Interestingly, besides the parent phenyl boronic acid ester, only derivatives carrying electron withdrawing substituents (CF3, NO2, COOEt) were applied. Noteworthy, the boronic acid ester can be used as crude material stemming from an Ir catalysed direct borylation.
The β-arylation of oxime ethers using diaryliodonium salts was developed for C(sp3)–H bond activation by Chen and co-workers (Scheme 56C).311 The arylated oxime ethers were not isolated but immediately hydrolysed to the corresponding ketones. The reaction worked for a wide variety of ketoximes and generally gave yields around 80%. Most interestingly, the method was also applied to the arylation of complex natural product derived molecules (steroids).
12.2. Alkylation
Only few alkylation reactions have been reported in the last three years. Li and co-workers reported Rh(III)-catalysed direct alkylation of arenes using commercially available alkyl trifluoroborates (Scheme 57A).312 The method is especially interesting, since it allows simple methylation reactions. Yields were good, between 64–90% but only electron withdrawing FGs were reported amongst the examples (CN, NO2, CF3, COOEt).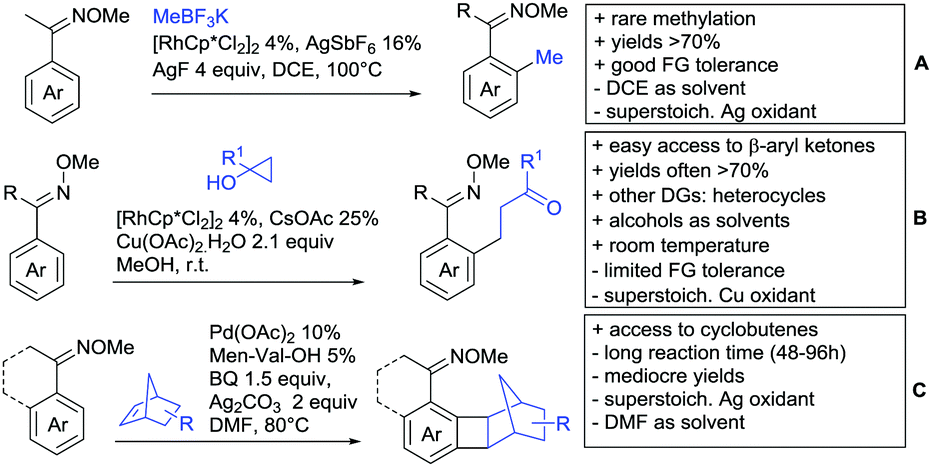 | ||
| Scheme 57 O-Methyl oxime-directed alkylation reactions. | ||
Via ring opening of cyclopropanols β-aryl ketones were made accessible by the group of Li.313 16 examples were presented but not much FG tolerance was explored (Scheme 57B).
An alkylation in the broader sense was the synthesis of benzocyclobutenes via a Pd catalysed reaction with norbornenes (Scheme 57C).314 Norbornene is a quite useful auxiliary in C–H activation chemistry, for example in the Catellani reaction.11,315 However, in the present transformation, the norbornene was not an auxiliary but a reaction partner and remained in the final products.
12.3. Alkenylation
ortho-Directed alkenylation reactions have been exploited extensively with a number of different DGs. As discussed in previous chapters, the alkenylation reaction was often immediately followed by a cyclisation towards a (hetero)cyclic ring system. Also with oxime derived DGs this behaviour was observed.However, the first example to be discussed was dedicated to an alkenylation reaction in which no further cyclisation occurred. The group of Zhao reacted acrylates with benzylic alcohols masked by acetone oxime ethers under Pd catalysis (Scheme 58A).316 The resulting alkenylated products were formed via a six- or seven-membered exo-acetone oxime ether palladacycle, in contrast to the often dominating 5-membered metallacycles. The reason why in this example no cyclisation occurred could be found in the architecture of the substrates. In this specific case, oxime benzyl ethers were the starting materials and olefination took place on the benzyl system. Hence, an intramolecular attack of the oxime nitrogen was disfavoured and the alkenylated products were stable.
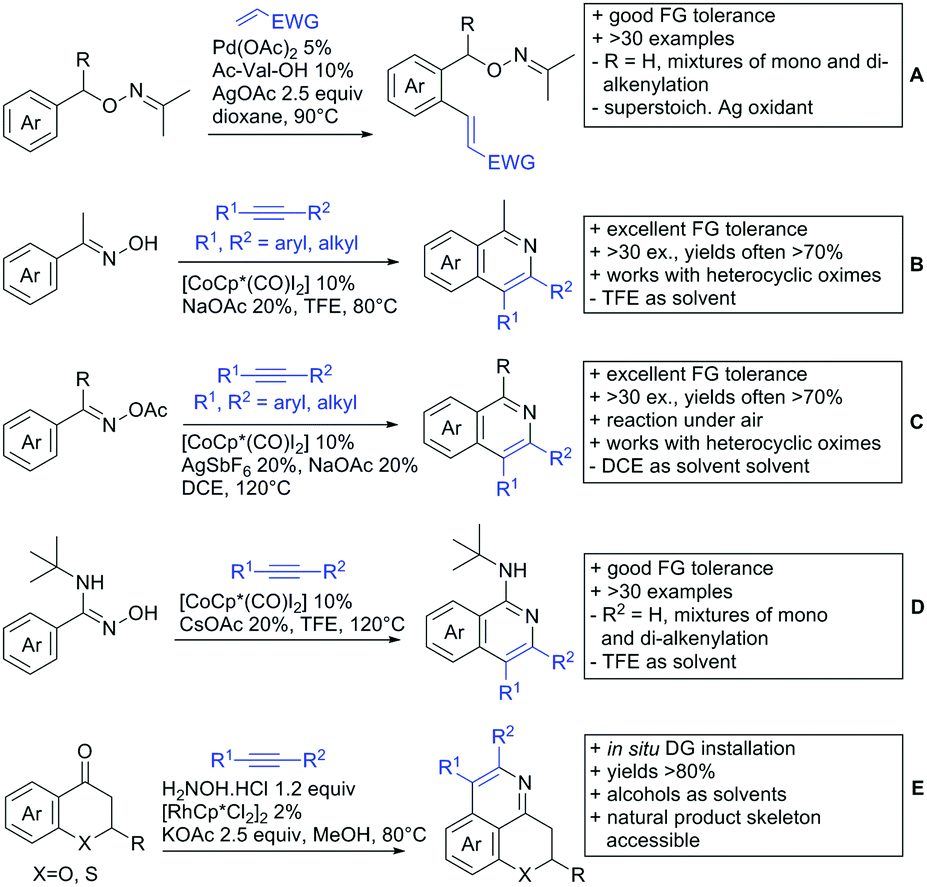 | ||
| Scheme 58 Alkenylation reactions and alkenylation–cyclisation sequences directed by oximes and derivatives. | ||
Several isoquinoline syntheses have been reported in recent years. Basically simultaneously, the groups of Sundararaju317 and Ackermann318 disclosed [Cp*Co(CO)I2]-catalysed alkenylation/cyclisation sequences of oxime derivatives and diaryl alkynes. In the Sundararaju protocol, free OH ketoximes were used as substrates (Scheme 58B), whereas in Ackermann's transformation O-acetylated oximes were used (Scheme 58C). Both moieties act as internal oxidant in the respective transformations. The reaction conditions were almost identical, with the difference that the Ackermann protocol required AgSbF6 as additive whereas in Sundararaju's procedure yields were better in its absence. Both transformations showed excellent FG tolerance and high yields.
An almost identical reaction was reported by Chen with the addition that besides oximes also N′-hydroxybenzimidamides were used as DG which then delivered 1-amino-isoquinolines as products (Scheme 58D).319
Hua and co-workers developed an alkenylation–cyclisation sequence in which the oxime DG is formed in situ via reaction of a carbonyl group with hydroxylamine hydrochloride (Scheme 58E).320 Starting from chroman-4-ones, tricyclic fused pyridines were easily accessible, which allowed rapid access to the skeleton of cassiarins, a family of alkaloids with antimalarial activity. Yields were typically >80% and a hydroxyl and bromine function were tolerated without any loss in efficiency.
12.4. Acylation
Several examples for O-Me ketoxime-directed acylation of π-systems, aromatic and olefinic ones, have been reported.The group of Jiao reported acylations of aryl systems using toluene under aerobic conditions as the acyl source, leading to benzophenone derivatives (Scheme 59A).321 This Pd(OAc)2 catalysed process is quite attractive since toluene is a simple, cheap, and stable starting material. The reaction required N-hydroxyphthalimide (NHPI) as additive which induced in presence of oxygen a radical process leading to the oxidation of toluene to a benzoyl radical, which could then be transferred to the ortho position of the oxime. Yields were in the 42–81% range and the most interesting FG tolerated was bromine, since that leaves a handle for further reactions.
 | ||
| Scheme 59 O-Methyl oxime-directed acylation and acylation/cyclisation reactions. | ||
The same group reported a similar approach, this time starting from benzylic alcohols or benzaldehydes, again with NHPI under aerobic conditions (Scheme 59B).322 This modification showed broader FG tolerance with similar yields and also examples of aliphatic aldehydes were reported, allowing also the synthesis of aryl alkyl ketones.
The acylation of α,β-unsaturated O-methyl ketoximes with benzaldehydes resulted overall in an interesting new method for the synthesis of highly substituted furan derivatives (Scheme 59C).323 Highly functionalised furans were synthesised from α,β unsaturated O-methyl oximes by using Co catalysed aldehyde addition and cyclative capture. Twelve examples were prepared tolerating several important FGs (e.g. Br, COOR) with moderate to good yields (25–84%).
12.5. C–O bond formation
A good amount of oxime-directed C–O bond formations have been disclosed in recent years. Acetoxylation, hydroxylation and alkoxylations have been reported, all using Pd catalysis and various oxidants. | ||
| Scheme 60 C–O bond forming reactions directed by oxime derivatives. | ||
Using substituted benzoic acids, Liao and Ji developed a mono-selective ortho-aroyloxylation protocol of O-benzyl acetoximes, again catalysed by Pd(OAc)2 (Scheme 60B).325 The benzoic acid as well as the benzyl part could carry common FGs and the transformation robustly gave 60–80% yield. As oxidant simple K2S2O8 could be applied. In two examples also aliphatic acids were used, which worked as well as the benzoic acid derivatives.
An alternative to commonly used oxidants in organic chemistry was used by the group of Mei. In their acetoxylation of the β-position of an O-alkyl ketoximes, they relied on electrochemical oxidation (Scheme 60C).326 They described a large set of examples and compared the yields with an alternative oxidant, namely NaNO3/O2. In most examples, the electrochemical oxidation proved to be superior, giving yields >70% in many cases. Eventually this report inspires more research in this direction since replacing typical strong oxidants can be quite useful.
Pd-Catalysed O-methyl oxime-directed ortho C–H hydroxylation of arenes was reported by the group of Jiao under neutral conditions (Scheme 60E).328 Simple oxone was used as oxidant and also the catalyst system was simple (Pd(OAc)2 + PPh3). Overall, more than 40 examples were reported demonstrating the excellent FG tolerance and the high yields (typically 70–90%) in this protocol.
12.6. C–N bond formation
Another interesting transformation developed by Jiao is the amination of oxime ethers with anthranil (Scheme 61A).329 Anthranil is here a quite special reagent, since it gets ring opened and overall delivers a 2-formyl aniline moiety into the ortho position of the substrate. So basically two FGs are introduced at once. The reaction proved to be quite robust with yields between 62–86%. Regarding FGs, besides MeO and halides no further moieties were investigated, but the oxime DG can be replaced also by N-heterocycles. | ||
| Scheme 61 C–N and C–Se bond forming reactions directed by O-methyl oximes. | ||
In recent years, 3-substituted-1,4,2-dioxazol-5-one has established itself as a good source for the amide group in C–H activation chemistry. The group of Li also made use of this reagent in the amidation of CH3 groups in carvone derived oxime methylether (Scheme 61B).330 The most interesting example was the late stage functionalisation of natural products, namely the oxime ether of (−)-santonin which worked with a number of 3-substituted-1,4,2-dioxazol-5-ones in 64–94% yield.
Besides acylations, the group of Jao disclosed also ortho nitration of aromatic oxime ethers using t-butyl nitrite as NO2 source under radical conditions (not shown).321 Acetophenone oximes (23 examples) bearing substituents such as Me, OMe, t-Bu, Br, and F were successfully converted into the corresponding ortho-nitrated products.
12.7. C–Se bond formation
Selenium-containing organic compounds are not very common and hence methods for introducing selenium are relatively rare. However, Wan and Li developed an aromatic oxime ether-directed Rh(III)-catalysed selenylation protocol of arenes (Scheme 61C). As selenylating reagents, electrophilic selenyl chlorides and diselenides were used. The catalytic system was highly efficient under mild conditions and delivered the corresponding products in typically >80% yields.33113. Hydrazine and hydrazone derivatives as DGs
Hydrazines and hydrazones are not frequently applied as DGs in C–H activation chemistry. In most cases, it is not the parent FG which acts as DG, but substituted (or protected) variants thereof.For hydrazines, few examples have been reported in which simple aryl hydrazines were the substrates. In the first one, the group of Cheng developed an alkenylation protocol in which the final products are ortho alkenylated anilines (Scheme 62A).332 The actual DG is a hydrazone, since 1.2 equivalents of ketone, namely 3-pentanone, transform the hydrazine in situ to a hydrazone. Only then ortho C–H insertion by Rh took place and the olefination occurred with styrene derivatives or acrylates in quite good yields (>70%). The catalyst was then also involved in the cleavage of the N–N bond and formation of the final aniline products.
 | ||
| Scheme 62 Hydrazine·HCl- and hydrazine-directed transformations. | ||
A similar olefination reaction has been disclosed by Zhu and co-workers, however in this example there was no ketone added and hence it is the hydrazine itself which exhibited the directing effect (Scheme 62B).333 With acrylates as coupling partners again ortho alkenylated aniline derivatives were the final products and also in this protocol yields were very good, typically >80%. Additionally, the FG tolerance was also significant. Alternatively, alkyl–aryl alkynes could be reacted under the same reaction conditions. In these reactions indoles were obtained as products via a cyclisation after the olefination step. Interestingly, the aryl group always ended up in position 2 and the alkyl group in position 3 of the indole derivative.
Furthermore, Cui and co-workers developed a synthesis of 1-aminoindoles via a Rh-catalysed intramolecular three component annulation (Scheme 62C).334 The aryl hydrazines were used as their HCl salts together with ethyl diazoacetoacetate and ketone using [Cp*RhCl2]2 as catalyst. According to the proposed mechanism, the hydrazine formed with the ketone the corresponding hydrazone and it was actually the sp2 hybridized nitrogen of the hydrazone which directed the Rh to close proximity to the ortho C–H bond. This allowed the transfer of the ethyl acetoacetate to this position and cyclisation with the NH group delivered the final products. The transformation showed broad FG tolerance with typical yields between 50–80%.
Reddy and co-workers reported a direct arylation protocol of 1-arylhydrazine-1,2-dicarboxylates with aryl iodides and Pd(OAc)2 as catalyst (Scheme 63A).335 The arylation worked quite well giving yields between 50–86% and some FG tolerance, most importantly a second iodine. The obtained products can be cyclised under oxidative conditions to the corresponding benzo[c]cinnoline derivatives.
 | ||
| Scheme 63 Miscellaneous transformations directed by hydrazine/hydrazide/hydrazone derivatives. | ||
Basically simultaneously to Cui's indole synthesis, the group of Zhu published another variant of this type of reaction. Starting from 1-aryl-2-Boc-hydrazines and diazodiesters or diazoketoesters 1-aminoindoles were obtained (Scheme 63B).336 The catalytic system was quite similar and a series of examples with high yields and good FG tolerance was reported. When they used N-Boc aromatic hydrazones as substrates, isoquinoline-3-ones were the products (Scheme 63C). Excellent yields were obtained using 0.5 equiv. AcOH as additive and slightly lower yields when LiOAc was used instead. More than 30 examples were reported with most yields in the 90% range.
Another indole synthesis starting from hydrazines, this time N-aryl, N′-Boc hydrazines, was reported by the group of Glorius under [Cp*Co(CO)I2] catalysis (Scheme 63D).337 Mainly symmetric diaryl alkynes were the coupling partners but in some cases also unsymmetrically substituted (e.g. aryl–alkyl) ones were used. The reaction showed good FG tolerance and also good to excellent yields, often >85%.
Using [Cp*RhCl2]2 benzoyl hydrazines have been transformed to isoquinolones via the reaction and subsequent cyclisation with diaryl alkynes (Scheme 63E). The resulting products have large conjugated π-systems and the authors also investigated their photophysical properties.338
The group of Zhu applied the starting material mentioned in Scheme 63C again, this time in a reaction with diaryl alkynes under [Cp*Co(CO)I2] catalysis (Scheme 63F).339Via this method, 3,4-diarylated isoquinolines were obtained as products. Again, this is a well working reaction with good FG tolerance. The group of Sun disclosed a similar reaction under Rh catalysis (not shown).340
So far all examples dealt with C(sp2)–H activation. Dong and co-workers could also realise a C(sp3)–H activation, namely a Pd(OAc)2 catalysed acetoxylation (Scheme 64A).341 As substrates served N2-protected hydrazones derived from 2,6-dimethoxybenzaldehyde and PhI(OAc)2 was the acetyloxy source. As N-protecting group 5 different sulfones were used and acetoxylation occurred in β-position. Quite a range of substrates were subjected to the reaction conditions and the typical yield was in the 50–70% range.
 | ||
| Scheme 64 Hydrazone and dibenzylidenehydrazine-directed transformations. | ||
Dibenzylidenehydrazines can be considered as a hydrazone derivative as well. Two transformations on this type of substrate have been reported since 2015, all under Rh catalysis. Zhu reported the reaction with vinyloxirane leading to unsaturated alcohols (Scheme 64B).342 Interestingly, only mono-functionalisation occurred with 1.2 equiv. vinyloxirane.
The second procedure used diazodiesters as coupling partners and with 2 equivalents, difunctionalisation on both aromatic rings was obtained in good to excellent yields (Scheme 64C).343
14. Bidentate DGs
In this section, recent advances in the astute use of bidentate DGs in C–H bond functionalisation will be depicted. Indeed, from the seminal work of Daugulis on the use of the amide derived from 8-aminoquinoline as a bidentate chelating auxiliary,344 the scientific community put a lot of efforts in the design and use of a large panel of bidentate groups considerably enlarging the current tool-box of C–H bond functionalisation reactions. Consequently, over the years, a myriad of transformations was developed playing with the unique properties of various bidentate DGs and were already abundantly described in the literature.57,108,345–350 Indeed, such bidentate DGs appear today as key tools to achieve efficient transition metal catalysed directed C–H bond activation and were even successfully employed in strategies involving transient DGs, which will not be depicted in this review.103,351–359 In this section, an overview of the major contributions made in the 2015–2017 period will be provided. In particular, this part will focus on the recent advances made in transition metal-catalysed bidentate group-directed functionalisation of both aromatic and aliphatic compounds. At first, transition metal catalysed C–H bond functionalisation involving N,N-bidentate DGs will be discussed. Then, a focus on the promising, albeit still less studied, N,S- and N,O-bidentate DGs will be reported.14.1. N,N-Bidentate DGs
Among all bidentate DGs, the N,N-bidentate ones represent the most commonly used auxiliaries for directed C–H bond activation.Note that in some cases the transition metal-catalysed functionalisation of 8-aminoquinoline itself (rather that the substrate) at the remote C5 position was observed when using these amides as DGs. These cases will not be discussed in this review.360–369 To overcome such an issue, though, a second generation of bidentate DGs was designed and the amides derived from 5-chloro-8-amino-quinoline and 5-methoxy-8-amino-quinoline were investigated (vide infra).
As the directing group often needs to be cleaved after the metal catalysed C–H bond functionalisation event, several strategies were developed leading to the corresponding carboxylic acid, amide or ester. Indeed, classical approaches relied on using basic or acid conditions to cleave the DG.370–378
If difficulties are met, additional steps such as N-methylation379–382 or N-Boc383–387 protection might be helpful in some cases for the deprotection of the DG under basic conditions. More recently, it has been demonstrated that oxidative cleavage388 (ozonolysis followed by hydrolysis) as well as Ni-389,390 and Co-catalysis387 were also good alternatives for instance although so far less commonly used compared to classical acid and basic conditions.
14.1.1.1. C(sp2)–H functionalisation.
14.1.1.1.1. Arylation. The arylation of C(sp2) centres has been extensively studied. In 2015, Babu reported a regiocontrolled arylation of C2-substituted thiophene or furan using aryl iodides as coupling partners.391 Good chemo-selectivity was observed, with tolerance of a bromide substituent for further post-functionalisations (Scheme 65A) and yields typically around 60%.
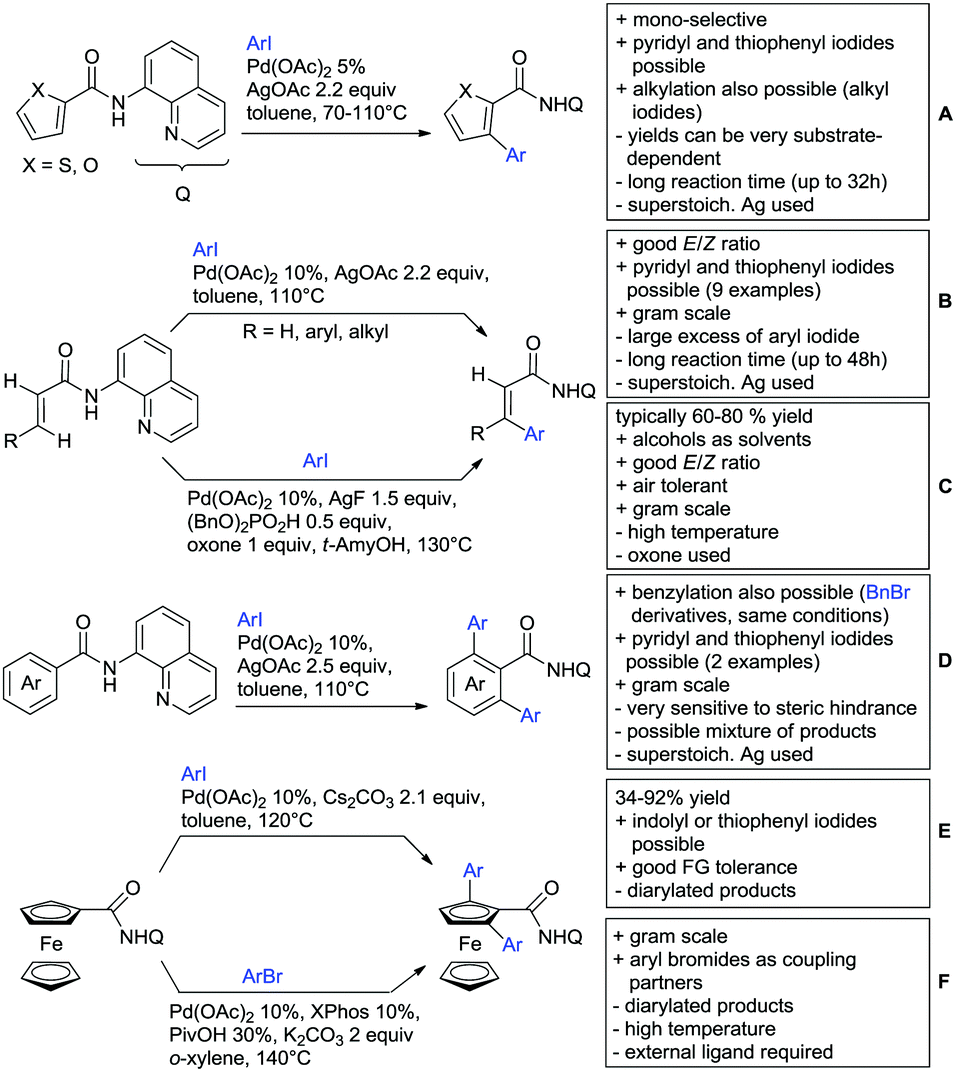 | ||
| Scheme 65 Pd-Catalysed arylation of C(sp2) centres with 8-aminoquinoline-derived amides as DG. | ||
Similar catalytic systems were applied for the arylation of acrylamides towards the synthesis of Z-cinnamamides by Babu372 (Scheme 65B) and Xue/Jiang (Scheme 65C).374 In the former case, a large scope of carboxamides was described with moderate to good yields, up to 88%. In the last case, attempts to extend the reaction towards the alkylation of acrylamides were performed, but with only low conversions.
The Pd-catalysed arylation of arenecarboxamides was reported in 2016 by Babu.392 Di-arylated products were usually obtained (Scheme 65D) with moderate to good yields (25–85%), except when 5-iodoindole and 4′-iodoacetophenone were used, leading either to the exclusive formation of mono-arylated product or a mixture of compounds, respectively. Depending on the substitution patterns on the arenecarboxamides, mono-arylation or a mixture of mono- and di-arylated compounds were also observed (46–63%). The replacement of aryl iodides by a benzyl bromide derivative enabled the mono-benzylation reaction in decent yields.
The arylation reaction was extended in 2016 to N-(quinolin-8-yl)ferrocenecarboxamide by Kumar.393 This Pd-based catalytic system gave access to di-arylated and mono- or di-alkylated ferrocenecarboxamides (Scheme 65E) and demonstrated a good FG tolerance on the aryl iodide partners (alkyl, CF3, OMe, NMe2, Cl, I, acyl, NO2). The efficiency of Ni(OTf)2 was also evaluated, but lower yields were obtained.
A study performed by Yang, Wu an Wu also investigated the arylation of ferrocene derivatives (Scheme 65F).394 The authors evaluated the potential of 8-aminoquinoline and 2-(2-pyridyl)-2-isopropylamine (PIPNH2) derived carboxamide as DGs to promote the transformation. Good yields, around 70–85%, were usually obtained with the 8-aminoquinoline-derived amide.
Other metals than Pd were also investigated. For instance, mechanistic studies were undertaken by Lan on the Ru-catalysed arylation of benzamide.395 Using Co(acac)2 as catalyst, 8-aminoquinoline-derived amides were shown to dimerize.375 An excellent regioselectivity was observed (Scheme 66A), but attempts to achieve the hetero-coupling of two different arenecarboxamides failed to get high selectivity. This reaction tolerated several common FGs (I, Br, NO2, OMe, CF3). Another example was reported in 2016, but required a stoichiometric amount of Co salt.396
 | ||
| Scheme 66 Arylation of 8-aminoquinoline-derived amides as DG. | ||
A mixed DG strategy adopted by Niu and Song allowed the Co-catalysed arylation of benzamides and arylpyridines, offering an access to valuable biaryl systems.397 This process required two parallel activations (SET and CMD pathways) for the formation of a high-valent Co intermediate coordinated to the coupling partners (Scheme 66B).
Three alternatives to the use of aryl halides were reported in the 2015–2017 period: cross coupling with arylboronic acids, a decarboxylative reaction with benzoic acids, and the use of an aryne precursor. Tan and coworkers published the arylation of arenecarboxamides with arylboronic acids mediated by Cu (Scheme 66C).379 A stoichiometric amount of Cu(OAc)2 is required, but a good selectivity as well as a good FG tolerance (F, Cl, Br, OMe, SMe, alkyl, aryl, CF3, CN) were observed. The corresponding products were generally obtained in moderate to good yields (48–79%). A considerable kinetic isotope effect (KIE) was observed with deuterium-labelled compounds, suggesting that C–H bond cleavage was involved in the rate-determining step of the mechanism. A similar Co-based system proved to be efficient in the same transformation and was also suitable for the arylation of acrylamide derivatives.380
Concerning the decarboxylative process, Hirano and Miura reported a Cu-mediated arylation of benzamides using ortho-nitrobenzoic acids.398 This reaction allowed the synthesis of biaryl compounds bearing a nitro group (Scheme 66D) with low to excellent yields (14–85%). By slightly changing the reaction conditions (addition of a base), access to phenanthridinones was possible (not shown).
The use of arynes was reported in 2017 by Zhang, with a Cu-based catalytic system.399o-(Trimethylsilyl)aryl triflates were employed as aryne precursors and the reaction offered access to phenanthridinones by means of an annulation process (Scheme 66E). The use of arynes is not common in C–H arylation processes, and this reaction is therefore a very interesting contribution to the field. A broad array of FGs was well tolerated on the amide partner (OMe, alkyl, CF3, Br, I, F, CN, ester). In order to avoid the extra DG installation step, Wan published in 2015 a multicomponent protocol, where the amide formation and the Pd-catalysed arylation occurred as a cascade process with moderate to excellent yields (47–95%).400 This step-economic approach directly employed acyl chlorides, aryl iodides and 8-aminoquinoline to obtain the 2,6-disubstituted products (Scheme 66F). The presence of a methyl group at the ortho- or meta-position of benzoyl chlorides led to the selective formation of mono-arylated products.
14.1.1.1.2. Alkylation. A selective Pd-catalysed mono-alkylation of 8-aminoquinoline-functionalised benzamides was reported by Chen in 2015 (Scheme 67A).376 Notably, the control of the reaction time and the amount of NaHCO3 added regulated the selectivity of the reaction, favouring either the mono- or the di-alkylated product with good yields with primary alkyl iodides (75–87%) and moderate to good yields with secondary alkyl iodides (31–88%).
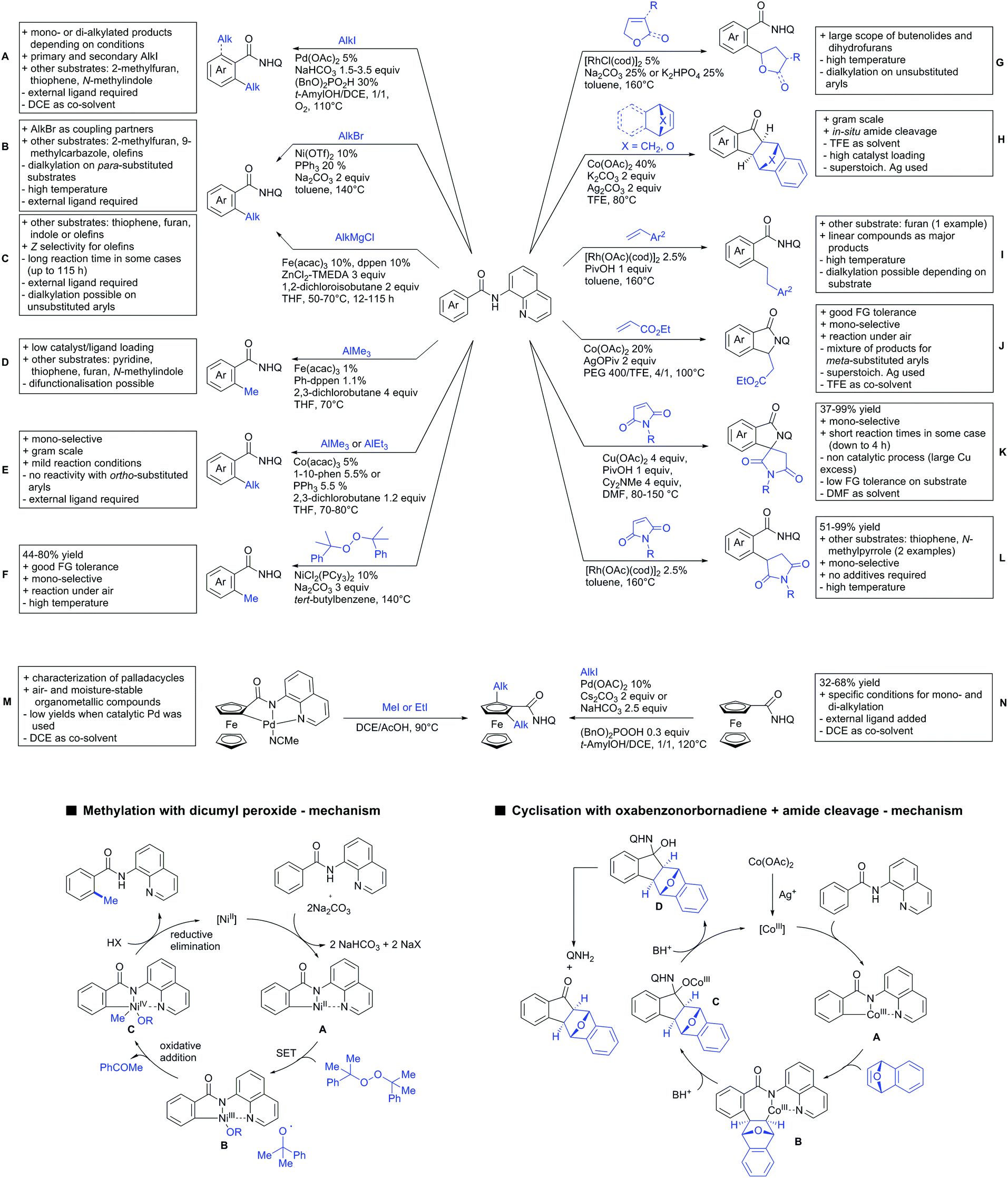 | ||
| Scheme 67 Alkylation of C(sp2) centres using 8-aminoquinoline-derived amides as DG. | ||
A Ni-catalysed system was also reported in the same period by Chatani.401 This protocol employed alkyl bromides with a wide range of substituted arenecarboxamides and acrylamide derivatives (Scheme 67B), resulting in good yields, typically >70%. An example with para-substituted arene carboxamide yielded the di-alkylated product. Benzylation and allylation reactions were also presented.
The Pd-based catalytic system described by Babu for arylation of thiophene and furan was also successfully applied with alkyl iodides (see Scheme 65A).391 An Fe-catalysed alkylation of acrylamides and arenecarboxamides using a broad range of Grignard reagents was established by Ilies and Nakamura in 2015 (Scheme 67C).402 A broad variety of substrates was used with excellent yields, mostly >90%. To avoid the homocoupling reaction of the alkyl partners and a possible β-H elimination, the stabilisation of an organoiron(III) species by the DG and an additional diphosphine ligand was crucial.
Other reagents were also suitable for the directed C–H alkylation. Indeed, the same group developed in 2015 a Fe(acac)3 catalytic system using AlMe3.403 Efficient with both 8-aminoquinoline- and picolinamide-derived DGs, this system was applied to a large scope of (hetero)arenecarboxamides (Scheme 67D) with moderate to excellent yields (48–99%), and was proven to be also active with AlEt3.
The combination of organoaluminium reagents with 2,3-dichlorobutane as oxidant was reported by Xu and Zhang with a Co-based catalytic system.404,405 An excellent mono-selectivity was observed and AlMe3 gave yields between 73–93%. Moderate to good yields (40–87%) were also obtained using AlEt3 as alkyl source (Scheme 67E). Importantly, one example of C(sp3)–H bond methylation was reported with a moderate yield.
Another original reagent, dicumyl peroxide, was reported as a methyl precursor by Chatani in 2016 for the methylation of arenecarboxamides.406 This selective methylation occurred selectively on the less hindered ortho-position (Scheme 67F), and was totally inhibited by the addition of radical scavengers, demonstrating the radical character of the process. The following mechanism was proposed: coordination of the Ni catalyst with the bidentate DG followed by formation of the corresponding complex A. Then, the intermediate Ni(III) B was obtained from peroxide along with an alkoxy radical via a SET process. After decomposition of the alkoxy radical into a methyl radical and acetophenone, the Ni(IV) species (C) was formed, which underwent reductive elimination/protonation steps to access the corresponding product and regenerated the Ni catalyst.
The ortho-alkylation of arenecarboxamides was also possible with butenolides and dihydrofurans using a Rh catalyst,409 generally providing mono-alkylated products (Scheme 67G) in synthetically useful yields (49–87%). Unfortunately, di-alkylation occurred with unsubstituted arenecarboxamides. In 2016, Cheng performed the reaction between the 7-oxabenzonorbornadiene and derivatives with arenecarboxamides bearing an 8-aminoquinoline pattern, with typically excellent yields (>70%, up to 93%).410 A [3+2] amide/alkene annulation took place, with cleavage of the 8-aminoquinoline group (Scheme 67H). A plausible mechanism was proposed: after coordination of the Co catalyst to the amide substrate, the formation of a metallacycle occurred, followed by the insertion of the bicyclic alkene, an intramolecular nucleophilic addition, a protodemetallation step and the elimination of the 8-aminoquinoline.
The Rh-catalysed directed alkylation with styrenes was reported by Chatani providing the corresponding linear products with excellent yields, mostly >80% (Scheme 67I).411 An electron-donating group on the arenecarboxamides accelerated the reaction, and a large panel of styrene derivatives were suitable.
The formation of isoindolin-1-ones by C–H bond alkylation/cyclisation with Co catalysis and electron-deficient alkenes was described by Ackermann in 2015.412 This approach demonstrated good FG tolerance and selectivity, except in the case of two meta-substituted arenecarboxamides (Me or F as substituents), for which a second isomer was obtained (Scheme 67J). A broad range of substituents was tested, including Cl, Br, I, F, OMe or NO2, and led to moderate to good yields (42–85%).
Another oxidative coupling, using maleimides as coupling partners and mediated by Cu(OAc)2 was published by Hirano and Miura.413 This method gave direct access to spirosuccinimide derivatives (Scheme 67K). A key role of Cy2NMe was suggested to promote the C–H bond cleavage and to increase the rate of the reaction. This process required a large excess of Cu, but a similar reaction was reported by Jeganmohan using only a catalytic amount of Co(OAc)2 in 2017.414 Maleimides were also used by Chatani, without intramolecular cyclisation, giving access to the corresponding alkylated products.415 This reaction was performed under Rh catalysis (Scheme 67L) and was also efficient under Ru-based conditions.
The alkylation of a C(sp2)–H bond of Cp rings was also examined in 2015. Methylation and ethylation of two palladacycles derived from ferrocene and a Co sandwich compound were realised (Scheme 67M) with moderate yields (31–66%).407 The Pd-catalysed system developed by Kumar for (hetero)arylation of ferrocenecarboxamides393 was equally efficient for the mono- and di-alkylation with alkyl iodides (Scheme 67N). The key to control the selectivity was the nature of the base: NaHCO3 promoted mono-alkylation while Cs2CO3 yielded di-alkylated products. A catalytic system using Co(acac)2 for alkylation of ferrocene derivatives was also studied, but only one example using an 8-aminoquinoline-derived amide as DG was reported, with 28% yield.408
14.1.1.1.3. Allylation. The introduction of allyl groups was also studied in recent years. In course of their studies regarding the alkylation reaction of amides, Chatani demonstrated that the same catalytic system might be used for the allylation reaction of arenecarboxamides in presence of an allyl bromide.401 For all examples, the presence of a substituent at the ortho- or meta-position allowed a steric control in order to give exclusively the mono-allylated products (Scheme 68A).
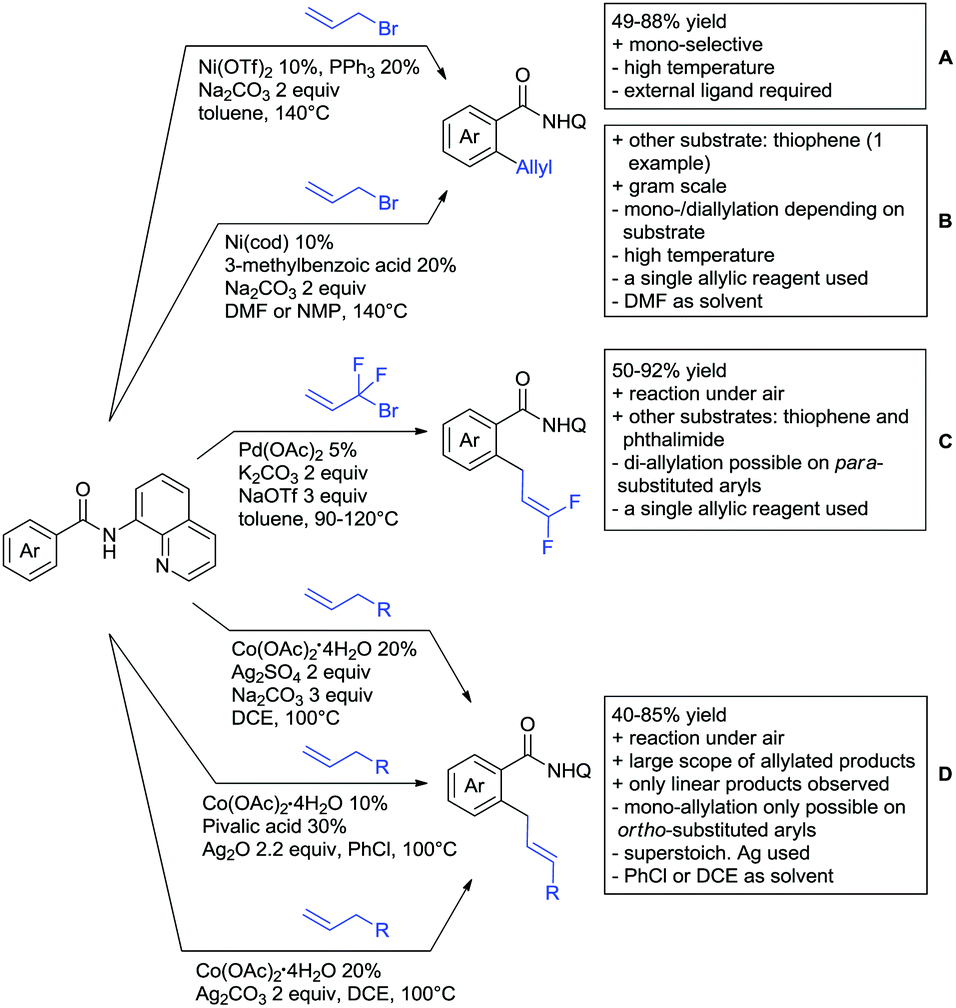 | ||
| Scheme 68 Allylation of C(sp2) centres using 8-aminoquinoline-derived amides as DG. | ||
Sundararaju published in 2015 another Ni-based catalytic system using allyl bromide.416 With ortho- and meta- substitution relative to the DG, mono-allylation occurred with moderated to good yields (40–87%) while di-allylation took place only with para-substituted or unsubstituted arene carboxamides (Scheme 68B). A Pd catalysed system was developed by Liu using 3-bromo-3,3-difluoropropene as coupling partner (Scheme 68C).383 The reaction tolerated a wide range of substituents on the arenecarboxamides (OMe, Br, F, CF3, alkyl, aryl).
Finally, three different groups (Maiti,417 Jeganmohan418 and Chatani419) concomitantly reported a Co-catalysed allylation of arenecarboxamides using aliphatic olefins. In all cases, in order to avoid a mixture of mono- and di-functionalised compounds in this cross-dehydrogenative reaction, the aromatic carboxamide partners were ortho-substituted and only linear allylated products were observed (Scheme 68D).
14.1.1.1.4. Alkenylation, alkynylation and annulation. The formation of isoindolinones was reported by Wei using readily available reagents such as carboxylic acids or anhydrides as coupling partners (Scheme 69A).420 In this process, the presence of an additional pivalic anhydride was required for the reaction with carboxylic acids.
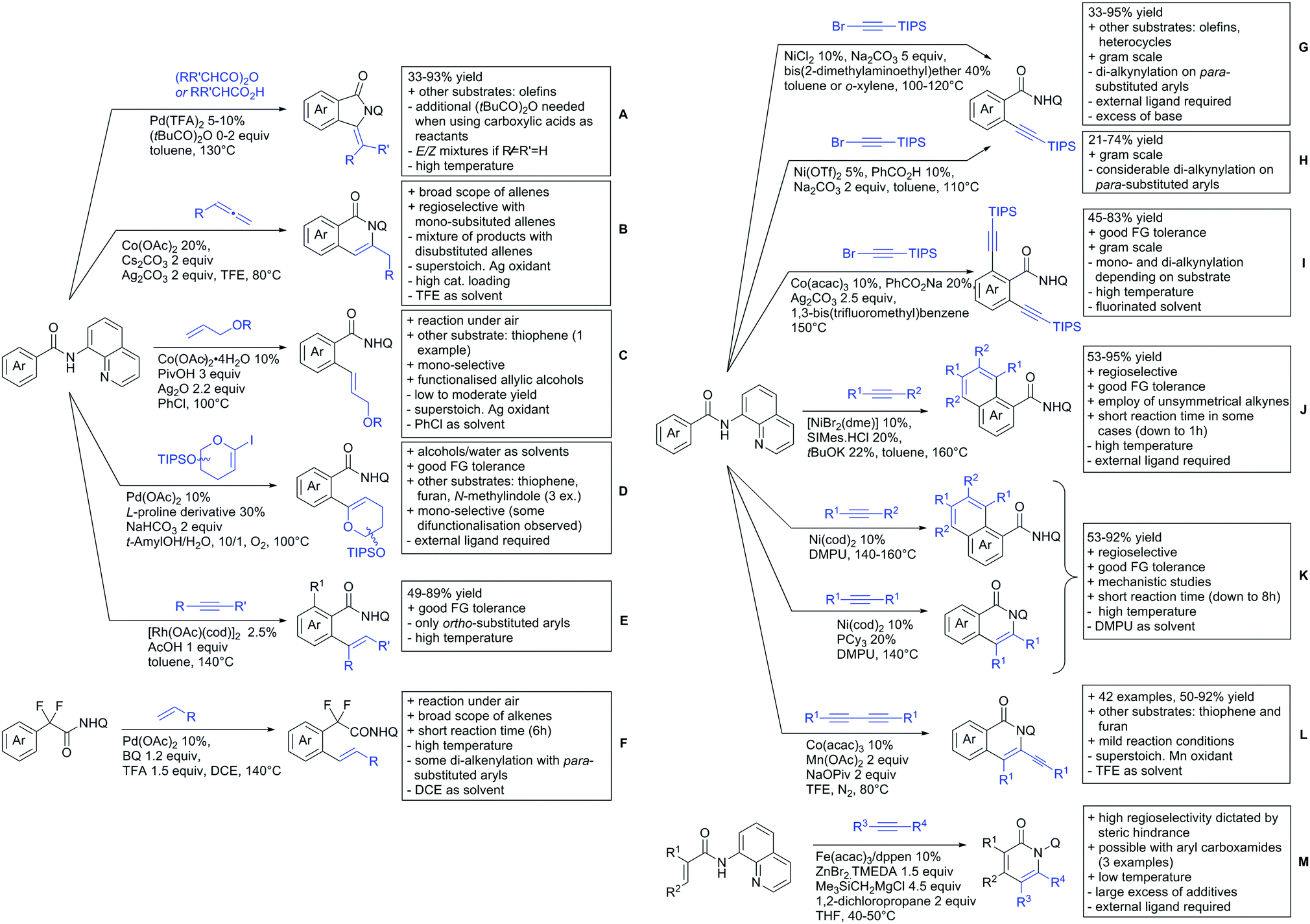 | ||
| Scheme 69 Alkenylation, alkynylation and annulation of C(sp2) centres using 8-aminoquinoline-derived amides as DG. | ||
Another original oxidative coupling using allenes was reported by Cheng in 2017.421 The reaction took place specifically on the terminal carbon of mono-substituted allenes (Scheme 69B) and yields between 30 and 83% obtained. Note that when unsymmetrical or cyclic allenes were used, mixtures of products were obtained.
The Co-based catalytic system developed by Jeganmohan, mentioned in the allylation part,418 was also efficient for alkenylation reactions. The addition was totally selective but only low to moderate yields (23–42%) were obtained. Surprisingly the acetate group was preserved during the process, and the reaction was applied to substituted allylic alcohols (Scheme 69C). It is worth to mention that a mechanistic study regarding the Pd-catalysed regioselective C–H olefination reaction with aliphatic alkenes was reported by Peng, Paton and Maiti.422
A Pd-based catalytic system was used by Wu and Ye to achieve the functionalisation of arenecarboxamides with 1-iodoglycals.423 An additional ligand derived from L-proline was required to obtain good to high yields (51–91%) and mono-selectivity (Scheme 69D), even if traces of diglycosylated compounds were generally detected. Various substituents on the amide partner were tolerated, such as a bromine atom particularly useful for further post-modifications.
In 2017, alkynes were also used by Chatani to perform alkenylation of ortho-substituted arenecarboxamides with alkynes relying on a Rh catalyst (Scheme 69E).424 This alkenylation reaction tolerated a broad array of substituents on the aromatic ring (Me, OMe, Cl, F, Br, Ac, NO2, aryl).
Zhang performed a Pd-catalysed alkenylation of aryldifluoroacetic acid derivatives bearing the 8-aminoquinoline scaffold, via a six membered palladacycle.425 The reaction showed excellent selectivity with only mono-substituted compounds detected with a large scope of styrenes and allylated reagents (Scheme 69F) with moderate to excellent yields (27–99%). However, small amounts of di-alkenylated compounds were observed with para-substituted arenecarboxamides, especially with F, COMe and SO2Me as substituents.
The alkynylation of C(sp2)–H bond was reported multiple times using a Ni-based catalytic system. In 2015, the groups of Li and Balaraman used (triisopropylsilyl)ethynyl bromide as coupling partner for the functionalisation of benzamides.426,427 In the former case, the reaction was also applied to acrylamide derivatives, but the use of a flexible bis(2-dimethylaminoethyl)ether (BDMAE) as ligand was required (Scheme 69G). The system developed by Balaraman required additional benzoic acid (Scheme 69H). In this case, important amounts of di-alkynylated products were observed with para-substituted arenecarboxamides.
In 2016, Balaraman published a Co-catalysed di-alkynylation of arenecarboxamides with an 8-aminoquinoline pattern as DG (Scheme 69I).428 Di-Alkynylated compounds were generally obtained except in few cases, and several common FGs were tolerated (Br, Cl, F, Me, OMe, CF3, NMe2).
Chatani and co-workers published an annulation reaction after a twofold C–H bond activation (Scheme 69J).429 The annulation reaction could be inhibited by addition of a proton source, such as pivalic acid, resulting in alkenylation of the aromatic ring in ortho position, and various FGs on the amide coupling partner were tolerated (alkyl, OR, SMe, CF3). A similar annulation reaction with alkynes was developed by Huang using Ni(cod)2 as catalyst without any additive (Scheme 69K). Otherwise, the use of an electron-rich phosphine ligand favoured a [4+2] annulation yielding isoquinolones.430
Other metals were also studied in the last three years.
In 2017, Nicholls disclosed a Co-catalysed alkenylation of arenecarboxamides with various 1,3-diynes to form alkynylated isoquinolone derivatives (Scheme 69L).431 Note that the remaining alkyne was able to undergo another C–H activation process to synthesize bis-heterocycles.
Similarly, Ilies and Nakamura reported an oxidative annulation of acrylamide derivatives with alkynes catalysed by Fe, leading to the formation of pyridones (Scheme 69M) in typically excellent yields (up to 99%).432 When arenecarboxamides were used, the corresponding isoquinolones were obtained albeit with a narrow scope. Due to the key role of the in situ formed silylmethylzinc reagent, slight modifications in the reaction conditions led to the formation of mono-alkenylated products.
14.1.1.1.5. Isocyanide insertion and reaction with nitroalkanes. The synthesis of 3-iminoisoindolinones was realized with various catalytic systems using amides derived from 8-aminoquinoline as DG. A Cu-catalysed formal [4+1] cycloaddition of benzamides with isonitriles was reported by Hirano and Miura in 2015 (Scheme 70A) giving moderate to good yields (21–85%).433 In 2016, Lei and Hao published a Ni(acac)2 catalysed reaction giving access to the same products (Scheme 70B) with typically good yields (>70%).434
 | ||
| Scheme 70 Reactions with isocyanide and nitromethane using 8-aminoquinoline-derived amides as DG. | ||
In 2016 and 2017, three protocols using a Co catalyst were studied by the groups of Hao,435 Ji437 and Sundararaju.436 In the former case (Scheme 70C), synthetically useful yields were obtained (30–85%) and a mixture of regioisomers was only observed for the arenecarboxamide bearing a fluorine as substituent at the meta position.
The protocol reported by Sundararaju provided the same selectivity (Scheme 70D), good yields (35–96% yield), and a mixture of regioisomers was only observed with meta-trifluoromethylated arenecarboxamides.436 An extension to acrylamide derivatives was possible, albeit the corresponding products were obtained only with poor to moderate yields (27–63%).
The system developed by Ji showed good tolerance to steric hindrance, for instance tolerating a mesitylene group attached to the isocyanide partner (Scheme 70E).437 Generally, aromatic isocyanides are known to be difficult coupling partners. The transformation was efficient with arenecarboxamides (typically >70% yield) and compatible with acrylamide derivatives giving moderate yields in the latter case (45–74%).
The use of nitromethane as a coupling partner was studied by the group of Tan.438 This reaction gave direct access to 3-hydroxyimino-1-isoindolinones (Scheme 70F). Note that 3-methylene-1-isoindolinones were obtained when other nitroalkanes were used.
14.1.1.1.6. Carbonylation. In the period 2015–2017, four protocols for carbonylation of C(sp2) centres avoiding the use of toxic CO gas were disclosed. The first, reported by Ge, employed DMF as a carbonyl surrogate and was applied to a wide range of amides, including arenecarboxamides (Scheme 71A) and aliphatic amides for the direct carbonylation of C(sp3) centres.439 A synergistic Ni/Cu system under oxygen was adopted, the Ni being coordinated to the DG and the Cu generating the electrophilic species from DMF.
 | ||
| Scheme 71 Carbonylation of C(sp2) centres using 8-aminoquinoline-derived amides as DG. | ||
A second Cu-catalysed carbonylation using 2,2′-azobisisobutyronitrile (AIBN) was reported in 2016 by Koley.440 With arenecarboxamides, only carbonylated compounds were observed with good yields (49–96%), but attempts to generalise this method to heteroarenecarboxamides gave only cyanated compounds (Scheme 71B), typically with excellent yields (mostly >90%).
Another carbonylation was reported in 2016 by Zhang, using diisopropyl azodicarboxylate (DIAD) as carbon monoxide surrogate.441 Various arenecarboxamides were used, including compounds substituted with F, Cl, Br, CN, NO2, CF3 or OMe and yields were typically around 70%. Several azodicarboxylates were also efficient for this reaction and three acrylamide derivatives were functionalised (Scheme 71C).
The last example was published by Daugulis in 2017, also using DIAD and a Co-based catalytic system, for the carbonylation of sulfonamide derivatives (Scheme 71D).442 This reaction gave easy access to 2-sulfobenzoic acid imides in moderate to good yields (47–73%).
14.1.1.1.7. Trifluoromethylation. Tan published in 2016 a Cu-mediated protocol for the trifluoromethylation of arenecarboxamides using the Togni reagent II under mild conditions (Scheme 72A). This selective method was robust (water as additive, air tolerant), efficient (36–80% yield) and tolerant to a large scope of substituents (alkyl, F, Cl, CF3, OR, CO2R, aryl).381
 | ||
| Scheme 72 Trifluoromethylation and halogenation of C(sp2) centres using 8-aminoquinoline-derived amides as DG. | ||
14.1.1.1.8. Halogenation. In 2016, Koley reported a Ni catalysed iodination of amides derived from 8-aminoquinoline.443 This process used molecular iodine and gave selectively mono-iodinated compounds (Scheme 72B) in good yield (typically >70%), with a good FG tolerance (alkyl, OR, CF3, Cl, Br, NO2, CN, aryl). Note that one non-aromatic amide was also reactive and was obtained in good yield.
14.1.1.1.9. C–N bond formation. Direct amination of arenecarboxamides was reported by Liu and Zhang using a Ni catalyst.444 Being efficient (48–81% yield) with several secondary amines (Scheme 73A), this reaction provided mono-functionalised products and mechanistic studies suggested the involvement of a NiI/NiIII catalytic cycle.
 | ||
| Scheme 73 Amination and nitration of C(sp2) centres using 8-aminoquinoline-derived amides as DG. | ||
A Cu-catalysed protocol was reported by Daugulis in 2016.445 The transformation was efficient with secondary amines, but the use of 1 equivalent of Cu was required in case of primary amines and aniline derivatives leading to the corresponding products in moderate to good yields (Scheme 73B). Note that slightly modified conditions were used for the amination of electron-poorer derivatives such as sulfonamides, which was also described in this study.
Another Cu-based system was developed by Jana for the amination of arenecarboxamides, using anilines and an excess of hydrated Cu(OAc)2 (Scheme 73C). A broad range of substrates with different substitution patterns was functionalised and the synthetic utility of this transformation was further demonstrated by the gram scale synthesis of a bioactive compound, namely mefenamic acid.446 Worth to mention that an extension to heteroaryls as coupling partners was reported by Punniyamurthy also relying on a Cu-mediated transformation.447
The synthesis of valuable triarylamines was performed in 2017 by Niu and Song by means of a Co-catalysed C–H bond activation strategy in the presence of an excess of aniline (Scheme 73D).448
Studying the properties of sulfoximines, Bolm used dibenzothiophene sulfoximine as an interesting ammonia surrogate in order to access valuable primary amines.449 The surrogate was recycled and mono-amination was observed in all cases (Scheme 73E), but unfortunately only few examples were high yielding (20–86% yield, mostly <60%)
In addition to amination, the nitration reaction was also investigated, and ortho-nitration of (hetero)arenecarboxamides was performed by a Cu mediated C–H bond activation, and tolerated common FGs (aryl, alkyl, F, Cl, Br, CF3, CO2R, OMe).450 By changing the solvent and the base and under higher reaction temperature, the selectivity of the reaction was switched from mono- to di-nitration (Scheme 73F).
14.1.1.1.10. Hydroxylation. A Cu-mediated ortho-hydroxylation of arenecarboxamides was reported by Jana and the chemoselectivity was controlled using pyridine as suggested by the author, probably by stabilizing a monomeric species and facilitating the reductive elimination (Scheme 74A). Indeed, in its absence, a dimerization process took place with some substrates.377 Note that, in comparison with the PIP DG, the amide derived from 8-aminoquinoline was less efficient to promote the related alkoxylation reaction.451
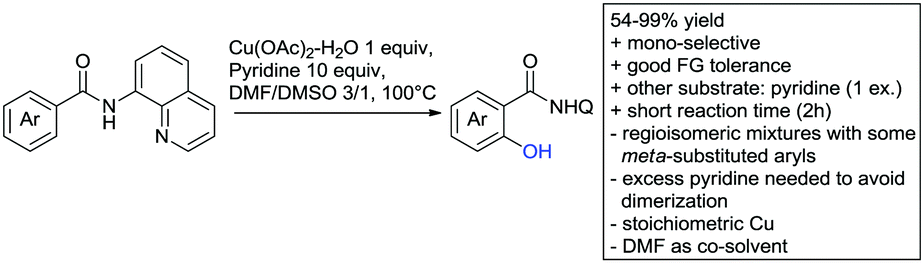 | ||
| Scheme 74 Hydroxylation of C(sp2) centres using 8-aminoquinoline-derived amides as DG. | ||
14.1.1.1.11. Thiolation and trifluoromethylthiolation. In 2015, a direct Ni catalysed ortho-thiolation was reported by Zhang with diaryl disulfides.452 Both, arenecarboxamides and acrylamide derivatives were suitable substrates, giving desired products in good yields (mostly around 80%) and the presence of TBAI and o-nitrobenzoic acid as additives was required (Scheme 75A).
 | ||
| Scheme 75 Thiolation and trifluoromethylthiolation of C(sp2) centres using 8-aminoquinoline-derived amides as DG. | ||
Kambe also studied Ni-catalysed thiolation and sulfonylation reactions with disulfides.453 Although the transformation was efficient (36–96% yield), a mixture of mono- and di-thiolated compounds was obtained with unsubstituted or meta- and para-substituted arenecarboxamides (Scheme 75B). Regarding the sulfonylation reaction, mono-sulfonylation of arenecarboxamides occurred (33–55% yield) along with traces of sulfonylation of the 8-aminoquinoline auxiliary with sulfonyl chloride under Ni-catalysis (Scheme 75C).
Another sulfonylation reaction was reported by Tan in 2015 using a Cu-mediated system and sodium sulfinates, prepared from sulfonyl chlorides.454 The reaction showed good tolerance to steric hindrance, moderate to good efficiency (46–80% yield) and a large variety of sulfinates proved to be good coupling partners (Scheme 75D).
An original reagent for C–S bond formation was used by Messaoudi in 2016, performing the thioglycosylation of arenecarboxamides.384 Such thioglycosides (Scheme 75E) obtained with moderate to good yields (25–88%) could mimic their O-glycosides equivalents, with enhanced stability. However, in case of arenecarboxamides unsubstituted at the ortho-position, mixtures of mono- and di-functionalised compounds were obtained.
In 2017, a diastereoselective trifluoromethylthiolation of acrylamide derivatives was reported by Bouillon and Besset, using PdCl2 as catalyst.455Z-SCF3-containing olefins were selectively synthesised in good yields (typically >70%), using the Munavalli reagent as both the SCF3 source and the oxidant (Scheme 75F). Mechanistic studies suggested that the C–H bond activation event was the rate-determining step.
Finally to avoid the installation of the DG, a one-pot procedure was reported in 2017 by Liu and Wei for the C–S bond formation via C–H bond activation.456 The initial conditions gave access to di-thiolated compounds, and mono-thiolated products were selectively prepared in moderate to good yields (53–81%) by changing the base and using a lower amount of thiol derivatives (Scheme 75G).
14.1.1.1.12. Selenylation. Two protocols for C–Se bond formation by C–H activation were reported in the 2015–2017 period. The first one, published by Baidya in 2016, reported the arylselenylation of arenecarboxamides catalysed by Cu.457 Selective di-functionalisation was observed and a wide range of FGs was tolerated on both the arenecarboxamides or the selenated coupling partners (Scheme 76A). Substitution of the 8-aminoquinoline auxiliary was also well tolerated.
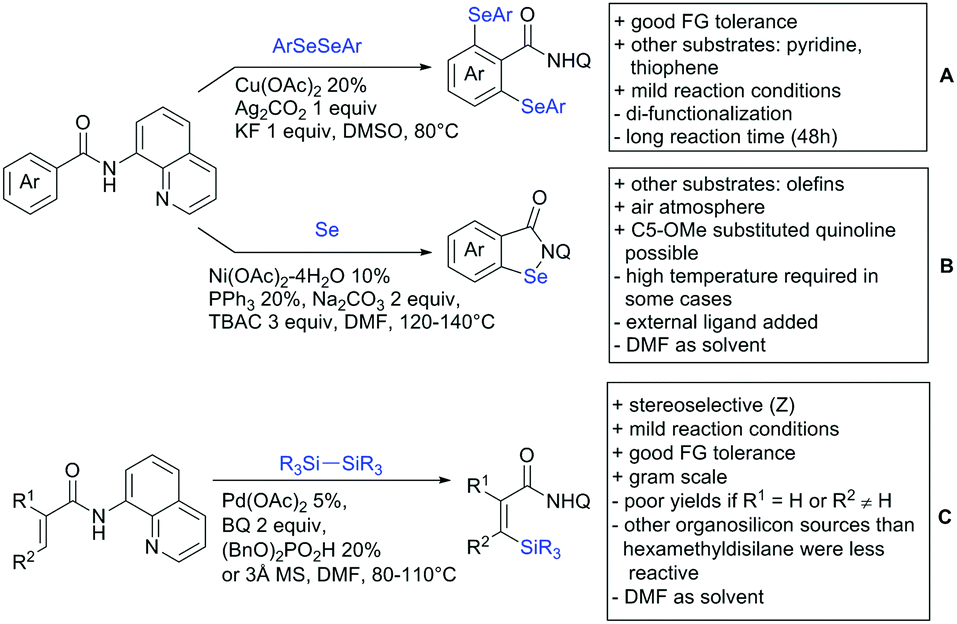 | ||
| Scheme 76 Selenylation and silylation of C(sp2) centres using 8-aminoquinoline-derived amides as DG. | ||
The second example was the Ni-catalysed synthesis of benzoisoselenazolone derivatives under air atmosphere, reported by Nishihara in 2017.458 Mechanistic studies suggested that the C–H bond cleavage is the rate-determining step of the reaction (Scheme 76B).
14.1.1.1.13. Silylation. A regio- and stereo-selective silylation of acrylamide derivatives was reported by Zhang in 2017.459 The reaction had a good FGs tolerance (Scheme 76C) and was efficient with α-substituted acrylamides. In contrast, only moderate yields were obtained with β-substituted derivatives. A stable palladacycle was characterized and explained the observed Z-selectivity.
14.1.1.2. C(sp3)–H functionalisation. The 8-aminoquinoline derived amides have been widely investigated in transition metal catalysed directed functionalisation of C(sp3)–H bonds and represent without any doubt a privileged bidentate DG for series of transformations.
14.1.1.2.1. Arylation. Pd-Catalysed arylation of unactivated aliphatic amides at the β-position constituted the most exemplified transformations with this DG. Since 2015, different coupling partners were used such as (diacetoxyiodo)arenes, the commonly used aryl halides (mainly iodide) and more scarcely polyfluoroarenes.460
Qin reported on Pd(II)-catalysed arylation of unactivated aliphatic amides using (diacetoxyiodo)arenes as arylation reagents via a Ag salt-free process, a real asset compared to other approaches (Scheme 77A).461 Note that in this transformation the presence of Cs2CO3 was crucial. The selective mono-functionalisation of secondary C(sp3)–H bonds was achieved, albeit in case of a primary C(sp3)–H, a mixture of mono- and di-functionalised products was obtained.
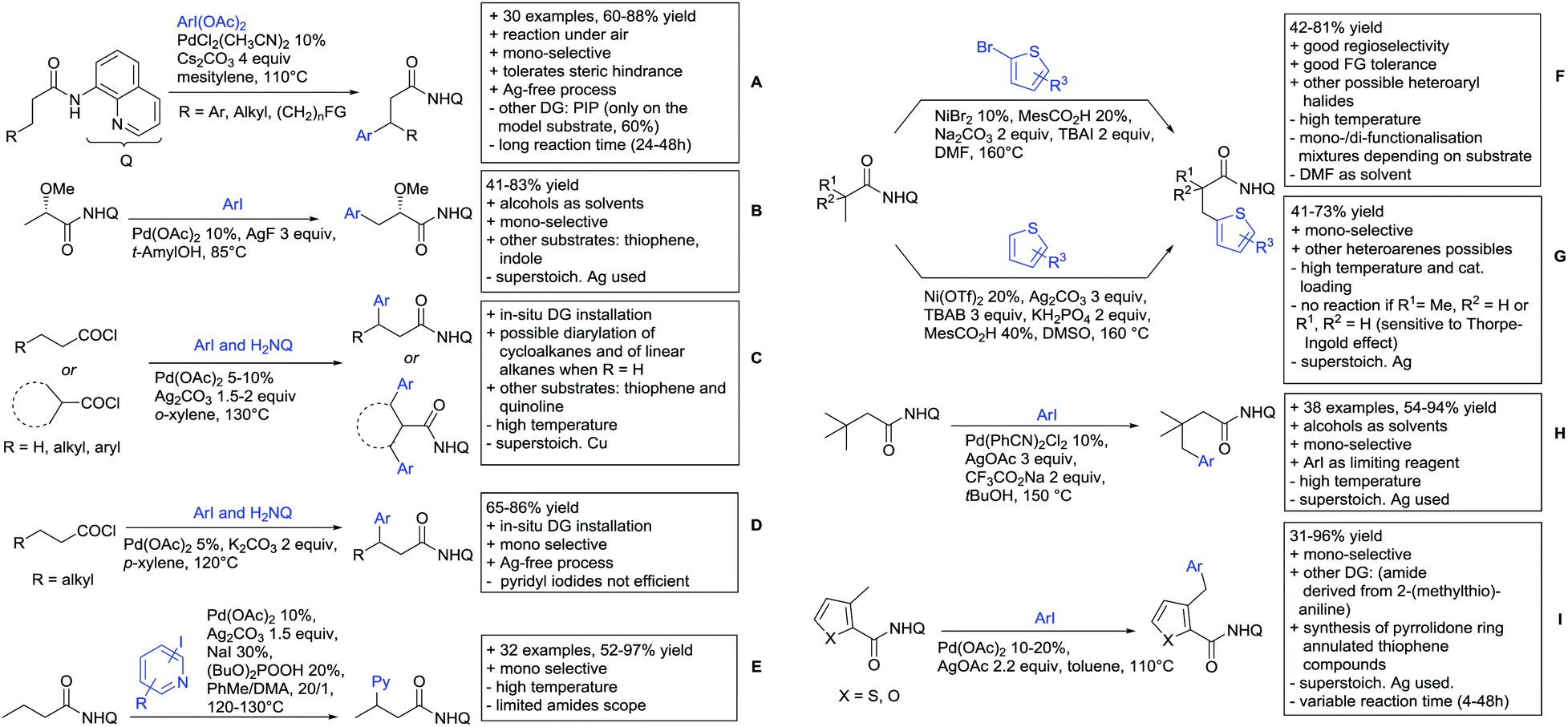 | ||
| Scheme 77 Arylation of C(sp3) centres with 8-aminoquinoline-derived amides as DG. | ||
Using aryl iodides as coupling partners, the Pd(II)-catalysed arylation of O-methyl lactic acids provided an efficient access to chiral β-aryl-α-methoxy acids under mild conditions (Scheme 77B).462 In this approach disclosed by Shi, no diarylation was observed. Note that the reaction was restricted to the functionalisation of primary C(sp3)–H bonds. One of the main limitations was the necessity to install the DG on the substrate. Consequently, as an improved protocol, the in situ generation of the bidentate DG and its use in a Pd-catalysed arylation reaction of unactivated aliphatic amides were independently studied by the groups of Babu463 and Liu.464 They developed a multicomponent reaction of acid chloride, aryl iodide and 8-aminoquinoline with a similar Pd-based catalytic system. In the former study, the monoarylation of linear aliphatic amides (22 examples, 50–97% yield) was reported. Note that in case of cycloalkanes, products resulting from a diarylation reaction (14 examples, 25–83% yield) was obtained (Scheme 77C). It is worth to mention that the Ag salt-free protocol reported by Liu was tolerant to various alkyl chains (Scheme 77D).
Moreover, the Pd-catalysed arylation of α-cyano aliphatic amides was also investigated, offering an access to useful and privileged more complex molecules.465 Extension of these reactions to the use of heteroaryl iodides as coupling partners was possible. Indeed, transition metal-catalysed heteroarylation of aliphatic amides were reported using iodopyridines as studied by Bach (Scheme 77E).466 Thiophene derivatives were also used by Xu and Qiu (Scheme 77F)467 as well as by Xia and Yin (Scheme 77G).468 In the last cases, the heteroarylation did tolerate several FGs on the heteroaromatic ring (Cl, Br, I, OR, C(O)R, CO2R, aryl) and was carried out under Ni catalysis.469,470
This bidentate DG was not restricted to β-functionalisation as the arylation at the γ position was also successfully achieved via Pd-catalysis by Maiti (Scheme 77H)471 and Babu (Scheme 77I),472 although so far restricted to specific substrates with blocked β-position.
Furthermore, cyclic saturated compounds were also functionalised. Pd-Catalysed arylation of symmetric cycloalkanecarboxamides at the β-methylene position was investigated by Chen (Scheme 78A).473 Derivatives of various ring sizes were studied giving efficient access to the corresponding mono-arylated products with high cis diastereoselectivity. Note that the efficiency of the reaction was dependent on the ring size of the substrate; 5-membered rings (24–68% of mono-arylated products and 2–5% of di-arylated products), 4 membered ring (25–32% mono- and 46–64% di-), 6 membered ring (33–54% mono- and 15–17% di-) and 7 membered ring (43% mono- and 5% di-). Surprisingly, the commonly used amide derived from pivalic acid was unreactive.
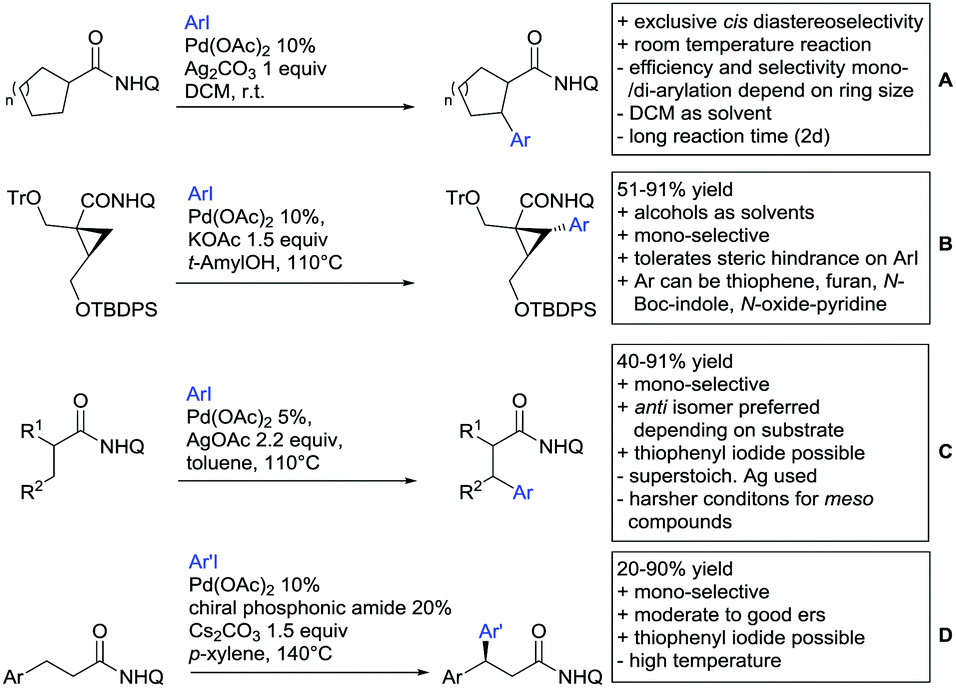 | ||
| Scheme 78 Diastereoselective or enantioselective arylation of C(sp3) centres with 8-aminoquinoline-derived amides as DG. | ||
Hoshiya and Shuto disclosed a new protocol for the synthesis of 1,1,2,3-type chiral tetra-substituted arylcyclopropanes by means of a diastereoselective Pd(II)-catalysed C–H bond functionalisation (Scheme 78B).474 The nature of the alcohol protecting group turned out to be crucial for the efficiency of this difficult reaction by influencing steric and electronic features of the cyclopropane. Noteworthy, the tri-substituted chiral cyclopropane used as starting material was prepared in 5 steps from (R)-epichlorohydrin.
In addition, functionalisation of cyclobutanes was also described in a process involving a Pd(II)-catalysed cascade reaction: (1) synthesis of cyclobutanes from 6-bromohexanamides via a Pd(II)-catalysed intramolecular methylene C–H alkylation followed by a subsequent (2) intermolecular C–H bond functionalisation (i.e. arylation, alkenylation, alkylation, alkynylation, allylation, benzylation and alkoxylation) with various RX coupling partners offering an access to quaternary carbon centered cyclobutanes (not shown).475
Only rare protocols investigated diastereoselective or enantioselective arylation of aliphatic amides. In 2015, Babu reported a Pd-catalysed diastereoselective arylation of aliphatic chains of a prochiral methylene group in β-position, with different substitutions patterns such as derivatives bearing a substituent at the α- or the γ-position and α- and β-disubstituted ones as well as (S)-2-phenylbutanamide for instance (Scheme 78C).371
The enantioselective arylation of secondary C(sp3) centers at the β-position of aliphatic amides was studied by Duan (Scheme 78D).476 The presence of chiral phosphoric acids and amides enabled controlling the enantioselectivity and was beneficial to the reaction rate.
Several groups investigated the arylation of unactivated secondary C(sp3)–H bonds of saturated heterocycles such as tetrahydrofuran- and 1,4-benzodioxanecarboxylic acid (Babu, Scheme 79A),370 proline (Liu and Zhang, Scheme 79B),477 furan-, N-protected piperidine- and THP carboxylic acid derivatives (Bull, Scheme 79C)378 as well as L-pipecolinic acid derivatives (Cao and Wu, Scheme 79D).478 In the last case, the method appeared to be robust with all excellent yields, typically around 80%, and tolerated a variety of FG such as free alcohol, nitrile and halogens. Moreover, using a similar catalytic system (Pd(OAc)2 (5 mol%), AgOAc (2 equiv.), toluene, 110 °C), the β-arylation of dipeptides was achieved offering the possibility to functionalise protected amino acids as well as at the C-terminus of peptides (not shown).479
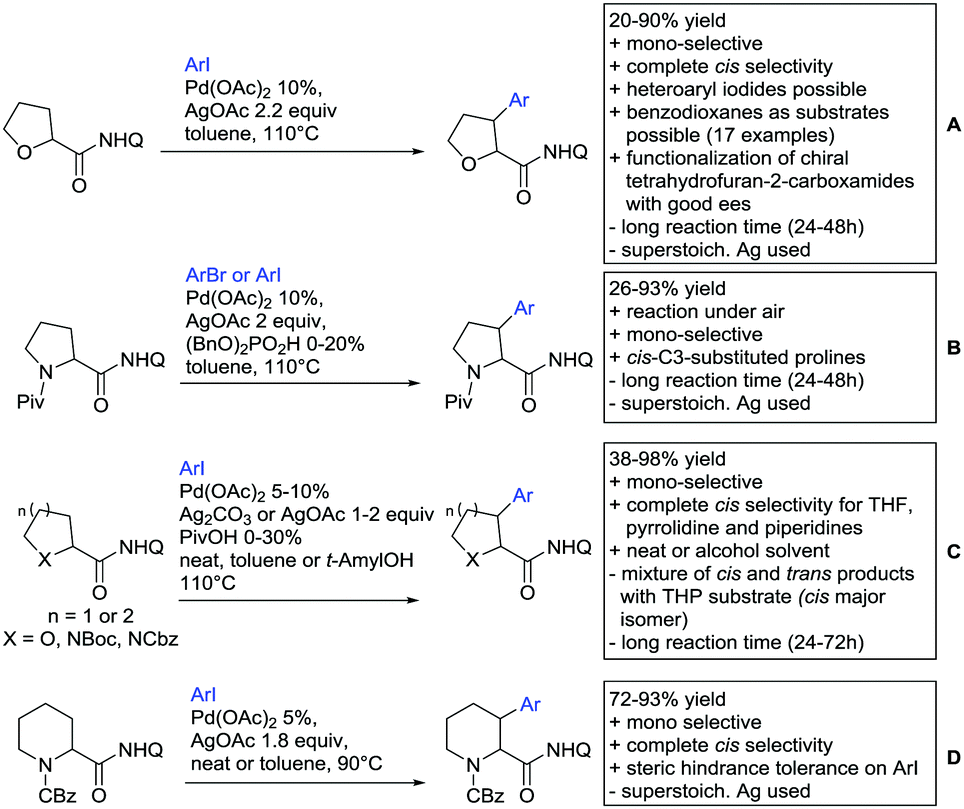 | ||
| Scheme 79 Arylation of C(sp3) centres of saturated heterocycles with 8-aminoquinoline-derived amides as DG. | ||
14.1.1.2.2. Alkylation. The Shi group reported a protocol based on a Co-catalysed intramolecular process leading to bicyclo[n.1.0]-alkanes in decent to good yields (32–77%) (Scheme 80A).480 The main drawbacks were a rather high catalyst loading and a relatively narrow FG tolerance (alkyl, Br, F). The same group developed an intermolecular process based on the Pd-catalysed functionalisation of O-alkylated lactic acid derivatives with alkyl iodides (linear or branched ones) to access chiral α-alkoxy acids in average (50–60%) yields (Scheme 80B).481 Only the mono-alkylated products were synthesised and the synthetic potential of this approach was demonstrated by a good tolerance to several FGs (halogene, amides, ester, protected alcohol and amine among others).
 | ||
| Scheme 80 Alkylation, cyanomethylation and alkoxycarbonylation of C(sp3) centres with 8-aminoquinoline-derived amides as DG. | ||
Alkylation of more rigid skeletons was possible as reported by Uenishi, who successfully developed a Pd-catalysed C–H bond functionalisation of a tertiary carbon of chiral cyclopropane derivatives with alkyl iodides and bromides (Scheme 80C) obtaining low to excellent yields (22–93%).482
In an interesting example, acetonitrile was used for Pd-catalysed cyanomethylation of unactivated aliphatic amides, by primary and secondary C(sp3)–H bond activation. The transformation displayed good β-selectivity, tolerated a broad scope of bulky aliphatic groups, and the methyl C–H bonds were preferentially functionalised in the presence of methylene C–H bonds (Scheme 80D).385
Phosphonoalkyl iodides were also suitable coupling partners as demonstrated by the Pd-catalysed alkylation of aliphatic chains from Yang, which offered access to valuable chiral γ-phosphono-α-amino acids using (iodomethyl)phosphonate (Scheme 80E) with yield typically around 70% and up to 97%.483
14.1.1.2.3. Alkoxycarbonylation. Shi investigated the alkoxycarbonylation reaction of both primary and secondary C(sp3)–H bonds via a Pd(II)/Pd(IV) catalytic cycle (Scheme 80F).484 The use of alkyl chloroformates as carbonyl sources constituted a real asset for this transformation. The quantity of Ag salt was important to prevent the intramolecular C–N bond reductive elimination as alternative reaction pathway, even though its exact role was not elucidated. Functionalisation at the β- and even γ-position was possible, albeit in lower yields in the last case.
14.1.1.2.4. Alkenylation. Transition metal catalysed alkenylation of aliphatic amides were investigated using different coupling partners. Rao reported a procedure based on a Pd-catalysed alkenlyation with vinyl iodides. Access to linear E-olefins from E-iodo alkenes and Z-isomers from Z-olefins was obtained, albeit with moderate E/Z ratios in the latter case (Scheme 81A).485
 | ||
| Scheme 81 Alkenylation and carbonylation of C(sp3) centres with 8-aminoquinoline-derived amides as DG. | ||
Internal alkynes turned out to be suitable coupling partners in the alkenylation of aliphatic chains under Ni-catalysis as demonstrated by the methodologies developed by You (Scheme 81B)486 and Maiti (Scheme 81C).487 Zhang used terminal alkynes (Scheme 81D).308 The system developed by Maiti was efficient with a broad scope of diaryl alkynes, and tolerant to common FGs (OR, CO2R, NO2, CF3). Alternatively, organoborate reagents were employed in an Fe-catalysed transformation, albeit restricted to few examples of alkenylated products (3 cases).488
A Co-catalysed reaction of aliphatic amides with terminal alkynes leading to the corresponding pyrrolidinones was reported by Zhang (Scheme 81E).489 Alkenylation of unactivated aliphatic C(sp3)–H bonds was also investigated by the same group, who developed a Cu(II)-catalysed and Ag(I)-mediated sequential alkynylation with alkynyl carboxylic acids and annulation via a decarboxylative process, offering access to pyrrolidinone derivatives (Scheme 81F).490
Shi also demonstrated that a Ni/Cu co-catalyst system might be used for the oxidative coupling of unactivated aliphatic amides with ethynyltriisopropylsilane as the terminal alkyne (not shown).491
14.1.1.2.5. Carbonylation. Carbonylation of aliphatic chains has attracted a lot of attention as demonstrated by recent contributions. In course of their investigations towards the carbonylation of aromatic amides by means of Ni/Cu catalysis, Ge extended this approach to the functionalisation of aliphatic amides using DMF as carbon source in an oxygen atmosphere (Scheme 81G) giving good yields in the 60–80% range.439 Transformations involving Co catalysts with carbon monoxide reagent were also efficient as depicted by the group of Gaunt (Scheme 81H) with moderate to good yields (49–89%) and a good FG tolerance with for instance presence of a phthalimide group.492 Similar systems were also reported by Sundararaju (Scheme 81I) with decent to good yields (15–87%)493 and by Lei (Scheme 81J) typically with yields around 70%.494
14.1.1.2.6. Halogenation. Only one report using the amide derived from 8-aminoquinoline was depicted for a chlorination reaction between 2015–2017. Besset studied the Pd-catalysed chlorination of unactivated aliphatic chains at room temperature in presence of NCS (Scheme 82A).495 The transformation was regioselective and even though both mono- and di-chlorinated products were obtained, they were easily separable by column chromatography. The reaction also tolerated the presence of common FGs such as halides, and gave typically a total yield between 60 and 70%. Worth to mention is that no chlorinated product at the C5 position of the 8-aminoquinoline was detected.
 | ||
| Scheme 82 Halogenation and C–O bond formation on C(sp3) centres with 8-aminoquinoline-derived amides as DG. | ||
In addition, to the reports from Yu496 and Shi497 who investigated the Pd-catalysed fluorination of amino acid derivatives at the β-position with other DGs, the Pd-catalysed directed fluorination of unactivated aliphatic chains with this bidentate DG was studied by the groups of Xu (Scheme 82B)498 as well as Wu, Zhang and Huang (Scheme 82C).499 In both cases, an electrophilic fluorinated reagent was used and a Pd(II)/Pd(IV) process was proposed to promote the reductive elimination step in this challenging transformation.
14.1.1.2.7. C–O bond formation. The Pd-catalysed C–O bond formation by C(sp3)–H activation was investigated by several groups. A protocol was proposed by Chen and Rao for the β-hydroxylation of aliphatic amides (Scheme 82D) with moderate to good yields (24–72%).500 In this catalytic system, water was used as the hydroxyl group source and the 1-hydroxy-1,2-benziodoxol-3-(1H)-one as oxidant. The reaction is moisture tolerant and in most cases, mono-hydroxylation occurred except for substrates bearing two methyl groups.
Acetoxylation reactions of methylene groups on aliphatic chains and benzylic positions were studied by Shi (Scheme 82E)501 under oxidative conditions (use of oxone/Ac2O) and gave the corresponding anti-β-hydroxy-α-amino acid amides in typically a yield around 60%, and up to 86%. Interestingly, these compounds were prepared in moderate to high diastereoselectivity via a sequential C–H functionalisation (mono-arylation or alkylation followed by an acetoxylation reaction). Note that to get a better understanding on the diastereoselectivity in Pd-catalysed alkoxylation of cyclic alkanecarboxamides, a study was reported by Rao (not shown).502
Stable nitroxyl radicals were used by the group of You to perform aminoxylation of C(sp3)–H bonds (Scheme 82F).503 This Ni-catalysed system gave fair to good yields (35–91%) and was tolerant to various FGs (halides, allyl, OMe, alkyl, aryl). Organosilanes were also suitable coupling partners allowing the aryloxylation/vinyloxylation of unactivated aliphatic chains as described by Zhang (Scheme 82G).149
14.1.1.2.8. C–N bond formation. The transition metal catalysed-C–N bond formation was investigated by several research groups over the 2015–2017 period. The access to β-lactams was achieved by intramolecular processes by Yang and You (Scheme 83A)504 and by Cao and Wu (Scheme 83B).505 In the first case, the use of Cu as catalyst was effective. In the second one, Pd was required. A large array of FGs was tolerated (alkyl, OR, F, Cl, Br, NO2, aryl) and excellent yields were typically obtained, mostly around 80–90%. Isoindolinones were also accessible thought a benzylic C(sp3)–H bond activation, as described by Hirano and Miura (Scheme 83C).506 Intermolecular processes were also disclosed, using simples alkylamines (Scheme 83D and E).507,508 Both systems are Cu-mediated, but in the first case the scope was limited to secondary cyclic alkylamines. However, a broad variety of substituent was tolerated on the amide partner (aryl or bulky aliphatic groups). In the latter case, both cyclic and acyclic dialkylamines were efficient.
 | ||
| Scheme 83 Amination of C(sp3) centres with 8-aminoquinoline-derived amides as DG. | ||
14.1.1.2.9. Sulfenylation, sulfonylation and trifluoromethylsulfenylation. Since 2015 several groups (Shi,509 Zhang,510 Shi511 as well as Qiu, Xu and Yin512) investigated the Ni-catalysed sulfenylation of aliphatic chains using generally disulfides or thiol derivatives (Scheme 84A–D). These protocols were complementary using either aromatic disulfides and even thiols (Scheme 84A and B) or both aromatic and aliphatic ones (Scheme 84C and D). Note that depending on the reaction conditions and the substrate skeleton, mono- and/or di-functionalised products were obtained. This approach was not restricted to the C–S bond formation, since an extension to a C–Se bond construction was also reported (Scheme 84B).
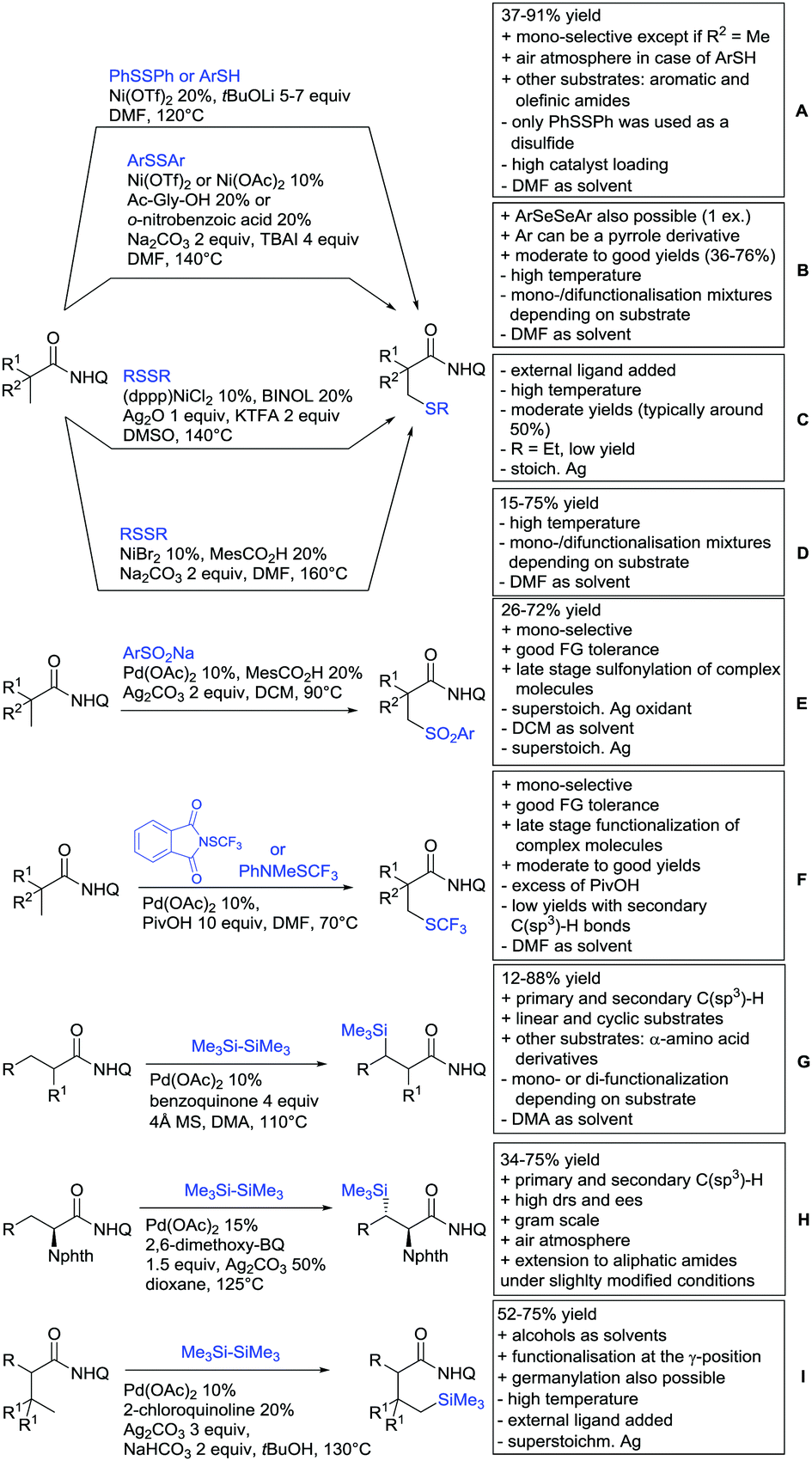 | ||
| Scheme 84 Sulfenylation, sulfonylation, trifluoromethylthiolation and silylation of C(sp3) centres with 8-aminoquinoline-derived amides as DG. | ||
A first example of a Pd-catalysed sulfonylation using sodium sulfinates was studied by Shi (Scheme 84E).513 Efficient with a large scope of sodium sulfinates, this reaction also tolerated the presence of common FGs on the amide partner (alkyl, aryl, F, Cl, CF3, Br). In addition, the Pd-catalysed direct introduction of a SCF3 group by C–H bond activation was developed by Besset, the sole report of the trifluoromethylthiolation of a primary C(sp3)–H bond reported so far (Scheme 84F).373 Decent yields were obtained (up to 53%) and this reaction was compatible with the presence of various common FGs. Besides, a large variety of aliphatic carboxamides were functionalised such as those derived from pivalic acid, α,α-dimethyl hydrocinnamic acid, phenylacetic acid and even more challenging substrates bearing a C–H bond at the α position of the carbonyl group. The synthetic utility of this transformation was further illustrated by the synthesis of analogues of bioactive molecules (ibuprofen and naproxen).
14.1.1.2.10. Silylation. Silylation of unactivated aliphatic chains, α-amino acid derivatives by means of Pd-catalysis at the β-position and even at the γ-one were studied by Zhang (Scheme 84G),514 Shi (Scheme 84H)515 and Maiti (Scheme 84I),516 respectively. Albeit an excess of the coupling partners was generally necessary, with these three protocols functionalisation at the β- and even at the γ-position when the β-position was di-substituted was possible and extension to germanylation was even possible as depicted by Maiti.
14.1.2.1. C(sp2)–H functionalisation. The amide derived from 5-chloro-8-aminoquinoline was employed as bidentate DG in the Ni(II)-catalysed sulfonylation of carboxamides as reported by Chatani (Scheme 85A).517 A mechanism involving a Ni(II)/Ni(IV) catalytic cycle was proposed, albeit the authors cannot rule out a radical pathway. Note that the 5-methoxy-8-quinolinyl derived amides led to the decomposition of the starting materials.
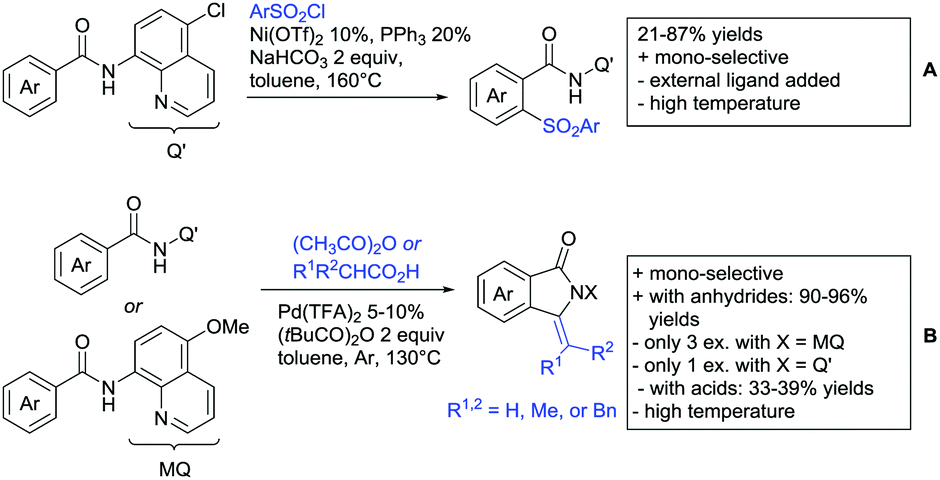 | ||
| Scheme 85 Functionalisation of C(sp2) centres using amides derived from 5-chloro-8-aminoquinoline (Q′NH2) and from 5-methoxy-8-amino-quinoline (MQNH2) as DGs. | ||
In the course of their studies regarding the synthesis of isoindolinones via Pd catalysis, Wei showcased that the amides derived from 5-chloro-8-aminoquinoline and from 5-methoxy-8-aminoquinoline were efficient DGs in Pd-catalysed C–H bond functionalisation of benzamides with anhydrides and carboxylic acids (Scheme 85B).420 Note that only four examples were reported.
14.1.2.2. C(sp3)–H functionalisation. It was reported that for arylation of C(sp3) centres, aryldiazonium salts were suitable coupling partners. Indeed, by combining Pd catalysis and photoredox, Polyzos achieved the arylation of aliphatic amides derived from Q′NH2 with α-quaternary centers in moderate to good yields (21–78%, Scheme 86A).518 The transformation was selective towards primary C(sp3) centres and even a benzylic methylene group remained unaffected.
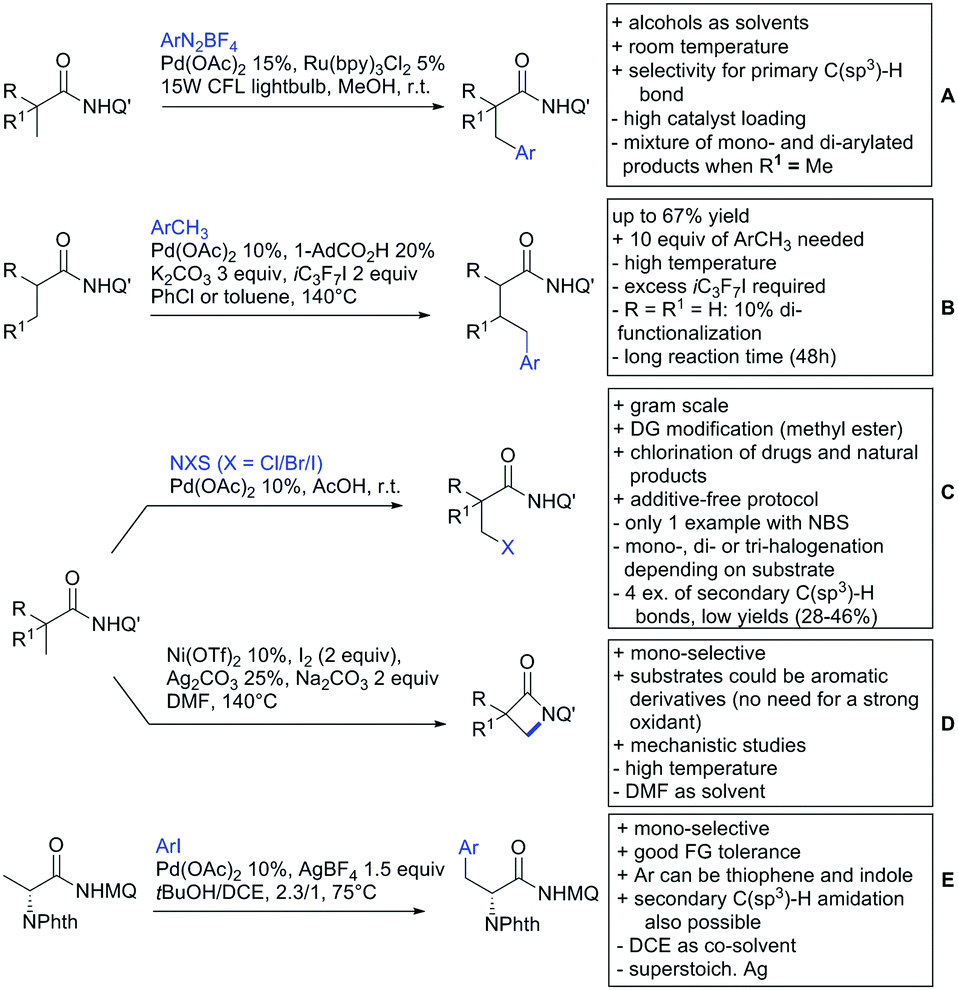 | ||
| Scheme 86 Functionalisation of C(sp3) centres using amides derived from 5-chloro-8-aminoquinoline (Q′NH2) and from 5-methoxy-8-amino-quinoline (MQNH2) as DGs. | ||
The Pd(II)-catalysed benzylation of aliphatic amides via a cross dehydrogenative coupling was disclosed by the group of Chatani (Scheme 86B).519 The transformation was selective for the β-position leading to the corresponding products in 40–75% yields and a real asset of this reaction was the use of toluene derivatives as reagent (10 equiv.) and not as a solvent. The use of this bidentate DG was not restricted to the C–C bond formation as the β-halogenation of unactivated aliphatic chains was also reported by Rao520 and Chatani.521 Rao developed a protocol enabling the halogenation of C(sp3) centres at room temperature via Pd catalysis (Scheme 86C). This bidentate DG was crucial for the success of the transformation since when the amide derived from 8-aminoquinoline was used, introduction of the chlorine atom at the C5 position was observed. Key features need to be highlighted such as (1) good FG tolerance (NO2, Br, F, acetyl, CF3), (2) the functionalisation of substrates bearing secondary, tertiary and quaternary centers at the α-position of the carbonyl groups (28–85% yields) and (3) the possibility to convert the C–X bond into various C–FG bonds.520 Note that depending on the skeleton of the amides, mono- or poly-halogenation occurred. Importantly, the amide derived from Q′NH2 was easily cleavable under acidic conditions (BF3·Et2O or concentrated H2SO4, 100 °C).520
Chatani showed that a Ni catalyst was suitable for the iodination reaction of aromatic and aliphatic amides derived from Q′NH2 with simple molecular iodine (I2), leading to the corresponding iodoarenes (44–83%) (not shown) or β-lactams (27–91%), respectively (Scheme 86D).521 When aromatic amides were used as substrates, mono- and di-iodinated products were obtained depending on the substitution patterns of the starting material (not shown), albeit in case of aliphatic chains, the corresponding iodinated products were not isolated affording a direct access to β-lactams.
Amides derived from 5-methoxy-8-aminoquinoline (MQNH2) were used in few reports involving Pd505 or Cu504,506 catalysed C–N bond formation and Pd-catalysed C–Cl bond formation.495 Indeed, although the methodologies were mainly developed for the amide derived from 8-aminoquinoline (QNH2) as DG, the good reactivity and the easy cleavage of the MQ-based DG in the presence of CAN495,504,505,521–523 or a demethylation/oxidation sequence506 towards the corresponding amides make this DG particularly attractive. Worth is to mention that only a small number of examples with MQ-based DG was depicted in these reports (1 to 3 max), hence only showcasing the potential of this DG. In addition, it was successfully applied in the C–C bond formation. Indeed, recently, Shi reported the Pd-catalysed β-arylation of alanine derivative (15 examples, 68–93% yields, Scheme 86E) and the subsequent amidation via an intramolecular process leading to the corresponding highly valuable α-amino-β-lactams.522 It is important to highlight that the cleavage of the DG was achieved for several products (6) in moderate to high yields (23–83%).
 | ||
| Scheme 87 Functionalisation of C(sp2) centres using sulfonamides featuring 8-aminoquinoline and amides derived from phenylenediamine as DGs. | ||
Then, Sundararaju reported a similar study involving Co-catalysis (45–90% yield) (Scheme 87B).525 Finally, a slightly different catalytic process was developed by the group of Yang (Scheme 87C).526 In that case, no Mn salt was required as NaClO3 was used as an alternative oxidant and PivOH played a key role. Aromatic and aliphatic terminal alkynes were suitable coupling partners as well as internal alkynes. When unsymmetrical aryl alkyl alkynes were used a mixture of regioisomers was obtained, the aryl group being at close proximity of the nitrogen atom as major isomer.
Very recently, a new N,N-bidentate DG based on a 2-aminophenyl moiety was designed by Watkins (Scheme 87D).527 It was successfully employed in a Pd-catalysed arylation of benzamide derivatives (>30 examples, up to 84% yield). Mono-arylated compounds were obtained and (hetero)aromatic iodides such as benzophenone, pyrimidines, and pyridine were suitable coupling partners. With this bidentate DG, the typical use of a Ag salt was no longer necessary as it could be replaced with Mn(OAc)2. The DG was efficiently removed under basic conditions (NaOH/EtOH under reflux).
14.1.4.1. C(sp2)–H functionalisation.
14.1.4.1.1. Arylation. Since 2015, several groups were interested in arylation and heteroarylation of carboxamides. Two reports from Shi focused on Ni catalysed arylation of (hetero)arene carboxamides using aryl boron reagents539 or arylsilanes530 as alternatives to the more classically used Ar2I+X− and ArX coupling partners. In the former case, a Ni(II)/Ni(III) catalytic pathway was proposed and the reaction proved to be efficient (40–85%), tolerant against several FGs (Cl, CF3, OMe, F) and steric hindrance (Scheme 88A).539 In the latter case, the use of non-toxic and safe to handle arylsilanes are key advantages and the transformation proceeded via a fluoride-promoted transmetallation in moderate to good yields (26–92% yields, Scheme 88B). The PIP based DG was removed after a nitrosylation/hydrolysis sequence affording the corresponding acid.530
 | ||
| Scheme 88 Transition metal catalysed functionalisation of C(sp2) centres using the amide derived from 2-(2-pyridyl)-2-isopropylamine as DG. | ||
A complementary approach relied on a Cu/Ag-mediated arylation of (hetero)arenecarboxamides using 2-thiophenecarboxylic acids as coupling partners (Scheme 88C).531 In this reaction, a protodecarboxylation/dehydrogenative coupling process was involved. The same authors reported that thiophenes were also suitable coupling partners. Indeed, they developed a Cu-catalysed oxidative dehydrogenative arylation of a panel of thiophene compounds and the reaction turned out to be tolerant to halogen and carbonyl groups for instance (Scheme 88D).540 To further demonstrate the synthetic utility of this bidentate DG, a Pd-catalysed PIP-directed arylation of C(sp3)–H bonds was successfully developed by Baudoin, offering an access to the aeruginosin family of marine natural products (not shown).541
Arylation reactions with this DG were not restricted to het(arenes) as ferrocenecarboxamides were functionalised with aryl iodides via Pd catalysis (30–81% yield) as developed by Yang, Wu and Wu (Scheme 88E).394 Note that the amide derived from 8-aminoquinoline was also a suitable DG in this transformation (see Section 14.1.1).
14.1.4.1.2. Alkylation. The alkylation of aromatic carboxamides bearing PIP as DG were reported by Lu and Li (Scheme 88F and G). Indeed, they developed two Co catalysed protocols. In one case, the dicumyl peroxide (DCP) was employed playing the role of both source of the methyl group and hydrogen acceptor via a radical process.542 In the second case, an oxidative cross dehydrogenative coupling with various alkylated derivatives (alkanes, (thio)ethers and toluene derivatives) was reported (47 examples, up to 85% yield).543
14.1.4.1.3. Alkynylation. The group of Shi investigated the alkynylation of aromatic and heteroaromatic amides. At first, a protocol using a Cu catalyst and terminal alkynes was developed, a clear advantage compared to the existing approaches (Scheme 89A).532 Then, they studied a modified process involving a low Ni catalyst loading with bromoalkynes as coupling partners (Scheme 89B).533 The efficient reaction turned out to be chemoselective towards the synthesis of mono- or di-alkynylated products depending on the amount of alkynes used, in 62–98% yield and 52–99% yield, respectively.
 | ||
| Scheme 89 Alkynylation of C(sp2) centres using the amide derived from 2-(2-pyridyl)-2-isopropylamine as DG. | ||
Additionally, a Ni(II)-catalysed dehydrogenative alkynylation using O2 as the sole oxidant was developed,544 a clear advantage compared to their previous report,532 in which a superstoichiometric amount of a Ag salt was necessary (55–95%, 28 examples, Scheme 89C). It is worth to note the key role of PIP as DG since all other bidentate DGs failed except the one derived from 8-aminoquinoline providing the expected product only in poor yield (37%).
14.1.4.1.4. Halogenation. Shi developed in 2016 the first Ni-catalysed halogenation of aromatic and heteroaromatic amides using inexpensive and readily available LiX salts as halide source (Scheme 90A).536 Note that a slight change of the catalytic system (NiCl2(PPh3)2 10%, LiCl 3 equiv., KMnO4 2.2 equiv., tBuCN, O2, 140 °C, 24 h), compared to the one used for the bromination and iodination reaction, allowed the formation of a C–Cl bond (not shown). Aromatic derivatives bearing various FGs (halogen, Ac, CF3) were functionalised in about 41–92% yield (20 examples) as well as heteroaromatic amides, albeit in somehow lower yields (30–76%, 9 examples). For unsubstituted and some para-substituted arenecarboxamides, mixtures of mono- and di-halogenated products were observed.
 | ||
| Scheme 90 C(sp2)–heteroatom bond formation using the amide derived from 2-(2-pyridyl)-2-isopropylamine as DG. | ||
14.1.4.1.5. Alkoxylation. C–O bond formation was also studied in recent years and two protocols for methoxylation (Scheme 90B)451 and acyloxylation (Scheme 90C)545via Cu catalysis were developed by Shi. In the first case, aromatic and heteroaromatic derivatives bearing various FGs (e.g. CF3, NO2, halogen) were methoxylated in moderate to good yields (25–96%). In the latter case, sterically hindered benzoic acids were crucial, as no expected product was obtained in presence of benzoic acid. The scope is pretty narrow (11 examples, 29–70%) and in several cases, mixture of products (mono- and di- as well regioisomers (one case) were obtained). The authors proposed a Cu(II)/Cu(III) catalytic cycle.
14.1.4.1.6. Sulfenylation and sulfonylation. The PIP bidentate based DG was particularly efficient for sulfenylation and sulfonylation reactions via Ni catalysis.
In 2014, seminal works on transition metal catalysed C–S bond formation were developed using Pd546 or Rh547 catalysts. One year later, key contributions in that field were reported by Lu and Shi using low cost metals, especially Ni catalysts. Indeed, using aryl disulfides as electrophilic sulphur sources, Lu developed a Ni-catalysed sulfenylation reaction; the use of a catalytic amount of benzoic acid was essential to promote the reaction in typically about 80–90% yields, except for three derivatives (Scheme 90D).537 Afterwards, Shi disclosed another Ni-catalysed protocol which did not require an additional oxidant, although in that case an extra ligand was required (up to 93% yield, Scheme 90E).548 For this transformation a Ni(II)/Ni(IV) catalytic cycle was proposed. Note that a complementary study was reported by Shi dealing with the Cu-mediated sulfenylation of heteroaromatic amides (37 examples, 83–99% yield, Scheme 90F).549
The sulfonylation reaction using sodium sulfinates was also studied by Shi534 (26 examples, up to 80% yield) as well as Gong and Song (33 examples, up to 90% yield),550 using a Cu- or Ni-catalyst, respectively (Scheme 90G and H).
14.1.4.2. C(sp3)–H functionalisation. The PIP-based bidentate DG was also successfully employed in transition metal catalysed C–O, C–N and C–F bond formation by activation of C(sp3)–H bonds. Regarding C–O bond formation, this DG was mainly employed in intramolecular processes for the functionalisation of secondary C(sp3) centers leading to the corresponding lactones and cyclic ethers as concomitantly studied by the groups of Shi551 and Ye,535 using a similar catalytic system (Scheme 91A and B).
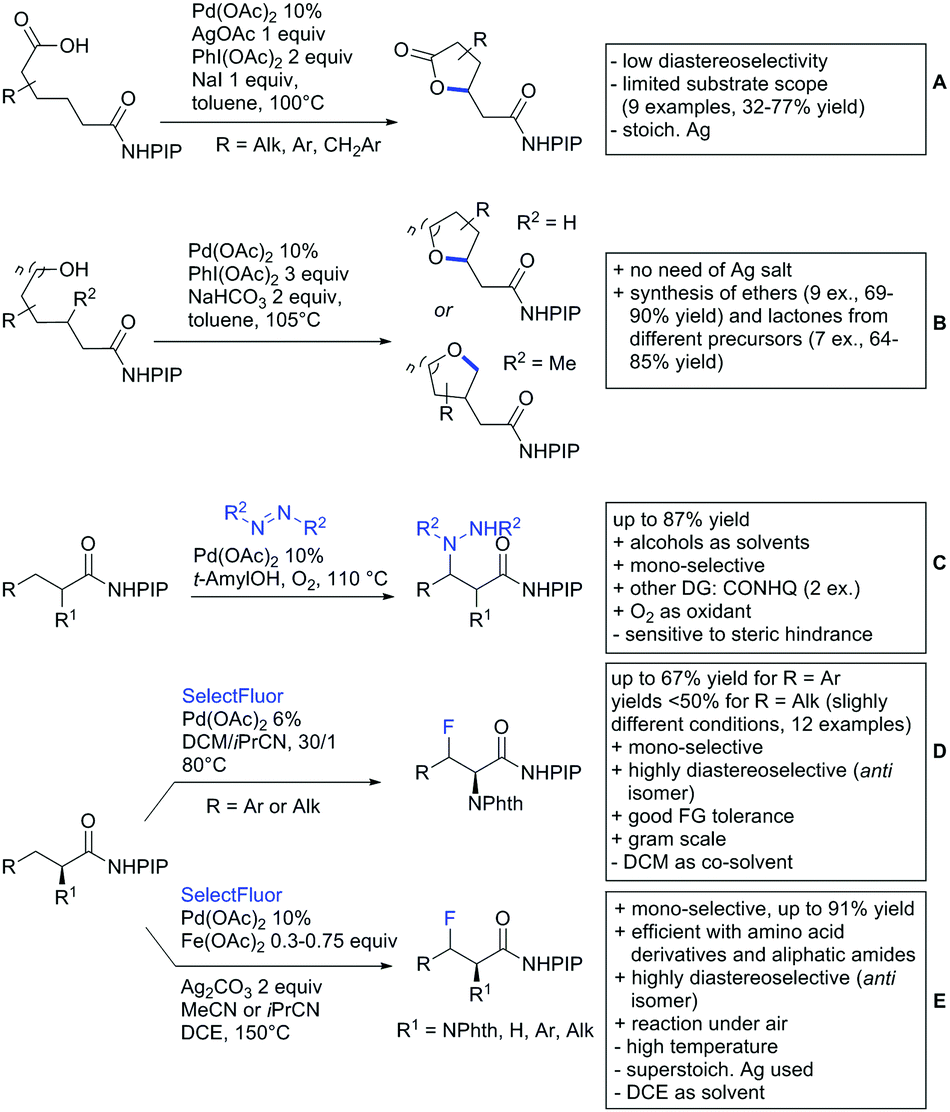 | ||
| Scheme 91 Pd-Catalysed functionalisation of C(sp3) centres using the amide derived from 2-(2-pyridyl)-2-isopropylamine as DG. | ||
14.1.4.2.1. Amination. Zhang studied Pd-catalysed direct amination of unactivated methylene C(sp3)–H bonds using azodiformates as the amino source, especially the diethyl azodicarboxylate, offering access to β-amino acid analogues (Scheme 91C).538 The transformation was achieved on primary and secondary C(sp3) centers and turned out to be sensitive to steric hindrance. Note that the amide derived from 8-aminoquinoline was also a suitable substrate even though providing the expected derivatives in lower yields.
14.1.4.2.2. Fluorination. The Pd-catalysed fluorination of amino acids at the β-position was independently reported by the groups of Shi497 and Ge552 using SelectFluor as the fluorine source. Shi reported the first example of the fluorination of aliphatic and benzylic methylene C(sp3)–H bonds via an inner sphere mechanism and the PIP amide group was cleaved leading to the corresponding esters (Scheme 91D).497 In the latter case, the functionalisation was extended to the fluorination of aliphatic amides and good yields were obtained on unactivated aliphatic amides with various skeletons (Scheme 91E).552
In 2016, He and Chen reported a Pd-catalysed arylation of phthaloyl alanines at the β-position using amides derived from PE as bidentate DG (Scheme 92A).553 With two sets of reaction conditions (conditions A, 16–90%; conditions B, 15–72%), they successfully employed sterically hindered ortho-substituted aryl iodides (e.g. OMe, NO2, Me, OAc, COMe, F), a significant synthetic challenge. The transformation was really selective as no product resulting from di-arylation or β-lactam byproducts were observed. However, when the arylation at the β-methylene position was concerned, mono-arylated products were obtained in rather lower yields (<48%). Note that PE-directed arylation of isoleucine derivatives at the γ-position was previously reported by the authors.554 In comparison, when PM was used as DG (Scheme 92B), the reaction was really sensitive to steric hindrance leading to poor yields (15–26%) with 2-substituted aryl iodides but the transformation was still efficient with para-substituted aryl iodides (62–86% yields).553
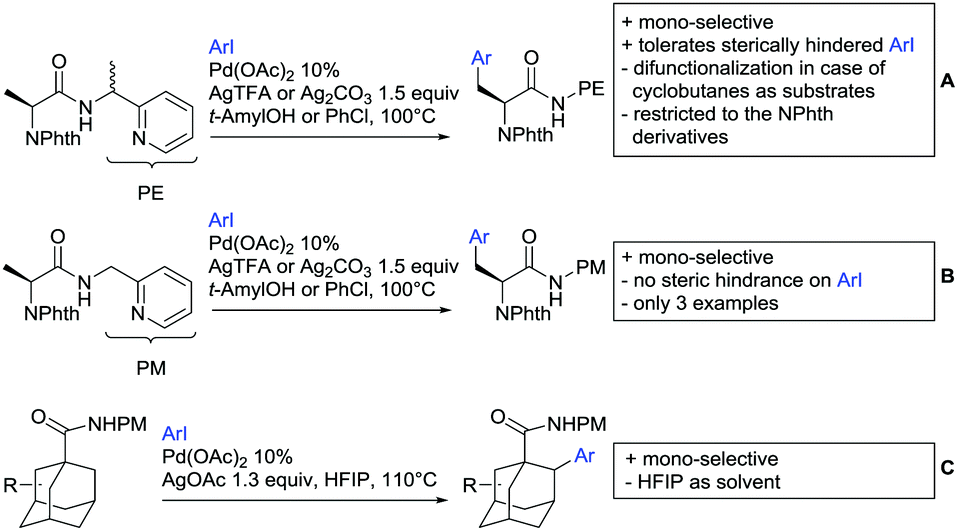 | ||
| Scheme 92 Functionalisation of C(sp3) centres using the amides derived from 2-(2-pyridyl)ethylamine (PE) and 2-(aminomethyl)pyridine (PM) as DGs. | ||
The PM DG was also employed in the Pd-catalysed arylation of diamondoids with aryl iodides (36–87%), a synthetic challenge due to the large number of possible C–H bonds to be functionalised (Scheme 92C).555
14.1.6.1. C(sp2)–H functionalisation.
14.1.6.1.1. Arylation and alkylation. Pd-Catalysed arylations of thiophene derivatives and allylamines were reported. In 2016, Babu and co-workers disclosed picolinamide-directed arylation of the C3-position of 2-(aminoethyl)thiophenes with aryl iodides and heteroaryl iodides in low to good yields and in a regioselective manner (Scheme 93A).568 The same group used a similar catalytic system for arylation of allylamines at the γ-position leading to the corresponding cinnamylamines with good to high E/Z ratios (up to 2
:98) in the favour of the challenging Z isomers (Scheme 93B).569 Note that substrates bearing a primary γ-C(sp3) center and a γ-C(sp2) center led to di-arylated products. Furthermore, Shao and He reported the Pd-catalysed cyclisation of alkenyl anilines via oxidative arylation (not shown).570
 | ||
| Scheme 93 Picolinamide-directed arylation and alkylation of C(sp2) centres. | ||
For the methylation of arenes, two reports were published using either AlMe3 or MeI as the methyl source. In 2015, Ilies and Nakamura developed a protocol allowing methylation of arenes via Fe catalysis, involving a plausible Fe(III)–Me intermediate (Scheme 93C).403 Although efficient, only a couple of examples were reported and the selectivity for mono-methylation vs. di-methylation depended on the substrate substitution pattern.
Another protocol from Zhang and Ma described the Pd-catalysed methylation of the amide derived from (S)-tyrosine analogues (Scheme 93D).559 It is worth to mention that only this DG was efficient for this di-methylation reaction and a scale up on ca. 20 g was successfully achieved with the complete retention of the chiral information.
14.1.6.1.2. Alkenylation. Two examples of alkenylation/annulation reactions have appeared since 2015. Rodriguez, Arrayás and Carretero reported a Rh(I)/Rh(III) catalysed functionalisation of aromatic picolinamide derivatives with internal alkynes (Scheme 94A and B).561 Depending on the choice of Rh catalyst (Rh(I) vs. Rh(III)), a nice divergent site-selectivity control was achieved via selective functionalisation of either the aromatic or the heteroaromatic part (the DG itself) of N-substituted picolinamide compounds. Depending on the reaction conditions, the synthesis of isoquinoline derivatives or the olefination of benzylamine and phenethylamine derivatives was achieved. Note that in the latter case, the di-olefinated products were obtained except on specific substrates. Symmetrical and unsymmetrical alkynes were used as coupling partners; the observed regioselectivity depended on the alkyne substitution. This study was strongly supported with several mechanistic studies and DFT calculations.
 | ||
| Scheme 94 Picolinamide-directed alkenylation and alkynylation of C(sp2) centres. | ||
Two years later, the same group reported a protocol for the synthesis of 1,4-dihydroisoquinoline compounds (56–96% yield) from benzylamines taking advantage of Co catalysis and using molecular O2 as oxidant (Scheme 94C).562 An interesting feature is that, in contrast to Rh catalysis, dialkyl alkynes were more efficient in that case as compared to diaryl ones. Additionally, terminal alkynes were suitable coupling partners as well. When unsymmetrical alkynes were used, the regioselectivity seemed to be controlled by electronic factors: 1-aryl-1-alkynes provided only one single diastereoisomer albeit no regiocontrol was observed with dialkylated alkynes.
The Co-catalysed alkynylation of benzylamine derivatives was studied by Balaraman and co-workers (Scheme 94D).571 The transformation was FG tolerant (NO2, Br, OMe) and importantly, primary, secondary, tertiary and even enantiopure benzylamines were suitable substrates. Depending on the substrate, mono-, di-alkynylated products or even a mixture was obtained.
14.1.6.1.3. Cyanation. Recently, the use of picolinamide as DG allowed the cyanation of naphthalene derivatives via Cu catalysis. Using this cost-attractive transition metal, the functionalisation of a panel of naphthalenes was possible using benzoyl cyanide as the cyano source (Scheme 95A).560 Although efficient, the transformation turned out to be sensitive to the substitution pattern of the substrate.
 | ||
| Scheme 95 Picolinamide-directed functionalisation of C(sp2) centres. | ||
14.1.6.1.4. Trifluoromethylation. Using an inexpensive Ni catalyst and the Langlois’ reagent, Zhang reported the trifluoromethylation of arylamines under mild conditions (50 °C, air atmosphere) with cheap oxidant leading to the corresponding trifluoromethylated products in moderate to good yields (Scheme 95B). It is worth mentioning that this transformation was carried out in water and the catalytic system was recycled and reused about eight times.572
14.1.6.1.5. Acetoxylation. In 2017, Zeng reported a Co-catalysed cross dehydrogenative coupling (CDC) between picolinamide derivatives with aliphatic and aromatic carboxylic acids (Scheme 95C).573 It is worth mentioning that this transformation was mainly restricted to naphthalene compounds and only two N-benzyl substituted 2-pyridinecarboxamides were functionalised as well, although in low yields (25 and 37%). Acetoxylation at the remote ε-C(sp2)–H bond was studied by Babu using this bidentate DG (not shown).574
14.1.6.1.6. Amination. The regioselective amination of 1-naphthylamine derivatives at the C8-position was independently reported by Yang, Wu and Wu563 as well as Punniyamurthy564 using a Pd or Cu catalyst, respectively (Scheme 95E and F). The Pd-catalysed mono-amination of naphthalene derivatives was achieved (up to 89% yield). Although the diversity in term of naphthalene derivatives was rather limited (4 different skeletons), several cyclic secondary aliphatic amines were suitable.471 Under Cu catalysis the introduction of indoles was possible in moderate to good yields (39–74%) and a large variety of FGs was tolerated on the indole skeleton (e.g. halogen, ester, vinyl, allyl) and even 7-azaindole.564 The approach was also applied to the introduction of azoles, albeit only 3 examples were reported (51–69%). Note that the arylation of the NH of the picolinamide part was also reported with boronic acids (not shown).575
Hirano, Miura and co-workers also investigated the Cu-promoted intramolecular amination reaction on phenethylamine derivatives. Indeed, in 2015, two sets of reaction conditions were established for the synthesis of indolines under microwave irradiation via a Cu-mediated or catalysed process using MnO2 as a terminal oxidant in the last case (Scheme 95F).576 In 2017, the same authors reported a DG controlled divergent process which allowed, in case of NHPA as DG, the selective access to indole derivatives starting from enamides via a Cu-catalysed intramolecular C–H bond amination reaction (Scheme 95G).577
14.1.6.2. C(sp3)–H functionalisation. The picolinamide DG has played an interesting role in the Pd-catalysed alkenylation of aliphatic amines at the δ-position with alkynes in a highly selective manner.567 Indeed, to date δ-C(sp3)–H bond functionalisation remains scarce and often occurred on substrates on which the γ-position was less accessible or sterically hindrance favoured a six-membered intermediate rather than the kinetically favoured five-membered metallacycle. Impressively, the functionalisation was completely site-selective even in presence of accessible γ-C(sp3)–H bonds. The reaction provided the corresponding products in moderate to good yields, and the role of 2,6-dimethylbenzoquinone (2,6-DMBQ) was crucial as both ligand and co-oxidant (Scheme 96A). Note that when unsymmetrical alkynes were used, mixtures of isomers were obtained, with moderate to good selectivity depending on the specific examples. Remote functionalisation was also successfully achieved by the Maes group, who developed a protocol for the remote C5-(hetero)arylation of 3-aminopiperidine via a Pd-catalysis (Scheme 96B).565 Indeed, starting from commercially available 1-Boc-3-aminopiperidine, they offered an access to cis-3,5-disubstituted piperidines in typically 60–70% yields (up to 82%). The mono-arylation only occurred in a regio- and stereo-selective manner. The nature of the nitrogen protecting group had a strong impact on the outcome of the reaction: a shutdown of the reactivity was observed with benzyl and carbamoyl protecting groups, while lower to good yields were obtained with pivaloyl, tosyl and alkoxycarbonyl groups. Albeit a large excess of (het)ArI was used, the unreacted reagent was easily recovery. Finally, very high yields were obtained with heteroaromatic compounds as coupling partners (two examples, 84 and 95% yields). In addition, the Pd-catalysed carbonylation of N-alkyl picolinamides was possible using this bidentate DG (Scheme 96C).566 With the methodology developed by Wang, not only γ-lactams and γ-amino acids were synthesized in high yields (60–90% yield) but this approach allowed the total synthesis of bioactive rac-pregabalin. Note that TEMPO was used as oxidant in the transformation.
 | ||
| Scheme 96 Picolinamide-directed alkenylation, arylation, carbonylation and esterification of C(sp3) centres. | ||
Finally, the regioselective Pd-catalysed esterification between two different alkylbenzenes was developed by Liu and Zhang (Scheme 96D) and products were obtained in a typical 50–70%.578 In this transformation, the role of picolinamide as DG was crucial to ensure the esterification reaction since the tested benzamide and acetanilide were completely inefficient; the oxygen from the benzyl esters comes from the O2 in the air.
14.1.7.1. C(sp2)–H functionalisation.
14.1.7.1.1. Alkylation. In 2015, the combination of Fe catalysis and TAM as bidentate DG allowed the methylation and ethylation of anilides (Scheme 97A).580 Using cheap FeCl3 as catalyst in presence of ZnCl2·TMEDA as additive, most likely involved in the transmetallation step, the methylation of arenes (71–96%) and heteroaromatic derivatives such as pyrrole, furan and thiophene was achieved (49–60% yield). The approach was also successfully extended to the functionalisation of the challenging olefinic C–H bonds leading to the selective formation of the Z isomers (57 and 90% yields).
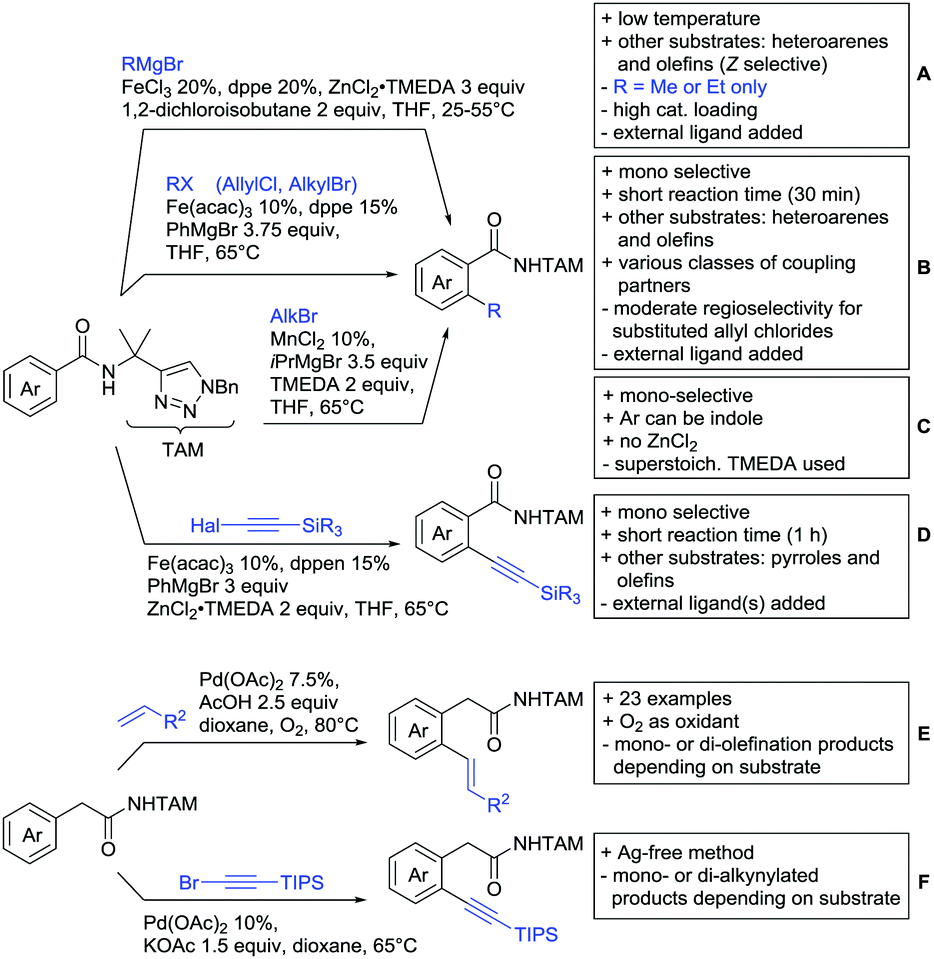 | ||
| Scheme 97 Alkylation, allylation, alkenylation and alkynylation of C(sp2) centres using triazolyldimethyl amide as DG. | ||
The same group further investigated the potential of the bidentate DG in Fe-catalysed C–H allylation and alkylation of amides (Scheme 97B).585 Using allyl chloride, the functionalisation of a broad range of substrates was possible including arenes, ferrocenes,408 heteroarenes and alkenes (47–92% yield). When substituted allyl chlorides were reacted, allylated benzamides were obtained with moderate to good regioselectivity (from 2:1 to 5.7:1) in favour of the branched products. Moreover, primary and secondary alkyl bromides were also suitable coupling partners. Note that such transformations were not restricted to Fe catalysis, since very recently Ackermann and co-workers reported a MnCl2-catalysed alkylation of amides with alkyl halides (up to 90% yield) (Scheme 97C).582 They developed a process involving a low-valent Mn catalyst, for which the presence of zinc additives and phosphine ligands were no longer required.
14.1.7.1.2. Alkynylation and alkenylation. In 2017, Ackermann and co-workers reported the Fe-catalysed alkynylation of aromatic amides using several silylated alkynyl halides (Scheme 97D).586 This is an efficient process, which allowed the synthesis of mono-alkynylated products in 44–71% yields under mild reaction conditions. This reaction was also successfully applied to heteroaromatic and olefinic compounds (54–59%, 4 examples). Note that the methodology was further applied to the synthesis of the corresponding isoquinolones via a two-step sequence (alkynylation/desilylation–cyclisation).
A TAM-DG promoted olefination of aromatic systems using activated olefins such as acrylates, vinyl phosphonate, or vinyl sulfones was reported by Shi and co-workers (Scheme 97E).587 Interestingly, O2 was suitable as oxidant. Although efficient (typically 80–90% yield), mixture of mono- and di-olefinated compounds were observed in case of meta- and para-substituted derivatives. In 2016, Chen and Shi, published a Pd-catalysed alkynylation reaction under mild reaction conditions (Scheme 97F).583 This reaction was tolerant to various FGs (e.g. NO2, CF3, F), provided the corresponding products in high yields (76–93%) and no Ag was required, a real asset.
14.1.7.2. C(sp3)–H functionalisation: alkylation and Alkynylation. In the course of their investigation of Fe-catalysed methylation of anilides,580 the group of Ackermann showcased the generality of their approach by an extension to the methylation of unactivated aliphatic amides (3 examples) in moderate to good yields (Scheme 98A). This bidentate DG also demonstrated its efficiency in the alkynylation of primary and secondary C(sp3)–H bonds (Scheme 98B). Various aliphatic derivatives (16 examples) including N-protected amino acids were functionalised in moderate to high yields (45–86%), moderate to good diastereoselectivity and di-functionalisation reactions were observed in some cases.583
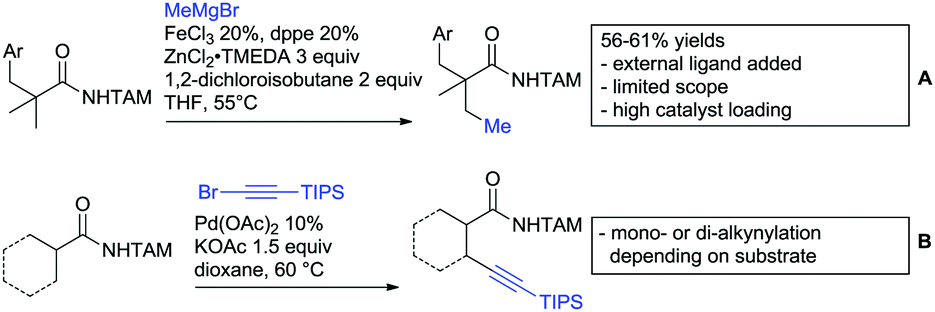 | ||
| Scheme 98 Alkylation and alkynylation of C(sp3) centres using triazolyldimethyl amide as DG. | ||
14.1.8.1. Amide-oxazoline.
14.1.8.1.1. Alkylation. Using the 2-(2-oxazolinyl)phenyl moiety on the amide nitrogen, the group of Yu developed a Cu-catalysed oxidative coupling of aromatic amides with malonate derivatives, offering a straightforward access to isoindolinone compounds in 40–75% yields (Scheme 99A).588
 | ||
| Scheme 99 Functionalisation of C(sp2) centres using 2-(2-oxazolinyl)phenyl amide as DG. | ||
14.1.8.1.2. Acetoxylation. In 2015, Ge and Li reported the Pd-catalysed acetoxylation of amide-tethered oxazoline. Indeed, via a six-membered bidentate complex with Pd(II), the di-acetoxylation of various aromatic and heteroaromatic amides was achieved in the presence of PhI(OAc)2 playing the role of both oxidant and OAc source in 32–73% yields (Scheme 99B).589 It turned out that depending on the substitution pattern of the substrate, mono-acetoxylation was achieved. Note that using very simple conditions, Yu also developed Cu-mediated hydroxylation of aromatic amides (Scheme 99C).590 The transformation was tolerant to various FGs (alkyl, CF3, F, I, Cl, vinyl moiety) and provided the products in 37–86% yields.
14.1.8.1.3. Sulfenylation. The formation of a C–S bond by making us of N-[2-(2-oxazolinyl)phenyl] amides was also studied by Manolikakes (Scheme 99D)591 as well as Liu and Chen (Scheme 99E)592via Cu mediated transformations using either sodium sulfinates (22–82% yield) or 2-mercaptobenzimidazoles as coupling partners (up to 84%). In both transformations, a good FG tolerance was noted (e.g. Cl, CF3, acetyl).
 | ||
| Scheme 100 Functionalisation using amides derived from other heterocyclic amines as bidentate DGs. | ||
Amide derived from 4-amino-2,1,3-benzothiadiazole (BTDNH2) was studied by Babu in Pd-catalysed arylation and acetoxylation (2 examples) of linear aliphatic carboxamides in yields 52–97% yields (Scheme 100B).594 In case of cyclic carboxamides, di-functionalisation generally occurred although in some cases, mixtures of mono- and di-functionalised products were obtained. Its cleavage was achieved under basic conditions (NaOH, ethanol, 85 °C) to access the corresponding carboxylic acid.
Furthermore, the amide derived from [2-(1-alkylbenzimidazolyl)]-2-isopropylamine offered a good alternative, in Cu-catalysed sulfenylation of aromatic and olefinic amides (47 examples, Scheme 100C).595 Note that the Pd-catalysed arylation of thiophene derivatives using pyrazine-2-carboxamide as DG was also reported (not shown). A two steps sequence (N-Boc) protection/hydrazinolysis (NH4I, NH2NH2·H2O) under microwave irradiation afforded the Boc-protected MBIPNH2 and the corresponding N-acylhydrazide in good yield.568
14.2. N,S-Bidentate DGs
An interesting alternative to N,N-bidentate DGs relies on the use of N,S-bidentate DGs, easily cleavable under acidic (HCl, MeOH, 100 °C)596 or basic597,598 (KOH, 80–85 °C) conditions. Although restricted to a handful of examples, these N,S-bidentate DGs were successfully applied in the arylation of alkanecarboxamides, especially on challenging cyclopropane derivatives.In 2015, Babu studied the arylation of thiophene- and furan-2-carboxamides by directed C–H bond activation using either the amide derived from 8-aminoquinoline or the 2-(methylthio)aniline.391 Using the bidentate N,S-DG, a selective Pd-catalysed C–C bond formation of thiophen- and furan-2-carboxamides at the C3 position was achieved using (hetero)aryl iodides as coupling partners in 25–81% yields (Scheme 101A). Then, the same group investigated the use of this N,S-DG in the functionalisation of C(sp3)–H bonds. Hence, they described the Pd-catalysed arylation of cyclopropanecarboxamides followed by ring opening of the cyclopropanes using the amide derived from 2-(methylthio)aniline as DG (not shown).472 Interestingly, di- or tri-arylation and the formation of a C–O bond was reported within a one pot process, leading to anti β-acyloxy amides in 10–86% yields, although an excess of Ag salt was necessary (Scheme 101B).599
 | ||
| Scheme 101 Arylation using N,S-bidentate DGs. | ||
Then, Wencel-Delord and Colobert employed amides derived from enantiopure sulfinyl anilines in the diastereoselective arylation of cycloalkanes (cyclopropanes and cyclobutanes) leading to the corresponding compounds in moderate diastereoselectivities (60/40 to 80/20), in 26–92% total yield of both diastereoisomers. It is worth to mention that both diastereomers were easily separated by column chromatography offering access to enantiopure compounds after cleavage of the DG (Scheme 101C).600
In 2017, Chen and He published a Pd-catalysed β-methyl and even more challenging β-methylene C–H arylation with sterically hindered aryl iodides by playing with the nature of the ortho-sulfinyl aniline auxiliaries (R3 = Me, C6H4Me) (Scheme 101D).601 The reaction showed a good FG tolerance (NO2, F, OMe, CO2Me) for ortho substituted aryl iodides and about 60–70% as typical yields. The same year, a diastereoselective transformation was developed by Wencel-Delord and Colobert (Scheme 101E), offering an access to arylated aliphatic amides at the β-methylene position (up to 9/1 dr), which was extended to one example of acetoxylation (91% yield, 7/3 dr).598
Miura and Shishido reported Ru-catalysed alkenylation of N-tosyl aromatic amides with internal alkynes followed by a cyclisation process affording the corresponding isoindolinones in 27–71% yields (not shown).602
14.3. N,O-Bidentate DGs
In this section the recent progresses involving N,O-bidentate DGs will be discussed. Indeed, a couple of them were investigated such as the bidentate amide derived from 2-pyridinamine-N-oxide, 1-aminoanthraquinone and the oxalyl amides.14.3.1.1. C(sp2)–H functionalisation. The pyridine-N-oxide moiety was used with first row eco-friendly transition metals such as Co and Ni catalysts. Besides its efficiency, a key advantage of this bidentate DG is its removal, which could be easily realized under basic conditions.603–608
14.3.1.1.1. Alkenylation. Such DGs were successfully applied in Co- and Ni-catalysed C(sp2)–H alkynylation/annulation reactions.
An interesting approach towards heterocycles was developed by the groups of Macgregor and Ackermann (Scheme 102A).609 Under mild conditions and using oxygen as the sole oxidant, the synthesis of 6-membered isoquinolone derivatives from aromatic derivatives was achieved using Co(II)-catalysis. The transformation turned out to be particularly efficient and highly regioselective and terminal as well as internal alkynes were suitable coupling partners. The regioselectivity of the reaction steered from electronic reasons as suggested by computational DFT investigations.
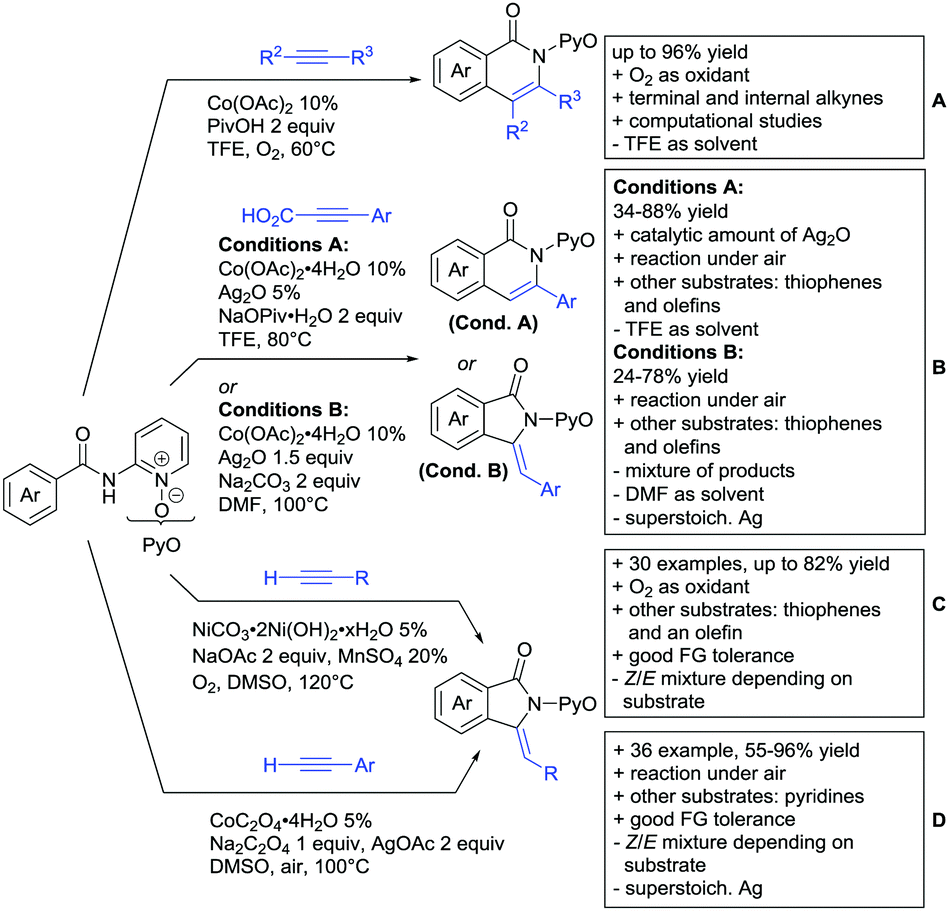 | ||
| Scheme 102 Alkenylation of C-(sp2) centres using 2-pyridine-N-oxide amide as DG. | ||
Niu and Song also investigated the synthesis of isoquinolone and isoindolinone derivatives by slightly changing the catalytic system via a Co-catalysed decarboxylative C–H activation using alkynylcarboxylic acids (Scheme 102B).610 The use of a catalytic amount of a Ag salt in the catalytic system to access isoquinolone compounds was a real asset of this approach, O2 playing the role of terminal oxidant.
Furthermore, Niu and Song reported the Ni-catalysed functionalisation of a panel of amides with terminal alkynes via a twofold C–H activation process leading to substituted 3-methyleneisoindolin-1-ones (Scheme 102C).611 The reaction demonstrated a good FG tolerance including halides and thiophene derivatives, and the stereoselectivity of the reaction was moderate to excellent (Z/E ratio from 52/48 to 100/0).
The same authors also developed a Co-catalysed alkynylation/annulation reaction of amides with terminal alkynes as coupling partners in presence of AgOAc as the terminal oxidant (Scheme 102D).612 The nature of the bidentate DG turned out to be crucial as other DGs were inefficient in that transformation. Both Co(II) and Co(III) might be used, CoC2O4·4H2O being the most efficient. The reaction was applied to a wide range of compounds (36 examples), although internal alkynes and aliphatic ones were not suitable coupling partners.
14.3.1.1.2. Alkoxylation, amination. Another example showcasing the unique properties of this bidentate DG was reported again by Niu and Song, who published Co-catalysed alkoxylation of aromatic and olefinic carboxamides under mild reaction conditions (Scheme 103A).604 Later on, the same group extended this approach to the amination of arenecarboxamides with alkylamines (Scheme 103B).605
 | ||
| Scheme 103 Alkoxylation and amination of C(sp2) centres using 2-pyridine-N-oxide amide as DG. | ||
14.3.1.2. C(sp3)–H functionalisation. This DG was also successfully applied in C(sp3)–H bond functionalisation. In 2015, Zeng and Lu reported a Pd catalysed pyridine-N-oxide-directed arylation of alkane carboxamides with aryl iodides (Scheme 104A).606 The functionalisation of both primary and secondary C(sp3) centres was achieved at the β- and even the γ-position on suitable substrates. Substitution on the pyridine part of the DG with halides, CF3 and NO2 groups was tolerated. Note that mono- and di-arylated products were obtained and the overall efficiency of the transformation was substrate-dependent.
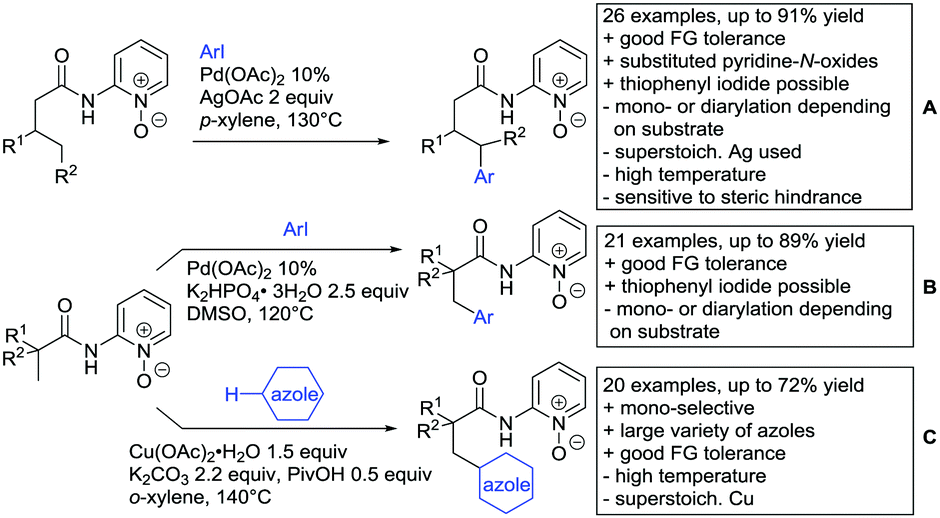 | ||
| Scheme 104 Arylation and amination of unactivated C(sp3)–H bonds using 2-pyridine-N-oxide amide as DG. | ||
A similar transformation was then further studied by the same authors, and aliphatic amides were functionalised with a large number of aryl iodides leading to mono- or di-functionalised products depending on the substrate (Scheme 104B).607
This DG was not restricted to the formation of C–C bonds, since very recently, the group of You demonstrated that this bidentate DG was successfully used in the oxidative amination of unactivated C(sp3)–H bonds with azoles via a Cu mediated process (Scheme 104C).608 Note that an extension of this transformation via Ni catalysis was also reported using in that case the amide derived from 8-aminoquinoline as DG.
14.3.2.1. C(sp2)–H functionalisation.
14.3.2.1.1. Arylation and alkenylation. Shi and Zhao reported the Pd-catalysed meta arylation of β-arylethylamine via a Catellani reaction using NHOA as a bidentate DG (Scheme 105A).619 Using norbornene as the transient mediator, the transformation was tolerant to various FGs, leading to the expected compounds in good yields and various aryl iodides bearing para, meta or ortho substituents were suitable. It is worth to mention that the mono meta arylation was possible on substrates bearing substituents at the ortho and meta positions. In contrast, when unsubstituted arenes were reacted, only meta di-arylated products were obtained. To further showcase the synthetic utility of their approach towards the synthesis of polyfunctionalised β-arylethylamines, the authors investigated the Pd-catalysed oxalyl-amide-directed functionalisation of meta arylated β-arylethylamines. This protocol turned out to be general and alkynylation, iodination, acetoxylation and amination reactions were achieved in moderate to good yields (42–82%), although some di-alkynylated products were produced in 17–30% yields (not shown).620
 | ||
| Scheme 105 Functionalisation of C(sp2) centres using oxalyl amide as DG. | ||
The same group also investigated Rh(III)-catalysed oxidative C–H/C–H cross-couplings of benzylamines and heteroarenes (Scheme 105B).614 When the ortho position was unsubstituted, mixtures of mono- and di-heteroarylated products were obtained, the ratio mono-/di-being dependent on the substitution pattern of the aromatic system.
In the course of their investigation for the Pd-catalysed arylation of 3-(aminoethyl)thiophene derivatives, the group of Babu showed that among the possible DGs for this transformation, the NHOA DG turned out to be efficient affording the products in 57 to 75% yields and the reaction was also performed on a gram scale (Scheme 105C).568
The NHOA DG was also used in Pd-catalysed olefination of β-arylethylamines (Scheme 105D).621 The reaction works with a broad array of functionalised olefins and a good selectivity for mono-olefination was observed in case of electron rich olefins such as allyl acetate, 3,3-dimethyl-1-butene and styrene derivatives. In contrast for more reactive olefins such as acrylamide, acrylonitrile, acrylaldehyde and vinyl sulfone, a mixture of mono- and di-olefinated products was obtained.
14.3.2.1.2. Carbonylation. Carbonylation of β-arylethyl- and benzylamines were achieved using different reagents. Indeed, in 2016, a first report from Huang and Zhao on the Pd-catalysed carbonylation using carbon monoxide as the CO source was reported (E).615 Then, Zhao and Shi developed an improved protocol based on a Ru-catalysed carbonylation reaction, which demonstrated interesting features such as the use of isocyanate as CO source and the absence of oxidants (F).618 21 derivatives were functionalised with up to 96% yields.
14.3.2.1.3. Silylation and germanylation. This bidentate DG was also employed for the regioselective and monoselective silylation of benzylamine and phenylethylamine compounds via Pd catalysis at the γ- and δ-positions, respectively (Schemes 106, 26 examples, 43–90%).616 This reaction was not restricted to aromatic derivatives since, using slightly modified reaction conditions, the silylation of 2-methylaniline derivatives was possible, albeit restricted to four examples in low to moderate yields (21–49%). It is worth to mention that the germanylation of benzylamines was also achieved by replacement of the hexamethyldisilane with Ge2Me6 (4 examples, 37–82%).
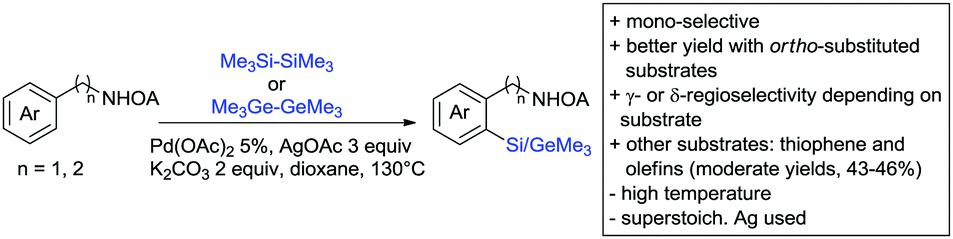 | ||
| Scheme 106 Silylation and germanylation of C(sp2) centres using oxalyl amide as DG. | ||
14.3.2.2. C(sp3)–H functionalisation. In 2015, the γ-arylation of aliphatic amines with aryl and heteroaryl iodides was investigated by Zheng, Huang and Zhao (Scheme 107A).622 Upon Pd-catalysis, mono- or di-arylated products were obtained (47–83%) depending on the substitution pattern of the substrates. When heteroaryl iodides were used as coupling partners, low to high yields (23 to 78%) were obtained. Later, the same group used a similar catalytic system for the functionalisation of amino acids (Scheme 107B).623 ArI and heteroaryliodides were suitable coupling partners leading to the corresponding products in 45–86% and 36–83% yields, respectively. The carbonylation of aliphatic amines via a Pd-catalysed C–H bond functionalisation under carbon monoxide atmosphere was reported by Yao and Zhao (Scheme 107C).617 Primary and secondary C(sp3) centers were functionalised in low to high yields (12–87%) and extended to the functionalisation of benzylamines (8 examples, 71–98%) (not shown). The reaction was even conducted on a gram scale. The authors suggested that 3-(trifluoromethyl)benzoic acid was important to stabilize the Pd intermediate.
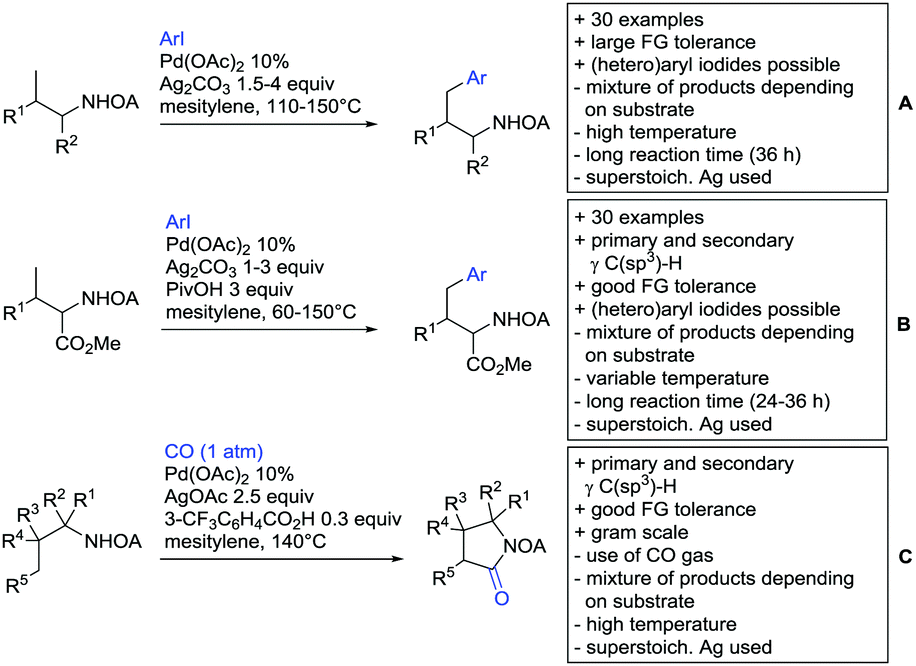 | ||
| Scheme 107 Functionalisation of C(sp3) centres using oxalyl amide as DG. | ||
 | ||
| Scheme 108 Acetoxylation of C(sp3) centres using amide derived from 1-aminoanthraquinone as DG. | ||
15. Heterocyclic DGs in C–H functionalisation
As for carbonyl-based and bidentate moieties, heterocycles are also a very common class of DGs for the functionalisation of C(sp2) and C(sp3) centres. The heterocycles that can be used as DGs are those featuring a C(sp2)-hybridised nitrogen atom, which act as the coordinating moiety for the formation of the metallacyclic intermediate essential to the C–H functionalisation. These DGs have attracted a lot of interest in the field for a number of reasons: (1) heterocycles are commonly found in bio-active and drug-like molecules, which allows late-stage modifications without requiring DG installation/removal; (2) if needed can be installed on the substrate in a number of ways; (3) despite being generally considered strong-coordinating DGs, their coordinating properties can be somewhat altered by substitution or by changing the heterocycle used for a specific transformation.Although DG cleavage can be sometimes tricky with monoheterocycles (i.e. pyridine), several methods have been developed during the years, thus increasing their synthetic utility. In the case of diheterocycles, cleavage (e.g. for pyrimidine) or modification of the DG (for example by ring opening) is intrinsically easier, although not often reported in publications involving less common heterocycles. The available cleavage/modification methods for heterocyclic DGs are discussed in each specific sub-section. In this section, yields have been specifically reported in case of particularly high yields (most examples >80–90%) or lower than average (most <70% or lower) results, and in case of strong substrate-dependence. If not reported, average yields of 70–80% for most examples is to be assumed (see discussion in the Introduction section).
15.1. Pyridine as DG
The use of pyridine (as well as other heterocycles such as pyrazole and pyrimidine) as a DG in C–H functionalisation chemistry dates back to the late 1990s–early 2000s, when it was pioneered by Murai/Chatani first, who reported Ru-catalysed alkylations and carbonylations on C(sp2) and C(sp3) centres,625–627 and then by Sanford628,629 and Daugulis,630 who extended its use to Pd-catalysed acetoxylations and arylations. Since then, pyridine is one of the most frequently applied DGs in C–H functionalisation chemistry. This is possibly due to its simplicity and the easy installation into aromatic rings or aliphatic chains via cross coupling631–633 or other methods.634 A great number of reactions have been reported with this DG, with a variety of protocols, reagents and reaction conditions. Recently this DG has also been commonly used for meta-selective C(sp2)–H functionalisation using Ru catalysts,635,636 which will not be treated in this review. Moreover, pyridine is the DG of choice for specific substrates, such as 2-pyridones, for which other DGs have proved ineffective.Although this DG is typically not removable when attached to the substrate via a C–C bond, the ubiquity of pyridine rings in many complex molecules (e.g. bioactive compounds), partially compensates this disadvantage, as its removal might not be required in all cases. The removal of this DG can nonetheless be achieved when attached to the substrate via a C-heteroatom bond (as is the case for N-(2-pyridyl)indoles, N-(2-pyridyl)anilines, or O-(2-pyridyl)phenols). These cleavage methods are discussed in Section 15.3.
15.1.1.1. Arylation. Many arylation procedures with pyridine as DG have been reported before 2014, and therefore are not included in this review. Nonetheless, several methods have been developed in the last few years. Szostak reported an unusual C–C cleavage/C–H functionalisation using activated benzamides as arylating agents (Scheme 109A),637 while boronic acids under Ru catalysis were utilised by Maheswaran (Scheme 109C).638 The hyper-valent iodine additive in this reaction was suggested to form a charge-transfer complex with the substrate, generating an aromatic radical cation639 which facilitates the formation of the ruthenacycle intermediate.
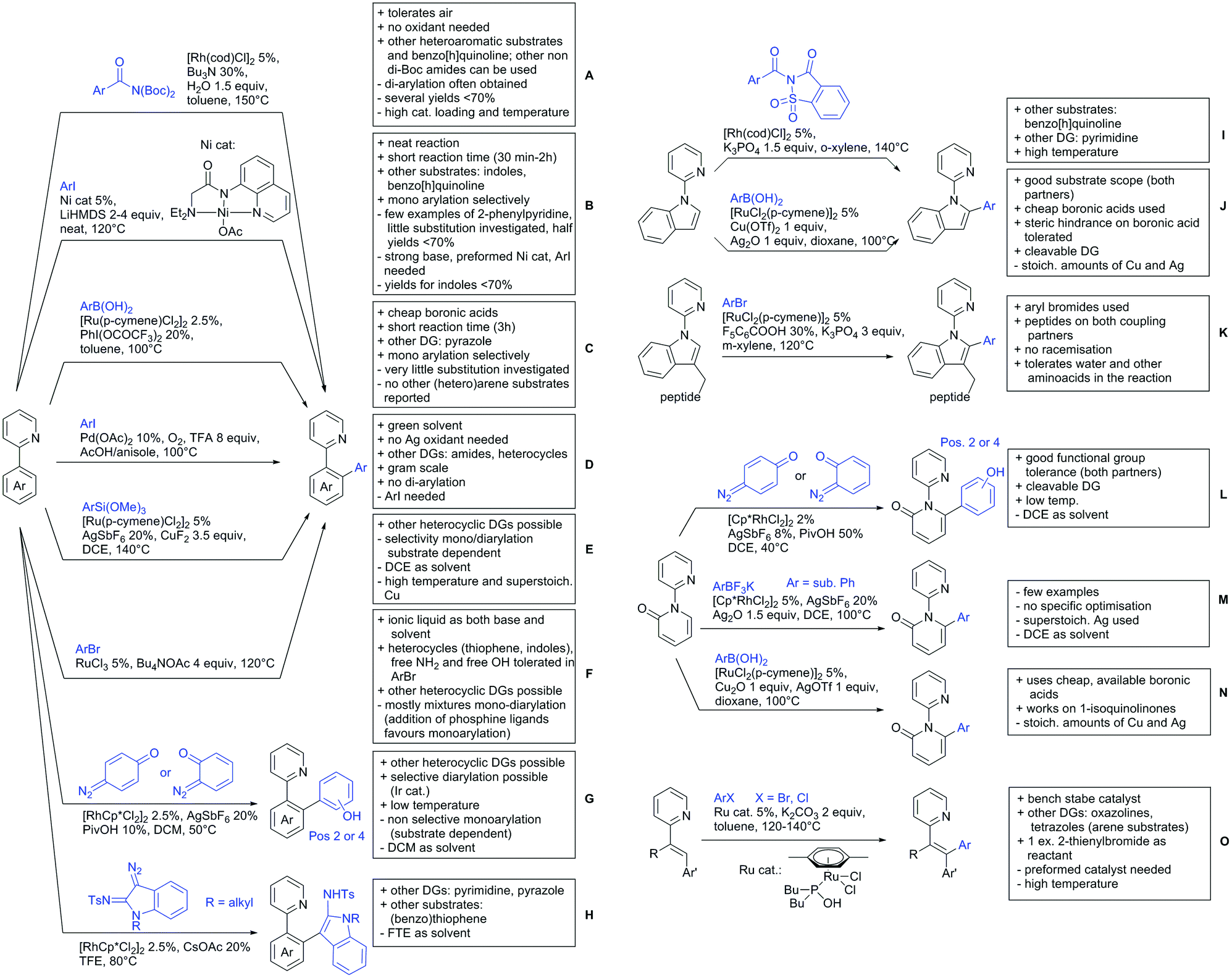 | ||
| Scheme 109 C(sp2)–H arylation using pyridine as DG. | ||
Punji and Wang reported the use of aryl iodides with Ni and Pd catalysis respectively (Scheme 109B and D).640,641 While the first required preformed Ni(II) catalysts and the use of strong base, the latter employed O2 as oxidant and green solvents, although a 10% Pd loading was required. Notably, both methods provide selective monoarylation, which is not always achieved. A coupling with aryl silanes was reported by Szostak (Scheme 109E).642 Although this procedure avoids the use of stoichiometric Ag oxidants, still a superstoichiometric amount of CuF2 was required, and a substrate-dependent selectivity (mono or diarylation) was observed. A recent paper by Kantchev demonstrated the use of the ionic liquid Bu4NOAc as both base and solvent in the RuCl3-catalysed coupling with aryl bromides (Scheme 109F).643 The reaction gave mostly mixtures of mono and diarylated products, but at the same time tolerates heteroaryls, free NH2- and free OH-substituted aryl bromides. In 2015 Wang reported the Rh (monoarylation) or Ir (diarylation) catalysed reaction with diazoquinones leading to phenol-functionalised arenes (Scheme 109G).644 The next year a related transformation, using 3-diazoindolin-2-imines as reactants was disclosed by Wang and Lu, giving 3-aryl-2-aminoindoles (Scheme 109H).645
Inspired by Szostak's arylation method using aromatic amides as arylation reagents (Scheme 109A), Zeng performed the arylation of indoles (and benzo[h]quinoline) using N-acyl saccarines as reagent under similar conditions (Scheme 109I).646 The use of boronic acids for indole arylation was instead reported by Kapur. This method provides good substrate scope and yields, although requiring stoichiometric amounts of both Cu and Ag (Scheme 109J).647 Ackermann recently reported the arylation of peptide-substituted indoles using aryl bromides. This procedure afforded good yields of arylated products, with no racemisation and good robustness. Importantly, peptides are tolerated on both coupling partners (Scheme 109K).648
The arylation of 2-pyridones with heteroaromatic compounds via cross dehydrogenative coupling was first reported by Miura and Hirano in 2014.649 Since then, further arylation procedures have been reported making use of diazoquinones and arylboron compounds (Scheme 109L–N).650–652
An example of stereoselective C–H arylation of olefins was reported by Ackermann in 2016 using a preformed phosphinous acid Ru(II) complex as catalyst (Scheme 109O).653 This method could also be applied to aromatic substrates using tetrazoles and oxazolines as DGs.
15.1.1.2. Alkylation. Alkylation can occur via different mechanisms: (i) addition to unsaturated moieties, (ii) cross couplings, and (iii) carbene insertions. Due to the many protocols reported for such functionalisation with pyridine as DG, these strategies are treated separately. Cascade alkylation/cyclisation involving the pyridine DG will also be reported separately due to the different nature of the products obtained.
15.1.1.3. Addition to double bonds. In 2015 Nishimura reported the Ir-catalysed regioselective (branched products) hydroarylation of vinyl, allyl and other alkenyl ethers, leading to alpha-arylated ethers.654,655 This reaction could be performed in a stereoselective manner using chiral biphosphine ligands (Scheme 110A).
 | ||
| Scheme 110 Pyridine-directed C(sp2)–H alkylations via addition to double bonds. | ||
The group-directed addition to aldehydes has been reported a number of times, and results in alpha-arylated alcohols. A Lewis or Brønsted acid additive in these reactions is necessary to activate the carbonyl group, and reduce the reversibility of the aldehyde insertion into the C–M bond. Zhou656 and Ding657 reported the Ru-catalysed addition to formaldehyde and electron-poor benzaldehydes (Scheme 110B and C). The scope in aldehydes was notably increased by Wang et al., using a Mn catalyst and a combination of ZnBr2 and Me2Zn. This allowed the use of (hetero)aromatic, aliphatic, and even unsaturated aldehydes as reactants; moreover, heteroaromatic and olefinic substrates could also be easily functionalised (Scheme 110D).658 The synthesis of 2-oxoalkyarenes was achieved by Kakiuchi using cyclic alkenylcarbonates, synthetic equivalents of enolates (Scheme 110E).659 Despite the originality of the protocol, its use with pyridine as DG was only demonstrated on relatively few examples, and required a long reaction time (48 h).
An interesting protocol for a directed cascade C–H functionalisation/cyclisation was reported by Ellmann, giving diastereoselective formation of 3 contiguous stereocentres (Scheme 110F and G).660,661 The benzylamine in the second protocol acts as an organocatalyst (enamine catalysis) to facilitate the intramolecular Michael addition, which is more difficult than in the first case.661 These are two of the relatively rare examples where the cascade cyclisation does not involve the DG itself, which is often the case (e.g.Scheme 112).
Park and Chang reported the effect of different DGs on the chemoselectivity of the reaction of aromatic substrates with activated alkenes.662 While with pyridine DGs, addition to the double bond was obtained selectively (Scheme 110H), the use of an amide DG in the same conditions resulted in selective olefination instead. This switch in selectivity was limited to the Ir catalyst used. The same reaction, catalysed by Re, was instead reported by Zhu and co-workers (Scheme 110H).177 This protocol, occurring under CO atmosphere (CO acts as ligand for Re), is one of the few Re-catalysed C–H activation processes, and proved very efficient for a variety of substrates, including olefinic and heteroaromatic ones. In 2015 Bolm reported the hydroarylation of unactivated bicyclic alkenes with Ru catalysis using O2 as oxidant (Scheme 110I).663
The functionalisation of indoles with activated ketones or aldehydes (alpha EWG-substituted) under Mn catalysis was reported by Ackermann (Scheme 110J).664 Although high loadings of Mn catalyst were used (up to 20% of the metal), the reaction gave good yields with a broad scope, and no additives were used (cf.Scheme 110B–D). It is worth noting that the C2 regioselectivity, despite the DG, was only observed with two very specific Mn catalysts ([Mn2(CO)10] and [MnBr(CO)5]), while all the other catalysts tested resulted in C3 functionalisation (no DG effect).664
A similar procedure for the addition to maleimides was reported recently by Song and Gong (Scheme 110K).665 The reaction was air tolerant and worked effectively in EtOAc. Importantly, both these protocols were applied to the functionalisation of other heterocycles (e.g. pyrroles, selectively mono-functionalised).
Recently, the Glorius group developed two important methods for the C–H propargylation and 2-(indolyl)methylation of indoles using substituted bromoallenes and 4-ethynyl benzoxazinanones (Scheme 110L and M).666,667 Propargylation is a challenging transformation, as the corresponding allenylation might instead occur more easily. For this Mn-catalysed process, the use of BPh3 as Lewis acid and a catalytic amount of H2O proved crucial for obtaining good yields and selectivity (with respect to the allene product). The reaction works well with a variety of functionalised allenes, including 1,3,3-trialkyl, 3,3-dialkyl (giving terminal alkyne products), 1,3-dialkyl and 1-monoalkyl allenes, and with other heterocyclic substrates such as pyrroles and (benzo)thiophenes (Scheme 110L).666 A robustness screening was also performed for this reaction, showing compatibility with functionalities such as aniline, furan, ketones, amides and acetals. The use of 4-ethynyl benzoxazinanones, still under Mn catalysis, results in the C–H (2-indolyl)methylation of indoles and other heterocycles via alkyne insertion into the intermediate manganacycle, release of CO2, formation of an allene–Mn intermediate complex, and cyclisation with the nitrogen nucleophile (Scheme 110M).667 Even in this case a robustness screening showed compatibility with a variety of FGs.
Pyridine-directed, Ni-catalysed alkylation of 2-pyridones using a variety of aliphatic alkenes and dienes (including cyclooctadiene) was reported by Miura and Hirano in 2017.668 Despite being one of the few examples of alkylation on such substrates in recent times, the reaction proved fairly sensitive to the substitution pattern on the pyridone and the identity of the aliphatic alkenes/dienes used.
15.1.1.4. Cross coupling and carbene insertion. A number of alkylation reactions based on cross coupling type of reactions have been reported. A very recent example by Aissa's group demonstrated the use of sulfoxonium ylides as a replacement reactant for diazo compounds, leading to α-arylated aromatic or aliphatic ketones in good yields (Scheme 111A).669 This was the first example of the use of such reactants, safer alternatives to diazonium compounds,670 in group-directed C–H functionalisation. Li and Liu reported very similar conditions for the alkylation of arenes and 2-pyridones respectively with alkyltrifluoroborates (Scheme 111B and C).651,671 The C6 alkylation of 2-pyridones was also performed via carbene insertion using diazo compounds (Scheme 111D).672
 | ||
| Scheme 111 Pyridine-directed C(sp2)–H alkylations via cross-coupling and carbene insertion. | ||
The Ni catalysed functionalisation of indoles using alkyl halides was achieved by Punji and co-workers (Scheme 111E).673 Although being a rare example of Ni-catalysed alkylation, this method required the use of a preformed Ni-catalyst, catalytic amounts of strong Li-amide bases, 2 equiv. of base and high temperatures to result in medium to good yields of functionalised indoles. The use of secondary alkyl halides was also possible, but 2 equiv. of KI as additive were required in this case.673
The functionalisation of non-aromatic C(sp2)–H bonds is considerably rarer, and much underdeveloped. Yoshikai reported the alkylation of cyclic alkenes with alkyl chlorides using Co catalysis (Scheme 111F).674 Despite having benefits such as being base metal-catalysed and allowing functionalisation at room temperature, the reaction requires the use of both a catalytic carbene and a superstoichiometric amine ligand (TMEDA), has a limited scope, and presents some regioselectivity issues for secondary alkyl chlorides, and therefore lacks generality.
15.1.1.5. Alkylation/cyclisation. The functionalisation of C–H bonds with unsaturated moieties can result in the subsequent involvement of the DG in a cyclisation process between the newly installed functionality and the nucleophilic nitrogen atom of the pyridine. In 2015 Glorius investigated the use of diazomalonates in combination with Co catalysis. Instead of obtaining the expected alkylation product, the corresponding fused heterocycles, derived from nucleophilic attack of the pyridine to the ester moiety of the alkyl group, were obtained (Scheme 112A).675 Later the same group reported the use of pyridotriazoles as carbene precursors for a similar reaction. In this case, Rh catalysis had to be used, as Co pre-catalysts did not produce any cyclised or alkylated product (Scheme 112B).676
 | ||
| Scheme 112 Pyridine-directed alkylation/cyclisation. | ||
15.1.1.6. Allylation. A large variety of allylation procedures for the functionalisation of indoles have been reported in recent years, with a number of different reactants and leading to variedly functionalised side chains. Two main different classes of reactants have been utilised: alkenes with (1) a leaving group, or (2) a strained ring in alpha to the double bond. In both cases most mechanistic proposals involve a migratory insertion of the double bond into the metallacycle initially formed, followed by a general β-elimination of a suitable leaving group (β-oxygen, β-fluoro, β-bromo), or the release of ring strain via a β-carbon cleavage. For the latter, the β-carbon elimination is favoured over β-hydride elimination due to the release of the ring strain via ring opening. Alternative mechanisms occurring via oxidative additions have been suggested in some instances.677
In 2016 Kapur reported the allylation of indoles with allyl alcohols (unsubstituted, 1-monosubstituted, 1,1-disubstituted), which selectively resulted in non oxygenated products (Scheme 113A).678 The chemoselectivity arises from a selective β-hydroxide elimination rather than β-hydride elimination step in the mechanism. Advantages are regio-, chemo-, and diastereoselectivity, and the cleavage of the DG in a number of compounds.678 Many papers have appeared using Mn and Co catalysis, in combination for example with vinyldioxazolones679,680 (Scheme 113B), vinylcyclopropenes679,681,682 (Scheme 113C, Mn cat. shown), or diazabicyclic compounds679 (Scheme 113D). Among these, notable are the fluorinated side chains introduced with Ackermann's (Mn,683Scheme 113E, and Co684) and Zhang's (Mn, Scheme 113F)685 procedures.
 | ||
| Scheme 113 Pyridine-directed allylations on C(sp2) centres. | ||
15.1.1.7. Alkenylation. Olefination reactions can occur via different mechanisms, mostly oxidative dehydrogenative coupling with olefins (Fujiwara–Moritani coupling, mostly with activated olefins), and hydrofunctionalisation of alkynes. Zhang reported the use of unactivated olefins for the functionalisation of arenes (Scheme 114A).686 The use of such alkenes as reactants is rare, as most C–H olefinations employ activated styrenes and acrylates. Li and Xu reported the use of acrylic acids as reactants, in combination with pivalic anhydride as additive (Scheme 114B).687 The formation of vinylic anhydride or mixed pivalic/vinylic anhydrides as intermediate results in a decarbonylative olefination process. The method shows good scope and generally excellent yields. Co catalysis was used for the hydroarylation of aromatic alkynes (Scheme 114C).688 The hydrofunctionalisation of alkynes bearing a leaving group in the propargylic position can proceed in two different ways. In case a Brønsted acid is added to the reaction, the olefination product is obtained, as recently reported for the olefination of indoles (Scheme 114D).689 In such a case the leaving group is maintained in the molecule and can be utilised successively. In the absence of acids the corresponding allene is obtained instead, as in Scheme 114E.689,690 Finally, noteworthy is the C7 functionalisation of indolines using styrenes, reported by Loh (Scheme 114F).691
 | ||
| Scheme 114 Pyridine-directed C(sp2)–H olefination. | ||
15.1.1.8. Alkenylation/cyclisation. The hydrofunctionalisation of dialkyl- or diarylalkynes in the presence of Rh(III), Ir(III), or Co(III) catalysts, a (super)stoichiometric oxidant and a non-coordinating anion source can result, instead of the olefinated product, in cyclisation products (extended π systems), interesting for their fluorescent properties and potential applications in material chemistry.692 This process results in the formation of a C–C and a C–N bond in a cascade manner (Scheme 115A). Several examples have been reported recently, making use of diaryl- or dialkylalkynes,693–695 including the isolation of a 7-membered cobaltacycle intermediate (see general mechanism in Scheme 115, bottom).696 Interesting one-pot protocols have been reported by Cheng and Jun: in one case the formation of the olefinic substrate from an alkyl chain occurred in situ via oxidation, and in the other case the reaction of allylamine with two equivalents of the alkyne resulted in the formation of the 2-pyridinylarene substrate first, followed by olefination/cyclisation (Scheme 115B and C).697,698 Another cyclisation product can be obtained by using acrylates or similar as the olefination agent. As reported by Cheng, the Rh-catalysed reaction with 2 equivalents of acrylates in water and using O2 as oxidant, results in the formation of pyridoisoindolium salts (Scheme 115D).699 The process was proposed to occur via ortho olefination, followed by intramolecular Michael addition of the pyridine to the activated olefin, deprotonation, and finally a second, intermolecular, nucleophilic attack to a second molecule of the acrylate.
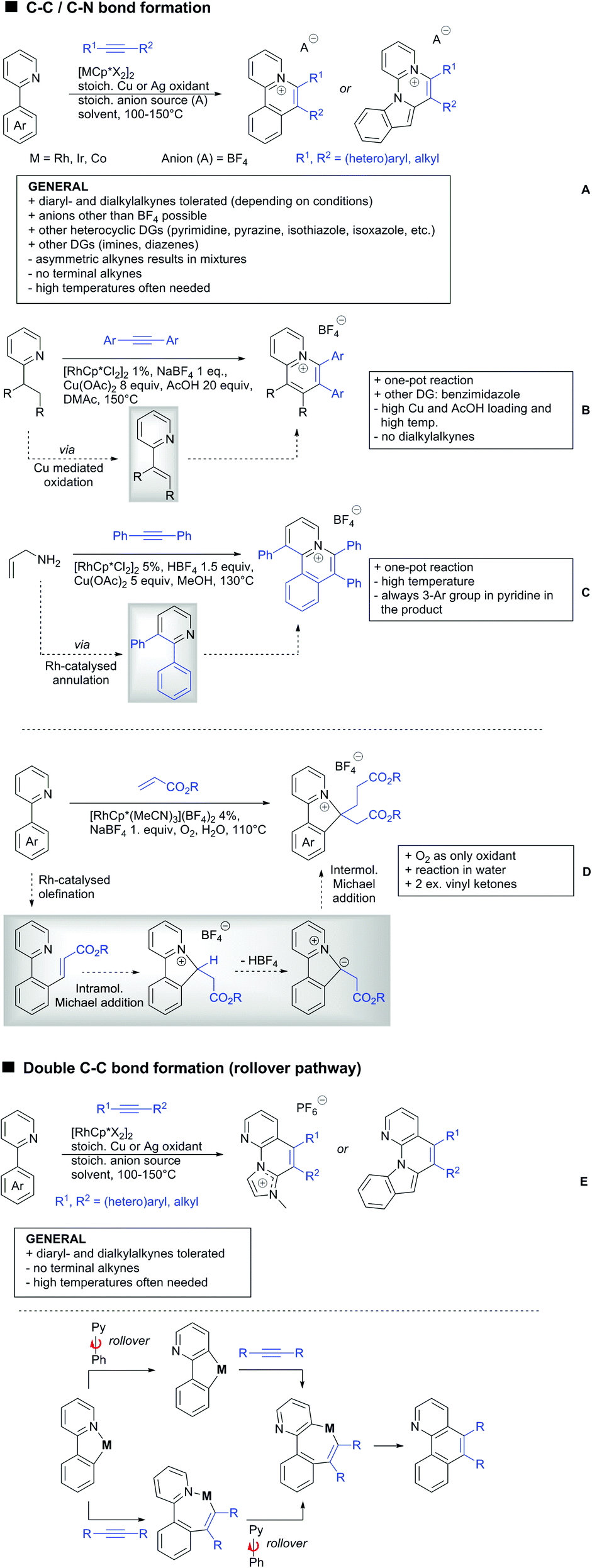 | ||
| Scheme 115 Pyridine-directed olefination/cyclisations via C–N bond formation or rollover pathways. | ||
Alternatively, the use of Rh might also result in the formation of two C–C bonds via a so-called “rollover cyclometallation” mechanism, where the nitrogen atom of the pyridine ring is not involved in the cyclisation, and can be used to promote a further C–H functionalisation (Scheme 115E). While the rollover mechanism is more common for doubly coordinating ligands, such as bipyridines,700 or in the case of a pyridine–carbene moiety like the one in Scheme 115E,701 its occurrence is far less common in other substrates (e.g. indole substrate in Scheme 115E),702 and is obviously facilitated by the absence of a non-coordinating anion in the reaction mixture.
15.1.1.9. Alkynylation. Generally speaking, alkynylation reactions in C–H functionalisation typically employ two main reactants: the hypervalent iodine compound TIPS-EBX (1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one), and TIPS-bromoacetylene. Variations include other silyl protecting groups instead of TIPS or, more rarely, aryl(bromo)acetylenes or alkyl(bromo)acetylenes. Recent additions to the reagent pool for this transformation include propargylic alcohols (see below). Although the silyl protecting group can be easily cleaved, and the resulting acetylene subsequently functionalised, this is still an intrinsic limitation of this type of functionalisation. The use of terminal alkynes was shown to result in the formation of considerable amounts of Glaser–Hay coupling products.703,704
Relatively few examples of alkynylation with a pyridine DG have been reported recently. In 2015 Li reported the C6-alkynylation of pyridones with TIPS-EBX with a Rh catalyst,705 while Ni-catalysed reaction of indoles with TIPS-bromoacetylene was reported by Punji706 (Scheme 116A and B). Wen reported the use of 1,1-dimethylpropargyl alcohols as alternative reactants for alkynylation reactions. This reactant avoids the use of halogenated compounds and only results in the formation of acetone as side product. The intermediate metal–alkyne complex is obtained via β-carbon elimination from the alcohol-ligated catalyst, as previously reported.707 Application of such propargylic alcohols to the functionalisation of indoles and carbazoles,703 although resulting in relatively low and substrate-dependent yields, allows a larger variety of alkynes to be used (alkyl and aryl substituted, Scheme 116C and D) than the classical reactants. Noteworthy in this case are the monofunctionalisation of carbazoles (carbazoles get usually doubly functionalised) and the necessity of a 6-Me pyridine DG for the indoles (without the 6-Me another transformation occurs704).
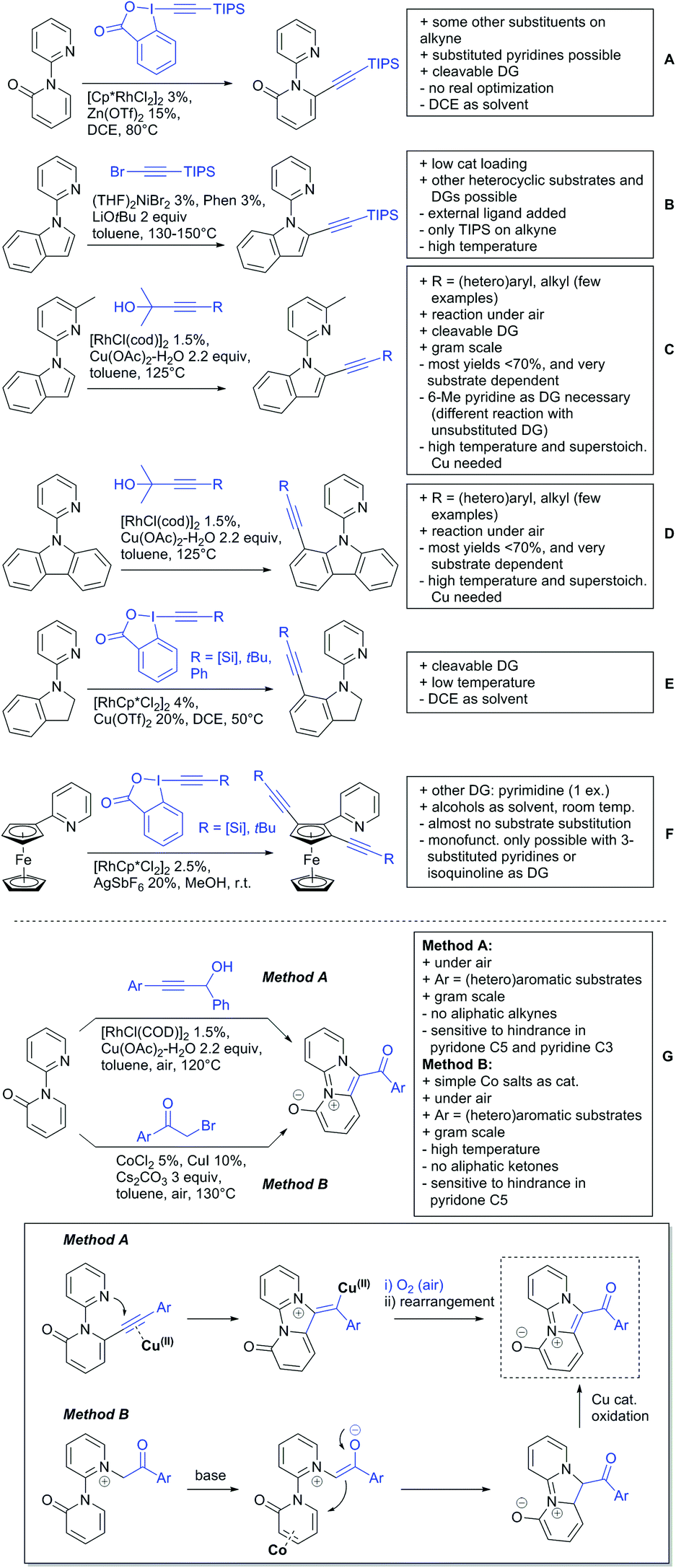 | ||
| Scheme 116 Alkynylation and alkynylation/cyclisation using pyridine-based DGs. | ||
Finally, alkynylation of indolines (C7) and ferrocenes have been reported by Loh and You using R-EBX (R = [Si], tBu) hypervalent iodine species (Scheme 116E and F).691,708
Cascade alkynylation–cyclisation reactions are also possible. For instance, two methods for the synthesis of dipyridoimidazolium compounds were reported by Li (Scheme 116G).709,710 Despite giving the same product, the two methods are mechanistically very different. While Method A is technically a cascade directed alkynylation/cyclisation (Rh–alkyne complex is obtained via β-carbon elimination from the alcohol-ligated intermediate707), Method B was proposed to involve pyridine alkylation first, followed by enolisation and nucleophilic attack of the enol to the Co-activated pyridone ring, and Cu-mediated oxidation.
15.1.1.10. Cyanation. Classical reactions for the cyanation of organic compounds are the Sandmeyer or the Rosenmund von Braun reaction, or cross coupling reactions between aryl halides and cyanide salts. All methods, in their classic version, require prefunctionalised substrates, and more importantly, the use of inorganic cyanides (very toxic) in the reaction. Although a number of (safer) reactants have been recently utilised for the cyanation of C–H bonds, still a common feature is the atom uneconomic nature of these reactants, where more than 50% of the reactant mass is actually wasted.
Co-catalysed reactions were reported by Ackermann711a and Chang711b in 2015, employing respectively N-cyano-N-phenyltosylamide (NCTS) and N-cyanosuccinimide (Scheme 117A and B). While the first protocol required a lower amount of additives and catalyst, the use of a more stable reactant, and the good FG tolerance in the second protocol are noteworthy. A third protocol, reported by Jiang and co-workers, employed ethyl(ethoxymethylene)cyanoacetate as a new reagent under aerobic oxidation conditions, but unfortunately stoichiometric Cu salts were required (Scheme 117C).712 Similar conditions (superstoichiometric Cu, air, 5% Pd cat.) were reported for the use of iminonitriles as cyanating agent (Scheme 117D).713 The generation of cyanide ions from these two reactants has not been yet fully understood, but it appears to occur through oxidative processes. The cyanation of indoles with NCTS was reported by Ackermann714 and Anbarasan715 using Mn/Zn and Rh catalysis respectively (Scheme 117E and F). Notable features are the functionalisation of tryptophans in the first case and the substrate scope of substituted pyrroles (often only explored as secondary substrates) in the second. Anbarasan also reported the cyanation of non-aromatic C(sp2)–H bonds in both cyclic and acyclic olefins using a similar procedure (Scheme 117G).716 This reaction was also applied to the formal synthesis of chlorpheniramine-based hystamine antagonists.
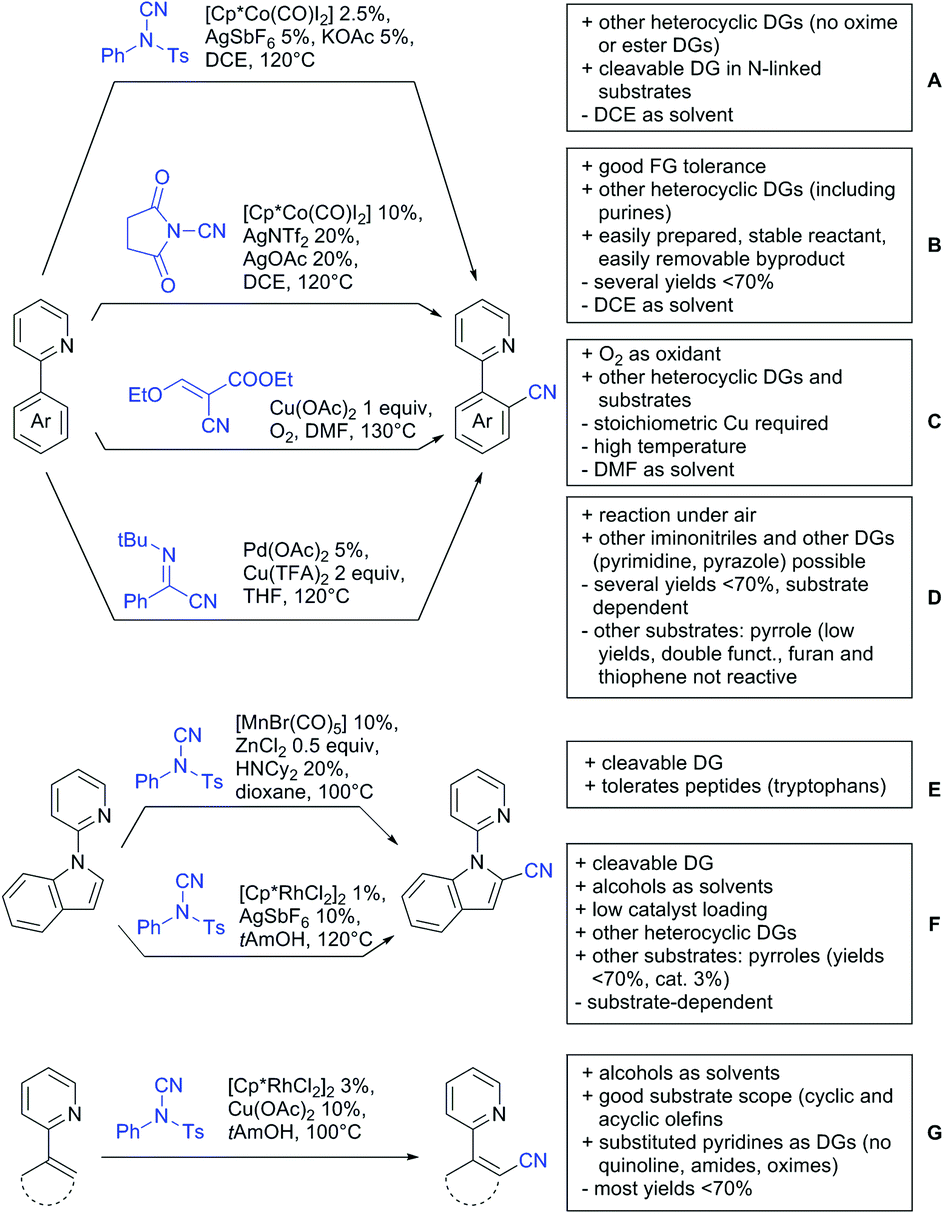 | ||
| Scheme 117 Pyridine-directed ortho cyanation of C(sp2)–H bonds. | ||
15.1.1.11. Acylation. This transformation is commonly achieved by using aldehydes or aldehyde precursors. An H-atom abstraction by an oxidant delivers the acyl radical, which then reacts with the cyclometallated Pd complex, giving a Pd(IV) intermediate (Scheme 118, bottom). A subsequent reductive elimination furnishes the acylated arene.
 | ||
| Scheme 118 C(sp2)–H acylation using pyridine as DG. | ||
Aldehydes were generated in situ by aerobic oxidation of toluene to benzaldehyde in Jiao's protocol.717 A heterogeneous version of this reaction was reported by Zhaorigetu, using tert-butylperoxybenzoate (TBPB) as oxidant (Scheme 118A).718 A decarboxylative procedure was instead developed by Liu, using mandelic acid derivatives as reactants (Scheme 118B).719 The use of aldehydes themselves was reported by Deng in water, using sodium dodecylsulphate (SDS) as a phase-transfer catalyst/surfactant and tert-butylhydroperoxide (TBHP) as oxidant (Scheme 118C).720
Exceptions to the general mechanism were reported by Li and Wang, where the acylation product was obtained by formal addition to ketenes (also applied to C(sp3)–H bonds)721 and nitriles (followed by hydrolysis)658 using Rh and Mn catalysis respectively (Scheme 118D and E).
Following the classical protocol with aldehydes, Dash demonstrated the functionalisation of carbazoles, resulting in double acylation (Scheme 118F).722
15.1.1.12. Alkoxy- and aminocarbonylation. Although other examples appeared with other DGs, only a few reports on pyridine-directed alkoxy- and aminocarbonylation were published recently. One example of the former was published by Wu in 2016, making use of Mo(CO)6 as CO surrogate, and requiring the superstoichiometric addition of benzoquinone (BQ), NaOAc and Ag2CO3 (Scheme 119A).723 This protocol worked for a variety of aliphatic alcohols, but not for other nucleophiles. The aminocarbonylation of similar substrates was recently achieved by Driver using Mn(CO)6 and nitroarenes (Scheme 119B).724 Nitrosoarenes, formed in situ by reduction by Mn, were identified in this case as the reactive species. In 2015 Ackermann reported a Mn-catalysed aminocarbonylation of indoles with isocyanates,725 one of the most commonly used reactants for this type of reactions.726 This reaction only required the catalyst and solvent, without the addition of any other additives or ligands (Scheme 119C).725
 | ||
| Scheme 119 Pyridine-directed alkoxy- and aminocarbonylation. | ||
15.1.1.13. C–S bond formation. Very recently, Matsunaga727 and Wang728 reported the first protocols for Co-catalysed trifluoromethylthiolation of (hetero)aromatic substrates using (PhSO2)2NSCF3 and AgSCF3 as reactants (Scheme 120A and B). In both cases, halogenated solvents had to be used and relatively low yields (<70%) were obtained.
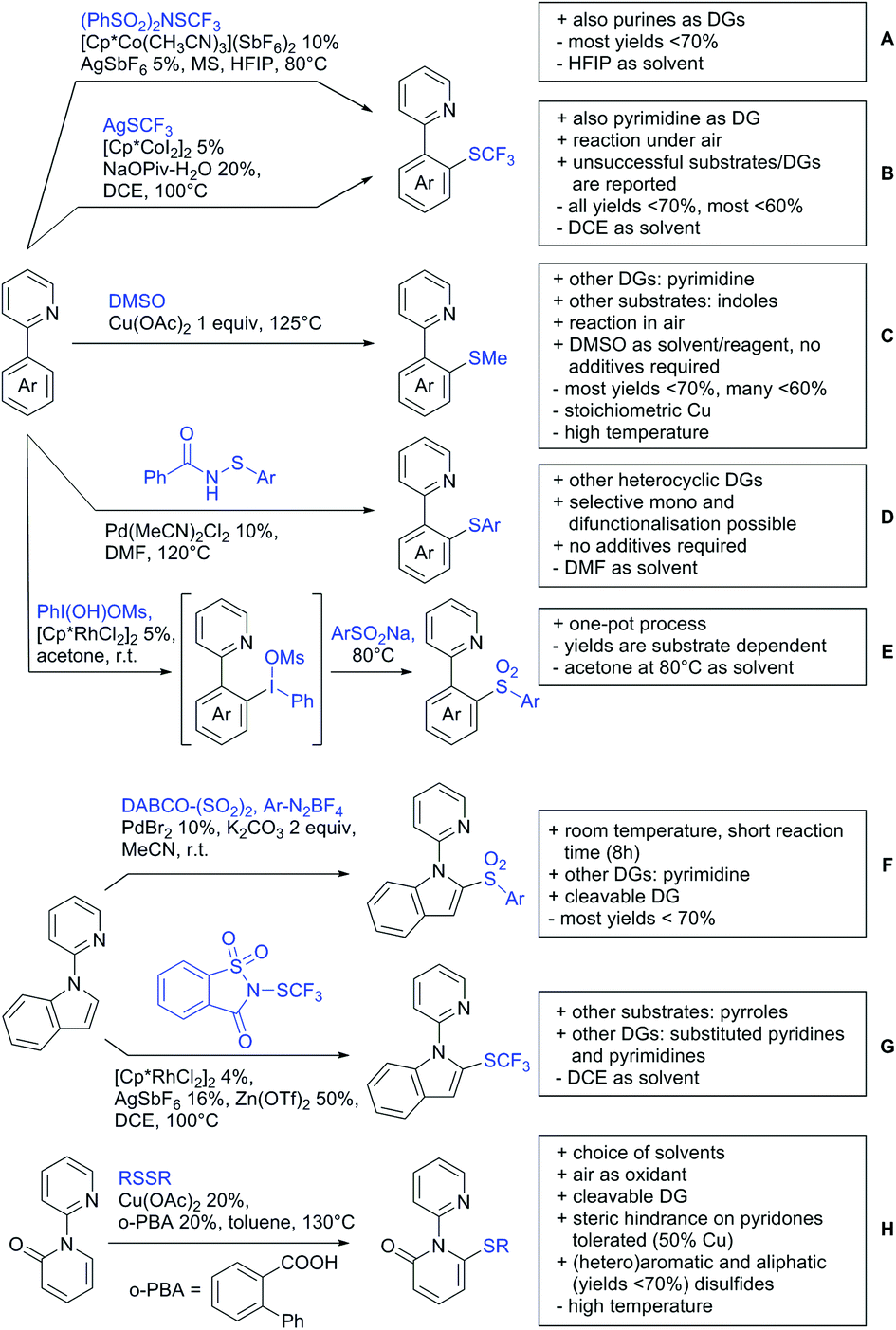 | ||
| Scheme 120 C(sp2)–S bond formation with pyridine DGs. | ||
The use of DMSO as reactant for the methylthiolation of similar substrates was reported by Jain via a Cu-mediated reaction (Scheme 120C).729 Arylthiolation was reported by Zhang using N-(arylthio)benzamides,730 while sulphonylation can be performed via an Umpolung reaction with hypervalent iodine compounds, followed by reaction with sodium sulphinates731 (Scheme 120D and E). Notably, the latter method is also suitable for the reaction with other nucleophiles.
The introduction of arylsulfonyl functionalities on indoles was recently reported with another method, using DABCO·(SO2)2 and aryldiazonium tetrafluoroborates in a multicomponent reaction catalysed by Pd(II) (Scheme 120F).732 The reaction is initiated by the formation of an arylsulfonyl radical, which is then introduced at the indole C2 position.
Trifluoromethylthiolation of indoles and pyrroles was reported by Li using N-(trifluoromethylthio)saccharin under Rh/Lewis acid catalysis (Scheme 120G).733 Interestingly, the functionalisation of 3-substituted indoles also occurred in the absence of Rh and Ag additive, suggesting that Lewis acids alone could promote the reaction on such compounds.
Finally, Liu demonstrated the aryl- and alkylthiolation of 2-pyridones with disulfides under Cu/Brønsted acid catalysis (Scheme 120H).734 It is worth mentioning that, although some of these reactions required an air atmosphere, an O2 atmosphere proved deleterious for the reaction,728,729,734 presumably due to sulphur oxidation in the reactants/products.
15.1.1.14. C–B and C–Si bond formation. Organosilanes and organoboranes have important applications in organic chemistry, for example in cross coupling reactions, and C–H borylation and silylation have therefore been considerably investigated even before 2015.31,735 Pyridine-directed C(sp2)–H borylation and silylation have been reported by a number of research groups. The compounds obtained in this way, which often contain an internal Lewis acid–base interaction as shown in Scheme 121, also have applications in material science, due to their fluorescent properties.736–739 Due to these non-covalent interactions, the C–H functionalisation process itself has been suggested to occur with two main different mechanisms. In the first one, the reaction is initiated by the C–H cleavage/formation of a DG-coordinated metallacycle (metal = catalyst, as for most of group-directed functionalisations), followed by insertion of the boryl/silyl group.740–743 The other mechanism assumes the Lewis acid–base interaction as the initial step, followed by catalyst coordination/C–H cleavage, on the basis of non-successful metallacycle formation or to early stage Lewis acid–base interactions observed spectroscopically.739,744
 | ||
| Scheme 121 Si–N and B–N non-covalent interactions during pyridine-directed functionalisations. | ||
Borylation employs diboranes or boranes as reactants; in the latter case, the addition of a hydrogen acceptor (generally an olefinic compound) to promote the reaction (as commonly used for silylation with hydrosilanes, vide infra) might instead result in complete inhibition due to hydroboration of the unsaturated moiety.739 Several examples of borylation of 2-phenylpyridines and analogues have appeared in the recent literature. Murata and Ackermann reported the use of HBpin or B2pin2 in combination with Ru catalysts (Scheme 122A and B),740,742 however only a few examples of C(sp2)–H borylation were shown (Ackermann's protocol was nonetheless also applied for C(sp3)–H borylation, see Scheme 136).
 | ||
| Scheme 122 Pyridine-directed borylation of C(sp2) centres. | ||
Kuninobu reported an interesting Fe-catalysed protocol using 9-BBN (9-borabicyclo[3.3.1]nonane) as borylating agent (Scheme 122C),739 and Miura/Hirano disclosed a Rh-catalysed borylation of 2-pyridones (Scheme 122D).741 The use of the reaction products as substrates in a Suzuki coupling nicely demonstrates the potential for applications in synthesis.
Typical reactants for silylation are disilanes or hydrosilanes, the latter generally requiring the addition of a hydrogen acceptor to be efficient (norbornene is often used for this role, in superstoichiometric amounts).735 Hydrosilanes/norbornene combinations have been used for the Ir-catalysed silylation of 2-arylpyridines by Oro/Iglesias and Kuninobu/Kanai (Scheme 123A and B).743,744 In the first case a preformed NHC–Ir(III) hydride complexes was used as catalyst for the silylation of 2-phenyl- and 2-thienylpyridines with a variety of silanes, ranging from triethyl to triphenyl and trialkoxy silanes. Interestingly, the catalyst also proved efficient for a variety of undirected C(sp2)–H silylations on (hetero)arenes.743 Kuninobu/Kanai's protocol was developed instead for diarylfluorosilanes, and was not effective for non-fluorinated reactants.744
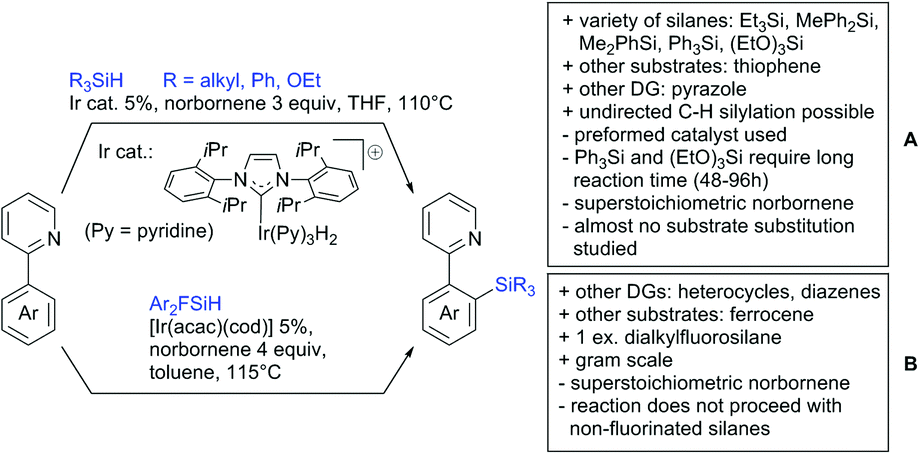 | ||
| Scheme 123 Pyridine-directed silylation of C(sp2) centres. | ||
15.1.1.15. C–N bond formation.
15.1.1.15.1. Amidation. While until 2014 a number of procedures were reported for C–H amidation using acyl azides as nitrene precursors, recently a number of different reactants have been employed for this transformation. Reactions with aminated hypervalent iodine reactants have been reported by Loh and DeBoef using catalytic Rh and stoichiometric Cu respectively, for the synthesis of N-aryl sulfonamides and phthalimides (Scheme 124A and B).745,746 A reaction with the electrophilic nitrogen source N-Boc-O-2,4-dinitrophenylhydroxylamine in water under Rh catalysis was developed by Lu (Scheme 124C).747 Notable features of this procedure are: the use of water as solvent, the good FG tolerance, and the possibility of functionalisation of purine-based nucleosides. Chang used a similar hydroxylamine reactant, N-Boc-O-acetylhydroxylamine, in the Co-catalysed amidation of arenes: in this case a variety of carbamates (other than N-Boc) could be employed in the reaction, and purines could be used as DG (Scheme 124D).748
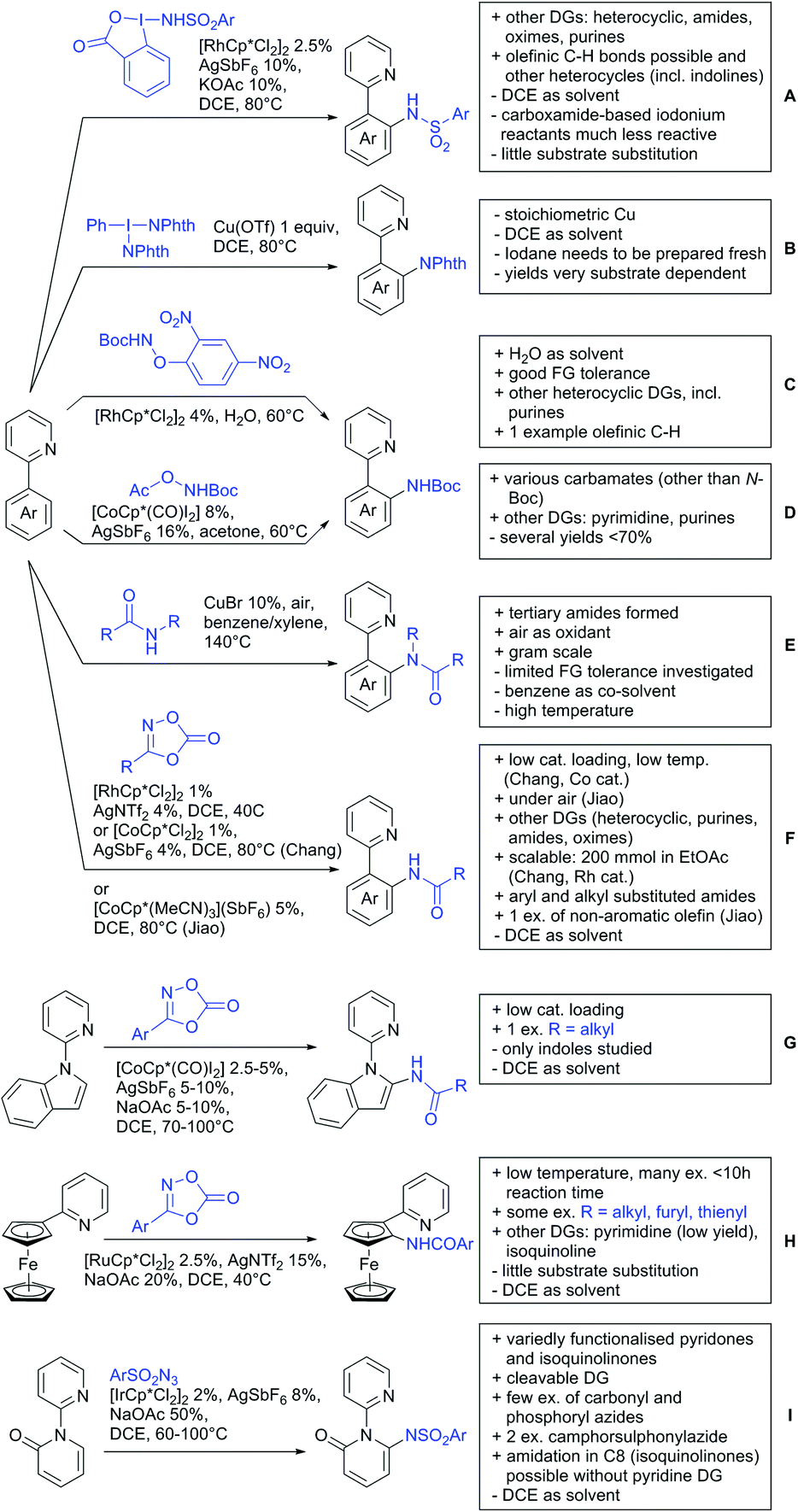 | ||
| Scheme 124 Amidation reactions with pyridine DGs. | ||
The Cu-catalysed dehydrogenative C–H amidation with secondary amides has been reported by Chen using air as the sole oxidant (Scheme 124E).749 Here the formation of tertiary amides is remarkable, as most protocols result in the formation of secondary amides instead.
In 2015 Chang introduced dioxazolones as reactants for C–H amidation. Dioxazolones are safer and more reactive than the corresponding acyl azides, due to their relative stability and stronger coordination to the metal centre, which promotes faster reactions. Moreover, these reactants are simple to prepare and to handle. Chang750–752 and Jiao753 reported their use in the directed amidation of arenes using Rh and Co catalysts, showing the reaction to be suitable for a number of different DGs (including purines, amides, and oximes, Scheme 124F). Notably, Chang reported the use of EtOAc as alternative solvent for a 200 mmol scale reaction.751 Ackermann subsequently applied a similar procedure to the Co-catalysed amidation of indoles,754 while You employed Rh for the functionalisation of ferrocenes755 (Scheme 124G and H).
Very recently, the amidation of pyridones (C6) and isoquinolinones (C3) was reported with sulphonyl azides under Ir catalysis (Scheme 124I).756 Interestingly, C8-functionalisation of isoquinolinones could be performed in similar conditions in the absence of the heterocyclic DG.
15.1.1.15.2. Amination. C–H amination reactions also typically involved nitrenes obtained in situ from azides. For example, Lu demonstrated the reaction of 2-phenylpyridines with aryl azides in water, also allowing for a double ortho-functionalisation with two different azides (Scheme 125A).757 The use of anthranils (2,1-benzisoxazoles) as alternative, more stable nitrene precursors was independently reported by Wang and Li.758,759 The reaction of indoles with anthranils using polar protic solvents (H2O or MeOH) and Rh catalysis resulted in a tandem amination–cyclisation process, leading to the formation of indoloquinolines (Scheme 125C).758,759 The same reaction was also shown to occur on 2-pyridones (Scheme 125D).759 Interestingly, the cyclisation did not occur when phenyl or thienyl substrates were investigated, and the reaction only resulted in the corresponding o-formylaniline-substituted product (Scheme 125B).758
 | ||
| Scheme 125 Amination and related reactions with pyridine DGs. | ||
15.1.1.15.3. Azidation. Narula reported in 2015 a protocol for the Cu-catalysed azidation of 2-phenylpyridines using benzotriazole sulfonyl azide (BtSO2N3) and K2S2O8 as oxidant (Scheme 126).760 NaN3 and 1-imidazole sulfonyl azide (ImSO2N3) also proved effective in these conditions.
 | ||
| Scheme 126 Azidation of aranes using pyridine as DG. | ||
15.1.1.16. C–O bond formation. Examples of C–O bond formation are somewhat limited in comparison to C–C and other C–heteroatom bond forming procedures, with yields often <70% or even <50%, possibly due to the strong coordinating ability of O, which might inhibit the catalysis by preventing reductive elimination or forming relatively stable complexes.761,762 Nonetheless, interesting advances are being made in terms of greener oxidants, cheaper base metal catalysts, and milder reaction conditions.
A number of papers on pyridine-directed C–O bond formation reactions on (hetero)arenes have been reported using Ru/PIDA,761 solid-supported Pd/PIDA763,764 or simple Pd salts/peroxides765,766 combinations, leading to ortho acetoxylated, hydroxylated or alkoxylated compounds (in case of PIDA as oxidant, when H2O or an alcohol are used as solvent, one-pot hydrolysis or alcoholysis of the acetoxylated product is obtained).
In the past few years, some reports appeared where O2 was used as oxidant. Shen reported a direct alkoxylation with alcohols (only 3-methylpyridine could be used as DG, Scheme 127A),767 while Guis used aldehydes/O2 as reagents in combination with Pd catalysis.768 In this case the reaction occurred via formation of an intermediate acyl peroxo-radical, followed by its reaction with the arene, ultimately resulting in hydroxylated compounds (Scheme 127B). Patel disclosed the benzoxylation of arenes using benzylethers and TBHP as oxidant under Cu catalysis (Scheme 127C).769 Although limited in yields and scope, this protocol is interesting as it shows different product selectivity from its Pd-catalysed counterpart (which gives acylated products).770 The formation of the ester product from the benzylic ether derives from Cu/TBHP mediated oxygenation to benzaldehyde and further to t-butyl arylperoxyester. The arylcarboxy radical generated by homolitic cleavage of this intermediate is finally introduced at the ortho position in the substrate.769 A very recent remarkable paper by Li and Xu demonstrated the acyloxylation of 2-phenylpyridines and analogues with carboxylic acids and Rh catalysis (Scheme 127D).234 Though using a superstoichiometric amount of Ag as oxidant, this protocol was applicable to a broad range of carboxylic acids: aliphatic (incl. saturated heterocyclic rings), (hetero)aromatic and vinylic reacted in good yields, often >80%. Moreover, a range of DGs proved effective in this protocol, and amides were used for the acylation of olefinic substrates, although in modest yields (<70%).234
 | ||
| Scheme 127 Pyridine-directed C(sp2)–O bond formation. | ||
Finally, the acetoxylation of indoles (2-substituted, C7 functionalisation) and carbazoles was reported by Miura using acetic acid, Ru catalysis, and Ag oxidant (Scheme 127E).771 It is worth noting that the functionalisation of unsubstituted indoles was not observed, either in C7 or in C2.
15.1.1.17. C–Halogen bond formation. C–H halogenation, as for arylation reactions, also suffered from the formation of mixtures of mono and di-halogenated compounds. In recent years, a few synthetic methods showing good selectivity were reported by a number of research groups and in different conditions. The use of simple and cheap NaX salts as reactants for the introduction of Cl, Br and I reported by Wang in 2015 is certainly a landmark (Scheme 128A).772 This method is the most atom economic reaction available in the series, although a limited substrate scope was investigated. The use of N-halosuccinimides as halogenating reactants using Pd supported on Cr- or Fe-based MOFS as heterogeneous catalysts was investigated by Martin-Matute (Scheme 128B).773 Although having a number of clear advantages, such as mild conditions and recyclability of the catalyst, the catalytic results are unfortunately considerably dependent on the substrate and DG employed, presumably due to the steric hindrance of the starting materials and the rigid structure of the MOFs. It is worth mentioning that, although the catalysts could be reused for several runs, some degradation of the solid structures was observed, as well as its halogenation as a side process. Similar reactions, using Pd@CNT (carbon nanotubes)763 or other Pd@MOFs (Zr-based)774 were reported by Ellis and Cohen.
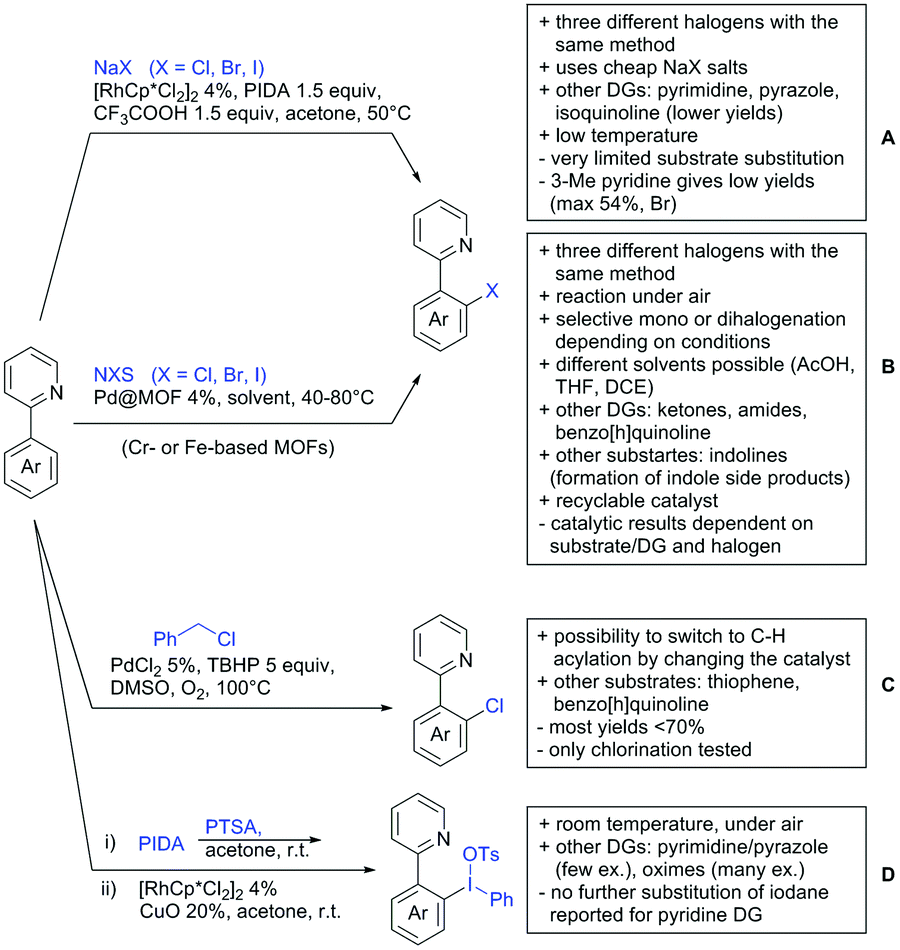 | ||
| Scheme 128 Pyridine-directed C(sp2)–H halogenation reactions. | ||
Wu reported the use of benzyl chloride as halogenating agent under oxidising conditions (Scheme 128C).775 While using PdCl2 as catalyst, and under O2 atmosphere, selective C–Cl bond formation was obtained, switching the catalyst to a preformed palladacycle with a ferrocenylimine ligand and adding a base under air, resulted in the selective acylation of the C–H bond (not shown).775 In the chlorination case, the intermediate tBuOCl is suggested as actual reactant, while in the second case the formation of an acyl radical is responsible for the reaction. In 2015 Li reported the synthesis of diaryliodonium salts starting from PIDA, PTSA (p-toluensulfonic acid) and a DG-equipped arene under Rh catalysis (Scheme 128D).776 These compounds can be used as intermediates for the formation of C–C, C–O, C–N, C–S and C–P bonds using a variety of nucleophiles (see also Scheme 120E). It is worth noting, however, that many procedures have been reported for the direct formation of such bonds with the same DGs, and the synthetic utility of this two-step procedure is therefore rather limited.
8-Methylquinolines are the most common substrate investigated, and its functionalisation is generally easier than that of 2-ethylpyridines, due to its benzylic nature. Typically, substitution on the aromatic rings is tolerated, although in some instances strong EWG substituents and steric hincrance in C7 or C2 can inhibit the reaction to various degrees. In general, apart from a few cases, substitution at the benzylic position (e.g. 8-ethylquinoline) strongly suppresses C–H activation.
2-Ethylpyridines (and in general any other flexible substrate), on the other hand, present the intrinsic issue of the flexibility of the alkyl chain, which makes the coordinating ability of the azine less efficient (metallacycle formation). In such substrates, monofunctionalisation may also pose some challenges, due to the lack of steric hindrance in close proximity. These issues are generally tackled by using branched alkyl chains instead of linear ones, to reduce flexibility/rotation (Thorpe–Ingold effect) and increase the steric hindrance around the desired C–H bond.
As C–C bonds connect the azine to the functionalisable C–H bonds, the DG is not cleavable in such substrates. Quinoline substrates can, however, be easily reduced to tetrahydroquinoline with NiCl2/NaBH4,781–783 H2/Pd@C,721 H2/PtO2,784 or NaBH(CN)3/BF3–Et2O,785 hereby enlarging the application scope. The same reduction is in theory possible for 2-ethylpyridine derivatives, to produce piperidines, but it has not been used in the context of C–H functionalisation.
15.1.2.1. Arylation. A few arylation procedures have been reported by Glorius, Shi/Xia and You under Rh/Ir catalysis, for the functionalisation of both 8-methylquinolines and 2-ethylpyridines. In 2015 Glorius reported the Rh-catalysed arylation of both substrates with aryl boroxines (Scheme 129A).786 Despite the use of up to 2 equiv. of boroxine (6 equiv. Ar) and superstoichiometric Ag as oxidant (due to boroxine homocoupling as a side reaction) the reaction gave good yields for both substrates. While only 8-benzylquinoline was functionalised for this class of substrates, both branched and linear 2-ethylpyridine analogues were tested in the reaction: branched substrates reacted to give selectively monoarylated products, while linear-chain substrates resulted in varying amounts of diarylated product.786
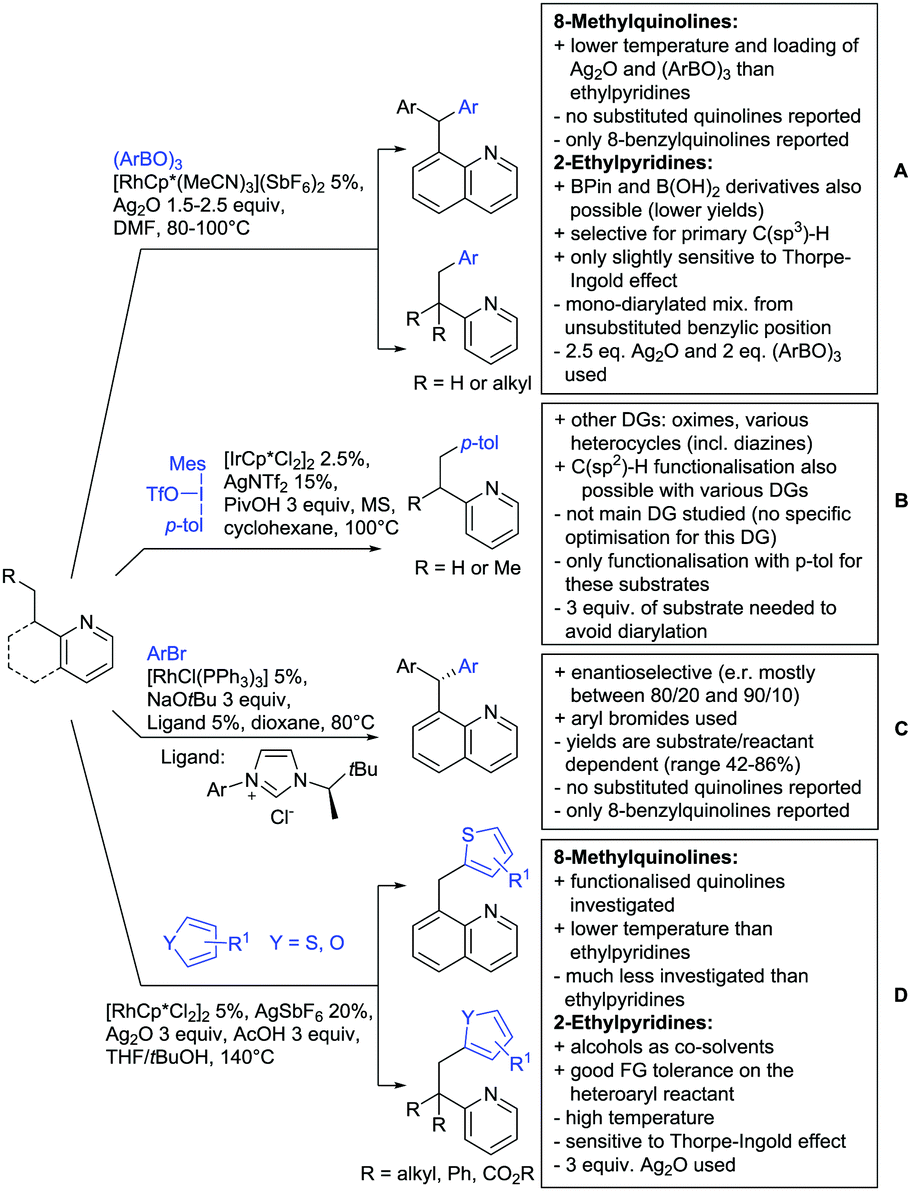 | ||
| Scheme 129 Arylation of 2-ethylpyridines and 8-methylquinoline derivatives. | ||
The arylation of 2-ethylpyridines with diaryliodonium salts was reported by Shi under Ir catalysis (Scheme 129B).787 This reaction, although requiring 3 equiv. of the substrate to avoid difunctionalisation, works well with a variety of azines (pyridines, pyrazines, quinolines, 73–95% yield) and azoles (pyrazole, isoxazole, 45–52%). The possibility to introduce aromatic rings other than p-tolyl was demonstrated using aliphatic oximes as substrates.787 In 2016 Glorius reported another protocol for the arylation of 8-benzylquinolines, this time with aryl bromides (Scheme 129C).788 The use of a chiral carbene ligand allowed the formation of enantio-enriched products (e.r. between 80/20 and 90/10). This reaction proved to be fairly substrate sensitive, with yields ranging between 42 and 86%, depending on the substituent on the benzylic ring of the substrate and the aryl bromide used.788
Finally, an interesting protocol from the You group specifically aimed at the heteroarylation of 2-ethylpyridines and 8-methylquinolines appeared in 2017 (Scheme 129D).789 Apart from the specific functionalisation with substituted thiophenes, benzothiophenes and furans, this is a challenging dehydrogenative coupling between a C(sp3)–H and a C(sp2)–H bond. Whereas the high temperature (140 °C), the 3 equiv. of oxidant (6 equiv. Ag overall) required, and the sensitivity to the Thorpe–Ingold effect can be seen as drawbacks, very notable features are: the use of alcohols as cosolvents for 2-ethylpyridines, the use of substituted quinolines as DGs, and the FG tolerance on the heteroarene partner, which includes esters, amides, aldehydes, halides and alkenes, furnishing yields in most cases >60%.789
15.1.2.2. Alkylation. For alkylation, 8-methylquinoline is the substrate of choice and, to our knowledge, no alkylation on 2-ethylpyridine derivatives have appeared in the last 3 years. Investigations using typical reactants such as α,β-unsaturated carbonyls and diazocompounds have been undertaken. A notable exception is the use of cyclopropanols as alkylating agents, reported by Li.790 In this case only two examples of C(sp3)–H alkylation were reported, thus this method is not covered in Scheme 130. Another interesting exception is the use of allylic alcohols as alkylating agents, reported in 2017 by Kim (Scheme 130A).783 Apart from the new reactant used, this reaction occurs in water under air and a range of substituted quinolines could be functionalised in good yields. The use of alcohols other than 3-buten-2-ol required a mixture H2O/PEG (polyethylene glycol) as solvent, and resulted in generally lower yields (<70%).783
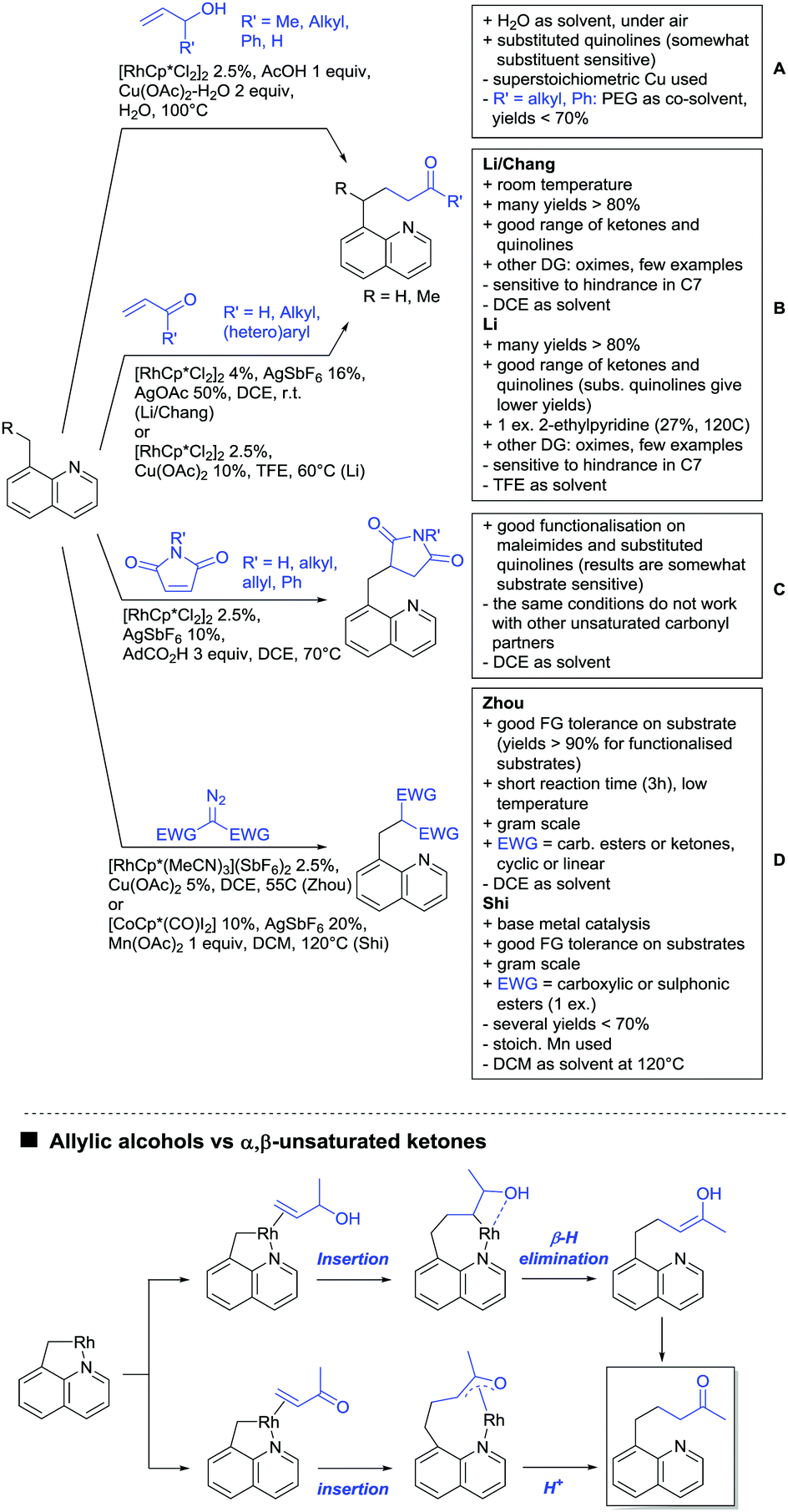 | ||
| Scheme 130 Alkylation of 8-methylquinoline derivatives. | ||
Two very similar protocols for the alkylation with α,β-unsaturated ketones were reported by Li/Chang and Li (Scheme 130B).781,791 Both protocols, using Rh catalysis in halogenated solvents at low/mild temperatures gave generally high yields of the desired alkylated quinolines. Somewhat lower yields were obtained with functionalised substrates, in particular sensitivity to steric hindrance in C7 was noted in both cases. The reactions proceeded well with aliphatic and (hetero)aromatic vinyl ketones.781,791 The formation of the same product starting from both vinyl alcohols and α,β-unsaturated ketones is due to different mechanisms. For the latter, the formal 1,4-addition product is obtained by migratory insertion of the double bond into the initial rhodacycle, followed by hydrolysis (protodemetallation). In the case of allylic alcohols, a migratory insertion/β-hydride elimination is the suggested pathway to the alkylated final products (Scheme 130, bottom).
The use of maleimides as coupling partners has been reported by Kim in 2016, leading to succinimide-functionalised methylquinolines (Scheme 130C).782 Although the reported conditions did not prove effective with other α,β-unsaturated carbonyl partners, the reaction proceeded in good yields with a range of N-functionalised maleimides and substituted quinolines (strong EWG substituents gave considerably low yields).
Two papers appeared recently on the alkylation of 8-methylquinolines making use of diazocompounds, one under Rh and the other under Co catalysis (Scheme 130D).784,785 Both protocols are characterised by a good FG tolerance on the quinoline substrate; for instance, strong EWG substituents can still lead to high yields in the Rh-catalysed process,784 while a variety of useful functionalisations are tolerated in the Co-catalysed process, such as alkenes, alkynes, free NH2 and Bpin.785
15.1.2.3. Alkenylation and alkynylation. Due to inherently poor reactivity and selectivity, olefination of C(sp3)–H bond is in general rare, and only a few papers have appeared in the literature specifically on the introduction of these functionalities on 8-methylquinolines. Sundararaju reported a Co-catalysed addition to internal alkynes, generating olefins in a stereoselective (syn) manner (Scheme 131A).792 The reaction with asymmetric alkynes resulted in varying degrees of regioselectivity, depending on the substituents. Interestingly, one example of addition to a terminal alkyne was also reported, albeit in 30% yield.
The reaction with geminal difluoroalkenes (F as leaving group) under Rh catalysis was developed by Li and Wang, and also occurred with good stereoselectivity for a variety of substituted 8-methylquinolines (Scheme 131B).793 Interestingly, a few examples of heteroaromatic (32–35% yield) and fluoroalkyl-substituted (94%) alkenes were demonstrated. Surprisingly, it was also shown that fluorine can act as a leaving group even in the presence of a tosylate group, which remained unaltered in the product.793
 | ||
| Scheme 131 Olefination and alkynylation of 8-methylquinolines. | ||
Only one example of alkynylation of 8-methylquinolines has been reported recently (Scheme 131C).794 A preformed Pd(II) catalyst was utilised for the coupling with TIPS-substituted acetylene bromide. A range of differently substituted quinolines (including alkene and ester substituents) could be coupled efficiently under these conditions. The Cu(OAc)2 in superstoichiometric amounts, despite being mentioned as oxidant, probably has another role, as no oxidant should be required for this transformation.
15.1.2.4. Acylation. Li reported two protocols for the C(sp3)–H acylation of 8-methylquinolines and 2-ethylpyridines, using relatively reactive ketenes or cyclopropenones with Rh catalysts (Scheme 132A and B).721,795 In both protocols substituted 8-methylquinolines were well tolerated, even at the C7 position. The acylation with cyclopropenones, although not particularly high-yielding, allows the introduction of enones with a single reactant, and can also be applied to 2-ethylpyridine derivatives.
 | ||
| Scheme 132 Acylation of 8-methylquinolines and 2-ethylpyridines. | ||
15.1.2.5. C–N bond formation. A relatively large number of examples of C(sp3)–N bond formation in 2-ethylpyridine or 8-methylquinoline derivatives have recently appeared in the literature. With a few exceptions, these mostly occur under Rh catalysis, using dioxazolones, amides, azides, hypervalent iodine compounds (amidation), anthranils (amination), and organic nitrites (nitration) as reactants.
15.1.2.5.1. Amidation. Amidation reactions have been studied with a range of nitrene precursors, such as azides, dioxazolones, and amidated hypervalent iodine compounds.
The reaction with sulfonazides works efficiently with a variety of substituted coupling partners (Scheme 133A).796 Notably, EWG substituents on the quinoline provided better results than EDGs, which rarely occurs. Li and Sundararaju employed dioxazolones for the amidation, using respectively Rh and Co dimers as precatalysts (Scheme 133B).797,798 Importantly, the reaction with Rh was demonstrated to occur even on 8-ethyl or 8-benzylquinolines, although substitution on the quinoline itself was not investigated. Notably, many examples of C(sp3)–H amidation, including more complex molecules, were reported with this protocol making use of oximes ad DGs.797 The protocol under Co catalysis, although being fairly sensitive to the substitution pattern in both coupling partners,798 is one of the rare examples of base metal-catalysed C(sp3)–H activation, and is therefore an important advancement.
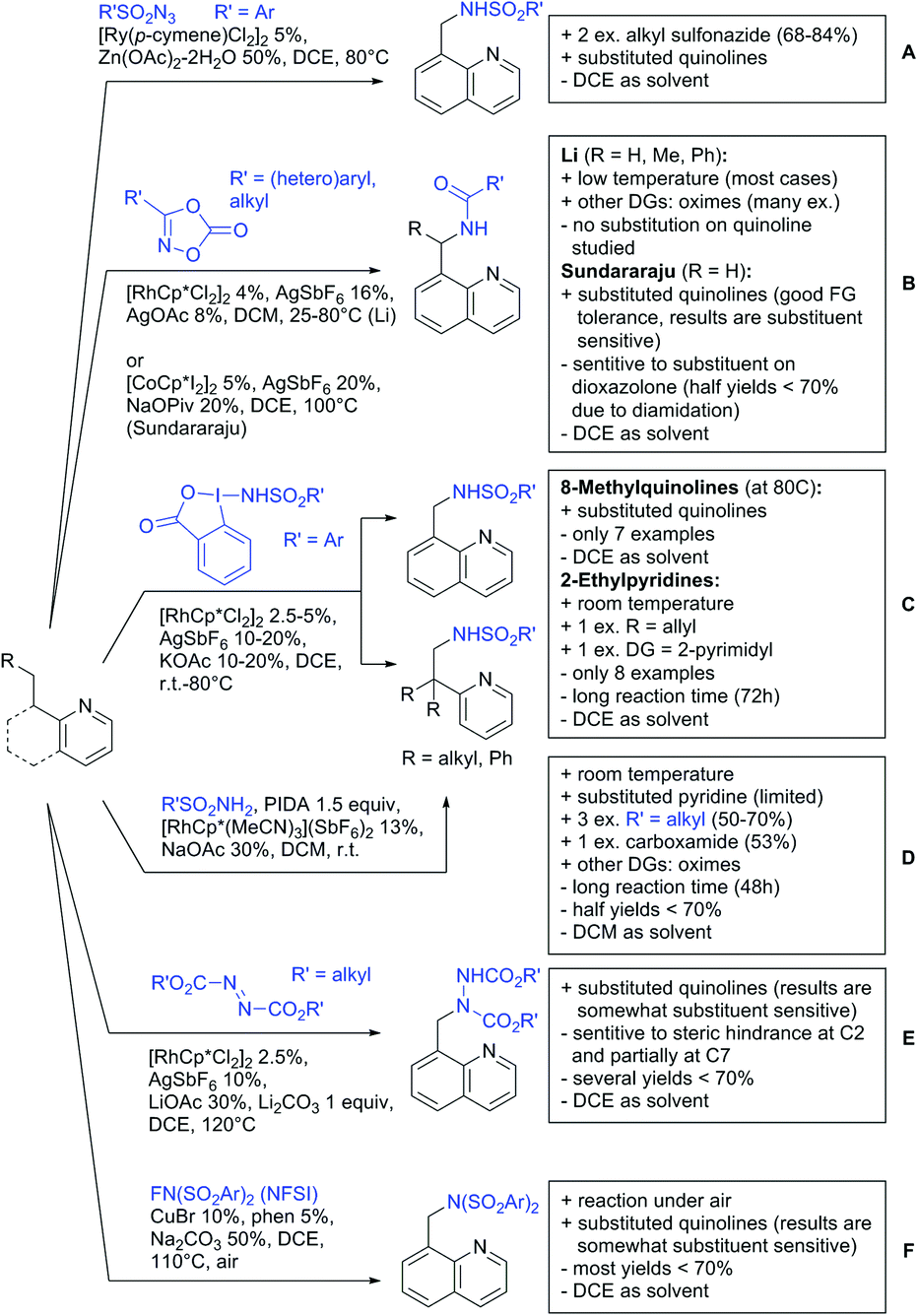 | ||
| Scheme 133 Directed amidation on 8-methylquinoline and 2-ethylpyridine derivatives. | ||
The use of amidated iodanes, either preformed or formed in situ, for the functionalisation of both C(sp2)–H and C(sp3)–H bonds was reported by Loh and You respectively under Rh catalysis (Scheme 133C and D).745,799 In particular, the amidation of 2-ethylpyridine derivatives can be performed at room temperature, although requiring a long reaction time (48–72 h). Interestingly, this functionalisation seems to require a quaternary carbon α to the functionalised C–H bond to be effective (Scheme 133C bottom and Scheme 133D). A couple of other procedures have been reported for the amidation of 8-methylquinoline, which proceed via a different mechanism (no nitrenes involved). Azodicarboxylates have been used by Kim and Park, while N-fluoroarenesulfonimides were utilised by Zheng under Cu catalysis (Scheme 133E and F).800,801 In both cases certain sensitivity to the substitution patterns on the quinoline was observed, resulting in some cases in poor yields.
15.1.2.5.2. Amination and nitration. Jiao and Li/Lan reported the amination of 8-methylquinolines and 2-ethylpyridines using anthranils under very similar conditions (Scheme 134A).802,803 A notable point, apart from various functionalities tolerated on both coupling partners, is the possibility to employ 8-ethylquinolines as substrates, which are generally not reactive.
 | ||
| Scheme 134 Directed amination and nitration on 8-methylquinoline and 2-ethylpyridine derivatives. | ||
The use of t-butylnitrite in combination with oxygen and a Pd catalyst for C(sp3)–H nitration was reported in 2015 by Liu (Scheme 134B).804 This protocol is interesting as it avoids the use of stoichiometric AgNO2 as reactant, often used in C–H nitration. Although proceeding relatively slowly (48 h reactions), variedly substituted 8-methylquinolines, including strong EWG-substituted, could be functionalised in good yields.
15.1.2.6. C–O bond formation. C(sp3)–O bond formation for this class of DG has not been much investigated. Apart from two isolated examples of methoxylation and acetoxylation of 8-methylquinoline (99 and 90% respectively) under heterogeneous Pd catalysis reported by Ellis in 2015,763 Chan demonstrated the coupling of 8-methylquinolines with methylketones catalysed by Cu.805 The proposed mechanism for this reaction does not actually involve coordination of the quinoline nitrogen to Cu and is therefore not entirely within the scope of this review. Nonetheless, it has been described in Scheme 135 for completeness. The reaction seems to be initiated by an acid-promoted aerobic oxidation, similar to a reaction developed in the Maes group,806,807 leading to the corresponding quinoline aldehyde. A subsequent (cascade) condensation with the methylketone results then in the formation of the enone.805
 | ||
| Scheme 135 C–O bond formation on 8-methylquinolines. | ||
15.1.2.7. C–B and C–Si bond formation. Few examples have been reported of C(sp3)–B bond formation, investigating cyclic and acyclic amines, rather than the typical 2-ethylpyridines and 8-methylquinolines.740,808 The functionalisation of cyclic amines is an interesting topic, and has been relatively little explored.809 The borylation of such compounds are therefore of interest. Ackermann's protocol utilises a preformed, bench-stable Ru–carboxylate catalyst, while Ir catalysis and a complex pre-ligand were used by Li (Scheme 136A and B).740,808 Although higher yields were obtained in this case, only three examples of C(sp3)–H functionalisation were shown, with the main focus of the paper being C(sp2)–H functionalisation with a number of different DGs.
 | ||
| Scheme 136 C(sp3)–H borylation and silylation. | ||
C(sp3)–Si bond formation has been investigated instead on 2-ethylpyridine derivatives (Scheme 136C).810 Despite clear disadvantages such as the use of 5 equivalents of silane and 3 of norbornene as additive (hydrogen acceptor), and the high temperature required, the process gave good yields for a variety of substrates, including substituted pyridines and unsubstituted alkyl chains, using a 2% Ru loading (total [Ru]).
15.2. Pyrimidine as DG
Pyrimidine is often tested as secondary DG in many protocols developed for pyridine-based strategies, and in many cases works as well in the same or very similar reaction conditions. Nonetheless, there are several cases in the literature where the pyrimidine DG works differently, and many transformations are specific for this DG. In particular, pyrimidine is often the DG of choice for the functionalisation of indoles (and pyrroles) in C2, and in most cases is used only for such substrates. Because of this, a separate section has been devoted to this DG. Protocols that are very similar to those reported for pyridine will not be reported again in this section (the interested reader can find many information in Section 15.1.1), and more selected literature will be discussed here.The cleavage of the pyrimidine ring is, as for pyridine, typically not possible when attached via a C–C bond, but it is easy when the DG is attached via a C–N bond (e.g. in indoles, Scheme 137). In this case no quaternisation is necessary (as it is with pyridine), and often simply heating with NaOEt in DMSO is enough to produce the pyrimidine-free substrate (side reactions may occur depending on R, for example in the case of silanes, Scheme 137). Unlike pyridine, however, pyrimidine commonly gives rise to rearrangements, instead of the desired cleavage. In particular, Smiles-type rearrangements (intramolecular nucleophilic aromatic substitution), resulting in 1,4-migration of the pyrimidine ring, have been observed several times when a nucleophilic site is available at a specific position, as shown in Scheme 137. These are specifically reported in the text whenever they were noted in the original publication.
 | ||
| Scheme 137 Common strategies for pyrimidine DG cleavage or migration. | ||
 | ||
| Scheme 138 Pyrimidine-directed arylation of indoles. | ||
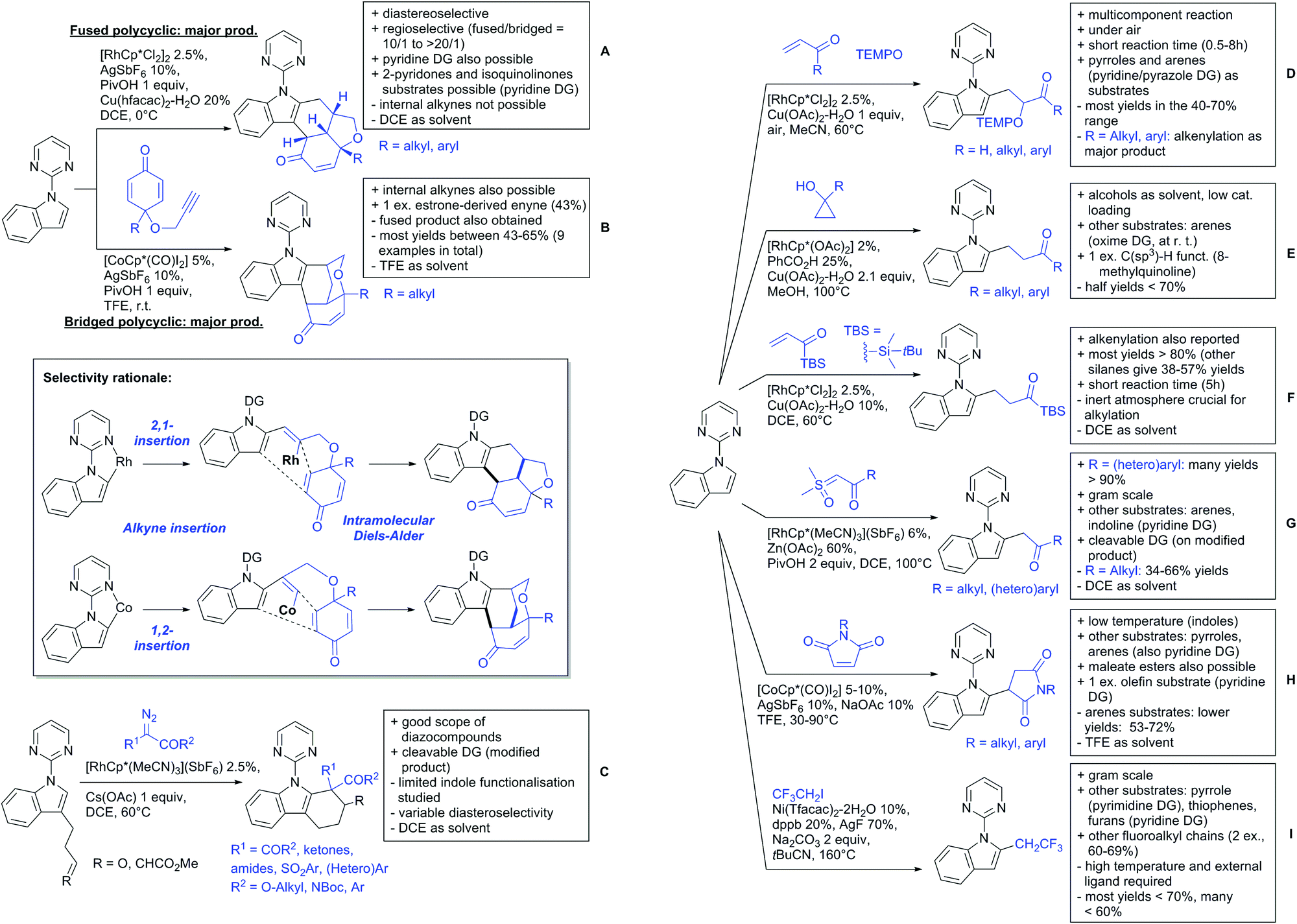 | ||
| Scheme 139 Pyrimidine-directed alkylation of indoles. | ||
Alkylation with diazo compounds, followed by in situ intramolecular aldol or Michael-type reactions was reported by Zhou. In this case, 2,3-fused indoles were obtained (Scheme 139C).818 Reactants featuring ester, amide, aromatic and heteroaromatic rings could be successfully employed in the reaction with good yields. Huang and Wang recently reported a multicomponent reaction between N-pyrimidylindoles, acrolein derivatives and TEMPO (Scheme 139D).819 The first step of this reaction is the addition of indole to the double bond of the acrolein (Rh-catalysed), followed by Cu-mediated introduction of the TEMPO radical in α to the carbonyl. Interestingly, MeCN as solvent proved crucial in suppressing other reaction pathways (e.g. β-hydride elimination and protonation), despite failing to do so when acrylic ketones were used instead of aldehydes. Apart from acyclic derivatives, Li introduced the use of cyclopropanols as new reactants for the introduction of carbonylethyl chains (Scheme 139E).790 Not being electrophilic compounds, their use constitutes a challenge and requires oxidising conditions (superstoichiometric Cu used). The reaction requires low Rh loadings (2%) and can be performed on arenes as well as indoles, but the yields obtained do not appear competitive in comparison to the use of the more common acrylates. Even in this case, the use of NaOEt in DMSO for pyrimidine cleavage did not result in the desired product, but in the migration of the pyrimidine from the indole to the alkyl chain.790
The use of acryloyl silanes was reported by Loh and Feng, and resulted in high yields of the corresponding alkylated products (Scheme 139F).820 Li and Ackermann/Li reported the use of sulfoxonium ylides (carbene precursors) and maleimide for the alkylation of indoles (Scheme 139G and H).821,822 These reactants have also been used with pyridine DGs, with somewhat different procedures (cf.Scheme 110K and Scheme 111A). In the case of Scheme 139H, again the use of NaOEt to cleave the DG resulted in pyrimidine migration. Finally, Shi reported an interesting Ni-catalysed trifluoroethylation of indoles using trifluoroethyl iodide as reactant. Unfortunately, not very high yields were obtained for a range of indoles and other heterocycles, with most yields remaining below the 70% threshold (Scheme 139I).823
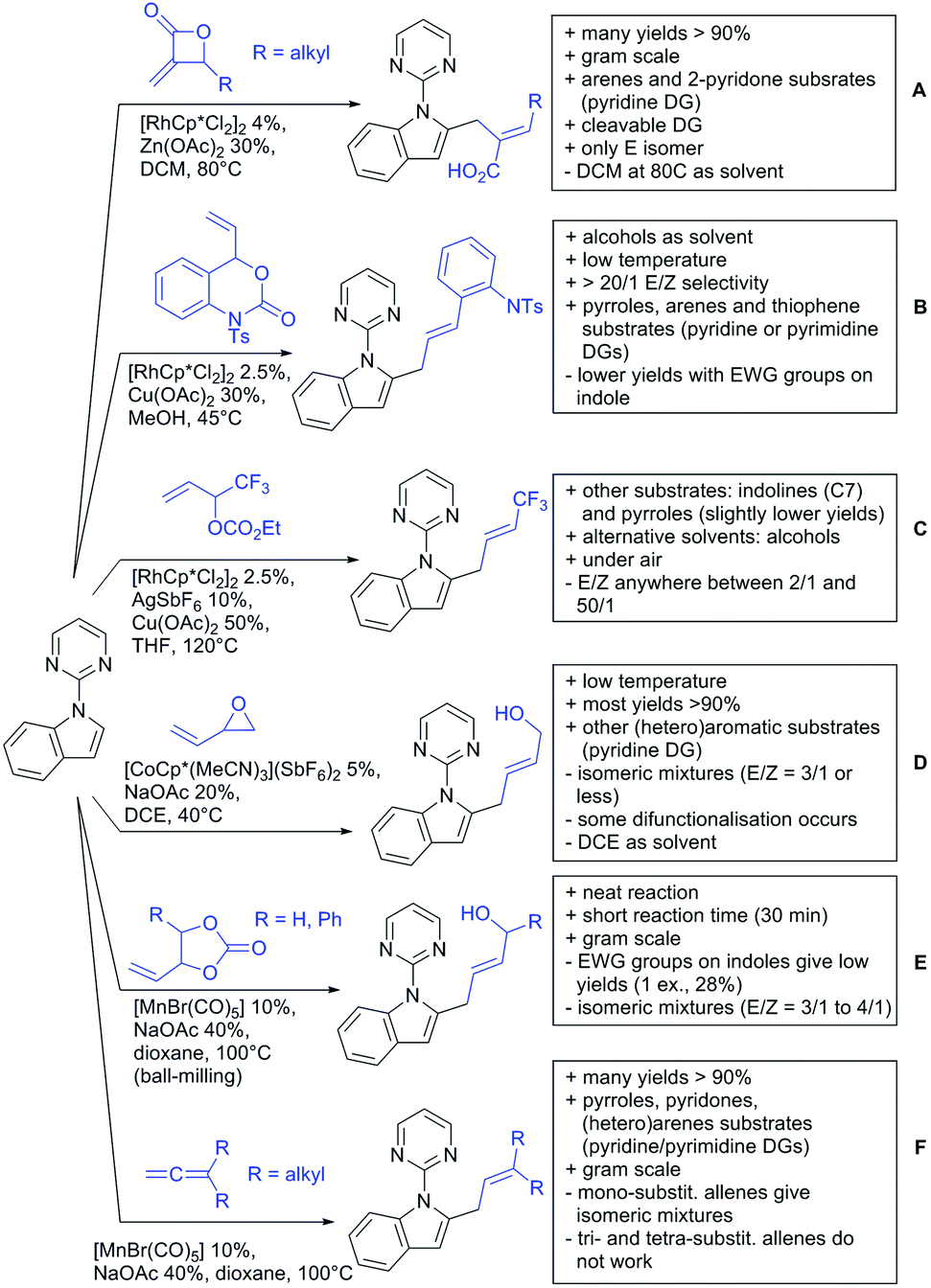 | ||
| Scheme 140 Pyrimidine-directed allylation of indoles. | ||
An interesting Mn-catalysed allylation reaction with 1,1-disubstituted allenes was reported by Wang (Scheme 140F).828 Very high yields were obtained for many examples, and the procedure is applicable to a variety of (hetero)arenes apart from indoles, using either pyrimidine or pyridine DGs. Unfortunately, monosubstituted allenes resulted in isomeric mixtures, and tri- and tetra-substituted ones proved ineffective in the reaction.
 | ||
| Scheme 141 Pyrimidine-directed olefinations. | ||
Li and Lan reported a rare selective 1,2-insertion of terminal alkynes using Co catalysis, giving α-gem-vinylindoles (Scheme 141B).829 The use of Co and TFE as solvent was crucial for the selectivity, due to the high steric sensitivity of Co and the H-bonding abilities of TFE. Propargylic alcohols, amines and silanes performed well when used as reactants, although yields and selectivity depended on their structure. The use of NaOEt for the cleavage of the DG resulted in the transfer of the heterocycle to the hydroxyl of the propargylic moiety.
A complementary procedure, giving the 2,1-insertion product with terminal alkynes was reported by Li and Lei using Mn as catalyst (Scheme 141C).830 Aryl- and alkyl-substituted alkynes proved efficient reactants in most cases, and the reaction could be performed on internal alkynes as well. A practical downside here is the use of Et2O at 80 °C.
Rueping and Wang/Li independently reported the Mn-catalysed olefination of indoles with allenic esters. The use of 1,3-disubstituted allenes (Rueping) resulted in the hydroarylation product (arylation at the C2 position of the allene) in a regio- and stereoselective manner (Scheme 141D).831 Interestingly, the reaction with tri- or tetra-substituted allenes (Rueping, Wang/Li) resulted in a cascade hydroarylation/DG migration via a Smiles-type rearrangement (see Scheme 137), furnishing the products in Scheme 141E.831,832 While Rueping's procedure831 resulted in higher yields (often >90%), but required 2 equiv. of NaOAc as additive, Wang/Li's protocol832 gave lower yields in several cases (although a much broader substrate scope was reported), but without the addition of any additive. A number of post-functionalisation reactions were reported by both authors, including: alkene reduction, amide hydrolysis (with consequent pyrimidine migration back to the indole), decarboxylative ring opening, and DG cleavage with LiAlH4 (concomitant carbonyl reduction).831,832
The Co-catalysed hydroarylation of ketenimines resulted in the olefination product depicted in Scheme 141F in generally good yields (lower yields on pyrroles, arenes and indolines).833 The use of NaOEt on these compounds resulted in a pyrimidine migration similar to that in Scheme 141E, via intramolecular nucleophilic attack of the enamine to the pyrimidyne ring.833 Finally, a couple of substitution procedures for the olefination of indoles were reported using alkenyl acetates or gem-difluoroalkenes. The reaction with cyclic and acyclic alkenyl acetates and similar electrophiles was reported by Ackermann under Co catalysis (Scheme 141G).834 The Mn and Co-catalysed reaction with difluoroalkenes was reported respectively by Loh/Feng and Li (Scheme 141H).835,836 While the substrate scope remains similar, the Co-catalysed process performed much better in terms of reaction temperature, yields and stereoselectivity (Z selective). In this reaction, Ca(OH)2 is suggested to act as a scavenger for the HF produced, pushing the reaction forward.836 It is worth noting though, that the Mn-catalysed reaction shows a certain preference for the less stable E isomer for certain substrates.835
 | ||
| Scheme 142 Pyrimidine-directed alkynylations. | ||
Li and Zhou disclosed a good example of C7 alkynylation of indolines using Ir catalysis (Scheme 142D).840 The protocol presents several advantages, such as the high yields, the use of ethanol as solvent at room temperature, and one example of reaction with tBu-EBX (yield 84%). Finally, Hong reported a Ru- or Rh-catalysed alkynylation of 4-quinolones and isoquinolinones with TIPS-EBX (Scheme 142E).841 This is a rare example of the use of pyrimidine as DG for the functionalisation of such type of compounds, usually performed using pyridine as DG. The reaction proceeds well in air, but no variation in the reactant (other silyl or carbon groups) was investigated. It is noteworthy that the cleavage of the pyrimidyl DG using basic conditions (NaOEt or NaOMe) when silanes are present in the final product results in the concomitant desilylation (see also Scheme 137), which might constitute an advantage for synthetic purposes.
 | ||
| Scheme 143 Pyrimidine-directed cyanation, acylation and aminocarbonylation of indoles. | ||
As for the pyridine DG, a number of publications on the use of Pd catalysis in combination with peroxides and acyl radical precursors (aldehydes, α-diketones, toluene) for the introduction of an acyl group onto N-pyrimidinylindoles have been reported.843–845 The general conditions are: 5–10% Pd(II) catalyst, 3–5 equiv. peroxide, apolar solvent (e.g. toluene, THF), at 90–120 °C, and the reaction follows the general mechanism shown in Scheme 118. From these conditions, Noel/Van der Eycken and Jana independently reported a protocol for the group-directed acylation of indoles with aldehydes using a dual photoredox-Pd catalytic strategy which allows functionalisation at room temperature.846,847 Noel/Van der Eycken reported both a batch and a flow protocol. Although only slightly higher or comparable yields were obtained in flow, and much higher amounts of aldehyde and TBHP were required, the reaction time was drastically decreased using this methodology (2 h instead of 20 h), as well as the photocatalyst loading (Ir) (Scheme 143B).846 Jana's protocol (batch), although requiring reaction times up to 48 h, was tested on more variedly functionalised indole substrates, giving overall comparable yields to the Noel/Van der Eyken protocol. An important advantage is the avoidance of the external ligand and the use of a lower excess of aldehyde/TBHP (Scheme 143C).847 Both protocols could be applied to benzylic alcohols (oxidised to aldehydes in situ), and the DG could be cleaved in basic conditions (NaOEt).
The photoredox catalyst in these procedures facilitates the reaction by promoting the formation of the tBuO radical first, needed to generate the acyl radical, and then the oxidation of the Pd(III) intermediate to Pd(IV), required for the reductive elimination step (see also Scheme 118, bottom). As these activations are not available in the simple Pd(II) catalytic reaction, high temperatures are required to push the reaction forward.
Two very similar procedures for the aminocarbonylation of indoles with aromatic or aliphatic isocyanates were reported by Kim and Van der Eycken in 2015 using a Rh dimer as catalyst (Scheme 143D).848,849 Both processes required 2.5 to 3 equiv. of isocyanates to perform decently, but the yields remain modest in both cases. Importantly for synthetic purposes, Kim observed the reaction with aromatic isocyanates to be reversible, thus explaining in these case the low yield obtained.848 Such reversibility had been observed before for the Rh-catalysed hydroarylation of imines.850 Both aliphatic and aromatic amide products could be treated with NaOEt to provide the deprotected indole. Interestingly, Kim showed that the use of EtOH as co-solvent in the process (standard solvent: DMSO) facilitates the reaction.848,851
Indole phosphoramidation was reported by Kanai and Matsunaga, notably under Co catalysis (Scheme 144A).852 Although the process presents some drawbacks, such as the long reaction times and the sensitivity to steric hindrance in the C3 position of the substrate, this is a rare base metal-catalysed example of a C–H phosphoramidation reaction. Substituted dioxazolones were instead employed for the carboxyamidation of indolines at the C7 position at room temperature (or 80 °C for several examples, Scheme 144B).853 The reaction proceeds giving generally good yields. Interestingly, the reaction proved efficient on tetrahydroquinoline as well, a not commonly studied substrate.
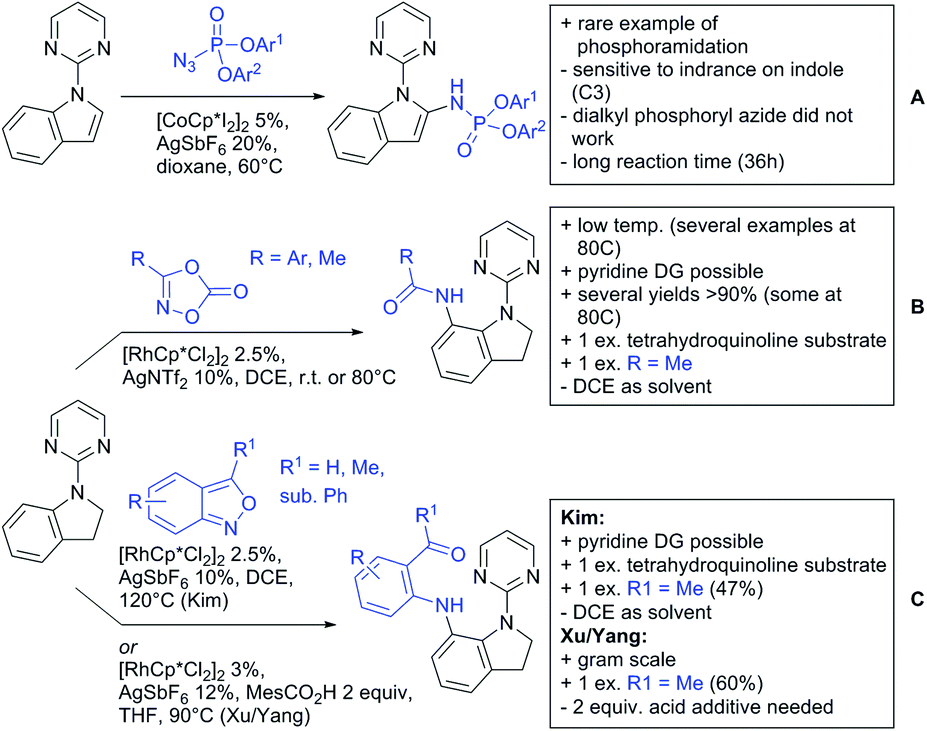 | ||
| Scheme 144 Phosphoramidation and amination on indoles and indolines. | ||
Concerning indoline amination, anthranils were used under Rh catalysis by both Kim and Xu/Yang (Scheme 144C).854,855 The substrate scope investigated and the results are comparable in the two cases. For these substrates, subsequent intramolecular cyclisation to quinoline fused indoline (as observed in indoles, cf.Scheme 125), did not occur in situ, but can be promoted by reaction of the formylaniline product (R1 = H) with TFA.854
 | ||
| Scheme 145 Directed C–S bond formation on indoles and indolines. | ||
15.3. N-, O-, S-, and Si-Linked (di)azines as DGs
The pyridine (and pyrimidine) DGs are not easily cleavable when attached via a C–C bond. Nonetheless, when attached via a C–heteroatom bond, the cleavage becomes relatively easy. This type of bond is found, apart from pyridine- or pyrimidine-functionalised indoles, indolines and pyridones, in a number of other substrates, where the DG is attached via a NH2-, O-, Si-, or SO/SO2 linker. Apart from their sometimes particular reactivity pattern, the cleavage of these DGs is a very important feature for synthetic purposes. Scheme 146 summarises the different cleavage/modification methods available in the literature for such DGs.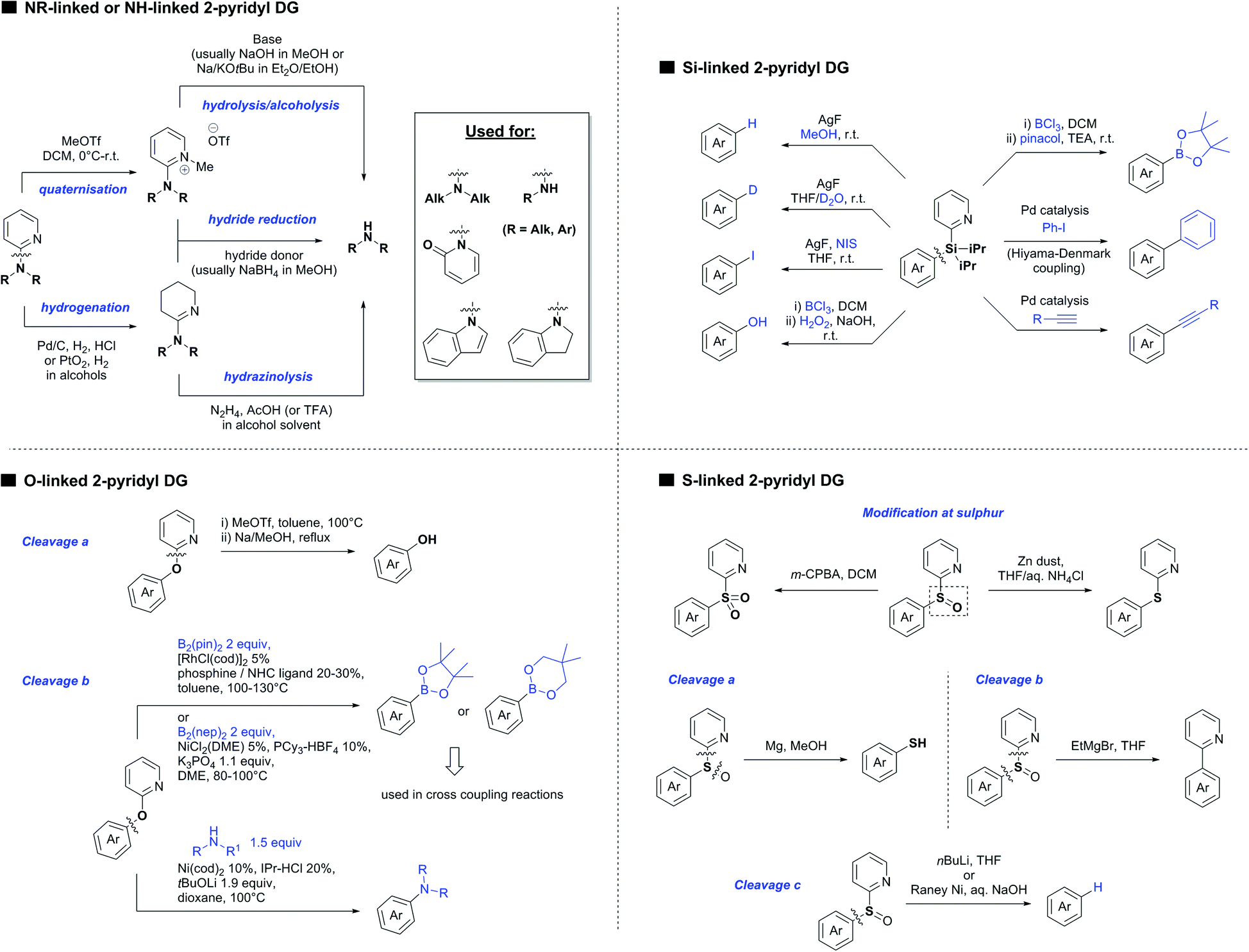 | ||
| Scheme 146 Protocols for the cleavage/modification of N-, O-, S-, and Si-linked pyridine DGs. | ||
The N-linked pyridine DG is by far the most explored among this family of DGs, and several methods have been developed with time for its cleavage, and routinely used. A common method utilises the “quaternisation-hydrolysis/alcoholysis” strategy,861 which involves methylation of the pyridine with MeOTf (no other methylating agent is generally used), followed by hydrolysis or alcoholysis. Other methods are the “quaternisation-hydride reduction”,862 where hydrolysis/alcoholysis is replaced by the reaction with a hydride donor. Alternative cleavage procedures involve a first hydrogenation step, leading to an unstable aminal. The cleavage of the latter can then occur using a hydride donor, or via hydrazinolysis.862–864 The quaternisation-based methods are most commonly encountered in the literature, with little variations from the conditions shown in Scheme 146, and are used for pyridine removal from indoles, pyridone, indolines, aliphatic amines and even anilines.
O-Linked 2-pyridyl DGs can also be easily cleaved (Scheme 146). A general procedure is the quaternisation of the pyridine ring, followed by treatment with NaOMe, resulting in the corresponding phenol.865 Apart from this traditional cleavage, a few cross coupling methods have been reported by Chatani and Wang to cleave the arene–O bond and introduce a boronic ester (which can be used in subsequent cross couplings)866,867 or an amino functionality868 in its place.
The use of a Si-linked 2-pyridyl and 2-pyrimidyl DG has the great advantage of being very easily cleaved at the arene–Si bond via substitution or cross-coupling reactions, as demonstrated in a seminal paper by the Gevorgyan group in 2011 (Scheme 146).869
The cleavage of S-linked 2-pyridyl DGs has been reported in a few papers, although it has not been extensively explored on a broad number of substrates. Interestingly, different possibilities are available for this linker, in particular modification (oxidation or reduction of sulfoxides) is straightforward,870,871 and cleavages at both the pyridine–S bond and the aryl–S bond have been reported (Scheme 146).870–872
15.3.1.1. Functionalisation without cyclisation. A number of functionalisations have been reported which do not involve tandem cyclisation at the free NH moiety. In the C–H functionalised product, the DG can be removed via cleavage of the NH-heterocycle bond, resulting in functionalised anilines (Scheme 146).
The alkylation with diazodiesters has been reported a number of times with Rh or Ir catalysts (Scheme 147A).873–875 Ackermann reported the Ni-catalysed ortho-alkylation and alkynylation of N-(2-pyrimidyl)anilines, using alkyl/alkynyl bromides (Scheme 147B and C).876–878 The same group also reported a Ni-catalysed thiolation with disulfides, leading mostly to difunctionalised products (Scheme 147D).878 Interestingly, a similar protocol using Rh catalysis and N-(2-pyridyl) or O-(2-pyridyl) as DG was shown to provide selectively the monofunctionalised phenols (Scheme 147G and Scheme 148I).879 Using N-(2-pyrimidyl), Cui reported the Ir-catalysed reaction with sulfonyl azides adding only catalytic amounts of AgSbF6 to the reaction. Although resulting in lower yields (max 63%), two examples of alkylsulfonyl amides were also reported, which adds value to the procedure (Scheme 147E).880 In this case, the use of aq. HCl on the reaction products results in the cleavage of both DG and amide, giving ortho-phenylenediamines. Two procedures for ortho nitration were reported with AgNO2 as nitro source. While Pd catalysis on N-(2-pyrimidinyl)anilines using K2S2O8 as oxidant resulted in regioisomeric mixtures in meta-substituted anilines (Scheme 147F),881 this was not observed on the N-(2-pyridinyl) analogues using aerobic Co catalysis (Scheme 147H).882 Noteworthy in this case is also the range of other N-linked heterocycles effective in the reaction, including pyrimidine, quinolines, pyridazine and benzothiazole. Also two different cleavage protocols were reported, leading to either the o-nitroaniline or the o-phenylenediamines.882 Finally, Schnürch reported the alkylation of benzylic C(sp3)–H bonds in N-(2-pyridinyl)benzylamines using Rh catalysis and tetraalkylammonium halides as terminal olefins surrogates (Scheme 147I).883 To our knowledge this is the only case of C(sp3)–H functionalisation with this DG type.
 | ||
| Scheme 147 C(sp2)–H and C(sp3)–H functionalisation with NH-linked pyridine and pyrimidine DGs. | ||
 | ||
| Scheme 148 Functionalisation with O-linked pyridine and pyrimidine DGs. | ||
15.3.1.2. Functionalisation with cyclisation. In these reactions the free NH of the DG reacts with an unsaturated moiety, leading to the formation of aza-heterocycles, particularly indole derivatives. In the final compounds the heterocyclic DG can be generally cleaved by the typical methods described in Scheme 146. The reaction with diazo-β-ketoesters or similar results in 3-alkoxycarbonyl-2-alkylindoles (Scheme 147J, cf. Scheme 147A),873–875,884 while the use of Meldrum's acid furnishes 1,3-dihydroindol-2-ones, although the product obtained strongly depends on the substitution pattern on the DG or the aryl ring (Scheme 147K).885 2,3-Diarylated indoles can be obtained by reaction with alkynes, of which Kantam recently reported a heterogeneous version using Pd supported on nanocrystalline Mg oxide [NAP–Mg–Pd(0)] (Scheme 147L).886 This heterogeneous catalyst could be recycled and reused for at least 4 runs without considerable changes in yields.
An interesting alternative, reported by Zeng, utilises arylalkynylcycloalkanols for the regioselective formation of 2-acyl-3-arylindoles, occurring via consequent ring opening of the propargylic alcohol employed (Scheme 147M).887 The formation of dihydroquinolinones via Rh-catalysed alkylation/cyclisation with unsaturated aldehydes was recently reported by Huang, and to our knowledge this is the only 6-membered ring formation reported for such systems (Scheme 147N).888 Finally, an Ir-catalysed amidation/condensation protocol was developed for the synthesis of functionalised benzimidazoles (Scheme 147O).889 Unfortunately, the cleavage of the DG in the final product did not succeed using previously reported methods. In all these functionalisations, the heterocyclic DG is maintained unaltered in the final product, and can be exploited for further functionalisation of the heterocycles, e.g. at the C7 in indoles or analogous positions.
The alkylation with diazocompounds under Rh catalysis occurs in good yields on a variety of substituted phenols, although monofunctionalisation was only possible in somewhat hindered substrates (Scheme 148A).890 When known cleavage methods for pyridine/pyrimidine DGs were applied, no cleavage was observed, instead a Smiles-type rearrangement took place (see Scheme 137, unusual for pyridines). Interestingly, this rearrangement proved consistent and occurred in good yields for a number of different products.890 Cui reported the aromatic homologation of O-(2-pyridinyl)phenols with internal arylalkynes to result in substituted 1-naphthols in good yields using tAmOH as solvent (Scheme 148B). Unfortunately, the reaction with alkyl or heteroarylalkynes did not occur, and asymmetric alkynes resulted in very poor regioselectivity.891 Acylation reactions were reported by Wu and Huo, using aldehydes and acyl chlorides as reactants. The first method (Scheme 148C)892 uses TBHP as oxidant and follows the mechanism depicted in Scheme 118, resulting mostly in monoacylation (difunctionalisation is substrate-dependent), giving low yields for strongly EWG-substituted phenols. The second method (Scheme 148D)893 is a group-directed Friedel–Crafts acylation, and does not require oxidants or additives, but 10% of expensive Pt catalyst was employed for the reaction. The use of PhCl as solvent results in the formation of difunctionalised phenols, while monoacylated ones could be obtained in hindered substrates.
Comparatively more protocols for C–heteroatom bond formation were reported with this class of DG, although suffering in several cases from low yields (<70%). The reaction with arylsulfonyl azides was reported by Cui (Scheme 148E),894 and C–O bond formation can occur via reaction with PIDA or carboxylic acids (Scheme 148F and G).895,896 The DG cleavage on the products obtained results in ortho-hydroxyanilines or in catechols. C–S bond formation has been reported making use of sulfonyl chlorides or disulfides (Scheme 148H and I),879,897 and C–Cl and C–F bonds are formed under Pd catalysis using NCS of NFSI as electrophilic halogen sources (Scheme 148J and K).898,899
Alkylation reactions were reported in 2016 by Gevorgyan (pyrimidylsilyl) and Zhang (pyridylsilyl), using very similar protocols: Pd catalysis, alkylboronic acids as reactants, superstoichiometric amounts of Ag salts as oxidant, an inorganic base and an aminoacid as ligand (Scheme 149A).900,901 Both protocols require long reaction times (48–72 h) and produce yields often in the range of 40–70%. Notable features are the smooth cleavage/substitution of the DG (Gevorgyan), the use of variedly functionalised alkylboronic acids (Zhang) and a single example of C(sp3)–H functionalisation (Zhang). Alkoxycarbonylation was reported by Gevorgyan using CO and hexafluoroisopropanol (HFIP, Scheme 149B).902 A good substrate scope and FG tolerance was obtained for this protocol; particularly noteworthy is the use of several heterocyclic substrates (dibenzofuran, thiophene, carbazole, indoline, and tetrahydroquinoline), featuring medium to good yields (47–77%). The same group also reported the C–O and C–halogen bond formation on the same starting materials. Diacyloxylation with hypervalent iodine reactant PhI(OPiv)2 proved effective on a number of para-substituted arenes in good yields, but required up to 120 h (Scheme 149C).903 C–halogen bond formation was achieved using NXS as reactants (X = I, Br, Cl), with different reaction conditions for each halogen (Scheme 149D).903
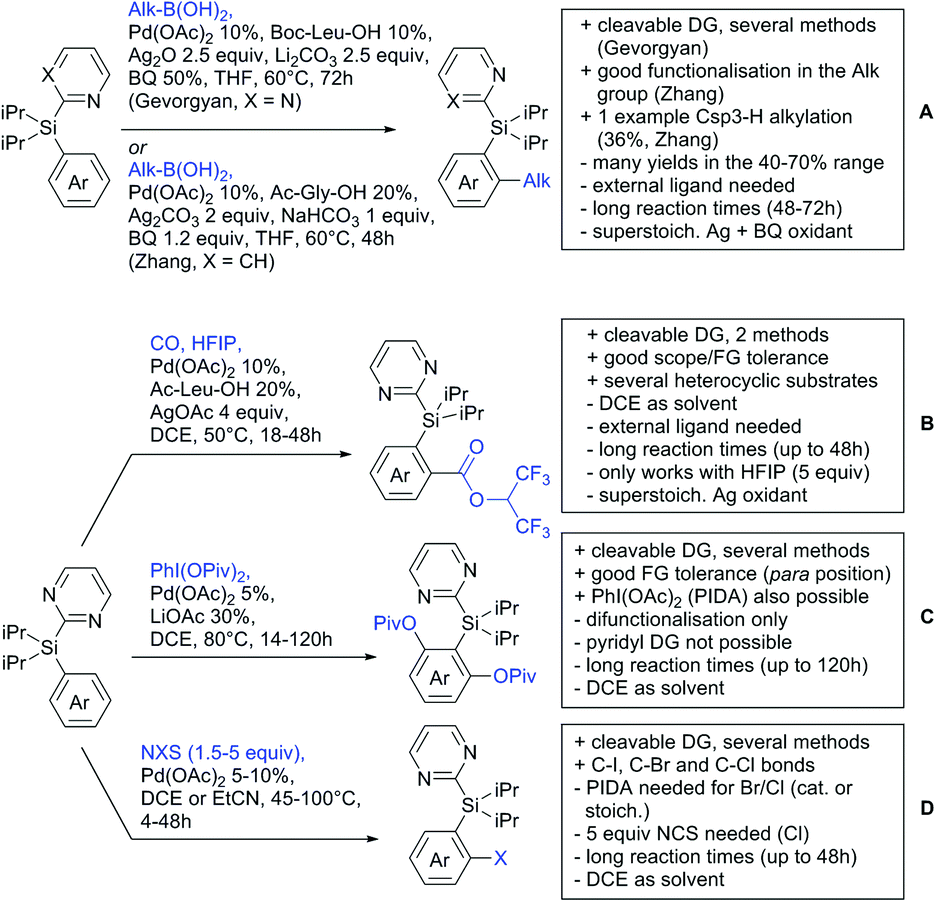 | ||
| Scheme 149 Functionalisation with Si-linked pyridine and pyrimidine DGs. | ||
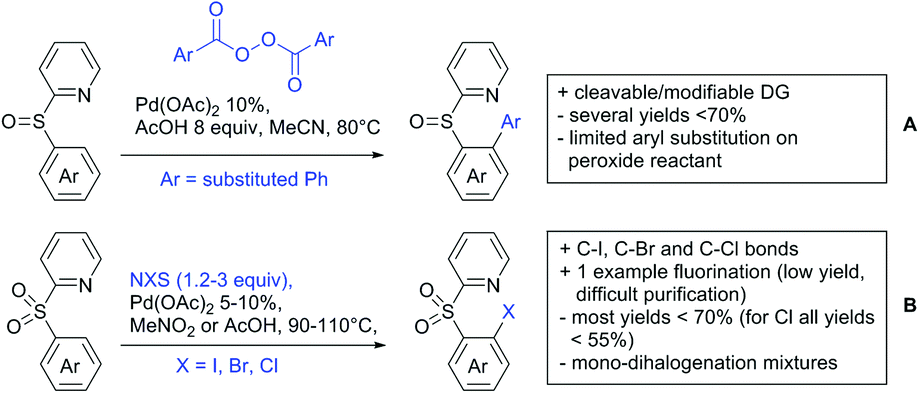 | ||
| Scheme 150 Functionalisation with S-linked pyridine DGs. | ||
15.4. Quinoline-N-oxide as substrate/DG
While the activation of C–H bonds (either metal-catalysed or metal-free) at the ortho position of pyridine-N-oxides has been known for long time, this activation is mostly due to electronic effects.905–907 These cases will therefore not be treated here. Instead, metal-catalysed reactions where the N-oxide is thought to have a coordinating and directing role will be discussed. It is important to mention that the N-oxide moiety is the DG here, and is therefore limited to the functionalisation of azines. Although, strictly speaking, the coordinating atom is not itself part of a heterocycle, this DG has been considered in the heterocyclic section for simplicity and immediate analogy to its heterocyclic precursors. For the use of different N-oxide moieties in C–H functionalisation see Section 14.3.1 and Section 16.9.The functionalisation of quinoline-N-oxides at the 8 position is most common in this area. Considering quinolines and their analogues are very common motifs in biologically-active molecules, the simple introduction (e.g. with peroxides) and cleavage (e.g. sulphur or phosphorus reagents)907,908 of the N-oxide moiety is very convenient for the selective functionalisation of these substrates. The facility of the N-oxide removal or the oxygen-transfer to the quinoline C2 (forming 2-quinolinones) or to an unsaturated moiety in the product (N-oxide as internal oxidant), make several of these procedures occur as cascade/tandem processes, leading to interesting compounds. These types of transformation have only been studied from 2014, and all the examples till 2017 have been reported in this section. Due to their limited number, all type or reactions are treated in one single section.
In 2014 Chang reported iodination and amidation with N-iodosuccinimide (NIS) and sulfonyl azides (Scheme 151A and B),909 also applied to the synthesis of Zinquin ethyl ester (a zinc indicator for biological systems). The only other report on the functionalisation with heteroatoms involved the use of selenyl chlorides for the formation of C–Se bonds. In this case, however, the N-oxide was not the main DG investigated and only 5 examples (45–67% yield) were included (Scheme 151C).910
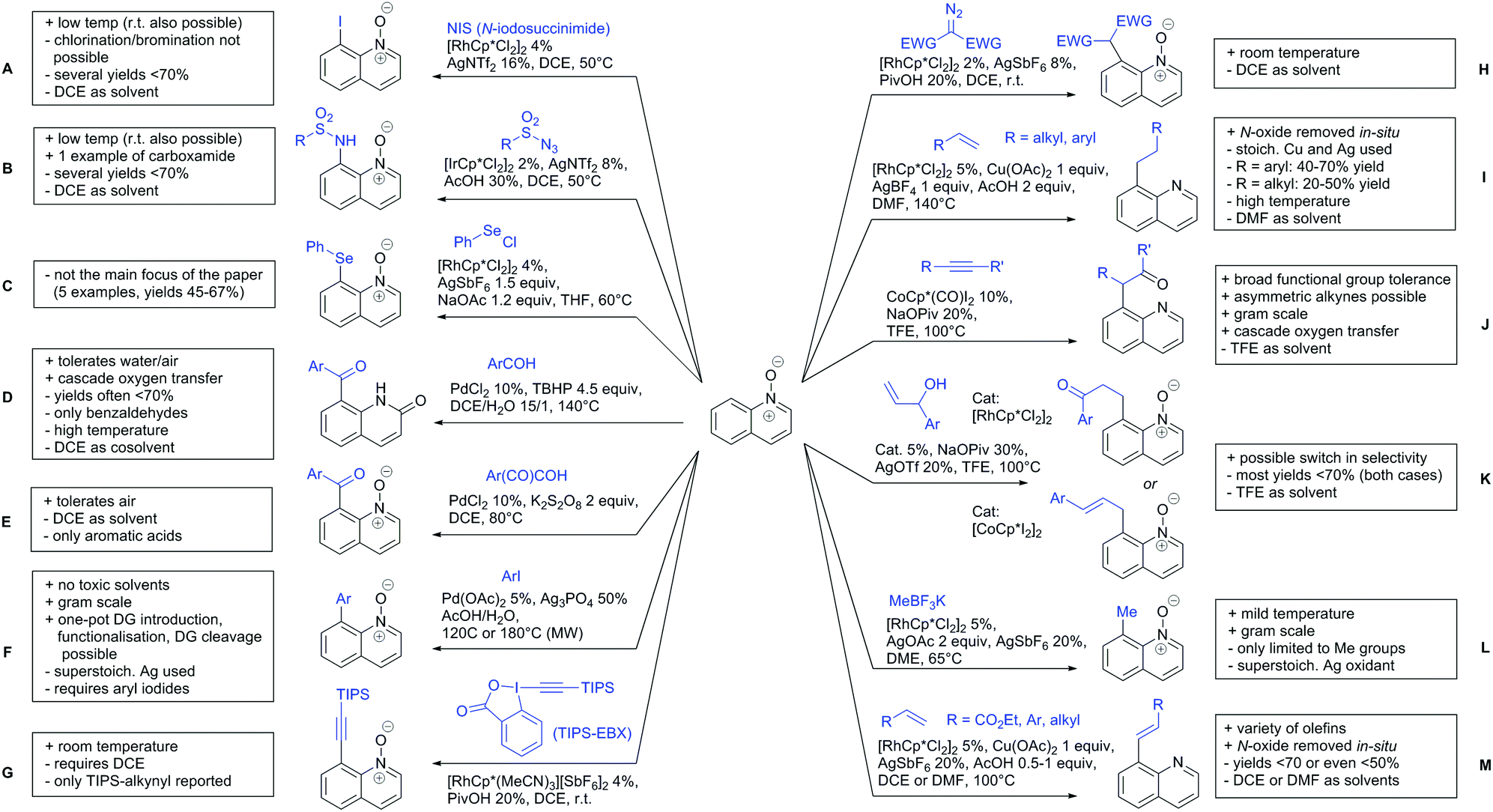 | ||
| Scheme 151 C8-Functionalisation of quinoline-N-oxides. | ||
Two acylation procedures were reported by Wu and Cui, both catalysed by PdCl2 in similar conditions. The use of TBHP (tert-butylhydroperoxide) with benzaldehydes resulted in the 8-acylation followed by in situ oxygen transfer from the N-oxide to the 2 position of the quinoline, producing 8-benzoyl-2-quinolinones (Scheme 151D).911 The use of α-oxocarboxylic acids as reagents in combination with K2S2O8 as oxidant produced instead 8-benzoylquinoline-N-oxides via decarboxylative coupling (Scheme 151E).912 A Pd-catalysed arylation with aryl iodides was reported by Larionov. Interestingly, this reaction performs best in AcOH/H2O, and no toxic solvents were required (Scheme 151F).913 On the other hand, high amounts of Ag3PO4 additive (1.5 equiv. total [Ag]) were used, presumably acting as both base and stoichiometric halide scavenger for the reactant. A similar procedure was later reported by the same authors for the dehydrogenative homocoupling of quinoline-N-oxides.914 8-Alkynylation can be achieved using TIPS-EBX (Scheme 151G).915 This reaction, although requiring DCE as solvent and the use of a wasteful reactant, proceeds with good yields at room temperature.
Formal alkylation at the 8 position can be achieved with a number of reactants. Chang reported the use of diazomalonates or similar reactants at room temperature (Scheme 151H).915 Very recently, alkylation via reaction with styrenes or aliphatic olefins was reported by Sharma (Scheme 151I).916 Although the reported yields are relatively low (<70% for styrenes and <50% for aliphatic olefines), even long aliphatic chains could be attached to the quinoline moiety, which is generally quite uncommon. The reaction was suggested to take place via the olefinic intermediate, then reduced in situ with formic acid, derived from the DMF solvent. The suggested mechanism (only partially) explains the requirement of stoichiometric amounts of both Cu and Ag for an overall non-oxidative coupling.916 This method was also applied to the total synthesis of an EP4 antagonist. In 2014 the reaction with internal alkynes under Rh catalysis was independently reported by Chang and Li. This reaction occurred via formal addition to the alkyne, followed by an intramolecular oxygen transfer from the N-oxide to the new functionality, resulting in the ketones shown in Scheme 151J.917,918 The same reaction was then reported by Sundararaju under Co catalysis,919 and showed considerable improvements in terms of coupling partners: diaryl-, dialkyl-, and arylalkyl–alkynes could be easily coupled with good yields. The reaction with allylic alcohols resulted in two different compounds depending on the catalyst. While the use of Rh produced aroylethyl-functionalised compounds, the use of Co in the same conditions only resulted in allylated compounds (Scheme 151K).920 A methylation procedure using MeBF3K was recently reported by Liu (Scheme 151L);921 the same procedure can be applied for C–H arylation using aromatic trifluoroborates, but very few examples with <40% yields were reported in this case.
Two examples of olefination have appeared from 2014. Shibata first reported the Rh(I)-catalysed reaction with internal alkynes, a reaction that was plagued by isomeric mixtures and yields often <70%.922 A second example was reported in 2015 by Sharma via reaction with terminal olefins. Although even in this case many yields were low (even <50%), olefination took place with a number of acrylates, styrenes and other linear olefins, also resulting in N-oxide cleavage in situ (Scheme 151M).923
15.5. Pyrazole as DG
Pyrazoles are the most common 1H-diazole DG type used, and an important pioneering publication by Chatani on the specific use of this DG dates back to 2003.924 As many other heterocycles, in recent years pyrazole DGs are often tested as secondary DGs in procedures mostly developed using pyridine-based groups. In general, in these cases pyrazole performs giving comparable or little lower yields than the corresponding pyridine-directed reaction, and only one or two examples are nowadays generally reported in most publications. It is worth noting that the lower yields obtained in these cases might just be due to a lack of specific optimisation for pyrazole DGs, and do not necessarily mean that pyrazole is a worse DG than pyridine. In fact, from a practical point of view, there are a few differences that might make pyrazole a much better choice than pyridine depending on the synthetic aim. Most commonly 1-aryl/alkyl-1H-pyrazoles are used as substrates, while 3-aryl/alkyl-1H-pyrazoles are much less explored. In the former substrates, the DG is linked to the substrate via a C–N bond, thus making DG introduction easier than with a 2-pyridyl DG requiring a C–C linkage. For example, 1-alkyl/aryl-1H-pyrazole can be easily obtained by: (i) metal-free (or metal-catalysed) nucleophilic substitution/cross coupling; (ii) reaction of functionalised hydrazines with 1,3-diketones or unsaturated carbon moieties, including in situ hydrazine formation from amines.925 Moreover, its cleavage/modification, although rarely reported, is easier than with pyridine for arenes or olefins (e.g. ozonolysis/reduction926,927 or elimination reactions,928Scheme 152), provided that there is FG compatibility with the required reagents.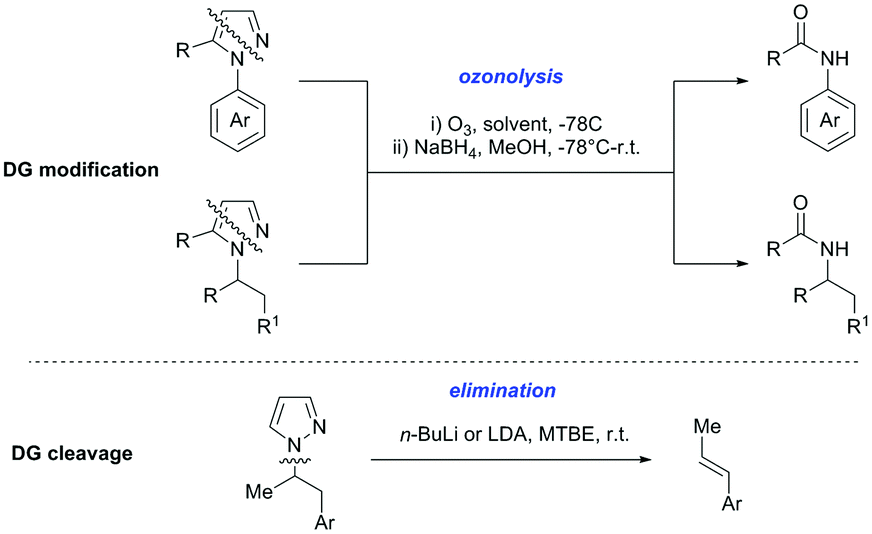 | ||
| Scheme 152 Examples of cleavage/modification of pyrazole DGs. | ||
Although not too many, several C–H functionalisation methods specifically developed for pyrazoles (or with more than 1 or 2 examples anyway) have been published recently. Greaney reported an elegant, atom economic one-pot DG introduction/C–H arylation using diaryliodonium salts. In this procedure, pyrazole is first reacted with the iodonium salt in metal-free conditions (or with catalytic CuI) to provide the 1-phenyl-1H-pyrazole substrate, then a Ru catalyst is added to the reaction to trigger the ortho C–H arylation with the aryl iodide formed in situ during the first step (Scheme 153A).929 The most notable feature is the possibility to use asymmetric diaryliodonium salts: N-arylation of the pyrazole occurs selectively with the less electron-rich arene, or to the more sterically hindered one. The same procedure can be applied to styryl(phenyl)iodonium salts, giving 1-styryl-1H-pyrazoles, and subsequent arylation of the olefin.929 A number of alkylations have been reported via addition to double bonds. Sundararaju demonstrated the Co-catalysed addition to acrylates (Scheme 153B),930 while diastereoselective protocols have been reported by Ellmann. A multicomponent/cascade reaction with 1-phenyl-1H-pyrazoles, α,β-unsaturated ketones, and aldehydes provide access to functionalised branched alkyl chains with two consecutive stereocentres, which could be obtained in good diastereoselectivity (above 90/10, Scheme 153C).931
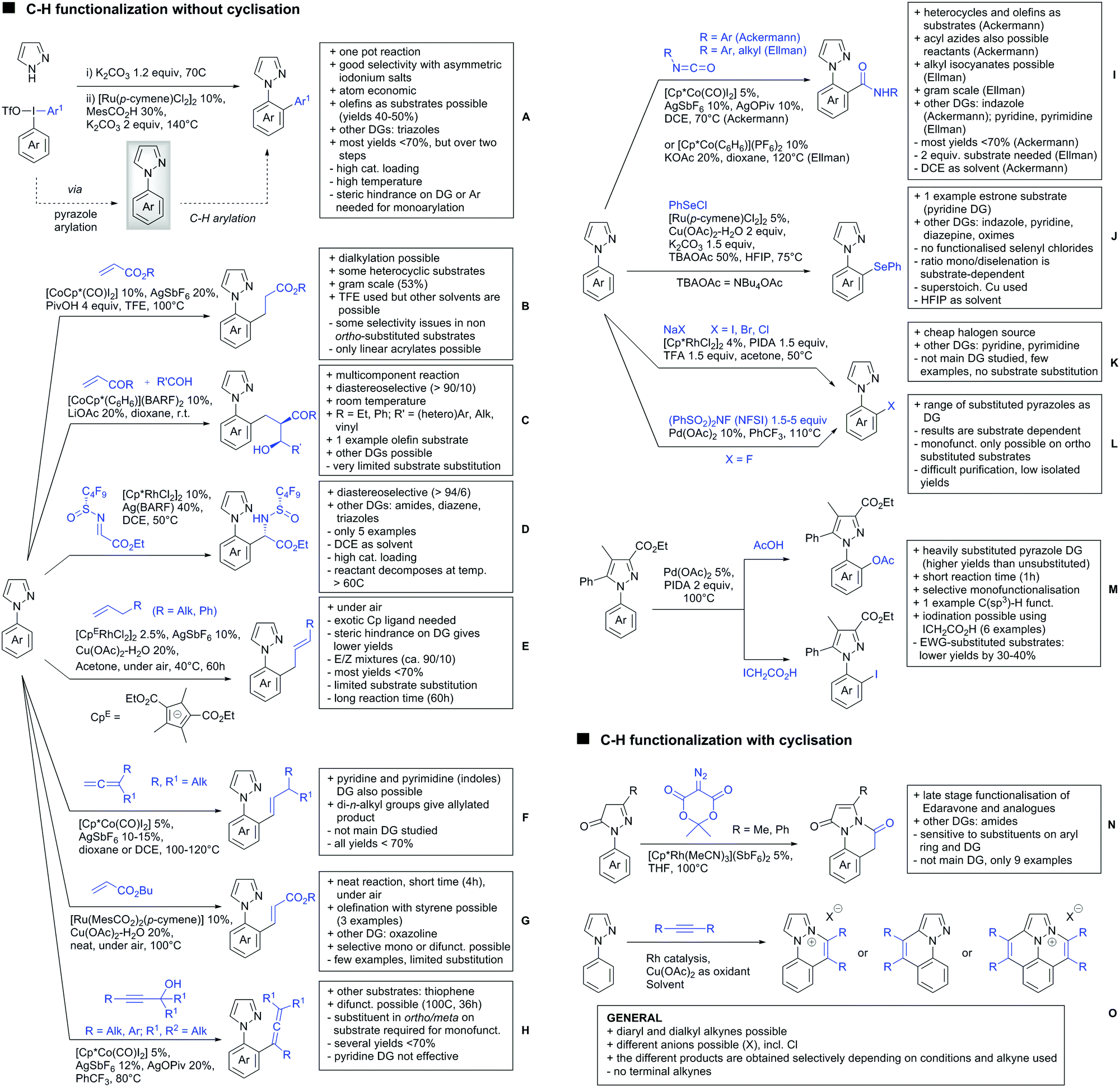 | ||
| Scheme 153 Pyrazole-directed C(sp2)–H functionalisation. | ||
The addition of 1-phenyl-1H-pyrazoles to chiral N-perfluorobutanesulfinyl imines also occurs with good diastereoselectivity, furnishing phenylglycine derivatives (Scheme 153D).932 It is worth mentioning that the same reaction does not work at all on the corresponding non-fluorinated substrates. A dehydrogenative allylation reaction was reported by Tanaka and Shibata using terminal olefins and Cu/air as oxidant (Scheme 153E). The reaction does not require leaving groups in the olefin, thus increasing the atom economy of the process. However, it has several disadvantages: (i) the exotic CpE ligand [1,3-bis(ethoxycarbonyl)cyclopentadienyl] is necessary for the reaction to proceed; (ii) a large excess of the substrate (3 equiv.) is required to prevent difunctionalisation in non ortho-substitutes substrates; (iii) many of the reported yields remain below the 70% threshold; and (iv) the reaction is slow (60 h, Scheme 153E).933
Addition to 1,1-dialkylallenes under Co catalysis provides olefinated arenes, although only medium yields were obtained using pyrazole as DG (Scheme 153F). Olefination was also performed via dehydrogenative coupling with acrylates with a Ru catalyst under aerobic conditions (Scheme 153G).934 Under these conditions, selective mono- or difunctionalisation was possible, only using different (small) excess of acrylates. Few examples were reported, but a similar reaction was demonstrated also for olefination with styrenes.934 Sundararaju published in 2017 a study on the reaction of 1-phenyl-1H-pyrazoles with propargyl alcohols, leading to the corresponding allenylated product (Scheme 153H).935 Whereas mono-functionalised products could be obtained selectively in good yields only on ortho/meta-substituted substrates, difunctionalisation was obtained on para-substituted arenes.
Two protocols for the aminocarbonylation of 1-phenyl-1H-pyrazoles, both exploiting Co catalysis and isocyanates as reactants, were reported independently by Ackermann and Ellmann in 2015 (Scheme 153I).936,937 Ackermann's protocol could also be applied to heterocyclic substrates (thiophene and furan) and to olefins, but only aromatic isocyanates were used. Ellman's protocol could instead be used with aliphatic isocyanates (2 examples), and generally provided higher yields, but 2 equiv. of the substrate were required.
Regarding C–heteroatom bond formation, selenation with phenylselenyl chloride was reported by Zhang (Scheme 153J),938 and halogenation by Wang and Hierso. Wang used Na halides and Rh catalysis for iodination, bromination and chlorination of a few 1-phenyl-1H-pyrazoles (Scheme 153K),772 while fluorination could be achieved using NFSI with Pd catalysis, as reported by Hierso (Scheme 153L).939 Notably, this functionalisation proved to be fairly substrate-dependent, requiring varying equivalents of NFSI or leading to mono or difluorinated products depending on the substitution pattern.
Acetoxylation and iodination were finally reported using a heavily functionalised pyrazole as DG. 4-Methyl-1,5-diaryl-1H-pyrazole-3-ethylcarboxylate was used as a model representing a class of pharmaceutically relevant substrates (Scheme 153M).940 Selective monoacetoxylation (due to steric hindrance) using Pd(OAc)2/PIDA/AcOH was achieved in good yields on a number of substrates, including aliphatic substrates (1 example). The corresponding iodinated product was obtained using iodoacetic acid instead under the same conditions.940 It is noteworthy that the presence of a EWG substituent on the substrate reduces the yields by 30–40% for both transformations.
Tandem C–H functionalisation/cyclisation processes similar to those reported for pyridine DGs (see Schemes 112 and 115) have also been developed for pyrazoles. Yi and Xu reported the late stage modification of the drug Edaravone via alkylation/cyclisation with Meldrum's acid diazo derivative (Scheme 153N).941 The reaction of 1-phenyl-1H-pyrazole with internal alkynes, apart from giving the typical olefination product, also results in neutral or ionic cyclisation products similar to those observed with pyridines and other heterocycles (Scheme 153O). In this case three different cyclisation products can be obtained, including ‘rollover’-derived products, strongly depending on the reaction conditions and the identity of the alkyne.942,943
Interestingly, a couple of C(sp3)–H bond functionalisation studies have been recently reported using pyrazole DGs. Using 3,4,5-trimethyl-1H-pyrazole as DG, Zhao demonstrated that both β and γ arylation is possible using Rh catalysis and aryl boroxines (Scheme 154A).928 While arylation at the β position is to be expected because of the formation of a 5-membered metallacycle intermediate, γ-functionalisation via 6-membered metallacycle selectively occurred when a tertiary or quaternary carbon in β was present.928 Another notable feature is the absence of any diarylation products, often observed when multiple functionalisable positions are available. More recently, Daugulis reported a similar arylation, but using Pd(OAc)2 and aryl iodides (Scheme 154B).926 A notable feature is the possibility to arylate secondary and even tertiary positions (1 example on adamantane substrate).
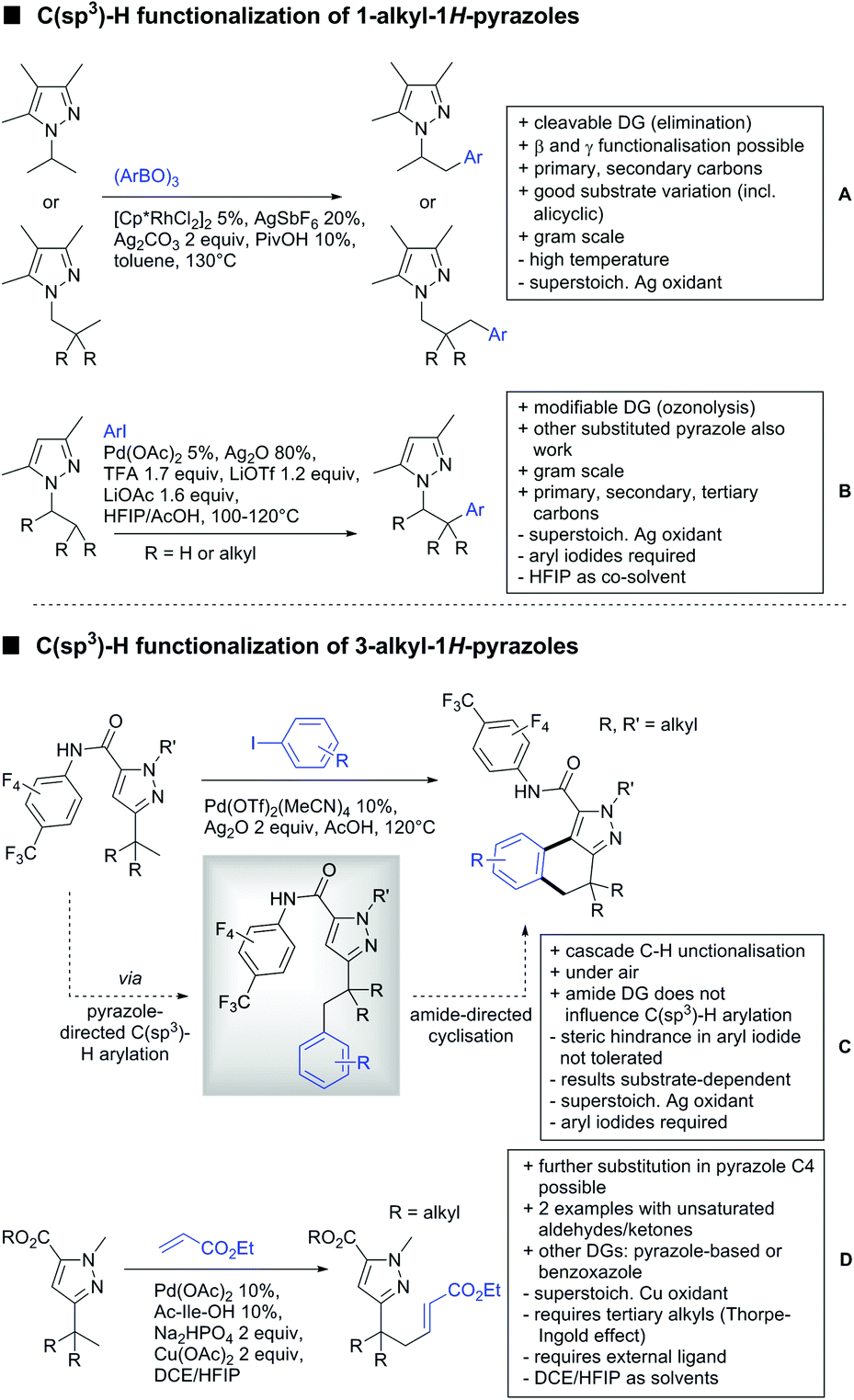 | ||
| Scheme 154 Pyrazole-directed C(sp3)–H functionalisation. | ||
Although a few papers have appeared in the literature about C(sp2)–H functionalisation of 3-aryl-1H-pyrazole derivatives,944,945 these are mostly related to olefination/cyclisation processes similar to those previously described, and will therefore not explicitly be discussed here. Nonetheless, two reports by Yu's and Stamos’ research groups on C(sp3)–H functionalisation have appeared. A domino process was developed where an initial C(sp3)–H functionalisation was promoted by a pyrazole DG, followed by an intramolecular, dehydrogenative, amide-directed C(sp2)–C(sp2) bond formation (Scheme 154C).946 The use of two different DGs for two different activations in cascade is a very innovative and useful concept for synthetic applications. The following year a C(sp3)–H olefination of 3-alkyl-1H-pyrazoles was reported by the same authors using ethyl acrylate and analogous reactants (Scheme 154D).947 To date this is the only example of pyrazole-directed C(sp3)–H olefination.
15.6. 1,2,3-Triazole as DG
Due to the easy access to triazoles, these heterocycles have also been used as DGs. Substrates can be attached to the DG at two different positions: (i) attached to the C4 atom of the DG (1,4-disubstituted triazoles, obtained by click chemistry), or (ii) attached to the N2 atom (2-monosubstituted triazoles, obtained via other methods). | ||
| Scheme 155 1,2,3-Triazoles as DGs. | ||
The selective functionalisation of the aryl ring in the C4 position of the triazole in the presence of a second aryl substituent in N1 (as for R = aryl in Scheme 155C–E) is suggested to be due to the higher electron density on the N3 in comparison to N2, which promotes selective coordination to this N atom and, consequently, selective functionalisation of the aryl ring in C4.950,953
Acylation in water using Pd/TBHP was reported by Jain (Scheme 155F).955 Interestingly, benzyl alcohols were used directly as reactant in this protocol, with oxidation to aldehyde (and formation of the acyl radical) occurring in situ. Importantly, heterocyclic-substituted (thiophene, furan) methylalcohols could also be used in this protocol. These substrates were also used in amidation reactions.
One protocol appeared making use of dioxazolones under Co catalysis. (Scheme 155G).956 Unfortunately, this reaction resulted in mixtures of mono- and difunctionalised products unless ortho or meta substituted substrates were employed. The same issue was observed using sulphonyl azides as reactants with Ru catalysts.957 In this respect, an interesting contribution came from Wu and Cui, who reported the use of triazole N-oxide as DG (under Ir catalysis) to suppress the undesired difunctionalisation due to the increased steric hindrance (Scheme 155H).958
15.7. 9H-Purines and analogues as DG
Purine-based compounds are an important class of biologically active molecules, and the modification of purine-containing starting materials has recently been explored in C–H functionalisation chemistry. The state of the art of the use of purines as both substrates and DGs in C–H functionalisation before 2015 has been summarised by Kapdi/Fairlamb.959 In this section the use of purine as heterocyclic DGs in the last few years is summarised. As most of the papers included in this section have already appeared in previous chapters (other heterocyclic DGs), the present section has mostly the purpose of collecting all the transformations together from a practical point of view for the reader, given the importance of the core structure, but little further discussion will be added. It is important to note that purines are mostly tested as secondary DG, therefore no specific optimisation and only few examples are typically reported. If not specified in Scheme 156, yields are around 60–70%; in case of considerably different results (higher or lower yields), these are specifically mentioned in the scheme. | ||
| Scheme 156 Purine-directed C–H functionalisations. | ||
Several types of functionalisation have been studied on 6-aryl-9H-purines, where the N1 atom of the pyrimidine ring acts as the coordinating moiety. Arylation with diazonium salts under photoredox catalysis was reported by Guo. The reaction worked nicely on a few purines and nucleosides, despite requiring 6 equiv. of the diazonium reactant and the limited substrate substitution investigated (Scheme 156A).960 Co-catalysed allylation and olefination were reported by Matsunaga/Yoshino and Yu/Chen respectively (Scheme 156B and C).688,961 Both these processes occurred in higher yields than the average 60–70% for these substrates, but only few examples were reported. Cyanation could be performed using N-cyanosuccinimide and Co catalysis, according to a protocol developed by Chang (Scheme 156D).711
Concerning the formation of C–heteroatom bonds in 6-aryl-9H-purines, three amidation methods were reported using electrophilic N-Boc O-acyl hydroxylamines or dioxazolones (Scheme 156E–G).747,748,753 This last procedure, in particular, furnished high yields, and offers the possibility of thiophene functionalisation (Scheme 156G). Finally, trifluoromethylthiolation727 and halogenation962 were reported by Matsunaga/Yoshino and Pawar, both under Co catalysis (Scheme 156H and I). A good substrate screening was investigated for the halogenation (especially for the iodination), allowing a comparison of the results. This shows that para EWG substituents on the aryl ring of the substrate generally result in much lower yields than para EDG substituents.962
A rare example of functionalisation on 9-aryl-9H-purines (coordinating atom: N3) was reported by Du. Alkoxylation was studied under Pd catalysis using alcohols as reactants/solvents and PIDA as oxidant. While methoxylation was successfully achieved on the 8-azapurine analogue (Scheme 156J, product A), ethoxylation did not occur on such substrate, and standard purines had to be used (Scheme 156J, Product B).963
15.8. 7-Azaindoles as DG
7-Azaindoles have been used several times as DGs, due to their presence in a number of biologically active compounds. An important aspect of this DG is the relatively broad functional group tolerance on its core, which ranges from halides, alkyl chains, olefins, carbonyls, to (hetero)arenes.A number of functionalisations have been reported using procedures similar to those for other DGs, exclusively under Rh (in one case Ir) catalysis. Among the alkylation and olefination with α,β-unsaturated moieties (Scheme 157A–C),964–966 an interesting cross-dehydrogenative olefination procedure which does not require the presence of a stoichiometric oxidant (as is usually the case), was reported by Dong (Scheme 157C).966 In this reaction the Rh(III) hydride intermediate formed at the final stage of the olefination process reacts with the acid, releasing H2. This is in turn suggested to react with the excess of unsaturated reactant (2 equiv.), which acts as a hydrogen acceptor.966 Another version of this transformation (not shown in scheme), was reported by Miura and Satoh. Despite the use of superstoichiometric Ag as oxidant and DME as solvent, this method allows fast reactions (3–6 h) under mild conditions (40–90 °C), giving generally good to excellent results.967
 | ||
| Scheme 157 C–H functionalisation directed by 7-azaindole moieties. | ||
Reactions with TIPS-EBX, N-cyano-N-phenyl-p-toluenesulfonamide, and anthranils result in ortho alkynylation, cyanation and amination as described in Scheme 157D–F.968–970 Notable is the use of heterocyclic substrates such as (benzo)thiophenes and furans in the cyanation protocol.969 One example of C–O bond formation was reported by Deb using PIDA and Rh catalysis. In contrast to many similar procedures employing Pd catalysis, the use of Rh allowed the reaction to run at a low temperature (40 °C) furnishing good yields often in a short time (Scheme 157G).971
The Dong group reported four procedures for the cascade alkylation–cyclisation or olefination–cyclisation, where a “rollover” mechanism after the initial C–H activation results in the formation of π-conjugated fused systems (see Scheme 115, bottom). The group-directed ortho-alkylation with diazocompounds or via addition to alkenyl esters, followed by intramolecular activation of the azaindole C2, and finally by dehydration or β-H elimination, results in the products in Scheme 157H and I.972,973 The reaction with alkynes, occurring as previously reported for other DGs, was also investigated (Scheme 157J and K).974,975 Interesting in this respect is the reaction with 3 equiv. of alkyne, which results in both ortho olefination–cyclisation and arene homologation, ultimately furnishing azahelicenes, interesting compounds for their fluorescent properties.975
Finally, the reaction with electron-deficient alkenes resulted in an oxidative olefination, followed by rollover and intramolecular Michael addition, giving the compounds shown in Scheme 157L.976 An interesting aspect is that steric hindrance at the C3 position of the azaindole DG prevents the final intramolecular cyclisation, selectively furnishing the olefination product.976
15.9. Oxadiazoles and benzisoxazoles as DGs
In the same way that oximes can act as internal oxidants when used as DGs (see Section 12), via cleavage of the N–O bond, also 1,2,4-oxadiazoles and 1,2-benzisoxazoles (cyclic oximes equivalents) have been used as DG/internal oxidant combinations, in particular for the synthesis of isoquinoline derivatives upon reaction with alkynes.Zhu and Miura independently reported in 2017 the use of 1,2,4-oxadiazoles, leading to 1-aminoisoquinolines and 1-acylaminoisoquinolines respectively, using Co and Rh catalysis977,978 (Scheme 158A and B). Either diaryl, dithiophenyl, or dialkyl alkynes could be used as coupling partners, while terminal alkynes seem unsuitable for this reaction.978 Interestingly, while in the Co-catalysed reaction the use of unsymmetrical alkynes gave rise to regioisomeric mixtures, under Rh catalysis only one regioisomer was obtained. The mechanism proposed for this transformation involves the insertion of the alkyne into the initial metallacycle formed, followed by a concerted or stepwise C–N bond formation/N–O bond cleavage, leading to the 1-acylaminoisoquinoline product. The presence of TFE in Zhu's protocol provides in situ hydrolysis and formation of the unprotected 1-amino compound.977
 | ||
| Scheme 158 Oxadiazoles and benzisoxazoles as DGs. | ||
Zhu also reported analogous reactions employing 1,2,4-oxadiazolones as DGs. In this case the N–O cleavage results in the elimination of CO2 from the reaction product.979,980
In 2017 the use of 1,2-benzisoxazoles as DGs for a similar reaction with alkynes, was disclosed by Miura. With this procedure, isoquinolines functionalised in C1 with a phenolic moiety were formed (Scheme 158C).981 The mechanism suggested is analogous to that proposed above.
15.10. Oxazolines and benzoxazoles as DGs
Oxazolines have been used a few times as DGs in C–H functionalisation. One of the reasons is that these functionalities can be easily introduced from the corresponding carboxylic acid, via condensation with 2-aminoalcohols, and can also be easily cleaved to return to the original carboxylic acid using a number of procedures: (i) strongly acidic or basic hydrolysis; (ii) activation via N-chlorination, sulphonylation or acylation, followed by hydrolysis (milder conditions). The oxazoline ring can also be reductively cleaved to aldehydes using: (i) N-quaternization, followed by reduction with NaBH4 and hydrolysis; (ii) reduction in the presence of NiCl2, followed by acidic hydrolysis.982 These and other methods are nicely summarised in a review by Meyers,983 and will therefore not be discussed in detail here. An interesting one-pot hydrolysis/cyclisation method, first discovered by Harrity, for ortho-amidated 2-aryloxazolines involves basic hydrolysis of the amide, and subsequent cyclisation with formamidine acetate, with consequent opening of the oxazoline ring (Scheme 159).754,984 | ||
| Scheme 159 Ring-opening/cyclisation involving oxazoline DGs. | ||
Sparse examples of oxazolines as DGs are generally reported in papers mostly dealing with other DGs, but a few specific investigations have been recently published. Arylation with aryl halides or aryl carbamates was reported by Ackermann and Chatani using Ru, Co or Rh catalysis (Scheme 160A–C),653,985,986 typically requiring preformed catalysts or the addition of a ligand to reach good results. An example of intramolecular alkylation leading to the formation of heterocycles was reported by Zhu (Scheme 160D).177 Interestingly, the intramolecular addition to an unsaturated moiety occurs regiospecifically in α-position to the carbonyl, instead of β, although the origin of for such selectivity was not investigated. Olefination by Fujiwara–Moritani coupling with acrylates and benzoxazoles as DG was reported by Hou under Rh catalysis (Scheme 160E).987 This reaction proved generally substrate- and DG-dependent, with yields ranging from 33 to 95%, and no monoselectivity could be achieved in non ortho-functionalised substrates. Nonetheless, this protocol was efficient for the coupling of different classes of olefins, including allylbenzene (unusual reactant for this type of reaction).987 Two protocols for amidation were reported by Harrity and Ackermann, using amides or dioxazolones respectively (Scheme 160F and G).754,984,988 A hydroxylation reaction on 2-arylbenzoxazoles was demonstrated by Chakraborti in 2015, where the hydroxy radical was generated in situ from 1,4-dioxane, used as solvent (Scheme 160H).989 Interestingly, this protocol only required 2.5% Pd loading, considerably lower than other analogous procedures. Finally, a C(sp3)–H silylation on 2-ethyloxazoline derivatives was reported by Murata and co-workers in 2016 (Scheme 160I).990 The reaction was performed at 180 °C with HSi(Me)(OSiMe3)2. This reaction did not require the use of a hydrogen acceptor, probably due to the high temperature used.
 | ||
| Scheme 160 Oxazolines and benzoxazoles as DGs. | ||
15.11. Benzothiazoles as DGs
A handful of papers on the use of benzothiazole DGs have appeared in the recent literature. The cleavage/ring opening of 2-functionalised (carbon chains/rings) benzothiazoles have been reported in a few cases,991–995 but these protocols have not been used in the context of DG cleavage in C–H functionalisation.Chakraborti investigated the use of benzothiazole DGs specifically for the olefination of heterocycles with alkynes using Ru catalysis (Scheme 161A).996 This protocol resulted in good yields of the desired olefinated products, and works efficiently on 2-thienyl, 2-furyl, 2-pyrrolyl and 3-indolyl substrates, and a few examples of regioselective olefination with unsymmetrical alkynes were demonstrated as well.996 Acylation of benzothiazole-equipped arenes was reported using classical Pd/oxidant/acyl radical precursor methods (mechanism in Scheme 118).762,997 Of note here, is the use of CAN (ceric ammonium nitrate) as a new alternative oxidant instead of peroxides or O2. Patel reported the use of CAN with α-ketocarboxylic acids for C–H benzoylation of 2-arylbenzothiazoles and a variety of other heterocyclic compounds (Scheme 161B).762 Another method sees the use of acylperoxycoumarins as acylating agents (Scheme 161C).998 Strangely, this reaction does not proceed via a (free) radical pathway, as demonstrated by radical trapping experiments, but instead occurs via a β-carbon elimination from an alkoxy-coordinated Pd species, which delivers the acyl groups directly to the metal, and a subsequent reductive elimination releases the acylated product.998 An example of Cu-catalysed C–H nitration was reported making use of Fe(NO3)3 as nitro group source (Scheme 161D).999 Although utilising a cheap nitro source, the reaction still requires 2 equiv. of Ag2O (4 equiv. of Ag in total). With a similar protocol to that shown in Scheme 161B, the benzoxylation of 2-aryl benzothiazoles occurs using benzoic acids (Scheme 161E).762 Mechanistically, a benzoxy radical is generated by CAN, and subsequently coordinates to the intermediate palladacycle, which ultimately results in its introduction onto the substrate.
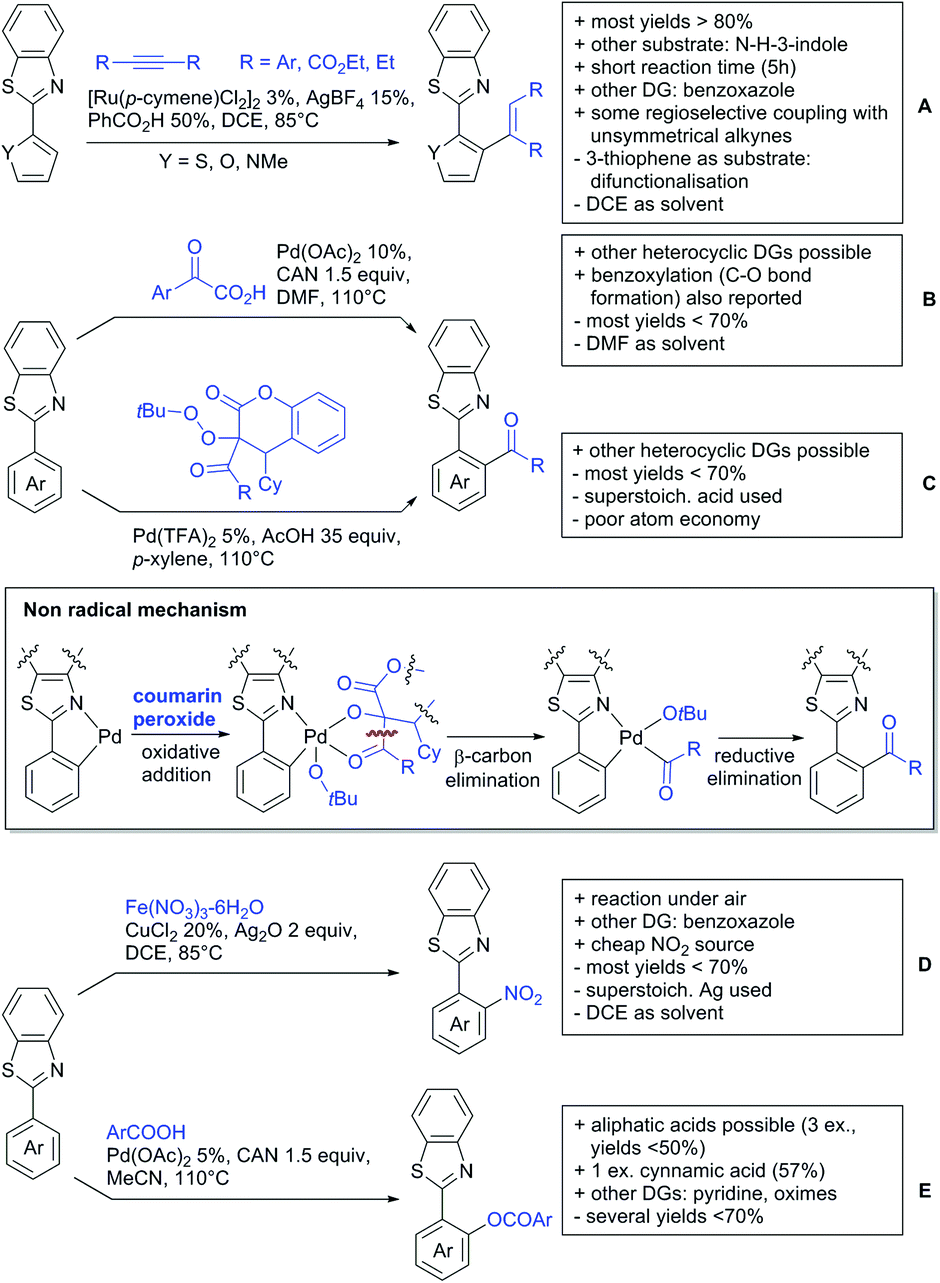 | ||
| Scheme 161 Benzothiazoles as DGs. | ||
16. Other DGs employed in C–H functionalisation
The synthetic value of C–H functionalisation chemistry is strongly related to the modularity of the DG used. Ideally, a DG should be traceless or easily convertible into an array of common functionalities. An alternative approach consists in direct utilisation of functionalities naturally present on a molecule of interest to play the role of DG. In such a case particularly sustainable transformations take place as installation and removal of the DG is not required anymore. Following these considerations, over the last decade intensive efforts have been focused on designing DGs based on heteroatoms such as P, S, Si, O, N, etc. Notably, these modern DGs are generally easily cleavable or may also remain present in the final product. An additional advantage of these scaffolds is their potentially stereogenic character, and thus diastereoselective C–H functionalisation reactions can be explored, even without the use of external chiral ligands. In the following sections, recent advances on the use of these increasingly important DGs are summarised. Due to their less common nature though, relative to the above reported DGs, important publications from before 2015 will also be included.16.1. Thioether as DG
Due to the strong coordination properties of the sulfur compounds, and in particular thioethers, this motif has been considered, for years, an inefficient DG, leading to catalyst poisoning. However, between 2012 and 2015 few reports have been published showing that the thio unit can be astutely employed in the context of directed functionalisation,1000–1003 and these pioneering contributions have already been summarized.17 More recently, due to a widespread presence of thio groups in natural products and biologically active scaffolds, several research groups reported complementary studies.In 2015, Urriolabeitia expanded the scope of previously known Rh-catalysed direct olefinations of benzylthioethers1000,1002,1003 by developing a related C–H hydroarylation catalysed by the [Ru(p-cymene)Cl2]2 complex (Scheme 162A).1004 Installation of the thioether motif at the benzylic position is crucial to allow efficient formation of a 5-membered ruthenacyclic intermediate. The optimization study revealed that choice of a protic but not too acidic solvent such as HFIP is essential for a high efficiency. Unfortunately, moderate selectivity between mono- and diarylation is observed, calling for exemplification of this methodology on ortho-substituted substrates. Interestingly, the reaction occurs smoothly under microwave heating (100 °C for 30 min), which enables to drastically reduce the reaction time (typically 24 h) required under conventional heating. The reaction well tolerates several aliphatic symmetric alkynes and the coupling occurs as a syn addition, delivering E-trisubstituted vinylic fragments (Scheme 162A).1004 Besides, a regioselective transformation was achieved when using 1-phenylpropyne coupling partner (9:1 isomeric ratio) although a significant drop in regioselectivity was witnessed for 2-hexyne. The reaction shows good FG tolerance on the substrate, both on the aromatic ring and the S-substituent (aliphatic and aromatic motifs are efficient), delivering the desired vinylic compounds in good to excellent yields (16 examples, 54–96% yield).
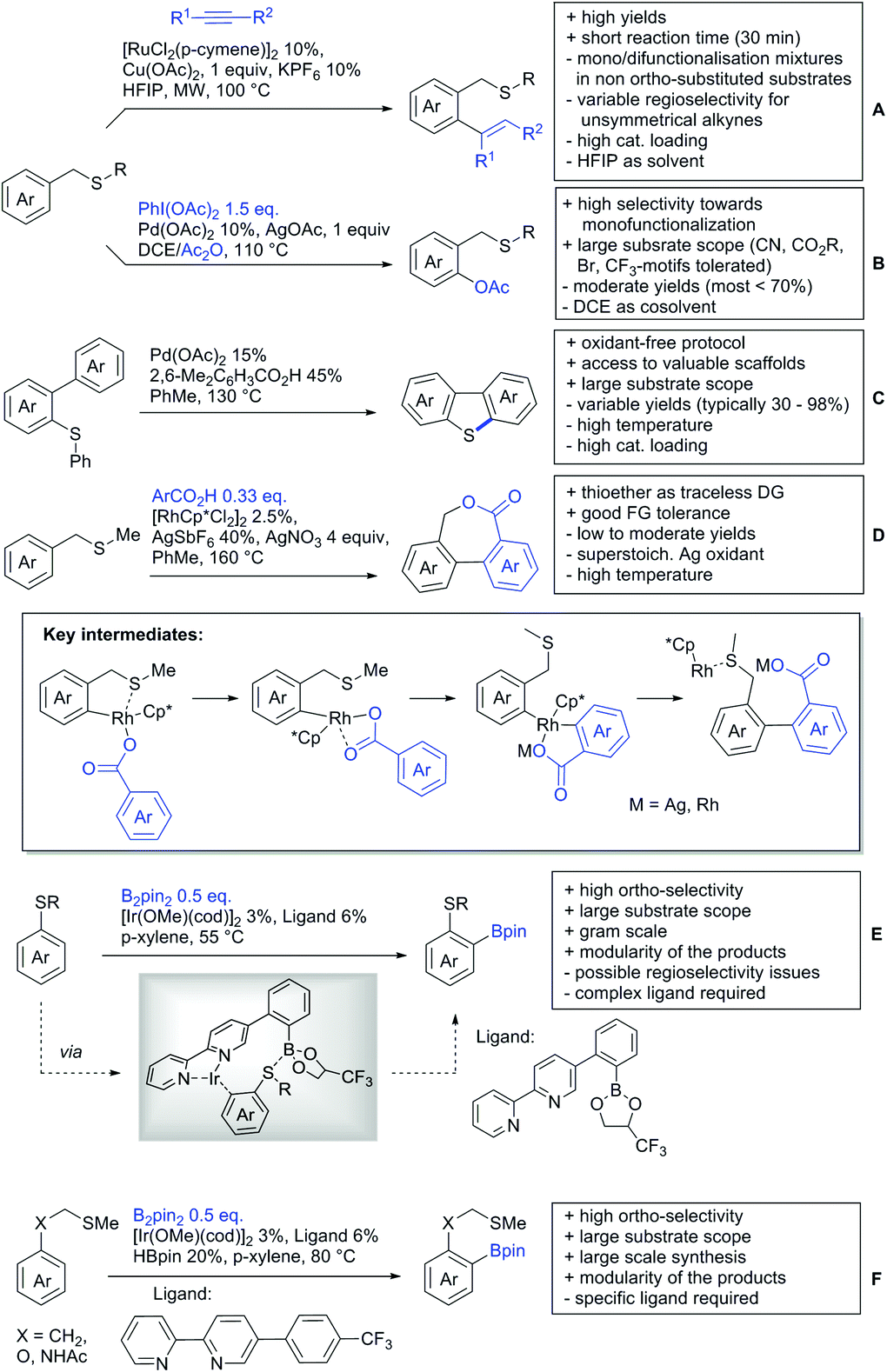 | ||
| Scheme 162 C(sp2)–H functionalisation using thioether-based DGs. | ||
To further expand the scope of molecular scaffolds accessible via thioether-directed C–H functionalisation Liu and Zhang reported a direct acetoxylation protocol (Scheme 162B).1005 As in the previous example, the reaction requires benzothiophene substrates but the DG may also be installed at homobenzylic position (formation of 6-membered metallacyclic intermediate). Using standard Pd(OAc)2 catalyst, in combination with Ac2O, PIDA, and AgOAc, the desired C–O bond formation takes place. Interestingly, this protocol permits selective mono-functionalisation and a remarkable array of FGs is well tolerated, including bromine and chlorine atoms, but also coordinating moieties such as nitrile, ester and OCF3 moiety. Yields are generally moderate to good (21 examples, 40–84% yield). Importantly, the DG may be removed from the final product either using RANEY® Ni to furnish the corresponding o-tolyl acetate, or by addition of m-CPBA to convert the thio-motif into an aldehyde.
Thiol-directed C–H activation can also be astutely used to access benzo-fused thiophenes via intramolecular C–H arylation, as illustrated in 2016 by Tobisu and Chatani (Scheme 162C).1006 The authors discovered that biphenylsulfide, in the presence of Pd(OAc)2 and under carboxylic acid assistance (2,6-Me2C6H3CO2H used) undergoes a tandem C–H cleavage/intramolecular C–S bond formation/S–C cleavage, delivering the desired heterocyclic products under oxidant-free conditions. This operationally simple and high-yielding protocol using readily accessible starting materials and reactants gives access to a large panel of unsymmetrical dibenzothiophenes, important scaffolds in advanced material science and bioactive molecules. It is noteworthy that this oxidant-free protocol allows the suppression of the usually observed reactivity for this type of compounds, i.e. C–H/C–H intramolecular arylation to give isomeric benzothiophene compounds.1007
In addition to this intramolecular thioether-directed arylation, Shi et al. developed a very interesting intermolecular coupling between benzylthioethers and carboxylic acids, yielding dibenzooxepinones (Scheme 162D).1008 This RhCp*-catalysed transformation involves initial C–H metallation of the thiophene substrate, followed by carboxylate-assisted second C–H activation and reductive elimination, and Rh- or Ag-assisted cyclisation and C–S cleavage. Accordingly, the thiophene moiety is clearly a traceless DG. Although rather harsh reaction conditions required (use of 4 equivalents of AgNO3 and reaction temperature of 160 °C) and the relatively low yields (28–75%), the reaction still well tolerates substituents such as Cl, Br, but also strongly electron withdrawing CF3 or ester moieties.
In 2017 Kuninobu and Kanai designed an interesting ortho-selective borylation of arylsulfides.1009 In this case the thioether DG is directly installed on the aromatic ring as, contrary to the previous examples, the S atom does not coordinate directly to the Ir catalyst, but generates a Lewis acid–base intermediate with a bipyridine-ligated boron species (Scheme 162E).1009 Such an unusual interaction directs the borylation highly selectively at the ortho-position. Fine-tuning of the catalytic system and ligand design allowed the development of a truly efficient and versatile transformation, occurring under mild reaction conditions and compatible with a range of functionalised substrates. Notably, the reaction is equally efficient at gram scale. As the newly introduced Bpin substituent is a handle for further modification of the molecular scaffolds, this transformation holds great promise for widespread applications. Few months later the authors further expanded the potential of this transformation by conceiving an ortho-borylation of phenol and aniline derivatives (Scheme 162F).1010 In this case the methylthiomethyl group installed on hydroxy- or amino-moieties allows, via the same type of interaction as above, directing the borylation at the ortho-position. As previously, the high regioselectivity observed, efficiency and possible post-modifications of the newly obtained scaffolds, render this coupling attractive, as further illustrated by its application in an expedient synthesis of a Ca receptor modulator.1010 The removal of the DG (i.e. deprotection of hydroxyl group) occurs smoothly using I2 in MeOH, furnishing the corresponding phenol in high yield.
Drawing inspiration from the standard thioether-directed C–H functionalisation, Satoh and Miura disclosed in 2015 the use of dithiane motifs (commonly used as aldehyde protecting group) as an effective DG for Rh-catalysed Fujiwara–Moritani couplings (Scheme 163A).1011 This C–C bond forming transformation, occurring under relatively mild conditions (60 °C, no need for acidic additives and use of Cu(OAc)2·H2O as mild oxidant) is not only highly monoselective but also compatible with an array of functionalised substrates bearing challenging motifs such as nitro, ketone, ether, thioether or halide substituents. The protocol is also compatible with a cyclic sulfide substrate. The synthetic value of this transformation arises from a straightforward post-modification of the dithiane-DG that can be either removed using RANEY®-Ni or converted into aldehyde in the presence of Dess–Martin periodinane.1011
 | ||
| Scheme 163 Rare examples of dithiane DGs. | ||
Very recently, Li et al. used the same DG to achieve an Ir-catalysed ortho-borylation (Scheme 163B).1012 This ligand-free coupling with B2pin2 is effective for the direct functionalisation of both aromatic and heteroaromatic moieties, like thiophene and ferrocene backbones. It is worth noting that the employment of the dithiane motif outcompetes thioether DGs as mono-functionalisation occurs selectively, whereas mixtures of mono- and biarylated products are typically generated when thioethers are used. The cleavage of the 1,3-dithiane was achieved using NaI, Fe(acac)3 in H2O2, delivering the corresponding aldehyde.
Further exploring the potential of the thioether motif as DG and drawing inspiration from the observation that benzylthioethers are effective in C–H activation reactions, Wang and Shi described an elegant example of disulfide-directed C–H hydroxylation (Scheme 164).1013 The optimized Cu-based catalytic system allows a sequential transformation involving: (1) thiophenol oxidation to disulfide, (2) disulfide-directed ortho-hydroxylation and (3) S-arylation using boronic acid coupling partner, affording otherwise difficult to access 1,2-thio-phenol derivatives. Remarkably, this complex transformation is rather high-yielding and shows good substrate scope, in particular with regard to halogenated substituents at both coupling partners. Besides, applying the same concept for a synthesis of thiophenol-substituted quinones further showcased the potential of this reaction. Few months later, Wang and Li reported a closely related Cu-catalysed disulfide-directed ortho-hydroxylation. In this case disulfide substrates were directly employed, and a Cu–bipy complex was selected as the optimal catalyst.1014
 | ||
| Scheme 164 Disulfide-directed C–H hydroxylation. | ||
16.2. Sulfoxide as DG
In addition to thioethers, higher oxidation state sulfur moieties also established themselves as powerful DGs. In particular between 2012 and 2014 several research groups employed with success sulfoxide DGs in Pd,1015–1017 Rh,1018 and Ru1019-catalysed C–H activation. Since 2015, few additional key contributions have been published,29 mainly relating to the use of enantiopure sulfoxides for stereoselective C–H activation.In 2013 Leroux and Colobert reported first example of diastereoselective sulfoxide-directed oxidative olefination of biaryl precursors leading to atropoenriched, axially chiral scaffolds.1020 This pioneering work clearly showcases the potential of enantiopure sulfinyl, accessible in a large scale from cheap menthol,1021 to play the double role of DG and chiral auxiliary. However, both efficiency and stereoselectivity of the catalytic system were rather moderate. A clear improvement was achieved in 2014 and 2016 by Wencel-Delord and Colobert, who discovered that a much more powerful and general transformation is achieved when changing the reaction medium from DCE to HFIP (Scheme 165A).1022,1023 Under such modified reaction conditions the Fujiwara–Moritani reaction may be conducted at room temperature and using a significantly lower amount of the Ag oxidant (2 equiv. vs. 6 equiv. initially required), delivering a large panel of axially chiral biaryls with excellent atroposelectivity and high yields. Besides, as the sulfoxide moiety undergoes exchange with Li bases, trapping of the resulting axially chiral Li species with an array of electrophiles could be performed, delivering variety functionalised biaryls without loss of stereopurity (introduction of functionalities such as aldehydes, Me, OH, CO2H, etc.).1022 This methodology was also further used to perform direct acetoxylation and iodination.1023 The synthetic potential of this methodology was further illustrated while designing a novel retrosynthetic route towards biologically active, axially chiral Steganacine scaffolds.1024 Remarkably, the synthesis based on this atropo-diastereoselective transformation delivered the direct precursor of the targeted enantiopure molecule in only 10 steps and in very high total yield (42%).
 | ||
| Scheme 165 Sulfoxide-directed C–H functionalisation. | ||
Very recently the concept of sulfoxide-directed atroposelective C–H functionalisation was employed to build up original tridimensional structures, i.e. ortho-orientated terphenyl scaffolds with two perfectly controlled chiral axis (Scheme 165B).1025 These molecules exhibiting “open clam shell architectures” could be obtained via direct arylation of the biaryl-sulfoxide precursors with ortho-substituted iodoarenes. During this process two chiral Ar–Ar axes, are induced in a single transformation with very high stereocontrol. Importantly, the traceless character of the sulfoxide, combined with the good FG tolerance of the catalytic system towards the iodoarene partner permits straightforward derivatization into unprecedented bidentate ligands.
Dibenzothiophenes are important molecular motifs in natural products, pharmaceuticals and functional materials. Accordingly, designing of C–H functionalisation-based strategies to access these molecular units has gathered the attention of several research groups. In 2013 Colobert described a Pd-catalysed intramolecular arylation of 2-bromodiarylsulfinyle, delivering dibenzothiophenes S-oxides.1026 Going one step further, in 2015 Huang and Lin reported an intramolecular two-fold C–H functionalisation of diarylsulfoxides (Scheme 165C).1027 Interestingly, under this Rh-based protocol deoxygenation of the newly formed dibenzothiophenes S-oxides occurs in situ. Unfortunately, the efficiency of the reaction is strongly affected by the nature of the starting material and harsh reaction conditions are required (140 °C).
16.3. Sulfones, sulfonamides & sulfoximines DG
In contrast to thioether and sulfoxides that have been regularly employed as DGs, the use of sulfones in this context has remained fairly unexplored till recently, despite the easy access to such molecules via electrophilic sulfonylation of aromatic rings. Initially, the sulfone moiety was used as a handle to install other DGs, as in the case to 2-pyridylsulfonate DG.1028 In this case the heterocycle enhances and dictates the metallation event whereas the SO2 moiety can be considered as a removable anchor (for examples of such transformations see Section 15.3.4). Satoh and Miura proved however in 2015 that sulfones may act as efficient DGs themselves, allowing ortho-selective alkenylation of aromatics (Scheme 166A).1029 The catalytic system based on Rh complexes, combined with adamantanecarboxylic acid additive gave good results with internal aromatic and aliphatic alkynes, delivering the desired vinylic products in moderate to good yields, but a high reaction temperature was generally required. | ||
| Scheme 166 Sulfone-directed C–H functionalisation. | ||
An additional attracting example of sulfone-directed intramolecular transformation was reported in 2015 by Zhou (Scheme 166B).1030 This cascade-type reaction involves sulfone-directed metallation, followed by base-assisted 1,5-H shift affording allene intermediates. Insertion into the C–Rh bond finally delivers seven-membered heteroaromatic products such as benzothiepines. This strategy constitutes an interesting pathway towards molecules of interest despite the complexity of the starting materials used.
Sulfonamide is amongst the most significant pharmacophores, frequently encountered in biologically active scaffolds. However, this weakly coordinating motif has attracted only limited attention from the scientific community working in the field of directed C–H functionalisation. In 2011 Yu et al. described a late stage modification of sulfonamide-containing motifs, via direct olefination, carboxylation, carbonylation, iodination, arylation and alkylation.1031 The variety of sulfonamide-directed transformations was enlarged in 2016 when Singh developed a Pd-catalysed direct arylation using iodoarenes as coupling partners (Scheme 167A).1032 This transformation is highly mono-selective, although the products are isolated in moderate to good yields (typically between 60–70%) and prolonged heating at 130 °C in acetic acid is necessary to promote this transformation.
 | ||
| Scheme 167 Direct functionalisation using sulfonamides as DG. | ||
In parallel, Kerr and Tuttle endeavored on designing a catalytic system to regioselectively install deuterium on aromatics (Scheme 167B).1033 Following this target they discovered that primary sulfonamides undergo highly selective H/D exchange, when using an Ir–NHC complex. Importantly, D2 may be employed as deuterium source and this transformation is compatible with an array of functionalities. Unfortunately, the level of deuterium incorporation is highly dependent on the substitution pattern of the substrates, possibly due to their different solubility in DCM. This protocol could however be successfully used to label pharmaceuticals, such as Celecoxib and Mavacoxib.1033 In 2017 the synthetic potential and field of application of this methodology was further expanded by conceiving a closely related deuteration of secondary and tertiary sulfonamides. Atzrodt and Derdau established a protocol based on an Ir catalyst bearing either a monodentate NHC or bidentate NHC/oxazoline ligand (commercially available Burgess catalyst), effective in chlorobenzene at higher temperature (typically 100–120 °C) (Scheme 167C).1034 However, as in the previous example, the outcome of the transformation might be difficult to predict in terms of deuterium incorporation and regioselectivity (in particular if other coordinating moieties are present on the substrate or when tertiary sulfonamides are employed).
Very recently, Yu and coworkers described an astute application for sulfonamide motifs installed at the benzylic position to promote meta-selective functionalisation (Scheme 167D).1035 The regioselectivity switch was reached using modified norbornene (NBE-CO2Me: 2-carbomethoxynorbornene) as a transient mediator, while addition of an external isoquinoline ligand was crucial for the efficiency of the catalytic system. Remarkably, both meta-arylation and meta-alkylations could be performed and the protocol features broad substrate scope as well as excellent FG tolerance. The synthetic value of this reaction was further illustrated by the modularity of the obtained products, which could be easily converted into the corresponding sodium sulfonates, sulfonate esters, other sulfonamides or olefins. Besides, a two-step procedure allows cleavage of the DG under Julia olefination conditions delivering the corresponding styrene.1035
Finally, sulfoximines further expand the portfolio of S-based DGs. In particular, heterocyclic molecules featuring the sulfoximine group are tempting for drug design. Hence, the development of modern, efficient and straightforward pathways to access them is of great importance. Accordingly, several research groups astutely used sulfoximine-directed C–H functionalisation/cyclisation protocols to access an array of benzothiazines congeners. Amongst pioneering contributions in this field, Bolm described in 2013 the Rh-catalysed oxidative annulation of sulfoximines and internal alkynes, delivering 1,2-benzothiazines with fully substituted heterocyclic cores.1036 Despite the synthetic value of this methodology, the use of unsymmetrical alkynes raised regioselectivity issues and mixtures of regioisomeric products were generally produced. To tackle this limitation, in 2015 the same research group combined Rh-catalysed sulfoximine-directed C–H alkylation with diazo compounds (Scheme 168A).1037 Rewardingly, the reaction between NH-sulfoximines and a large variety of alkyl- and phenyl-containing diazo esters occurred smoothly in presence of a Rh catalyst and NaOAc additive, delivering structurally diversified 1,2-benzothiazines in generally excellent yields. Besides, a scale-up of this reaction was achieved and the newly obtained scaffold could be straightforwardly converted into several added-value congeners.
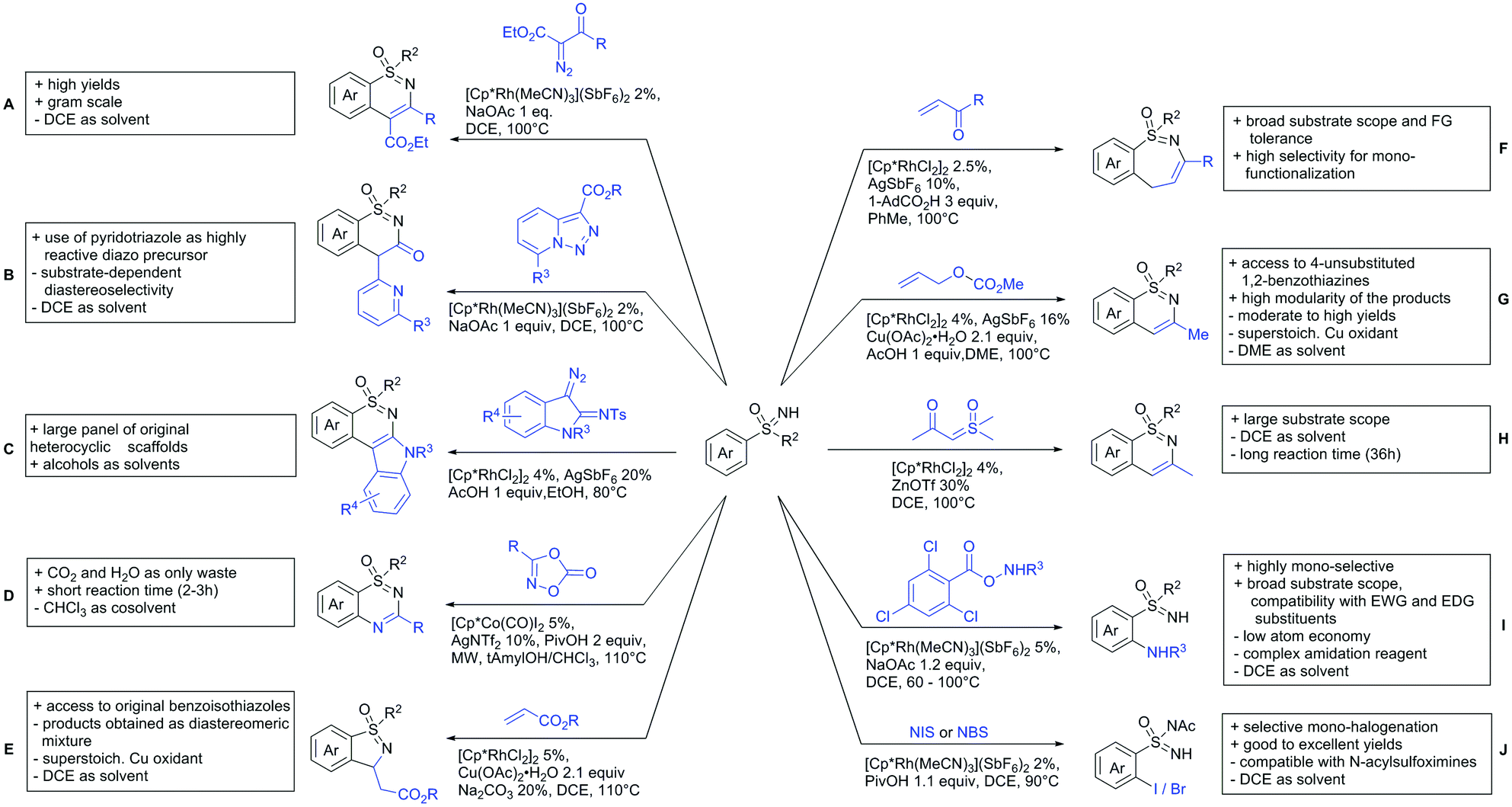 | ||
| Scheme 168 (Free-NH)-sulfoximine-directed C–H functionalisation. | ||
Following this idea, Lee and coworkers developed an alternative synthesis of 1,2-benzothiazine derivatives (Scheme 168B).1038 Using pyridotriazoles as highly reactive diazoimine precursors (see also Scheme 112), pyridine-substituted 1,2-benzothiazines were afforded via a domino C–H functionalisation/cyclisation/elimination sequence in good to excellent yields regardless of the substitution pattern of the aromatic substrate. Few months later the same group further expanded the panel of related transformations, now employing 3-diazoindolin-2-imines as coupling partners.1039 Under Rh catalysis the desired direct coupling/annulation sequence occurred producing a indolo-1,2-benzothiazines with various substituents at both, the substrate and the indole core (Scheme 168C).1039
An efficient cyclometallation event for sulfoximines substrates combined with the high reactivity of the resulting metallacyclic intermediates towards carbene migratory insertion encouraged further elaboration of innovative synthetic routes towards original heterocycles. Accordingly, in 2017 Chen performed direct C–H amidation of sulfoximines using a Co catalyst and 1,4,2-dioxazol-5-ones as coupling partners (Scheme 168D).1040 Under optimized microwave assisted conditions, the expected aza-heterocycles were delivered, generating only CO2 and H2O as waste.
The metallacyclic intermediates can be generally considered as nucleophiles and may hence participate in reactions with various electrophiles, such as Michael acceptors for example. Such reactivity was surveyed by Li and Dong, who synthesized benzoisothiazoles via Rh-catalysed coupling between sulfoximines and activated olefins (Scheme 168E).1041 Unfortunately, although the original 5-membered heterocyclic products are generally afforded in good yields (typically 70–90%), poor stereoselectivity was observed (usually 1:1 ratio), clearly hampering the synthetic potential of the reaction. A complementary reaction delivering original heterocyclic scaffolds was achieved by combining C–H activation and coupling with unsaturated ketones (Scheme 168F).1042 The reaction sequence involving formal metal-catalysed functionalisation and subsequent [4+3] annulations produced 7-membered 1,2-benzothiazepines 1-oxides. Broad substrate scope observed for this transformation, quite high efficiency and compatibility with cheap 3d-metal catalysts render this transformation appealing in drug-design science. Finally Bolm illustrated that benzothiazine may also be build-up while performing a Rh(III)-catalysed domino allylation/oxidative cyclisation between sulfoximines and allyl carbonates (Scheme 168G).1043 This transformation is a synthetic route of choice to prepare 4-unsubstituted 1,2-benzothiazines, scaffolds difficult to access when employing diazo-type coupling partners. Noteworthy, both electron rich and electron poor substituents may be introduced on the aromatic ring of the sulfoximine, and several different alkyl S-substituents are well tolerated. Thus obtained benzothiazine may also be rapidly diversified into few molecules of interest for pharmaceutical industry. An alternative route towards 4-unsubstituted 1,2-benzothiazines involves coupling between the metallacyclic intermediate and sulfoxonium ylides, used now as novel C2 synthons (Scheme 168H).1044 This RhCp*-catalysed transformation was accelerated in presence of Zn(OTf)2 additive, is of particular interest if benzothiazines bearing various, aliphatic and aromatic substituents at 3-position are targeted as an array of sulfoxonium yield performed very efficiently in this transformation.
Finally, it should be mentioned that using a standard RhCp*-catalyst, in combination with NH-Boc oxycarbamates, direct amidation of sulfoximines occurs smoothly for both, electron-poor and electron-rich aromatics (Scheme 168I).1045 Furthermore, as the newly accessed ortho-amidated sulfoximines may be converted into few steps into thiadiazine 1-oxides. In addition, still benefitting from efficient metallation of sulfoximine substrates in presence of RhCp*-catalysts, Bolm et al. reported direct C–X coupling (Scheme 168J).1046 Indeed, in presence of NIS or NBS as halogenating agents sulfoximes may be directly converted into ortho-halogenated congeners in high to excellent yield. This reaction allows therefore late-stage installation of amenable motifs for further diversifications.
A further key contribution to the field of sulfoximine-directed C–H activation was made by Sahoo who designed a S-methyl-S-phenylsulfoximine (MPS) DG.16 This motif, straightforwardly installable on carboxylic acids, easily removable (under basic conditions using NaOH in MeOH/H2O or acidic conditions using aq. HCl at 80 °C) and recyclable, demonstrated excellent reactivity under Ru- and Pd-catalysis, undergoing efficiently direct hydroxylation1047,1048 or amidation1049 reactions. In 2014 the versatility of this chemistry was further spread by devising a protocol allowing ortho- and chemoselective C–H alkenylations (Scheme 169A).1050 This oxidative Heck reaction catalysed by Ru(p-cymene) complex was compatible with an array of sulfoximine substrates but activated olefines (typically acrylates) were required. Following this exciting research field Sahoo discovered that MPS-equipped substrates, in presence of the same Ru-complex and N-protected phthalimides, undergo ortho-selective C–N bond formation (Scheme 169B).1051 Interestingly, the reaction protocol tolerates several functionalised aromatics (including halo-substituted and N-heteroaroyl ones) and, in presence of a large excess of the phthalimide, double ortho,ortho′-C–H activation occurs. Subsequently, intramolecular direct heteroarylation of MPS-sulfoximines was disclosed. Sahoo and coworkers installed on an aromatic substrate a judiciously selected olefin tether (Scheme 169C).1052 After the Ru-catalysed C–H cleavage, spacial proximity between the metallacyclic intermediate and an external double bond enhanced precoordination of a double bond by Ru, to favour migratory insertion into the C–Ru bond and final protodemetallation liberating a cyclised product. Quite remarkably, the same approach may also be used for two-fold unsymmetrical functionalisation. In such a case after initial intramolecular hydroarylation the Ru-catalyst re-enters a new catalytic cycle, inserts into the C–H bond at ortho′-position and final intermolecular amidation delivers a large panel of highly decorated scaffolds. MPS-bearing vinylic substrates are equally appealing precursors for this chemistry.1053 Indeed, one-pot double annulation of MPS-acrylamides occurred smoothly using a standard Ru(p-cymene)-catalyst in combination with internal alkynes, afforded pyrido-fused isoquinolone scaffolds (Scheme 169D). In course of this reaction both, symmetrical alkynes and two distinct unsymmetrical coupling partners may be employed. Besides, heteroaromatics substituted by sulfoximine motifs are equally efficient via a closely related protocol, delivering π-conjugated polycyclic amides in high yields.1054 Noteworthy, the MPS auxiliary is a traceless DG that can be readily removed after the C–H functionalisation (under basic conditions) or is directly cleaved during the annulation events.
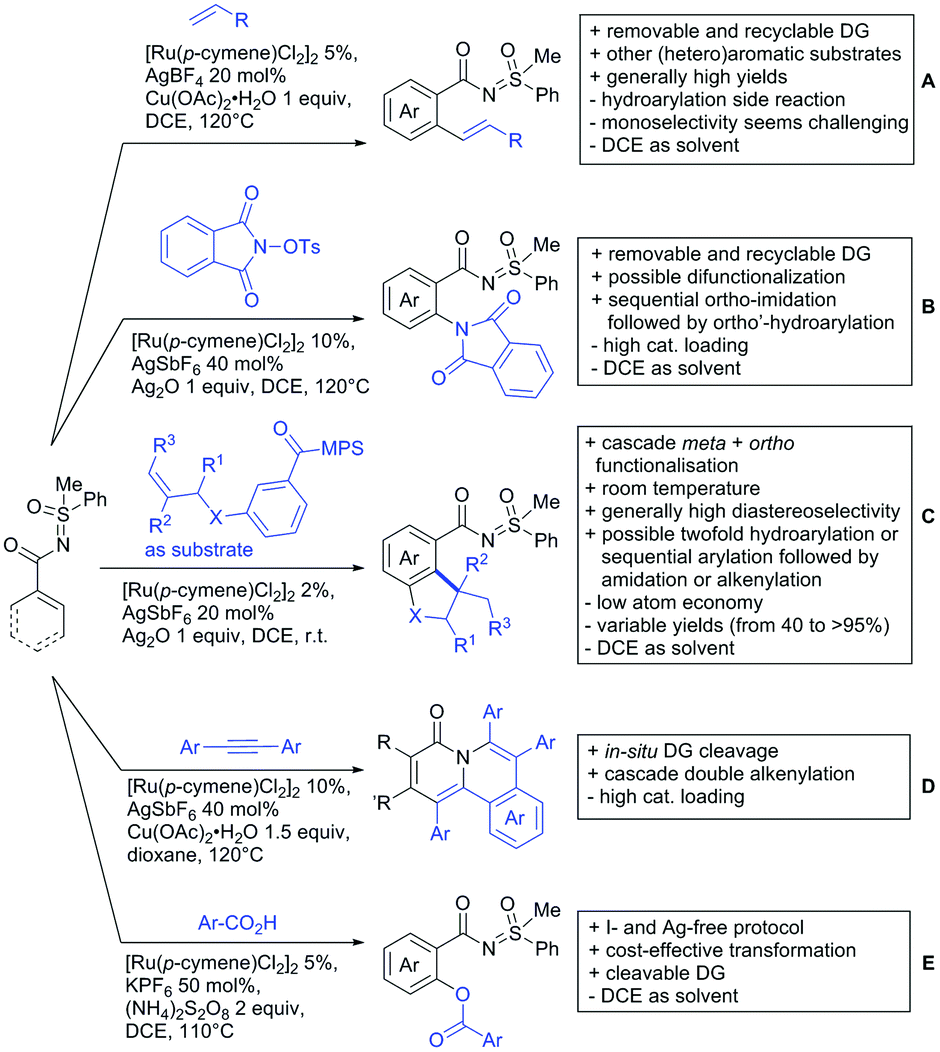 | ||
| Scheme 169 (N-Benzoyl)sulfoximine and related DGs. | ||
In parallel, Ackermann designed a C–H oxygenation reaction of sulfoximines. As for previous examples, Ru(p-cymene) is an optimal catalyst and after fine optimization, an iodine(III) and Ag(I)-free protocol was established to allow direct C–O coupling between sulfoximine benzamides and aromatic carboxylic acids (Scheme 169E).1055 This highly monoselective transformation well tolerates both electron-donating and electron-withdrawing substituents on the carboxylate, delivering the desired products in moderate to good yields (typically 50–70%). Notably, the functionalised sulfoximines may be readily converted into salicylic acids under acidic conditions.
16.4. Thioamide, thioketones, alkoxythiocarbonyls and thiocarbamates as DG
Another possibility of employing sulfur in a coordinating group relates to the use of thiocarbonyl motifs. Pioneering work in this challenging field was reported in 2015 by Satoh and Miura who disclosed Rh-catalysed direct coupling of benzothioamides with olefines and alkynes.1056 In this case N,N-diisopropylthioamide was used as DG and the expected functionalisation performed well under standard conditions. Importantly, oxidative Heck reaction with acrylates and styrenes delivers ortho-olefinated products while intramolecular cyclisation and desulfurization occur in presence of an alkyne partner, delivering indenone-derived products (Scheme 170A). Both couplings are high yielding and support well both electron-rich and electron-poor substrates. Few months later Yu and co-workers showcased that installation of a thiocarbonyl moiety on heterocycles such as pyrrolidine, piperidine, azapines or N-methyl amine is an efficient approach for α-arylation.1057 The corresponding thioamides undergo smoothly Pd-catalysed metallation and following coupling with boronic acids affords functionalised amine-derivatives (Scheme 170B). Remarkably, this protocol well tolerates a large panel of boronic acids, including sterically demanding ortho-substituted ones as well as heteroaromatic partners. High levels of diastereoselectivity are generally reached when substituted pyrrolidine substrates are used and complete diastereoselectivity is observed when one pot, sequential diarylation is performed. Importantly, this thiocarbonyl motif can be removed with methyl lithium as base and the generated amine is protected as the Boc-carbamate. Alternatively, the corresponding amide may also be accessed by oxidation with silver salts. In 2017 the same group reported an enantioselective version of this transformation.1058 Chiral induction (up to 98/2 e.r.) was accessed in presence of a phosphoric acid ligand (Scheme 170C). This arylation reaction is compatible with both cyclic and acyclic amines and as cleavage of the DG might be achieved (2-steps procedure including reduction of the thiocarbonyl in presence of NiCl2 and NaBH4, followed by BCl3-mediated debenzylation) it can be considered as an interesting strategy to access stereogenic amines.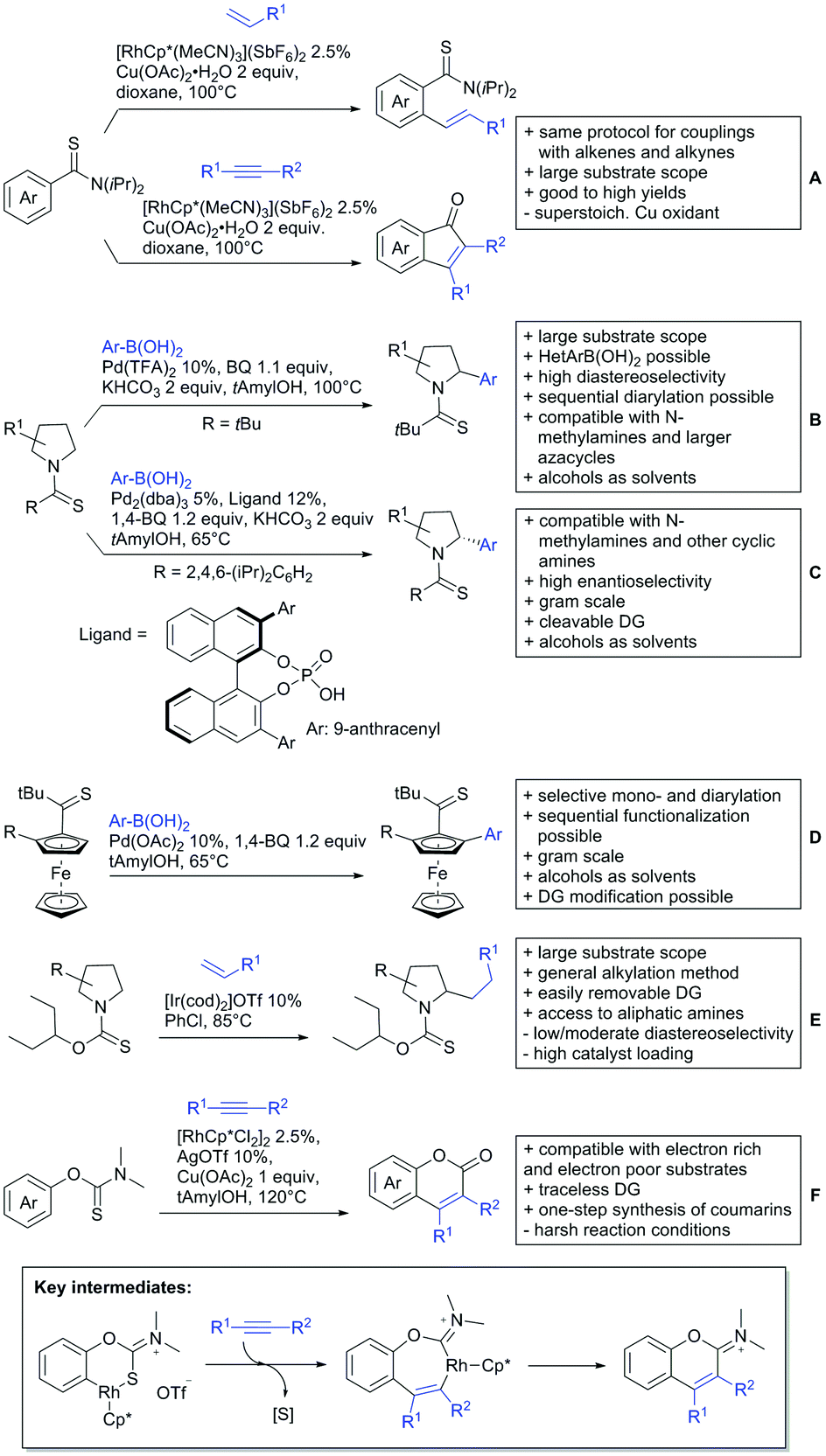 | ||
| Scheme 170 Thioamide, thioketones, alkoxythiocarbonyls and thiocarbamates as DG. | ||
Further potential of the thiocarbonyl moiety to enhance C–H activation was illustrated by You who disclosed thioketone-directed arylation of ferrocenes (Scheme 170D).1059 Under reaction conditions close to the one developed by Yu, palladation of thiocarbonylferrocene occurs straightforwardly, delivering either mono- or bi-arylated ferrocenes in generally high yields. The protocol is also applicable for sequential diarylation. The final product could be converted either into the corresponding ketones or a RANEY®-nickel mediated reduction delivered a benzylated congeners.
To further improve the traceless character of a thiocarbonyl based DG, Yu designed in 2017 an alkoxythiocarbonyl auxiliary.1060 This novel sulfur-based DG is particularly appealing in Ir-catalysed C–H activation, affording alkylated compounds in presence of non-activated olefin coupling partners (Scheme 170E). Thanks to a large scope of this coupling and convenient removal of the auxiliary (treatment with ac. solution of TFA followed by N-protection) an array of original aliphatic amines may be build-up efficiently.
An alternative C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S type directing group, thiocarbamate, was disclosed by Xia.1061 Remarkably, in this case the DG group is truly traceless; in presence of an internal alkyne coupling partner, the C–H activation step and subsequent C–C bond formation event are followed by annulation, delivering coumarins (Scheme 170F).
S type directing group, thiocarbamate, was disclosed by Xia.1061 Remarkably, in this case the DG group is truly traceless; in presence of an internal alkyne coupling partner, the C–H activation step and subsequent C–C bond formation event are followed by annulation, delivering coumarins (Scheme 170F).
16.5. Phosphine oxides and sulfides as P-based DG
Organophosphorus molecules are key structural motifs in a large panel of pharmaceuticals, agrochemicals and organic materials. Straightforward access to such scaffolds hence presents an appealing synthetic challenge for organic chemists and therefore it is not surprising that diversification of phosphorus-containing molecules by means of C–H activation has been attracting a great deal of attention. The pioneering key contribution in this field were already elegantly summarized elsewhere.17 The use of auxiliary phosphorus-containing DGs, such as monophosphonic acid phosphonate, presented however a serious drawback related to a low modularity of these DGs. Accordingly, the improvements that were expected in this domain concerned: (1) design of transformations implying generation of the functionalised P-containing molecules where the auxiliary is voluntarily presented in a final product skeleton; (2) conception of innovative, readily transformable or traceless DGs.Amongst phosphorus-based DGs, phosphine oxides are particularly interesting motifs. Indeed biaryl phosphines are key ligands for both non-asymmetric and stereoselective transformations and thus synthesis of (chiral) biarylphosphine oxides is highly appealing and much work has been already done in this field.1062 This coordinating group exhibits a modular activation mode as coordination of the metal catalyst can be achieved either via oxygen or phosphorus atoms, depending on the (1) hard/soft properties of the catalyst used and (2) geometrical considerations and size of a metallacyclic intermediate formed. Accordingly, if functionalisation of biaryl substrates bearing the phosphine oxide-DG is targeted, Pd-based catalytic systems seem particularly suited as a precoordination of this soft metal by the soft P-atom is expected. In accordance with these predictions, a variety of Pd-catalysed functionalisations of phosphine oxides biaryls have been achieved. More recently, this approach was valorized by synthesis of axially chiral phosphine ligands. Initially, a diastereoselective approach has been devised using a chiral phosphine based DG, bearing a menthol substituent as chiral source (Scheme 171A). Rewardingly, direct functionalisation of the corresponding substrates occurred smoothly, delivered atropisomerically highly enriched products via C–C, C–O and C–I couplings.1063a Subsequently, a closely related acylation of biaryl substrates bearing now a P-chiral DG was disclosed.1063b,c This methodology was further complemented with an enantioselective transformation. Indeed, Yang et al. discovered that if a simple diphenylphosphine oxide DG is installed on biaryl substrates, Fujiwara–Moritani coupling catalysed by Pd(OAc)2 in presence of a chiral amino acid ligand, takes place efficiently in ortho′-position with an excellent enantioselectivity (Scheme 171B).1064 The extensive ligand screening revealed that structurally simple Boc-L-Val-OH ligand is optimal in terms of chiral induction, delivering the atropisomeric olefin/phosphineoxides in typically 90% ee. The reaction occurs under relatively mild reaction conditions (60 °C) and probably implies a dynamic transformation such as dynamic kinetic resolution and/or dynamic catalytic asymmetric transformation.
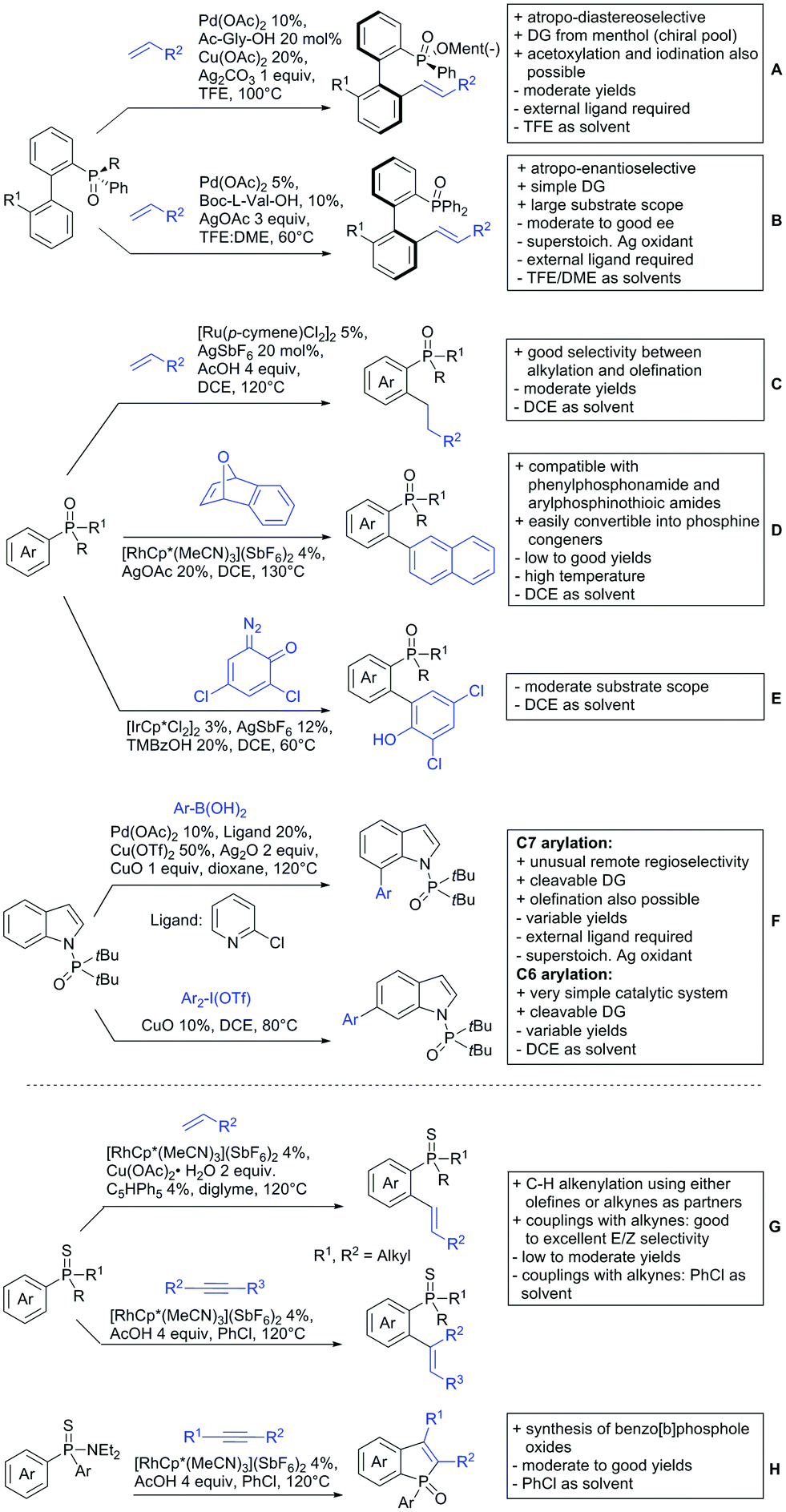 | ||
| Scheme 171 Phosphine oxide- and sulfide-directed transformations. | ||
Aside from Pd-catalysed transformations, phosphine oxide DGs are also compatible with Ru, Rh and Ir-based catalysts. Likewise sulfoxide-DGs, as phosphine oxide coordinate these rather hard catalysts via the O-atom, a 5-membered metallacyclic intermediates is hence generated when this DG is installed directly on an aromatic ring. Following this analysis, Soulé reported recently direct alkylation of arylphosphine oxides in presence of a Ru-based catalyst (Scheme 171C).1065 This monoselective C–C coupling occurs with moderate to high yields, but the newly accessed scaffolds can be considered as original phosphoros ligand.
Straightforward ortho-metallation of arylphosphine oxides may also be performed in presence of a RhCp*-catalyst, as illustrated by Satoh and Miura (Scheme 171D).1066 The authors discovered that heterocyclic compounds are also suitable coupling partners, allowing direct arylation. This strategy is an interesting alternative to commonly used iodoarenes as coupling partners, delivering biaryl compounds in variable yields ranging from 30 to 91%. Metallacyclic Ir/phosphine oxide intermediates are also prone to undergo migratory insertion of metal carbenes (Scheme 171E).1067 This synthetic route delivers original biaryl scaffolds up to gram scale and their diversification via FG interconversion was illustrated.
Outstanding application of the phosphine oxide DG was reported in 2016 by Shi.1068 The authors installed this coordinating motif on the N-atom of indole substrates and observed unexpected regioselectivity during a Pd-catalysed coupling with boronic acids. Indeed, selective arylation at C7, over generally observed C2-functionalisation could be attained in synthetically useful yields when installing bulky phosphine oxide DGs, such as P(O)(tBu)2 (Scheme 171F). Due to steric reasons, the O-atom is perfectly oriented to perform C–H activation at this specific C7 position. In addition, bulky tBu-substituents on P-atom prevent N–P bond rotation further favouring the observed selectivity. Few months later the same research group succeeded in designing a complementary Cu-catalysed, C6-selective transformation. Drawing inspiration from Gaunt,1069 they reacted the same di-tBu-phosphine oxide substituted indole with iodanes.1070 In line with the expectations, C7 vs. C6 regioselectivity shift occurred via transfer of the aromatic ring bound to Cu (resulting from oxidative addition of Cu into iodine) to C6 position via the Heck-type four-membered-ring transition state. Accordingly, using the same indol substrate with a judiciously selected DG the functionalisation could be triggered at unusual position, by simple changing the catalyst used and the nature of the arylating agent. Importantly, the DG can be removed by treatment with lithium aluminum hydride to give the unprotected indole derivatives.
In addition to phosphine oxide, phenylphosphine sulfides may also act as efficient DG in RhCp*-catalysed transformations. Such a PS DG was used in 2014 by Satoh and Miura to provide new protocols for direct alkenylation occurring either via oxidative Heck reaction or redox-neutral coupling with internal alkynes (Scheme 171G).1071 Also thiophosphinamides exhibit a somehow related reactivity as illustrated Satoh and Miura who used this P-DG in Rh-catalysed coupling with heterobicyclic alkenes.1066 The same substrates may also undergo ortho-alkenylation and subsequent cyclisation delivering arylthiophosphinamides,1072 compounds of interest due to their intriguing electronic and optical properties (Scheme 171H).
Phosphonic acids constitute another sub-category of P-based DGs. The originality of this DG arises from the presence of an OH-free group on the phosphorus atom and accordingly precoordination of a metal by this motif is expected. Several interesting examples of C–H activation reactions directed by this moiety have been reported before 2015 and they were highlighted elsewhere.17 More recently, the group of Lee focused on synthesis of phosphaisocoumarins via C–H activation strategy directed by P-containing DG (Scheme 172A). The first successful protocol relates to a phosphoannulation reaction of aryl- and benzylphosphonic acids with unactivated alkenes via direct C–H coupling/C–O cyclisation step sequence.1073 This reaction is efficient for both, aryl-and benzylphosphonic acids. The olefination reaction delivers products in moderate to good efficiency. Few months later the same research group provided an alternative approach towards dihydrophosphaisocoumarins via C–H activation followed by C–O cyclizing bond formation (Scheme 172B).1074 In this case 1,3-dienes are used as coupling partners and the regioselectivity of this transformation may be arguably attributed to a formation of Pd π-intermediates. Unfortunately, the reaction does not allow diastereocontrol and the products are obtained as mixture of diastereomeric products albeit in high yields.
 | ||
| Scheme 172 Other P-based DGs in C–H functionalisation. | ||
Targeting design of transformable/traceless P-containing DGs, several research groups focused on auxiliaries installed on an aromatic substrate via a heteroatom like O, or NH motif. Following this idea, enol phosphate DGs have been designed1075–1078 and employed them in direct functionalisation of aromatics. In 2015 Loh expanded this reactivity by developing a catalytic system able to functionalise vinyl C–H bonds (Scheme 172C).1079 Cationic RhCp* catalyst in combination with Cu(OAc)2 and pivalic acid allowed selective alkenylation of phenylvinyl phosphates. The reaction well tolerates an array of alkene substrates and the diene products are generally obtained in high yields (typically 70–90%) and good E/Z selectivity (approx. 90:10). Besides, when using vinyl ketones as coupling partners selective hydroalkenylation occurs and also unsymmetrical alkynes may be used in this C–C coupling. Finally, in accordance with the general target of devising traceless DGs, efficient removal of this motif using DIBAL-H or TBAF based protocols. In addition, the O-tethered DG may also be considered as a reactive site to perform Ni-catalysed C–C coupling delivering an arylated product.
Besides O-tethered P-containing DGs, use of N-linked directing groups also attracted scientific curiosity. In 2013 Lee developed phosphoramidite-directed arylation.1076 More recently, Oestrich et al. designed phosphinic amides as novel handle for Fujiwara–Moritani reaction (Scheme 172D).1080 Both, NH and NMe anilline derived phosphonic amides performed well. Besides, the oxidative olefination with this DG occurred smoothly at temperature as low as 40 °C, vs. 90 °C required for the activation of substrates bearing an NH-anilide. The transformation well tolerates few electron-donating substituents on the aromatic ring of the substrate as well as few activated olefins.
Alternative, as P-chiral molecules are intriguing motifs, in particular for asymmetric synthesis, design of stereoselective C–H activation reactions directed by P-motifs and delivering chiral molecules is highly appealing. Following this idea Han surmised that if symmetrical phosphinamides are used, a direct activation of one of the aromatic substrates would result in a desymmetrisation of the product, hence delivering P-stereogenic molecules (Scheme 172E).1081 As expected, when using a Pd-catalyst in combination with mono protected amino acid ligands, desymmetrizative arylation of phosphinamides occurred, delivering the corresponding P-chiral molecules in moderate to good yields and good enantiomeric excess of approx. 90%. Notably, the post-diversifications were performed with little or no loss of chiral information.
In 2017 Cramer reported a complementary catalytic system based on the use of chiral RhCp-derived catalyst bearing an atropisomeric Cp-type ligand (Scheme 172F).1082 During this directed alkynylation intramolecular annulation takes place delivering P-chiral cyclic phosphinamides. The reaction shows large scope, good tolerance towards substrates bearing a variety of electron-withdrawing and electron-donating substrates and very high regioselectivity with respect to unsymmetrical alkynes. The yields are moderate to good end ees of roughly 90% were obtained. Subsequently, this chemistry was employed to functionalise racemic dissymmetric phosphinic amides. The same catalytic system was now employed to perform direct stereoselective C–C coupling via kinetic resolution.1083
Finally, the panel of P-based DGs is complemented by using simple phosphines as coordinating motifs. This approach seems highly challenging and counterintuitive as phosphines are excellent ligands and their use in a stoichiometric amount as DG could lead to “catalyst poisoning”. In addition, oxidative conditions frequently applied in C–H activation might lead to the oxidation of this DG. However, in 2014 Clark reported Ir-catalysed phosphine-directed ortho-borylation of aromatics (Scheme 172G).1084 Like in a case of thioether (see Section 16.1), the phosphine is installed at the benzylic position to allow generation of 5-membered metallacyclic intermediates. This reactivity could also be extended to diaryl and heteroaryl compounds where a PR2-motif directs functionalisation at ortho′-position. Noteworthy, in situ protection of the phosphine as the borane complex takes place, enabling convenient isolation of otherwise air-sensitive boronate esters, and deprotection in presence of Et2NH can be conducted when requested.
Following this seminal work, in 2017 Shi discovered that direct C–H functionalisation may be applied as an efficient handle to diversify privileged biaryl-monophosphine ligands. Indeed, a PR2-motif is able to transiently coordinate a Rh(I)-catalyst and via formation of a metallacyclic intermediate direct the metallation step at the ortho′-position and further promoting C–C coupling with arylbromides (Scheme 172H).1085 Use of an equimolar ratio between a phosphine substrate and Ar–Br partner permits selective mono functionalisation while an increased amount of the coupling partner favours formation of diarylated congeners. Remarkably, this protocol is compatible with an array of commercially available monophosphine ligands such as CyJohnPhos, DavePhos and cataCXium auxiliaries like PinCy or POMeCy. Noteworthy, with enantiopure substrates such as binaphthylphosphine H-MOP or Tang's ligand, the arylation occurs with no loss of the chiral information. Accordingly, this reaction is a unique tool for the late stage diversification of ligands.
Noteworthy, the phosphine motif can also be applied in Cu-mediated alkynylation reactions, delivering, after a cyclisation step, an array of phosphindolium salts.1086 Unfortunately, although this protocol gives straightforwardly access to important heterocyclic motifs, need for 2 equivalents of the metal salt limits its synthetic attractiveness.
16.6. Si-Based DGs
One of the major limitations of directed C–H activation relates to the need for preinstalled coordinating motifs, prompt to precoordinate a metal catalyst and thus enhance regioselective metallation. Although over the last years particular attention has been focused on devising C–H activation reactions benefitting from the functionalities inherently present on a desired product that may play a role of a DG, many catalytic systems still call for the use of non-modular and/or difficult to remove DGs that become static entities. In such a case synthetic value of the C–H activation methodology is clearly reduced. To obviate this serious limitation, the concept of “traceless” DGs emerged recently, enhancing conception of innovative coordinating motifs that are easy to install but also readily cleaved or transformed under mild reaction conditions after the C–H activation step.1087 In this context use of silicone moieties is particularly appealing due to: (1) relative stability and easy access to silicone derivatives; (2) availability of cheap silicone precursors; (3) straightforward Si–X bond cleavage allowing removal of these tethers under mild and often chemoselective protocols, such as the Tamao–Fleming oxidation1088 or the Hiyama coupling.1089 Besides, in intramolecular transformations, if a SiR1R2H motif is used, this moiety can play a dual role of both DG and reagent.Following this general concept several research groups entered the field of Si-based DGs in C–H functionalisation.735,1090,1091 Already more than one decade ago several research groups questioned the potential of “SiR2H” to play a double role of a DG to drive the C–H activation event at one specific position and to provide, in an intramolecular fashion, a silicone-based coupling partner. The potential of such reactions holds also a major synthetic value as the newly formed C–Si bonds might be further interconverted into several functionalised motifs.
Following this concept, before 2015 several catalytic systems allowing direct silylation of aromatics and heteroaromatics were reported whereas direct functionalisation of aliphatic substrates remained a less developed field.1092–1094 Besides, design of stereoselective transformations was a challenging goal. Following several pioneering key contributions, Murai and Takai reported in 2015 an improved Rh-based catalytic system for intramolecular silylation of unactivated C(sp3)–H bonds (Scheme 173A).1095 The authors discovered that replacement of the previously employed dppp diphosphine ligand by an axially chiral, structurally well defined SEGPHOS auxiliary in combination with an hydrogen acceptor (such as an olefin) drastically accelerates the desired reaction, enhancing smooth conversion of the starting materials into products at a temperature as low as 50 °C vs. 180 °C previously requested. Importantly, use of a chiral ligand and milder reaction conditions permitted a stereoselective outcome of this transformation, however only moderate chiral induction was observed (ee up to 40% was reached using the BINAP ligand). Highly enantioselective silylation of aliphatic substrates was achieved two years later, in 2017. Hartwig et al. discovered that switching from Rh–diphosphine catalyst towards an Ir complex bearing a chiral N,N-donor ligand, such as indane-fused oxazoline, clearly improved this challenging C–H activation reaction that now delivers the silylated compounds in high yields and good enantioselectivities (ee of approx. 90%) (Scheme 173B).1096 Importantly, the newly generated stereoenriched dihydrobenzosiloles (accessible also on gram scale using a largely decreased catalyst loading) are a unique platform to access an array of chiral molecules via chemoselective functionalisation of each C–Si bond.
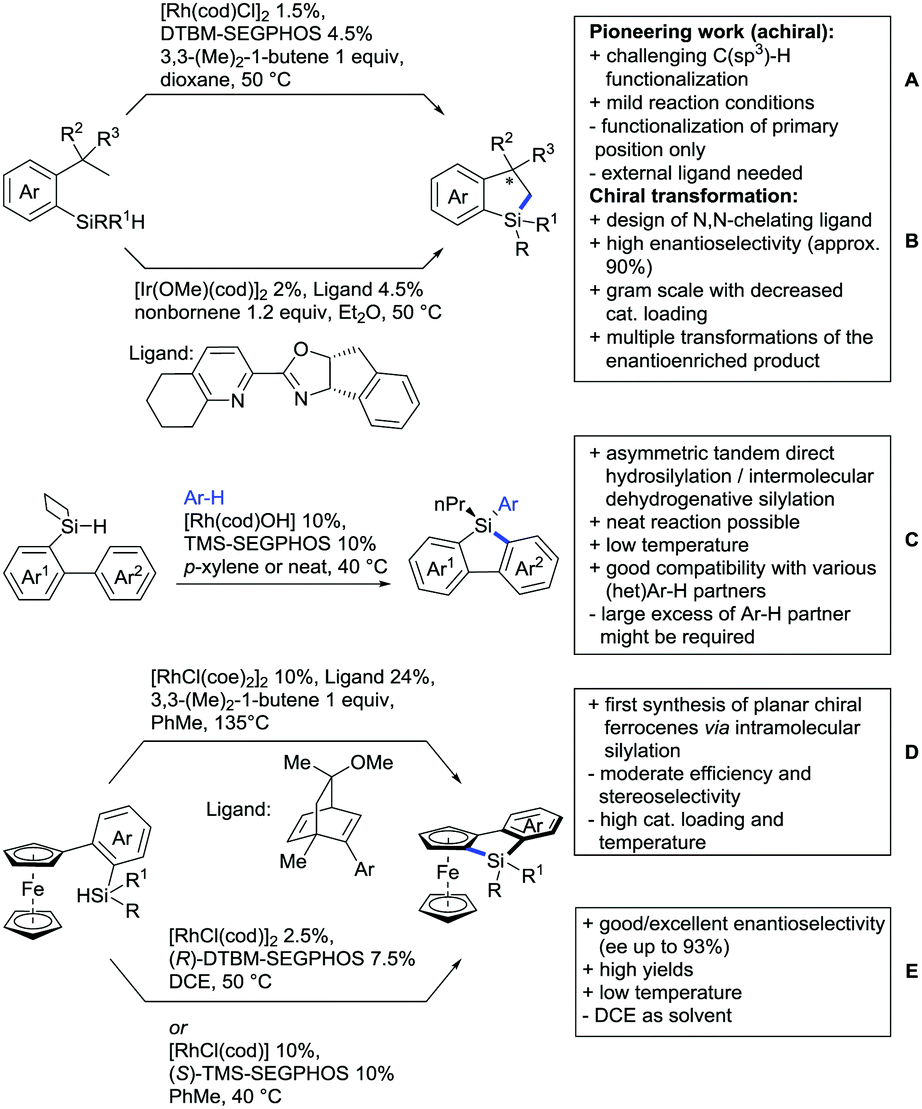 | ||
| Scheme 173 Silanes as DGs (intramolecular silylation). | ||
In addition to stereoselective silylation of unactivated C(sp3)–H bonds, several research groups applied the same concept to synthesize chiral molecules via functionalisation of C(sp2)–H bonds of prochiral substrates. The pioneering key contribution by Takai and Kuninubu provided access to original enantiomerically pure spirosilabifluorenes via two-fold intramolecular silylation.1097 Expanding this methodology, He reported in 2017 a tandem transformation involving C–H desymmetrizating silylation, followed by intermolecular dehydrogenative silylation (Scheme 173C). Such a transformation, catalysed by a Rh–SEGPHOS complex, allows the interconversion of silacyclobutanes into chiral tetraorganosilicons in moderate to good yields and ees between 65 and 93%.1098
Intramolecular stereoselective C(sp2)–H silylation was also astutely used to access planar chiral benzosiloloferrocenes. The groups of Shibata,1099 Murai/Takai1100 and He1101 reported simultaneously but independently intramolecular silylation of metallocenes. Although when employing the first protocol (Scheme 173D) based on a Rh-catalyst and a chiral diene ligand only moderate chiral induction was observed (up to 86% ee),1099 clearly more appealing ees were obtained in presence of Rh–SEGPHOS catalyst (Scheme 173E).1100,1101 Notably, using the second protocol either reaction temperature could be decreased to 40 °C or significantly lower catalyst loading (2.5 equiv.) was employed.
An alternative approach to push even further the synthetic value of the above described methodology consists of installing the silicone moiety as a temporary tether such as a silyl ether group. This elegant concept, developed in 2010 by Hartwig, implies in situ generation of silyl ethers from ketones or aldehydes via Ir-catalysed hydrosilylation, followed by direct intramolecular silylation catalysed by a second Ir-catalyst.1102 Importantly, thus accessed scaffolds may now be chemoselectively converted into an array of functionalised molecules via selective O–Si or C–Si cleavage reactions using standard protocols such as Tamao–Fleming oxidation or Hiyama coupling. A further key step forward in this field consisted of designing a related stereoselective transformation. Following this target, Hartwig described in 2015 a (hydrido)silyl-directed desymmetrisation reaction of benzophenone derived substrates.1103,1104 The authors discovered that while conserving the Ir-catalyst for the first step, i.e. ketone hydrosilylation, high reactivity and excellent chiral induction for the C–Si bond formation are reached in presence of Rh(I)-catalyst coordinated diphosphine ligand, in particular catASium or Walphos-derivatives and nonbornene as a hydrogen acceptor. The reaction takes place under mild reaction conditions (50 °C). Accordingly, a large panel of highly enantioenriched diarylmethanols is accessed. Noteworthy, when dissymmetric enantiomerically pure diaryketones are used as substrates high catalyst-controlled site-selectivity is observed. Very recently, the same research group designed an alternative catalytic system based on Ir(I) bearing an N,N-bidentate quinoline-oxazoline ligand.1105 The reaction is equally efficient and enantioselective as the Rh-based protocol but even lower reaction temperature, i.e. 35 °C, could be used. In addition, kinetic resolution took place when submitting racemic silyl ethers to the standard reaction protocol, affording the silylated products with high selectivity (Scheme 174).
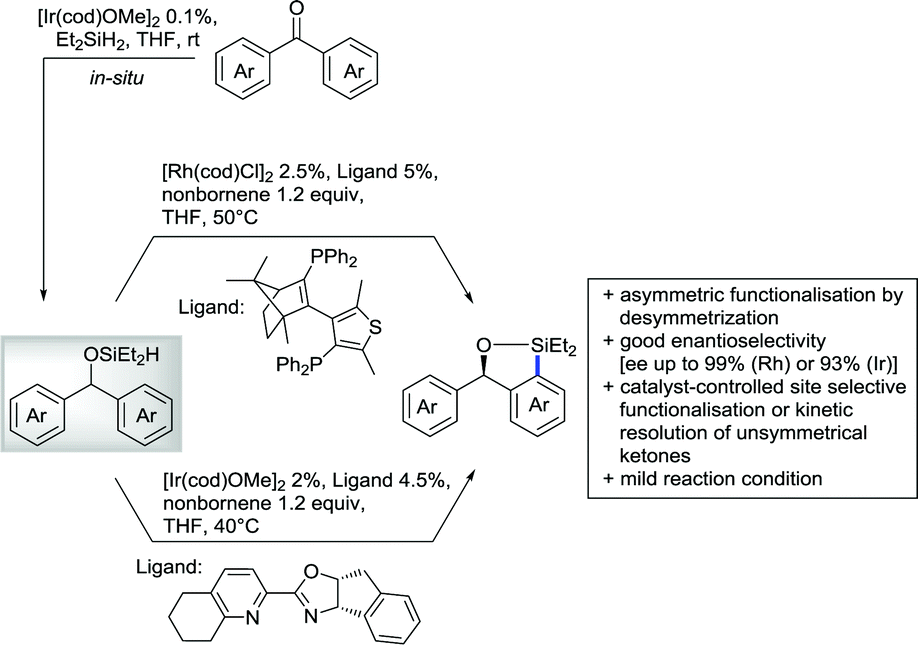 | ||
| Scheme 174 In situ formed silyl ethers as DGs (C(sp2)–H intramolecular silylation). | ||
In addition to direct C(sp2)–H activation under mild reaction conditions, the concept of intramolecular silylation using a silyl ether moiety could be expanded to the functionalisation of simple aliphatic substrates. Such a transformation holds great promise for synthetic chemists as, considering high modularity of both C–Si and O–Si bonds, new synthetic routes towards diols could hence be envisioned. Following this inspiration Hartwig described in his pioneering work in 2014 hydroxy-directed silylation of secondary C(sp3)–H bonds (Scheme 175A).1106 When a (hydrido)silyl ether (obtained via dehydrogenative coupling between a ketone and diethylsilane) is reacted with the Ir-catalyst supported by phenanthroline type ligand, formation of a 6-membered metallacyclic intermediate operates furnishing 5-membered oxasilolane. Finally, smooth oxidation of the newly generated oxasilolanes using TBHP, in combination with CsOH monohydrate and tert-butyl ammonium delivers the expected 1,3 diols. Accordingly, the overall transformation allows conversion of aliphatic ketones into 1,3-diols, fully validating the initial hypothesis of the project. Very recently, design of protocols devoted to a synthesis of 1,2 and 1,4-diols spread the outlook of this methodology further. The regioselectivity switch towards more distal, δ-position was possible by means of catalyst design (Scheme 175B).1107 Hartwig discovered, in accordance with his previous work, that while using a Rh-catalyst with previously employed phenanthroline or common diphosphine ligands, such as BINAP or SEGPHOS, silylation occurs selectivity at 3-position. However, quite unexpectedly, addition of large bite angle Xanthpos ligand drastically altered the outcome of this C–H activation, directing the metallation event at 4-position and thus providing 6-membered oxasilolanes in good yields (around 70%). Accordingly, a 3-step procedure for direct conversion of aliphatic alcohols into 1,4-diols was devised.
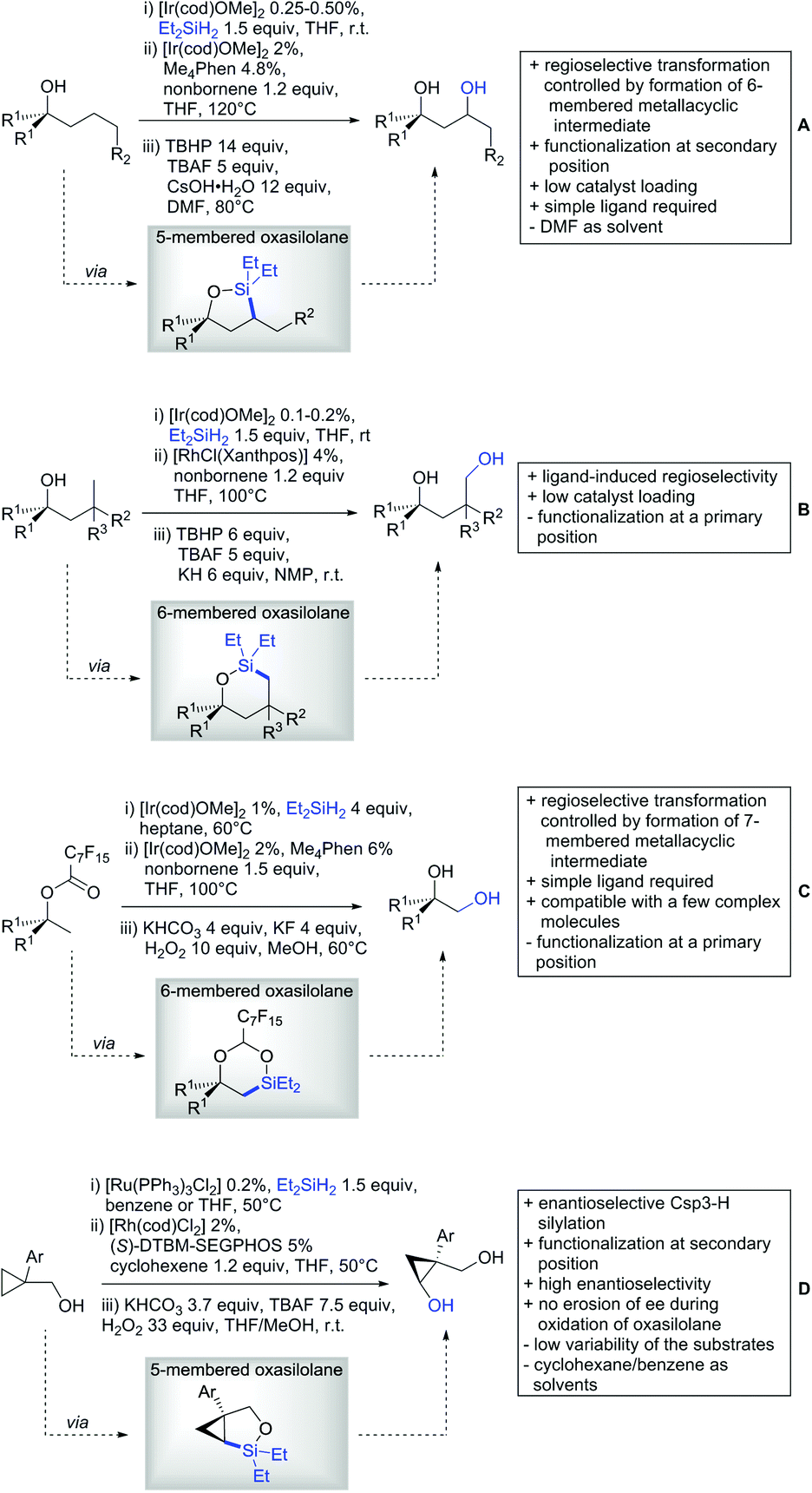 | ||
| Scheme 175 In situ formed silyl ethers as DGs (C(sp3)–H intramolecular silylation). | ||
In contrast, 1,2-selectivity for this direct silylation reaction was possible thanks to the substrate designed (Scheme 175C).1108 Accordingly, an easy to install and readily cleavable linker needs to be placed between an oxygen atom of a substrate and the silyl-moiety. Detailed investigations revealed that perfluoroacetal is perfectly suited to deliver the silyl DG in close proximity to the targeted δ-position. Accordingly, a reaction sequence including hydrosilylation of a perfluoroalkyl ester, followed by silylation of a C(sp3)–H bond and final Tamao–Fleming oxidation at the C–Si bond provides 1,2-diols. The protocol requires use of the Ir-catalysts in two initial steps and harsher reaction conditions are necessary to favour this short-distance functionalisation.
An additional challenge to be addressed in this field concerns stereoselectivity issues and development of an enantioselective directed silylation. The first step towards this goal was achieved in 2016 when the same group disclosed enantioselective synthesis of cyclopropane derivatives (Scheme 175D).1109 The authors hypothesized that the hydrosilyl ether of 1-arylcyclopropylmethanol may be an effective substrate under Rh(I)-based protocol. Despite significant steric hindrance, the expected C–Si coupling occurs at the more activated (presence of a phenyl substituent), tertiary carbon. Interestingly, the previously employed SEGPHOS type ligand outcompeted other axially chiral diphosphines such as BINAP, MeOBIPHEP and GARPHOS derivatives, delivering the expected cyclised products with high optical purity of approx. 90% yet in moderate to good yields. The reaction well tolerates an array of aromatic substituents on the cyclopropane substrate. Besides, oxidation of the oxasilolanes occurred with no erosion of the enantiopurity.
In his seminal work in 2008 Hartwig reported for the first time the Ir-catalysed ortho borylation of aryldimethylsilanes (Scheme 176A).1110 In this case, the Si moiety acts as a traditional DG, rather than a silylating agent, as in the previous cases in this section (i.e. the Si only coordinates to the metal, rather than being involved in a Si–C bond). The desired C–H functionalisation required Ir(I)–dtbpy complex and B2pin2 supplemented with a catalytic amount of HBpin, to provide the expected ortho-functionalised compounds. This methodology could be further expanded to silyl phenol-ethers. A sequential transformation thus permits direct conversion of phenols into 2-phenyl trifluoroborate salts (Scheme 176A, box). Besides, the structurally related 5-membered metallacyclic intermediates may also be generated when C–H activation occurs at the benzylic position of 2-alkylarylsilanes.1111
 | ||
| Scheme 176 Silane-directed borylation reactions. | ||
In addition, following the same concept, original 7-selective functionalisation of indoles was performed (Scheme 176A). In this example the silyl DG was installed in situ on the N-atom and drives the metallation event at C7, suppressing the inherent C2 reactivity of such substrates.1112 Accordingly, simple indoles were readily converted into 7-Bpin substituted heterocycles using [Ir(cod)Cl]2 catalyst in combination with dtbpy ligand.
A further major challenge in this field was taken up in 2016, when Hartwig devised a slightly modified catalytic system, sufficiently active to promote directed functionalisation of simple aliphatic substrates (Scheme 176B).1113 The strategy proposed by the authors consisted in (1) straightforward installation on the Si-based DG on an olefin via alkene hydrosilylation generating highly stable compounds; (2) hydrosilyl-directed Ir-catalysed direct borylation; (3) diversification of the accessed molecules via chemoselective cleavage and conversion of the C–Si and C–B bonds. The optimization study revealed that efficient and reproducible C–B coupling occurs when using an isolated Ir-complex bearing a mesityl-substituted phenanthroline ligand in combination with highly reactive Et3SiBpin boron source, in isooctane and at 80 °C. Via this protocol, selective mono-borylation occurs selectively at a secondary γ-position generating trans-adducts with high stereoselectivity. Noteworthy, in case of cyclic substrates, geometrical considerations (size of the metallacyclic intermediate formed) override steric issues, as the functionalisation of a secondary carbon at γ-position is favoured over borylation of a primary δ-position.
In 2017, the first example of an enantioselective silyl-directed C–B coupling was disclosed. Drawing inspiration from their previous work on desymmetrizative silylation of diarylketones, Hartwig and Shi developed directed borylation of diarylmethylsilanes (Scheme 176C).1114 As Ir-catalysed borylation are generally milder, the intensive efforts were focused on designing a chiral ligand able to promote this challenging transformation at room temperature. Although enantiopure ligands such as BINAP, various bisoxazolines and bipyridine derivatives failed, the desired coupling took place in presence of a quinolyl oxazoline auxiliary. Further fine-tuning of the ligand structure led to the design of the optimal chiral inductor, allowing the desired C–H activation to occur smoothly even at 0 °C and with high chiral induction. As previously mentioned, this catalytic system is robust and the reaction performed at 5 mmol scale delivered the product in comparable yield and ee, albeit significantly decreased catalyst loading (0.75% vs. 3% used for small scale reactions). Besides, the enantioenriched borylated products could be further diversified via selective FG interconversions.
A complementary application of silyl moieties in the context of C–H activation consists in using these motifs as temporary tethers allowing installation of proper strongly coordinating DGs, like heteroaromatics, or a remote nitrile function. This type of DG is presented in Section 10.2. Alternatively, Si-atom may also be used as linker connecting two moieties of a molecule for an intramolecular coupling initiated by oxidative addition of Pd into a C–X bond.1115,1116 Finally, inspired from the recent regain of interest in using weakly coordinating DGs, concerns use of silanols and effective precoordination of a metal via hydroxy motif.
Silanols have established themselves as appealing weakly coordinating DGs at the beginning of this decade,1117 mainly based on key contributions by Gevorgyan1118,1119 and Ge.1120 Accordingly, an OH group directly installed on an aromatic ring allowed efficient and selective Pd-catalysed ortho-alkenylation1118 and oxygenation.1119 More recently, silanol-directed C–H carboxylation has been used to prepare salicylic acid derivatives (Scheme 177A).1121 Indeed, this new methodology implying: (i) conversion of an phenol into silanol, (ii) direct C–C bond formation using CO as a coupling partner and (iii) final deprotection of the OH in presence of TBAF delivers an array of salicylic acids in high yields, almost regardless the substitution pattern of the starting material (an array of FGs is well tolerated).
 | ||
| Scheme 177 Silanols and siloxanes as DGs. | ||
The potential of silanols has been further expanded by Cui and Xu who astutely tethered a second silicone unit on this DG (a siloxane moiety), thus targeting intramolecular C–H silylation (Scheme 177B).1122 Rewardingly, using an Ir-based protocol in combination with phenanthroline ligand they established a general protocol for direct C–Si bond formation. Notably, the reaction is compatible not only with arylsilanols, but also benzylsilanols and aryloxysilanols are efficient substrates. The design of the substrates allows installation of different substituents on the distinct silicone atoms hence chemoselective cleavage of C–Si and C–O bond occurs, ensuring remarkable post-modularity of the unsymmetrical cyclic siloxanes obtained.
16.7. OH- and OR-Based DGs
Although initially it was commonly recognized that metal-catalysed C–H functionalisation reactions require strongly coordinating DGs to enhance formation of stable metallacyclic intermediates, key contributions by Yu33,248 and others1123 clearly showed the potential of weakly coordinating DGs. This discovery encouraged the scientific community to explore the potential of many different “naturally occurring” DGs, amongst which the hydroxy group has attracted particular attention.1124 Indeed, between 2010 and 2015 many innovative direct C–H activation reactions have been reported, relating mainly to the functionalisation of phenols, benzylic alcohols or biarylphenols and they are already elegantly summarized elsewhere.17 In 2016 Cheng et al. illustrated that OH-directed C–H activation may also be achieved when using CoCp*-catalyst (Scheme 178A).1125 The authors astutely used the hydroxy-motif to enhance direct metallation of a vinylic C–H bond, located ortho to the DG and subsequent reaction with an allene, acting as a one-carbon coupling partner allows [5+1] annulation to occur delivering 2H-chromenes. The reaction occurs under very mild reaction conditions, delivering the targeted heterocyclic products in moderate to good yields. High regioselectivity is attaint when using a simple, mono-substituted allene, however mixtures of products were generally formed when structurally more complex coupling partners were employed. | ||
| Scheme 178 Hydroxy- and methoxy-directed transformations. | ||
Alternatively, the OH motif was also used as thether to install temporarily a second, strongly coordinating DG. In this context many examples of O-Py and O-heteroaromatic moieties have been disclosed and these examples are already shown in Section 15.3. Recently, an interesting example of in situ conversion of the OH moiety into a potent DG was disclosed by Maleczka, Singleton and Smith (Scheme 178B).1126 These research groups reported an ortho-borylation of phenols, a reaction elegantly complementing other C–H borylation protocols, for which regioselectivity is usually imposed by steric factors. The unusual selectivity of this reactions results from in situ borylation of the hydroxy group and subsequent electrostatic interactions between the OBpin motif and the bipyridine ligand coordinating the Ir-complex driving the metal catalyst at the ortho position. While excellent ortho-selectivity was observed for the functionalisation of para-substituted phenols (para-substituent larger than CN or F) with B2pin2, use of less sterically demanding B2eg2 was required to induce total regioselectivity for arenes not biased by para-substitution.
Although the quite widespread use of the hydroxy motif as DG, ethers are only rarely employed to precoordinate a catalyst and drive the metallation event.1127,1128 Recently, Echavarren addressed this challenge by designing a RhCp*-based catalytic system promoting highly efficient ortho-alkynylation of benzyl ethers (Scheme 178C).1129 When TIPS-protected bromoacetylene is used as a coupling partner, a panel of benzylethers is available, as the reaction well tolerates several substituents on the aromatic ring (mainly halogens). However, for para-substituted substrates mono- vs. difunctionalisation issues arise. Unfortunately, the reaction is efficient only for methyl-benzylethers. It is important to highlight that other weakly coordinating DGs, such as sulfoxides, thioethers, sulfones, dialkylamines, sulfones, etc also perform well under a very similar protocol.
Another interesting example for the use of a methoxy-group as coordinating motif was disclosed by Hou. The authors devised a catalytic system based on half-sandwich rare earth catalysts to enhance unusual simultaneous chain-growth polymerization (by continuous CC insertion) and step-growth polymerization (C–H addition of anisole units to vinyl groups).1130 The same catalyst was further exploited in a copolymerization process between 1,4-dimethoxybenzene and 1,4-divinylbenzene or norbornadiene, delivering original polymeric materials (Scheme 178D).1131
16.8. Amine-based DGs
Amines occupy a particular position in the field of C–H activation. This motif was amongst the first ones to be explored as a DG while studying stoichiometric cyclometallation reactions.1132 Not surprisingly, dialkyl amines have thus been used with some success in catalytic reactions.17 In recent years, dialkylamine DGs were astutely used to enhance direct functionalisation of the ferrocene scaffold. In presence of Pd-based catalysts in combination with a chiral ligand, powerful protocols for stereoselective arylation, alkynylation/dehydrogenative annulation and olefination have been designed. These works were already elegantly summarized elsewhere1133–1135 and will not be detailed in this review.Aside from ferrocene chemistry, the amine moiety is also a handy tool to promote regioselective, direct diversification of biaryl scaffolds. Indeed, much attention has been focused on 2′-functionalisation of biaryl-2-amines as, likewise biarylsulfoxide substrates for example, coordination of a metal catalyst by the N-atom generates a 6-membered metallacyclic intermediate. This general approach was used for both intramolecular couplings (synthesis of carbazoles for instance)1136–1139 and intermolecular transformations, like domino olefination/cyclisation,1140,1141 direct carbonylation delivering heterocyclic compounds1142,1143 or C–H arylation.1144,1145
In 2015 Wang and Ji spread the portfolio of such biaryl functionalisations by developing 2′-selective acylation (Scheme 179).1146 The optimized protocol allows selective mono-acylation of the Ts-protected biaryl-2-amines, under Pd-based catalysis and in presence of aldehydes as coupling partners and peroxide oxidants (TBHP). The reaction well tolerates several different FGs, both electron-withdrawing and electron-donating, on the biaryl substrates and a large panel of aldehydes is compatible with this protocol. Besides, in presence of a larger excess of the coupling partner and replacing the AcOH additive by PivOH, difunctionalisation is favoured.
 | ||
| Scheme 179 Aromatic amines as DGs. | ||
Amine DGs are also a unique handle for one-step carbazole synthesis.1136–1139,1147–1149 Biaryls bearing an NH2-motif in the ortho position are indeed potent substrates for Ir-catalysed C–H activation followed by intramolecular C–N bond formation, delivering the heterocyclic product (Scheme 179B).1150 This generally high-yielding protocol is compatible with an array of either electron-rich or electron-poor substrates, straightforwardly furnishing dissymmetric carbazoles.
A conceptually different strategy towards amine-directed C–H activation has been developed over the last three years by Gaunt and coworkers. This group recognized a lack of precedents in direct functionalisation of aliphatic substrates, and in particular they were interested in devising transformations showing complementary regioselectivity with respect to commonly observed γ-C–H direct couplings. Following this target, they discovered that aliphatic amines may undergo metallation at unusual β-position (via formation of an unprecedented 4-membered metallacycle intermediate) and in presence of an oxidant, such as PIDA, aziridine-type products are generated (Scheme 180A).1151 This unusual reaction is possible thanks to the steric hindrance of the amine that prevents formation of inactive bis(amine)–Pd complexes but consequently this transformation is mainly limited to structurally complex secondary cyclic amines, typically aminolactone congeners. Importantly, aziridine ring opening can be performed efficiently on the isolated products, delivering a panel of complex amine congeners, including quaternary amino acids. Moreover replacement of the hypervalent iodine oxidant with carbon monoxide results in a clean carbonylation reaction delivering β-lactams, albeit higher reaction temperatures are necessary to obtain synthetically useful yields (120 °C vs. 70–80 °C required for the aziridation process). Following this pioneering work, in 2017 the same research group reported a stereoselective version of this transformation. High chiral induction was achieved using BINOL-derived phosphoric acid ligands.1152 Another major step forward in this field was achieved by modifying the oxidation system.1153 Indeed, replacement of the previously used PIDA-type hypervalent iodine by benziodoxole tosylate in combination with AgOAc alters regioselectivity of the transformation, yet favouring azetidine formation (Scheme 180B). This regioselectivity switch is attributed to the influence of the OTs group. Formation of a 4-membered product is believed to occur via a two-step process including initial γ-tosylation followed by internal displacement of OTs by the amine moiety. The broad range of original azetidine compounds could hence be accessed and derivatization of these scaffolds delivers nitrogen-containing original compounds such as diols, amides, hydroxyl acids and hemiaminals.
 | ||
| Scheme 180 Functionalisation of C-sp3 centres directed by aliphatic amines. | ||
Following this seminal work, Gaunt expanded also the field of amine-directed C(sp3)–H carbonylation reactions (Scheme 180C). Firstly, a general catalytic system promoting highly selective β-functionalisation of primary carbons (even in presence of protons at γ-position) was devised thanks to the addition of hindered carboxylic acid derivatives in combination with a quinone oxidant.1154 This modified protocol shows exceptional tolerance towards an array of secondary aliphatic amines so that a large panel of 4-membered amides was formed in good to excellent yields, including few biologically active scaffolds. Few months later it was discovered that addition of an external diphosphine ligand and replacement of the Cu-based oxidant by Ag salts generates a much more reactive catalytic system, prompt now to activate methylene β-C–H bonds of aliphatic amines.1155 The reaction is highly regioselective and compatible with an ample panel of substrates, bearing not only electron-donating and electron-withdrawing substituents, but also structurally complex motifs such as heterocycles. The reaction proceeds smoothly even when sterically congested α-tertiary amines are selected as starting materials.1156 Accordingly, a large panel of β-lactams may now be accessed from simple, non-protected and non-prefunctionalised aliphatic amines. In addition to the carbonylation reactions, Gaunt and coworkers described in 2017 alkenylation reactions (Scheme 180D). This formal dehydrogenative C–C coupling is assisted by an amino acid ligand (under this protocol the metallation step is reversible yet promoting and facilitating the targeted reactivity) and hence the C–H coupling followed by the annulation step leads to formation of complex heterocyclic structures.1157 Also a closely related arylation using arylboronic esters as coupling partners takes place under ligand-assistance (Scheme 180E).1158 To further illustrate the synthetic value and versatility of the above-presented projects, Gaunt proposed a new retrosynthetic route towards K-252 (stauroeporinone) implying two amine-directed C–H activation events, i.e. Cu-catalysed ortho-arylation of dibenzyl para-toluidinearyl iodane, followed by aromatic carbonylation/intramolecular C–C bond formation.1159
In addition to this elegant work on amine-free directed C–H activation, several examples of C–H transformations based on the use of N-protected DGs have been reported. In particular, nosyl-protected phenylethylamine, benzylamines and 2-aryl aniline derivatives could be successfully used in Catellani-inspired1160meta-selective arylation (Scheme 181A).1161 The catalytic system combining Pd-catalyst and a pyridine-derived ligand promotes meta-functionalisation thanks to the participation of norbornene in a catalytic cycle. The scope of this transformation is rather remarkable, as not only three substrate scaffolds are effective, but also a diversity of both electron-rich and electron-poor aryliodanes may be used, although the products are generally isolated in rather modest to good yields.
 | ||
| Scheme 181 Amine-directed remote functionalisations. | ||
Quaternary ammonium salts have also met growing interest in the context of direct C–H functionalisation. The cationic nature of this moiety renders it particularly appealing in devising alternative, supramolecular strategies for C–H activation. In 2016 Phipps focused on direct C–B coupling catalysed by Ir-bipyridine type complex (Scheme 181B).1162 This research group stipulated that a quaternary ammonium moiety present on a substrate should participate in ion-pairing interactions with a modified bipyridine ligand bearing an anionic substituent. Such DG–ligand interactions should thus direct the Ir-catalyst to a more distal position, yet allowing remote C–H activation. Rewardingly, it was found that an Ir-catalyst coordinated by a bipyridine ligand with an appended sulfonate tether participates in strong ionic interactions with a quaternized benzylamine DG, consequently validating the initial hypothesis. This catalytic system allowed formation of the expected meta-borylated aromatics in high yields and generally high regioselectivity. The reaction is particularly efficient if ortho-substituted substrates are used and for unsubstituted precursors double borylation might be expected. In addition, as the ammonium moiety can be further valorized via Pd-catalysed C–C couplings, the newly obtained products are highly modular. The concept of ion pair-directed C–H functionalisation is also effective for meta-selective borylation of benzylamine, phenylethylamine and phenylpropylamine-derived amides.1163
An alternative approach based on supramolecular recognition between a cationic amine-type DG and a catalyst was disclosed recently by Olivo, Di Stefano and Costas (Scheme 181C). In order to achieve remote and site-selective functionalisation of aliphatic chains, a bioinspired catalyst, i.e. Mn complex equipped with 18-benzocrown-6 ether receptors was designed. Reversible preassociation of the crown ether with the protonated aliphatic primary amines approaches C8 and C9 positions of the aliphatic chain to the Mn-atom, yet enhancing regioselective oxidation. Remarkably, the length of the aliphatic chain does not impact the reaction outcome. This conceptually unprecedented transformation is, for the moment, limited to simple, non-substituted substrates and the regioselectivity level still needs improvement.1164
Finally we would like to highlight a recent contribution by Suzuki and Yamashita who designed original aluminabenzene–Rh and Ir complexes. Catalytic activity of this new Ir-based complex was evaluated studying C–H borylation of aliphatic tertiary amines and N,N-diethylbenzylamine.1165
16.9. Nitrone as DGs
In 2013, the panel of coordinating motifs employed as DG was further expanded, as a potential of nitrones to precoordinate a metal catalyst yet directing its insertion into a particular C–H bond was evidenced by Li.1166 Following this pioneering report, several examples of nitrone-directed couplings with alkenes, alkynes, diazo compounds and arenes were reported till 2015. More recently, several research groups astutely used this coordinating motif to build up heterocyclic compounds, and in particular indole derivatives. In 2015 Liu and Lu discovered that nitrone may play the role of an oxidizing DG in RhCp*-catalysed direct coupling with alkynes (Scheme 182A).1167 This redox-neutral C–H annulation occurs in absence of an external oxidant affording an array of 2,3-disubstituted indoles with moderate to high yields. Substituents such as CN, F, Br or OMe are well tolerated on the aromatic ring and both aryl–aryl and alkyl–aryl disubstituted alkynes are effective. Targeting more reliable and industrially appealing transformations, Ackermann and collaborators designed a closely related Co-catalysed transformation (Scheme 182B).1168 This redox-neutral coupling between nitrones and alkynes, proceeding via C–H/N–O functionalisation proceeds smoothly when a cationic CoCp*-catalyst is used in combination with either sodium acetate or an amino acid ligand. The reaction displays an excellent site- and regio-selectivity with unsymmetrical nitrones and alkynes, shows outstanding FG tolerance and is easily scalable.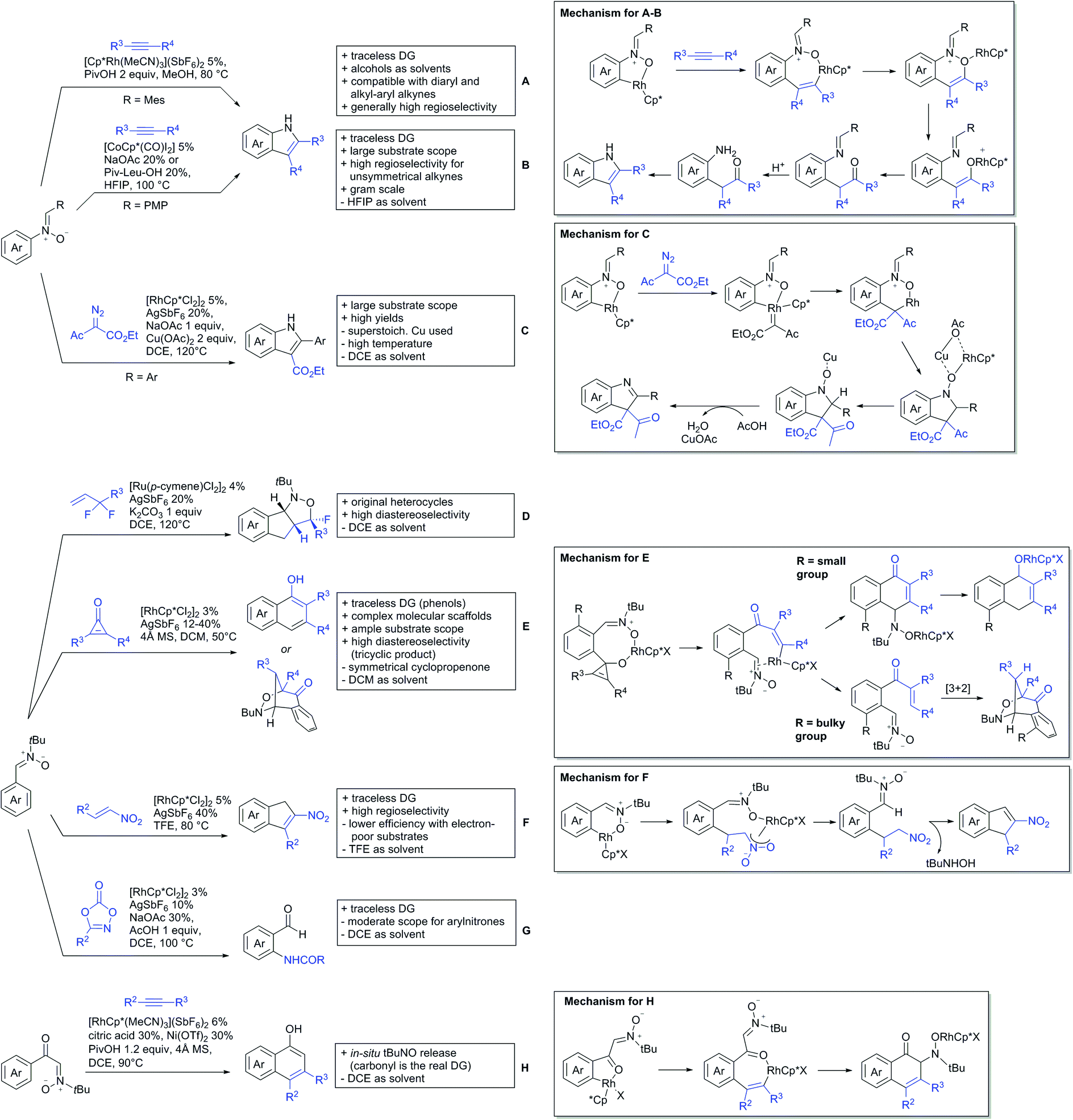 | ||
| Scheme 182 Nitrone-based DGs. | ||
Alternatively, indole synthesis may also be achieved using a RhCp*-catalyst in combination with diazo compounds as coupling partners (Scheme 182C).1169 In contrast to the previous examples, this transformation requires a substoichiometric amount of Cu(OAc)2 as oxidant but the substituent present on the nitrone is conserved in a final product (as C2-substituent of the indole scaffold). Accordingly, this strategy is of particular interest if indoles bearing an aromatic moiety at C2 position and an ester group at C3 are targeted.
In 2018 Li astutely used nitrone DG to construct fused isoxazolidines (Scheme 182D).1170 The corresponding regio- and diastereoselective C–H coupling of nitrones with perfluoroalkylolefines delivers rare heterocyclic compounds with three continuous stereogenic centers and features remarkable atom economy. The reaction proceeds under Ru(II)-based catalysis and, although rather harsh reaction conditions are required (temperature of 120 °C) it is compatible with an array of functional motifs, delivering the desired products in moderate to good yields.
A traceless character of the nitrone moiety is an additional key advantage, of particular importance for the C–H activation field. This purpose was elegantly illustrated by Li when conceiving a new synthetic route towards 1-naphthols, based on RhCp*-catalysed coupling of N-tert-butyl nitrones with cyclopropenones (Scheme 182E).1171 In this original reaction strained cyclopropenones are used as C3-type coupling partners and the catalytic cycle is believed to be completed by in situ removal of the nitrone moiety delivering 2,3-disubstituted naphthols. Besides, an alternative scenario is operative for ortho-substituted nitrones; a C–H acylation step is followed by [3+2] dipolar cyclo addition pathway, affording complex bicyclic structures. Noteworthy, the reaction occurs under oxidant free conditions and compounds otherwise difficult to access may now be constructed efficiently and straightforwardly. Naphthols may also be obtained via direct coupling between α-carbonyl nitrones and alkynes (Scheme 182H).1172 This redox-neutral RhCp*-catalysed transformation also implies in situ cleavage of the tert-butyl nitrone moiety. However, contrary to the previous example, symmetric 3,4-disubstituted naphthols are generated. It should be noted however that in this particular case the carbonyl moiety is rather expected to precoordinate the catalyst and direct the metallation event into the ortho-position.
In 2017, a nitrone-directed C–H activation protocol was used to construct indenes (Scheme 182F).1173 This reaction implies a direct coupling with internal nitroalkenes and, likewise previous examples, the N-tert-butylnitrone moiety is released via a Henry-type reaction delivering an ample selection of nitro-functionalised indenes, convertible via reduction into the corresponding ketones.
To complement the portfolio of direct functionalisation reactions compatible with nitrone DGs, Li and Wang developed site-selective amidation (Scheme 182G).1174 This RhCp*-catalysed transformation implies use of 1,4,2-dioxazol-5-ones as coupling partners and the DG decomposes at the end of the reaction releasing ortho-functionalised aldehydes. The optimized protocol allows installation of various amide moieties and several functionalities are also tolerated on the aromatic backbone furnishing the desired products in rather high yields.
17. Conclusions and outlook
It can be seen that the field of DG assisted catalytic C–H functionalisation has come a long way since its beginnings more than two decades ago. Most of the FGs you can think of have been used in one or the other example as DG in one or more transformation. This allows making the statement that most of the compounds which occur in a multistep sequence carry a FG which can be exploited as DG in C–H functionalisation chemistry, at least in principle. The question remains why the methodology is still not explored more frequently in complex synthesis or in R&D processes. The following critical outlook discusses the shortcomings of C–H functionalisation, which need to be addressed in the future.C–H functionalisation chemistry is very often said to be the future technology replacing cross coupling reactions because of its inherent potential for greenness in comparison to the latter. As green chemistry is a major concern for industry, considerations related to its future application in process chemistry need to be made. Nowadays cross coupling reactions can be performed in a variety of (green) solvents, under air, with catalyst loadings lower than 0.5%, catalytic amounts of ligands and stoichiometric bases. These reactions are now routinely used in chemical development and production of fine chemicals. C–H functionalisation, instead, still requires very often specific cocktails of a number of compounds (as discussed in the introduction), often under inert atmosphere, and additionally necessitate installation and cleavage of a DG, unless already present in the starting materials or found in the target product. The demonstration of the cleavage of a DG was specifically highlighted throughout this review since in most examples DG cleavage has not been investigated. Consequently, progress in the area of cleavable or traceless DGs is highly desirable in the near future.
Apart from these issues, less obvious limitations appear evident from the analysis of the literature in the field of C–H functionalisation. For example, a large number of protocols make use of toxic solvents, in particular DCE and other halogenated solvents are widely used. Although other solvents might be used as replacements in some cases, these are normally not thoroughly investigated, and often result in much lower yields. The efficiency of the reaction in more sustainable solvents is therefore not guaranteed. In a typical process the solvent accounts for 50–60% of the entire mass,1175 and the use of greener media is therefore of major importance in chemical development programs, although still less studied in academia. In case a C–H functionalisation protocol would be of interest to industry, the process chemists will have to spend considerable efforts in finding a valid replacement solvent before even considering using such a protocol. While investigations on green solvents are nowadays common for many type of reactions, this is not yet the practice for C–H functionalisations. Nonetheless, several examples were reported recently of functionalisations in alcoholic media, and some in alkyl acetates. Both these classes of compounds are listed as preferred solvents in solvent guides.1176,1177 In particular, the latter type of solvents has been used in a variety of transition metal-catalysed reactions,908,1178–1182 and EtOAc has for instance been demonstrated by Chang to be an effective replacement of DCE in a larger scale (200 mmol) C–H amidation protocol.751 This will hopefully inspire further research in this direction.
Regarding the metal catalysts which dominate the field, we still see Pd, Rh, and Ru as the three major players. In a society ever more aware of environmental issues, it is highly desirable to substitute these metals for more abundant ones. Consequently, the number of examples using Fe, Co, Ni, Cu, and Mn are increasing. Not only are these metals less toxic (with the exception of Co, see below), but also their extraction from soil is much simpler and requires less energy or less hazardous chemicals. Hence, as in all fields of metal catalysis, the quest for switching from precious metals to more abundant ones is ongoing and one of the big driving forces of innovation.
Sticking to the issue of the catalyst another important aspect is the often high catalyst loading necessary for C–H functionalisation reactions. Although many protocols utilise lower amounts, it is not uncommon to find a loading of 10% [total metal] in Pd, Rh, Ru or Ir catalysed reactions. While such loadings are still acceptable when using base metals, efforts should be devoted to lowering of these amounts when using noble metals. Aside from matters of cost, the accepted residual levels in e.g. formulated drugs, based on permitted daily exposure (PDE), are typically higher for base metals (e.g. Cu and Fe) than for noble metals.1183 On the other hand, although base metal catalysis is developing quickly in this field, in many cases up to 20% of the metal is used, and several protocols are also reported making use of stoichiometric amounts of metal (e.g. for Cu). Catalyst recycling is not often considered in academic publications, but a few interesting examples of heterogeneous catalysts for C–H functionalisation have been reported, which allow easy catalyst recycling and reuse.1184 Research in this direction, involving both heterogeneous and homogeneous catalysts, should be developed further.1185 Concerning base metals, a very important note has to be made: base metals are not by definition less toxic. In fact, despite the growing use of Co catalysis in C–H functionalisation, Co is not a low-toxicity metal, and the permitted daily exposure for this metal is actually lower than for noble metals used in catalysis such as Pd, Rh, Ru and Ir.1183
Concerning the regioselectivity of C–H functionalisation reactions, most examples of DGs listed in this review are ortho-DGs, with respect to aromatic C–H functionalisation. This has been identified as a shortcoming and several groups have started to address this issue. A handful of meta- and para-DGs have been reported and this area will without doubt see more examples in the near future.
Other selectivity issues have also been identified. It is true that regioselectivity, for example on an aromatic substrate, is achieved using a DG, but monofunctionalisation is often challenging. Many of the protocols included in this review produce in several cases mixtures of mono and difunctionalised compounds. This is common, for example, in arylation and olefination reactions, while with other functionalities this is less of a problem. In the less selective cases, monofunctionalisation is generally achieved by blocking (substitution) one of the reactive positions, or of a contiguous one (e.g. in arenes substituted in the 5 position, the DG in position 1 often selectively promotes functionalisation in position 2 based on steric reasons). Although this might be considered an acceptable solution in academia, this will be more important to tackle when these protocols are used in the synthesis of specific target molecules, where this specific substitution pattern might not be required/desired. Another strategy to deal with difunctionalisation found in the academic literature is the use of an excess of the DG-equipped substrate to avoid difunctionalisation. Although this might not be a big problem per se (as the substrate can be easily recovered), the excess substrate is often the most complex and expensive molecule in the mixture (e.g. it contains the DG), and therefore this is also not the ideal solution as efficient separation avoiding column chromatography might be tricky.
Furthermore, in some transformations, the applied coupling partners to date leave significant room for improvements. As an example, arylation reactions are often performed with the use of aryl iodides, which the fine chemical industry typically avoids, while relatively few examples currently involve the use of cheaper, widely available and less wasteful aryl chlorides.
At a more general level, stereoselective transformations for the formation of chiral quaternary centres are still quite limited. Enantioselectivity is here achieved using a chiral ligand, and several examples demonstrate that good to excellent selectivity is possible. The mechanism-guided stereoselective formation of alkenes (e.g. after Fujiwara–Moritani reactions or hydrofunctionalisation of alkynes) is often not achieved and mediocre E/Z ratios are obtained in many cases. As alkenylation is one of the most studied transformations, we believe more emphasis should be put in the development of selective strategies. Similarly, the hydrofunctionalisation of unsymmetrical alkynes usually results in regioisomeric mixtures.
Important advancements are being made concerning the use of aerobic conditions for oxidative couplings. Despite the safety issues related to the use of air/O2 in a chemical process (especially in development and production scale),1186 the use of oxygen results in higher atom economy and cheaper processes, especially in comparison to the use of other (super)stoichiometric Ag and Cu salt oxidants (utterly common for group 8–10 metal catalysis, see also introduction). Sometimes O2/air is used in combination with Cu salt allowing for their use in catalytic amounts. The variety of reported oxidants is anyway limited.
While a huge variety of C(sp2)–H functionalisation protocols have appeared, C(sp3)–H functionalisation still lags behind, due to higher inherent difficulty in activating these bonds. Nonetheless, important developments have been brought forward by a number of groups, mostly using bidentate amides as DGs. These transformation in many cases require external ligands (such as monoprotected amino acids, MPAA), and conformational restraints (aliphatic rings as substrates or Thorpe–Ingold effect) to be efficient. This field is however very promising, due to the many different compounds potentially obtained via this strategy.
It has been stated that many of the most frequently occurring FGs have been explored as DGs in C–H activation chemistry. This offers on the one hand great potential in applications, but on the other hand it also raises questions regarding selectivity, in cases a substrate carries more than one potential DG. Hence, research focused on exploring such substrates and eventually developing orthogonal sets of conditions in which first DG1 and then DG2 exhibits its directing potential would be highly interesting. However, such reports are scarce192,1031,1187–1190 and much more work in this direction would be highly desirable, since it would increase the number of cases in which C–H functionalisation could be practically applied.
This very critical discussion is not meant to discourage the use or development of this type of chemistry. Quite the contrary in fact! We hope that by critically analysing the weak points of this technology, research groups around the world would realise where important advancements need to be made, and focus their research not just in developing higher-yielding reactions, changing the metal for catalysis, or developing new reagents/reactants, but also in making these protocols more general, selective, and actually greener and industry-appealing.
List of abbreviations
| @ | Supported on |
| acac | Acetylacetonate |
| Ac2O | Acetic anhydride |
| AcOH | Acetic acid |
| AdOH | Adamantane carboxylic acid |
| AIBN | Azobisisobutyronitrile |
| Ala | Alanine |
| ampaphos | tBu2PCH2P(o-C6H4OMe)2 |
| APAQ | Acetylprotected aminoethyl quinolone |
| ArF | Pentafluorophenyl |
| BADME | Bis(2-dimethylaminoethyl)ether |
| B2eg2 | Bis(ethylene glycolato)diboron |
| B2pin2 | Bis(pinacolato)diboron |
| BarF4- or BARF | Tetrakis[3,5-bis(trifluoromethyl)phenyl]borate |
| 9-BBN | 9-Borabicyclo(3.3.1)nonane |
| bipy | 2,2′-Bipyridine |
| Boc | tert-Butoxycarbonyl |
| BP | Benzophenone |
| BQ | 1,4-Benzoquinone |
| Brij 35 | Poly(oxy-1,2-ethanediyl), α-dodecyl-ω-hydroxy-polymer |
| BTP | 2-Bromo-3,3,3-trifluoropropene |
| CAN | Ceric ammonium nitrate |
| CDC | Cross dehydrogenative coupling |
| cod | Cyclooctadiene |
| Cp* | 1,2,3,4,5-Pentamethylcyclopentadienyl |
| CpE | Diethyl-2,4,5-trimethylcyclopentadienyl-1,3-dicarboxylate |
| m-CPBA | m-Chloroperbenzoic acid |
| CPME | Cyclopentyl methyl ether |
| Cy | Cyclohexyl |
| cymene | p-Cymene |
| cym | p-Cymene |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| dba | Dibenzylideneacetone |
| DBE | Dibutylether |
| DCB | 1,2-Dichlorobenzene |
| 2,3-DCB | 2,3-Dichlorobutane |
| DCDMH | 1,3-Dichloro-5,5-dimethylhydantoin |
| DCE | Dichloroethane |
| DCP | Dicumyl peroxide |
| DCM | Dichloromethane |
| dcpm | Bis(dicycloheylphosphine)methane |
| DG | Directing group |
| DIAD | Diisopropyl azodicarboxylate |
| DIBAL-H | Diisobutylaluminium hydride |
| DIOP | O-Isopropyliden-2,3-dihydroxy-1,4-bis(diphenylphosphino)butan |
| DIPEA | N,N-Diisopropylethylamine |
| dippz | 1,2-Bis(diisopropylphosphino)benzene |
| DMAc or DMA | Dimethylacetamide |
| 2,6-DMBQ | 2,6-Dimethoxy-1,4-benzoquinone |
| DME | 1,2-Dimethoxyethane |
| DMF | Dimethylformamide |
| DMI | 1,3-Dimethyl-2-imidazolidinone |
| DMPU | 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone |
| dppb | 1,4-Bis(diphenylphosphino)butane |
| dppe | 1,2-Bis(diphenylphosphino)ethane |
| dppen | trans-1,2-Bis(diphenylphosphino)ethene |
| dr | Diastereomeric ratio |
| drs | Diastereomeric ratios |
| DTBEDA | N,N′-Di-tert-butylethylenediamine |
| dtbpy | 4,4′-Di-tert-butyl-2,2′-dipyridyl |
| EBX | Ethynyl-1,2-benziodoxol-3(1H)-one |
| EDG | Electron donating group |
| ee | Enantiomeric excess |
| EWG | Electron withdrawing group |
| FG | Functional group |
| Gly | Glycine |
| HBpin | Pinacolborane |
| hfacac | Hexafluoroacetylacetone |
| HFIP | Hexafluoroisopropanol |
| HODmb | 2,2-Dimethylbutyric acid |
| Ile | Isoleucine |
| IPr-HCl | 1,3-Bis(2,6-diisopropylphenyl)imidazolium chloride |
| LDA | Lithium diisopropylamide |
| LED | Light-emitting diode lamp |
| Leu | Leucine |
| LiHMDS | Lithium bis(trimethylsilyl)amide |
| Men | Menthyl-OC(O) |
| Me2-TP | 4-(Bis(2-(diphenylphosphanyl)phenyl)phosphanyl)-N,N-dimethylaniline |
| Mes | Mesitylene |
| MOF | Metal–organic framework |
| MPS | Methyl phenylsulfoximine |
| MTBE | Methyl tert-butyl ether |
| MS | Molecular sieves |
| o-NBA | o-Nitrobenzoic acid |
| nbd | Bicyclo[2.2.1]hepta-2,5-dienyl (= norbornadiene) |
| NBE | Norbornene |
| NBS | N-Nromosuccinimide |
| NCTS | N-Cyano-N-phenyl-p-toluenesulfonamide |
| nep | Neopentyl glycolato |
| NFSI | N-Fluorobenzenesulfonimide |
| NHC | N-Heterocyclic carbene |
| NHPI | N-Hydroxyphthalimide |
| NIS | N-Iodosuccinimide |
| NMO | N-Methylmorpholine-N-oxide |
| NMP | N-Methyl-2-pyrrolidone |
| Nphth | Phthalimide |
| NXS | N-Halosuccinimide (X = halogen) |
| ODCB | 1,2-Dichlorobenzene |
| o-PBA | o-Phenylbenzoic acid |
| OTf | Trifluoromethanesulfonate |
| PCC | Pyridinium chlorochromate |
| PE | 2-(2-Pyridyl)ethylamine |
| PEG | Poly ethylene glycol |
| PEPPSI-IPr | [1,3-Bis(2,6-diisopropylphenyl)imidazol-2-ylidene](3-chloropyridyl)palladium(II) dichloride |
| 1,10-phen or phen | 1,10-Phenanthroline |
| Phe | Phenylalanine |
| PIDA | Phenyliodine diacetate |
| pin | Pinacolato |
| PivOH | Pivalic acid |
| PM | 2-(Aminomethyl)pyridine |
| PTSA | p-Toluenesulphonic acid |
| PyO | Pyridyl-N-oxide |
| rt | Room temperature |
| SDS | Sodium dodecylsulfate |
| SIXyl·HCl | Bis(2,6-dimethylphenyl)imidazolinium chloride |
| SPGS-550-M | β-Sitosterol methoxypolyethyleneglycol succinate |
| TAM | Triazolyldimethyl |
| tAmOH | tert-Amylalcohol |
| TBAA or TBAOAc | Tetrabutylammonium acetate |
| TBAB | Tetrabutylammonium bromide |
| TBAF | Tetra-n-butylammonium fluoride |
| TBAI | Tetrabutylammonium iodide |
| TBHP | tert-Butylhydroperoxyde |
| TBPB | tert-Butyl peroxybenzoate |
| TBS | tert-Butyldimethylsilyl |
| TCC | Trichloroisocyanuric acid |
| TCP | 1,2,3-Trichloropropane |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| TFA | Trifluoroacetic acid |
| TFE | 2,2,2-Trifluoroethanol |
| THF | Tetrahydrofurane |
| THP | Tetrahydropyran |
| TIPS | Triisopropylsilyl |
| TMBzOH | 2,4,6-Trimethylbenzoic acid |
| TMEDA | Tetramethylethylenediamine |
| TMS | Trimethylsilyl |
| TON | Turn over number |
| Tween20 | Sorbitan, monododecanoate, poly(oxy-1,2-ethanediyl) derivatives |
| Val | Valine |
| XPhos | 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an Excellence of Science (EOS) project of the Research Foundation Flanders (FWO) under Grant no. G0H0918N and by INSA Rouen, Rouen University, CNRS, EFRD, Labex SynOrg (ANR-11-LABX-0029) and Région Normandie (Crunch Network). CS is grateful to the University of Antwerp for funding (BOF fellowship). TB and RB thank the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 758710). All authors would like to acknowledge the contribution of the COST action CA15106 (CHAOS – C–H Activation in Organic Synthesis). The authors acknowledge the TU Wien University Library for financial support through its Open Access Funding Program.References
- T. Swamy, B. V. Subba Reddy, R. Gree and V. Ravinder, ChemistrySelect, 2018, 3, 47–70 CrossRef.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef PubMed.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef PubMed.
- W. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435–1467 RSC.
- N. K. Mishra, S. Sharma, J. Park, S. Han and I. S. Kim, ACS Catal., 2017, 7, 2821–2847 CrossRef.
- X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef PubMed.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef PubMed.
- V. P. Mehta and J.-A. Garcia-Lopez, ChemCatChem, 2017, 9, 1149–1156 CrossRef.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef PubMed.
- F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275–296 CrossRef.
- N. Della Ca, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef PubMed.
- D. Pla and M. Gomez, ACS Catal., 2016, 6, 3537–3552 CrossRef.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- D. Wei, X. Zhu, J.-L. Niu and M.-P. Song, ChemCatChem, 2016, 8, 1242–1263 CrossRef.
- J. Wencel-Delord and F. Colobert, Synlett, 2015, 2644–2658 CrossRef.
- M. R. Yadav, R. K. Rit, M. Shankar and A. K. Sahoo, Asian J. Org. Chem., 2015, 4, 846–864 CrossRef.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- M. S. Khan, A. Haque, M. K. Al-Suti and P. R. Raithby, J. Organomet. Chem., 2015, 793, 114–133 CrossRef.
- J. Mo, L. Wang, Y. Liu and X. Cui, Synthesis, 2015, 439–459 Search PubMed.
- U. Sharma, A. Modak, S. Maity, A. Maji and D. Maiti, RSC Catal. Ser., 2015, 21, 551–609 Search PubMed.
- J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70–76 RSC.
- D. A. A. Wilton, C–H Activation of Heteroaromatics, in C–H Activation in Organic Synthesis, ed. J. J. Li, CRC Press, 2015, pp. 267–304 Search PubMed.
- X.-F. Wu, Chem. – Eur. J., 2015, 21, 12252–12265 CrossRef PubMed.
- M. Schnuerch, N. Dastbaravardeh, M. Ghobrial, B. Mrozek and M. D. Mihovilovic, Curr. Org. Chem., 2011, 15, 2694–2730 CrossRef.
- J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef PubMed.
- K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC.
- R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef PubMed.
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef PubMed.
- A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842–9860 CrossRef PubMed.
- Y. Yang, K. Li, Y. Cheng, D. Wan, M. Li and J. You, Chem. Commun., 2016, 52, 2872–2884 RSC.
- A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC.
- C. Zhu, R. Wang and J. R. Falck, Chem. – Asian J., 2012, 7, 1502–1514 CrossRef PubMed.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef PubMed.
- J. Maes and B. U. W. Maes, in Adv. Heterocycl. Chem., ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2016, vol. 120, pp. 137–194 Search PubMed.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef.
- R. Bandichhor, A. Bhattacharya, L. Diorazio, P. Dunn, K. Fraunhoffer, F. Gallou, J. Hayler, M. Hickey, B. Hinkley, D. Hughes, L. Humphreys, B. Kaptein, S. Mathew, L. Oh, P. Richardson, T. White and S. Wuyts, Org. Process Res. Dev., 2014, 18, 863–874 CrossRef.
- R. Bandichhor, A. Bhattacharya, A. Cosbie, L. Diorazio, P. Dunn, K. Fraunhoffer, F. Gallou, J. Hayler, M. Hickey, B. Hinkley, L. Humphreys, B. Kaptein, L. Oh, P. Richardson, S. Roberts, T. White, S. Wuyts and J. Yin, Org. Process Res. Dev., 2015, 19, 1924–1935 CrossRef.
- R. Ely, P. Richardson, A. Zlota, A. Steven, D. P. Day, R. Kargbo, C. C. Nawrat, A. Ramirez and J. Knight, Org. Process Res. Dev., 2017, 21, 675–688 CrossRef.
- M. Jean and P. van de Weghe, Transition metal C–H activation: application to stereoselective synthesis of natural products and drugs, in Stereoselective Synthesis of Drugs and Natural Products, ed. V. Andrushko and N. Andrushko, John Wiley & Sons, Inc., 2013, vol. 1, pp. 667–686 Search PubMed.
- M. Anada, H. Nambu and S. Hashimoto, CSJ Curr. Rev., 2011, 5, 175–191 Search PubMed.
- D. Y. K. Chen and S. W. Youn, Chem. – Eur. J., 2012, 18, 9452–9474 CrossRef PubMed.
- Y. Qiu and S. Gao, Nat. Prod. Rep., 2016, 33, 562–581 RSC.
- P. B. Brady and V. Bhat, Eur. J. Org. Chem., 2017, 5179–5190 CrossRef.
- L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC.
- R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef PubMed.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1119–1126 CrossRef.
- Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2009, 5820–5831 RSC.
- J. Vana, J. Hanusek and M. Sedlak, Dalton Trans., 2018, 47, 1378–1382 RSC.
- O. Baudoin, Acc. Chem. Res., 2017, 50, 1114–1123 CrossRef PubMed.
- M. Fultz, Rhodium-Catalyzed C–H Activation, in C–H Activation in Organic Synthesis, ed. J. J. Li, CRC Press, 2015, pp. 95–112 Search PubMed.
- G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007–1020 CrossRef PubMed.
- G.-F. Zha, H.-L. Qin and E. A. B. Kantchev, RSC Adv., 2016, 6, 30875–30885 RSC.
- A. Molnar and A. Papp, Curr. Org. Chem., 2016, 20, 381–458 CrossRef.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef.
- G. Pototschnig, N. Maulide and M. Schnurch, Chem. – Eur. J., 2017, 23, 9206–9232 CrossRef PubMed.
- A. C. Williams, Nickel-Catalyzed C–H Activation, in C–H Activation in Organic Synthesis, ed. J. J. Li, CRC Press, 2015, pp. 113–144 Search PubMed.
- L. C. M. Castro and N. Chatani, Chem. Lett., 2015, 44, 410–421 CrossRef.
- J. Yamaguchi, K. Muto, K. Itami and K. Itami, Top. Curr. Chem., 2016, 374, 55 CrossRef PubMed.
- S. Prakash, R. Kuppusamy and C.-H. Cheng, ChemCatChem, 2018, 10, 683–705 CrossRef.
- N. L. Snyder and E. J. Medici, Cobalt-Catalyzed C–H Activation, in C–H Activation in Organic Synthesis, ed. J. J. Li, CRC Press, 2015, pp. 217–250 Search PubMed.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef.
- N. Yoshikai, ChemCatChem, 2015, 7, 732–734 CrossRef.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef PubMed.
- T. K. Hyster, Catal. Lett., 2015, 145, 458–467 CrossRef.
- J. G. Kim, K. Shin and S. Chang, Top. Organomet. Chem., 2016, 55, 29–51 CrossRef.
- J. Wencel-Delord, F. W. Patureau and F. Glorius, Top. Organomet. Chem., 2016, 55, 1–27 Search PubMed.
- N. M. Ahmad, Copper-Mediated C–H Activation, in C–H Activation in Organic Synthesis, ed. J. J. Li, CRC Press, 2015, pp. 175–216 Search PubMed.
- K. Hirano and M. Miura, Chem. Lett., 2015, 44, 868–873 CrossRef.
- J.-L. Hu, J. Li and Y.-F. Zhou, Curr. Green Chem., 2015, 2, 170–191 CrossRef.
- G. Cera and L. Ackermann, Top. Curr. Chem., 2016, 374, 1–34 CrossRef PubMed.
- N. B. Ambhakir, Iron-Catalyzed C–H Activation, in C–H Activation in Organic Synthesis, ed. J. J. Li, CRC Press, 2015, pp. 145–174 Search PubMed.
- W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef.
- W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef PubMed.
- C. Wang, Synlett, 2013, 1606–1613 CrossRef.
- Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764–7786 RSC.
- Y. Ogiwara, M. Miyake, T. Kochi and F. Kakiuchi, Organometallics, 2017, 36, 159–164 CrossRef.
- B. Zhang, H.-W. Wang, Y.-S. Kang, P. Zhang, H.-J. Xu, Y. Lu and W.-Y. Sun, Org. Lett., 2017, 19, 5940–5943 CrossRef PubMed.
- C. Zhang and Y. Rao, Org. Lett., 2015, 17, 4456–4459 CrossRef PubMed.
- M. Weitzberg, E. Abu-Shakra, A. Azeb, Z. Aizenshtat and J. Blum, J. Org. Chem., 1987, 52, 529–536 CrossRef.
- D. E. Ames and A. Opalko, Tetrahedron, 1984, 40, 1919–1925 CrossRef.
- D.-B. Zhou and G.-W. Wang, Adv. Synth. Catal., 2016, 358, 1548–1554 CrossRef.
- D. J. Paymode and C. V. Ramana, J. Org. Chem., 2015, 80, 11551–11558 CrossRef PubMed.
- K. R. Bettadapur, V. Lanke and K. R. Prabhu, Org. Lett., 2015, 17, 4408–4411 CrossRef PubMed.
- S. H. Han, S. Kim, U. De, N. K. Mishra, J. Park, S. Sharma, J. H. Kwak, S. Han, H. S. Kim and I. S. Kim, J. Org. Chem., 2016, 81, 12416–12425 CrossRef PubMed.
- S. Pan, H. Jiang, Y. Zhang, D. Chen and Y. Zhang, Synlett, 2016, 1997–2002 Search PubMed.
- N. Kimura, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2017, 139, 14849–14852 CrossRef PubMed.
- R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2016, 138, 10132–10135 CrossRef PubMed.
- Y. Kommagalla, K. Srinivas and C. V. Ramana, Chem. – Eur. J., 2014, 20, 7884–7889 CrossRef PubMed.
- K. Srinivas, Y. Dangat, Y. Kommagalla, K. Vanka and C. V. Ramana, Chem. – Eur. J., 2017, 23, 7570–7581 CrossRef PubMed.
- X.-X. Chen, J.-X. Wang, J.-T. Ren, H. Xie, Y. Zhao, Y.-M. Li and M. Sun, Synlett, 2017, 1840–1844 Search PubMed.
- S. Yu, S. Liu, Y. Lan, B. Wan and X. Li, J. Am. Chem. Soc., 2015, 137, 1623–1631 CrossRef PubMed.
- S. Zhou, J. Wang, L. Wang, C. Song, K. Chen and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 9384–9388 CrossRef PubMed.
- G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef PubMed.
- M. Bakthadoss, P. V. Kumar and T. S. Reddy, Eur. J. Org. Chem., 2017, 4439–4444 CrossRef.
- F. Hu and M. Szostak, Chem. Commun., 2016, 52, 9715–9718 RSC.
- V. Lanke, K. R. Bettadapur and K. R. Prabhu, Org. Lett., 2016, 18, 5496–5499 CrossRef PubMed.
- X. Li, G. Wu, X. Liu, Z. Zhu, Y. Huo and H. Jiang, J. Org. Chem., 2017, 82, 13003–13011 CrossRef PubMed.
- P.-Y. Lee, P. Liang and W.-Y. Yu, Org. Lett., 2017, 19, 2082–2085 CrossRef PubMed.
- K. Raghuvanshi, D. Zell, K. Rauch and L. Ackermann, ACS Catal., 2016, 6, 3172–3175 CrossRef.
- P. Shi, L. Wang, K. Chen, J. Wang and J. Zhu, Org. Lett., 2017, 19, 2418–2421 CrossRef PubMed.
- F. Wang, L. Jin, L. Kong and X. Li, Org. Lett., 2017, 19, 1812–1815 CrossRef PubMed.
- P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 Search PubMed.
- Q. Zhao, T. Poisson, X. Pannecoucke and T. Besset, Synthesis, 2017, 4808–4826 Search PubMed.
- X. Liu, X. Li, H. Liu, Q. Guo, J. Lan, R. Wang and J. You, Org. Lett., 2015, 17, 2936–2939 CrossRef PubMed.
- J. Luo, S. Preciado, S. O. Araromi and I. Larrosa, Chem. – Asian J., 2016, 11, 347–350 CrossRef PubMed.
- S. Chen, B. Feng, X. Zheng, J. Yin, S. Yang and J. You, Org. Lett., 2017, 19, 2502–2505 CrossRef PubMed.
- M. Pichette Drapeau and L. J. Goossen, Chem. – Eur. J., 2016, 22, 18654–18677 CrossRef PubMed.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- J. Schwarz and B. Konig, Green Chem., 2018, 20, 323–361 RSC.
- Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef PubMed.
- M. Font, J. M. Quibell, G. J. P. Perry and I. Larrosa, Chem. Commun., 2017, 53, 5584–5597 RSC.
- A. Modak and D. Maiti, Org. Biomol. Chem., 2016, 14, 21–35 RSC.
- L. Huang, D. Hackenberger and L. J. Goossen, Angew. Chem., Int. Ed., 2015, 54, 12607–12611 CrossRef PubMed.
- Z. Xu, T. Yang, X. Lin, J. D. Elliott and F. Ren, Tetrahedron Lett., 2015, 56, 475–477 CrossRef.
- C. Zhu, Y. Zhang, J. Kan, H. Zhao and W. Su, Org. Lett., 2015, 17, 3418–3421 CrossRef PubMed.
- M. Simonetti, D. M. Cannas, A. Panigrahi, S. Kujawa, M. Kryjewski, P. Xie and I. Larrosa, Chem. – Eur. J., 2017, 23, 549–553 CrossRef PubMed.
- A. Biafora, T. Krause, D. Hackenberger, F. Belitz and L. J. Goossen, Angew. Chem., Int. Ed., 2016, 55, 14752–14755 CrossRef PubMed.
- L. Huang and D. J. Weix, Org. Lett., 2016, 18, 5432–5435 CrossRef PubMed.
- R. Mei, C. Zhu and L. Ackermann, Chem. Commun., 2016, 52, 13171–13174 RSC.
- H. Gong, H. Zeng, F. Zhou and C.-J. Li, Angew. Chem., Int. Ed., 2015, 54, 5718–5721 CrossRef PubMed.
- H. Zhang, G.-J. Deng, S. Li, C.-J. Li and H. Gong, RSC Adv., 2016, 6, 91617–91620 RSC.
- X. Qin, X. Li, Q. Huang, H. Liu, D. Wu, Q. Guo, J. Lan, R. Wang and J. You, Angew. Chem., Int. Ed., 2015, 54, 7167–7170 CrossRef PubMed.
- N. Dastbaravardeh, T. Toba, M. E. Farmer and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 9877–9884 CrossRef PubMed.
- L. Huang and D. J. Weix, Org. Lett., 2016, 18, 5432–5435 CrossRef PubMed.
- D. Sun, B. Li, J. Lan, Q. Huang and J. You, Chem. Commun., 2016, 52, 3635–3638 RSC.
- X. S. Zhang, Y. F. Zhang, Z. W. Li, F. X. Luo and Z. J. Shi, Angew. Chem., Int. Ed., 2015, 54, 5478–5482 CrossRef PubMed.
- H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879–9884 CrossRef PubMed.
- S. Mochida, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 3024–3033 CrossRef PubMed.
- J. Luo, S. Preciado and I. Larrosa, Chem. Commun., 2015, 51, 3127–3130 RSC.
- C. Arroniz, J. G. Denis, A. Ironmonger, G. Rassias and I. Larrosa, Chem. Sci., 2014, 5, 3509–3514 RSC.
- A. J. S. Johnston, K. B. Ling, D. Sale, N. Lebrasseur and I. Larrosa, Org. Lett., 2016, 18, 6094–6097 CrossRef PubMed.
- X. Qin, D. Sun, Q. You, Y. Cheng, J. Lan and J. You, Org. Lett., 2015, 17, 1762–1765 CrossRef PubMed.
- Y. Zhang, H. Zhao, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 3817–3821 CrossRef PubMed.
- N. Y. P. Kumar, T. Rogge, S. R. Yetra, A. Bechtoldt, E. Clot and L. Ackermann, Chem. – Eur. J., 2017, 23, 17449–17453 CrossRef PubMed.
- J. L. Yu, S. Q. Zhang and X. Hong, J. Am. Chem. Soc., 2017, 139, 7224–7243 CrossRef PubMed.
- J.-L. Yu, S.-Q. Zhang and X. Hong, J. Am. Chem. Soc., 2017, 139, 7224–7243 CrossRef PubMed.
- A. Mandal, H. Sahoo, S. Dana and M. Baidya, Org. Lett., 2017, 19, 4138–4141 CrossRef PubMed.
- K. R. Bettadapur, V. Lanke and K. R. Prabhu, Chem. Commun., 2017, 53, 6251–6254 RSC.
- T. A. Davis, C. Wang and T. Rovis, Synlett, 2015, 1520–1524 Search PubMed.
- R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2016, 138, 10132–10135 CrossRef PubMed.
- D. A. Loginov and V. E. Konoplev, J. Organomet. Chem., 2018, 867, 14–24 CrossRef.
- M. Simonetti and I. Larrosa, Nat. Chem., 2016, 8, 1086–1088 CrossRef PubMed.
- S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepötter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513–5517 CrossRef PubMed.
- E. Kudo, Y. Shibata, M. Yamazaki, K. Masutomi, Y. Miyauchi, M. Fukui, H. Sugiyama, H. Uekusa, T. Satoh, M. Miura and K. Tanaka, Chem. – Eur. J., 2016, 22, 14190–14194 CrossRef PubMed.
- R. Mandal and B. Sundararaju, Org. Lett., 2017, 19, 2544–2547 CrossRef PubMed.
- M. Zhang, H. J. Zhang, T. Han, W. Ruan and T. B. Wen, J. Org. Chem., 2015, 80, 620–627 CrossRef PubMed.
- W. J. Han, F. Pu, J. Fan, Z. W. Liu and X. Y. Shi, Adv. Synth. Catal., 2017, 359, 3520–3525 CrossRef.
- X.-G. Liu, H. Gao, S.-S. Zhang, Q. Li and H. Wang, ACS Catal., 2017, 7, 5078–5086 CrossRef.
- J. Zhang, H. Chen, B. Wang, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 2768–2771 CrossRef PubMed.
- R. Prakash, K. Shekarrao and S. Gogoi, Org. Lett., 2015, 17, 5264–5267 CrossRef PubMed.
- H. Miura, K. Tsutsui, K. Wada and T. Shishido, Chem. Commun., 2015, 51, 1654–1657 RSC.
- A. Bechtoldt, C. Tirler, K. Raghuvanshi, S. Warratz, C. Kornhaaß and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 264–267 CrossRef PubMed.
- Y.-Q. Zhu, T.-F. Han, J.-L. He, M. Li, J.-X. Li and K. Zhu, J. Org. Chem., 2017, 82, 8598–8603 CrossRef PubMed.
- N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929–6932 CrossRef PubMed.
- J. Zhang, R. Shrestha, J. F. Hartwig and P. Zhao, Nat. Chem., 2016, 8, 1144–1151 CrossRef PubMed.
- A. Biafora, B. A. Khan, J. Bahri, J. M. Hewer and L. J. Goossen, Org. Lett., 2017, 19, 1232–1235 CrossRef PubMed.
- L. Huang, A. Biafora, G. Zhang, V. Bragoni and L. J. Goossen, Angew. Chem., Int. Ed., 2016, 55, 6933–6937 CrossRef PubMed.
- H. Chen, C. Lin, C. Xiong, Z. Liu and Y. Zhang, Org. Chem. Front., 2017, 4, 455–459 RSC.
- K. Kim, D. Vasu, H. Im and S. Hong, Angew. Chem., Int. Ed., 2016, 55, 8652–8655 CrossRef PubMed.
- C. Chen, P. Liu, J. Tang, G. Deng and X. Zeng, Org. Lett., 2017, 19, 2474–2477 CrossRef PubMed.
- C. Chen and X. Zeng, Eur. J. Org. Chem., 2017, 4749–4752 CrossRef.
- E. Tan, A. I. Konovalov, G. A. Fernández, R. Dorel and A. M. Echavarren, Org. Lett., 2017, 19, 5561–5564 CrossRef PubMed.
- R. Mei, S.-K. Zhang and L. Ackermann, Org. Lett., 2017, 19, 3171–3174 CrossRef PubMed.
- Y. Liu, Y. Yang, Y. Shi, X. Wang, L. Zhang, Y. Cheng and J. You, Organometallics, 2016, 35, 1350–1353 CrossRef.
- X.-Y. Shi, K.-Y. Liu, J. Fan, X.-F. Dong, J.-F. Wei and C.-J. Li, Chem. – Eur. J., 2015, 21, 1900–1903 CrossRef PubMed.
- Y.-Q. Zhu, Y. Liu, H. N. Wang, W. B. Liu and C.-J. Li, Org. Chem. Front., 2016, 3, 971–974 RSC.
- D. Lee and S. Chang, Chem. – Eur. J., 2015, 21, 5364–5368 CrossRef PubMed.
- A. Mandal, S. Dana, H. Sahoo, G. S. Grandhi and M. Baidya, Org. Lett., 2017, 19, 2430–2433 CrossRef PubMed.
- Y. Zhu, X. Chen, C. Yuan, G. Li, J. Zhang and Y. Zhao, Nat. Commun., 2017, 8, 14904 CrossRef PubMed.
- G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, J.-Q. Yu, Y. Hsiao and C. L. Joe, Angew. Chem., Int. Ed., 2017, 56, 1506–1509 CrossRef PubMed.
- K. K. Ghosh and M. van Gemmeren, Chem. – Eur. J., 2017, 23, 17697–17700 CrossRef PubMed.
- B. M. Trost, K. Imi and I. W. Davies, J. Am. Chem. Soc., 1995, 117, 5371–5372 CrossRef.
- S. Motohiro, K. Fumitoshi, K. Asayuki, C. Naoto and M. Shinji, Chem. Lett., 1996, 109–110 Search PubMed.
- S. Sueki, Z. Wang and Y. Kuninobu, Org. Lett., 2016, 18, 304–307 CrossRef PubMed.
- G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef PubMed.
- J. Hu, M. Guan, J. Han, Z.-B. Huang, D.-Q. Shi and Y. Zhao, J. Org. Chem., 2015, 80, 7896–7904 CrossRef PubMed.
- H. Jin, Z. Zhu, N. Jin, J. Xie, Y. Cheng and C. Zhu, Org. Chem. Front., 2015, 2, 378–382 RSC.
- L. Kong, X. Yang, X. Zhou, S. Yu and X. Li, Org. Chem. Front., 2016, 3, 813–816 RSC.
- W. Yu, W. Zhang, Z. Liu and Y. Zhang, Chem. Commun., 2016, 52, 6837–6840 RSC.
- A. Banerjee, S. K. Santra, P. R. Mohanta and B. K. Patel, Org. Lett., 2015, 17, 5678–5681 CrossRef PubMed.
- J. Mo, L. Wang and X. Cui, Org. Lett., 2015, 17, 4960–4963 CrossRef PubMed.
- G. Wang, L. Liu, H. Wang, Y.-S. Ding, J. Zhou, S. Mao and P. Li, J. Am. Chem. Soc., 2017, 139, 91–94 CrossRef PubMed.
- M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, J. Am. Chem. Soc., 2017, 139, 7745–7748 CrossRef PubMed.
- D.-D. Li, T.-T. Yuan and G.-W. Wang, J. Org. Chem., 2012, 77, 3341–3347 CrossRef PubMed.
- P. Gao, L. Liu, Z. Shi and Y. Yuan, Org. Biomol. Chem., 2016, 14, 7109–7113 RSC.
- P. Gao, W. Guo, J. Xue, Y. Zhao, Y. Yuan, Y. Xia and Z. Shi, J. Am. Chem. Soc., 2015, 137, 12231–12240 CrossRef PubMed.
- E. Kianmehr and A. Tanbakouchian, Tetrahedron, 2017, 73, 5337–5343 CrossRef.
- B. P. Mathew, H. J. Yang, J. Kim, J. B. Lee, Y.-T. Kim, S. Lee, C. Y. Lee, W. Choe, K. Myung, J.-U. Park and S. Y. Hong, Angew. Chem., Int. Ed., 2017, 56, 5007–5011 CrossRef PubMed.
- V. Kondapalli, X. Yu, Y. Yamamoto and M. Bao, J. Org. Chem., 2017, 82, 2288–2293 CrossRef PubMed.
- Y. Jaiswal, Y. Kumar, R. Thakur, J. Pal, R. Subramanian and A. Kumar, J. Org. Chem., 2016, 81, 12499–12505 CrossRef PubMed.
- M. H. Daniels, J. R. Armand and K. L. Tan, Org. Lett., 2016, 18, 3310–3313 CrossRef PubMed.
- W. Yang, S. Ye, D. Fanning, T. Coon, Y. Schmidt, P. Krenitsky, D. Stamos and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 2501–2504 CrossRef PubMed.
- T. Gensch, S. Vasquez-Cespedes, D.-G. Yu and F. Glorius, Org. Lett., 2015, 17, 3714–3717 CrossRef PubMed.
- H. Dai, C. Yu, C. Lu and H. Yan, Eur. J. Org. Chem., 2016, 1255–1259 CrossRef.
- T. Sato, T. Yoshida, H. H. Al Mamari, L. Ilies and E. Nakamura, Org. Lett., 2017, 19, 5458–5461 CrossRef PubMed.
- J. Li and L. Ackermann, Org. Chem. Front., 2015, 2, 1035–1039 RSC.
- Y. Xia, Z. Liu, S. Feng, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 223–236 CrossRef PubMed.
- D. C. Simko, P. Elekes, V. Pazmandi and Z. Novak, Org. Lett., 2018, 20, 676–679 CrossRef PubMed.
- P. Keshri, K. R. Bettadapur, V. Lanke and K. R. Prabhu, J. Org. Chem., 2016, 81, 6056–6065 CrossRef PubMed.
- B. L. Toth, S. Kovacs, G. Salyi and Z. Novak, Angew. Chem., Int. Ed., 2016, 55, 1988–1992 CrossRef PubMed.
- D. Xing and G. Dong, J. Am. Chem. Soc., 2017, 139, 13664–13667 CrossRef PubMed.
- T. J. Coxon, M. Fernandez, J. Barwick-Silk, A. I. McKay, L. E. Britton, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2017, 139, 10142–10149 CrossRef PubMed.
- P. Patel and G. Borah, Chem. Commun., 2017, 53, 443–446 RSC.
- P. Bai, Y.-Q. Li and Z.-Z. Huang, Org. Lett., 2017, 19, 1374–1377 CrossRef PubMed.
- G. S. Kumar, P. Kumar and M. Kapur, Org. Lett., 2017, 19, 2494–2497 CrossRef PubMed.
- X. Chen, X. Hu, S. Bai, Y. Deng, H. Jiang and W. Zeng, Org. Lett., 2016, 18, 192–195 CrossRef PubMed.
- H.-J. Zhang, W. Lin, F. Su and T.-B. Wen, Org. Lett., 2016, 18, 6356–6359 CrossRef PubMed.
- L. Zhang, C. Chen, J. Han, Z.-B. Huang and Y. Zhao, Org. Chem. Front., 2016, 3, 1271–1275 RSC.
- L. Guo, Y. Chen, R. Zhang, Q. Peng, L. Xu and X. Pan, Chem. – Asian J., 2017, 12, 289–292 CrossRef PubMed.
- H. Ikemoto, M. Kanai, R. Tanaka, T. Yoshino, S. Matsunaga, K. Sakata, K. Sakata, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2017, 56, 7156–7160 CrossRef PubMed.
- J. A. Boerth and J. A. Ellman, Angew. Chem., Int. Ed., 2017, 56, 9976–9980 CrossRef PubMed.
- D. Wang, X. Yu, X. Xu, B. Ge, X. Wang and Y. Zhang, Chem. – Eur. J., 2016, 22, 8663–8668 CrossRef PubMed.
- F. Li, C. Yu, J. Zhang and G. Zhong, Org. Lett., 2016, 18, 4582–4585 CrossRef PubMed.
- K. Meng, J. Zhang, F. Li, Z. Lin, K. Zhang and G. Zhong, Org. Lett., 2017, 19, 2498–2501 CrossRef PubMed.
- Q. Zhao, V. Tognetti, L. Joubert, T. Besset, X. Pannecoucke, J.-P. Bouillon and T. Poisson, Org. Lett., 2017, 19, 2106–2109 CrossRef PubMed.
- L. Kong, S. Yu, X. Zhou and X. Li, Org. Lett., 2016, 18, 588–591 CrossRef PubMed.
- Q. Lu, S. Vasquez-Cespedes, T. Gensch and F. Glorius, ACS Catal., 2016, 6, 2352–2356 CrossRef.
- N. S. Upadhyay, V. H. Thorat, R. Sato, P. Annamalai, S.-C. Chuang and C.-H. Cheng, Green Chem., 2017, 19, 3219–3224 RSC.
- K. Fukuzumi, Y. Unoh, Y. Nishii, T. Satoh, K. Hirano and M. Miura, J. Org. Chem., 2016, 81, 2474–2481 CrossRef PubMed.
- R. N. P. Tulichala, M. Shankar and K. C. K. Swamy, J. Org. Chem., 2017, 82, 5068–5079 CrossRef PubMed.
- S. Ruiz, C. Carrera, P. Villuendas and E. P. Urriolabeitia, Org. Biomol. Chem., 2017, 15, 8904–8913 RSC.
- L. Song, G. Tian, Y. He and E. V. Van der Eycken, Chem. Commun., 2017, 53, 12394–12397 RSC.
- Y. Lu, H.-W. Wang, J. E. Spangler, K. Chen, P.-P. Cui, Y. Zhao, W.-Y. Sun and J.-Q. Yu, Chem. Sci., 2015, 6, 1923–1927 RSC.
- O. Tischler, Z. Bokanyi and Z. Novak, Organometallics, 2016, 35, 741–746 CrossRef.
- K. Jing, J.-P. Yao, Z.-Y. Li, Q.-L. Li, H.-S. Lin and G.-W. Wang, J. Org. Chem., 2017, 82, 12715–12725 CrossRef PubMed.
- Z. Xiong, D. Liang and S. Luo, Org. Chem. Front., 2017, 4, 1103–1106 RSC.
- S. Sharma, S. H. Han, Y. Oh, N. K. Mishra, S. H. Lee, J. S. Oh and I. S. Kim, Org. Lett., 2016, 18, 2568–2571 CrossRef PubMed.
- P. S. Aparna, A. Vijayan, S. P. Raveendran, E. Suresh, R. L. Varma and K. V. Radhakrishnan, Synlett, 2017, 572–576 Search PubMed.
- F. Lied, A. Lerchen, T. Knecht, C. Mueck-Lichtenfeld and F. Glorius, ACS Catal., 2016, 6, 7839–7843 CrossRef.
- R. Das and M. Kapur, J. Org. Chem., 2017, 82, 1114–1126 CrossRef PubMed.
- V. Pascanu, F. Carson, M. V. Solano, J. Su, X. Zou, M. J. Johansson and B. Martin-Matute, Chem. – Eur. J., 2016, 22, 3729–3737 CrossRef PubMed.
- L. Yang, S. Li, L. Cai, Y. Ding, L. Fu, Z. Cai, H. Ji and G. Li, Org. Lett., 2017, 19, 2746–2749 CrossRef PubMed.
- Z.-W. Yang, Q. Zhang, Y.-Y. Jiang, L. Li, B. Xiao and Y. Fu, Chem. Commun., 2016, 52, 6709–6711 RSC.
- C. Chen, Y. Pan, H. Zhao, X. Xu, J. Xu, Z. Zhang, S. Xi, L. Xu and H. Li, Org. Chem. Front., 2018, 5, 415–422 RSC.
- K. Seth, M. Nautiyal, P. Purohit, N. Parikh and A. K. Chakraborti, Chem. Commun., 2015, 51, 191–194 RSC.
- J. Park, J. Lee and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 4256–4260 CrossRef PubMed.
- H.-W. Wang, Y. Lu, B. Zhang, J. He, H.-J. Xu, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 7449–7453 CrossRef PubMed.
- G. Chen, W. Gong, Z. Zhuang, M. S. Andrae, Y.-Q. Chen, X. Hong, Y.-F. Yang, T. Liu, K. N. Houk and J.-Q. Yu, Science, 2016, 353, 1023–1027 CrossRef PubMed.
- Q.-F. Wu, P.-X. Shen, J. He, X.-B. Wang, F. Zhang, Q. Shao, R.-Y. Zhu, C. Mapelli, J. X. Qiao, M. A. Poss and J.-Q. Yu, Science, 2017, 355, 499–503 CrossRef PubMed.
- J. He, H. Jiang, R. Takise, R.-Y. Zhu, G. Chen, H.-X. Dai, J.-Q. Yu, T. G. M. Dhar, J. Shi, H. Zhang and P. T. W. Cheng, Angew. Chem., Int. Ed., 2016, 55, 785–789 CrossRef PubMed.
- S. L. Yedage and B. M. Bhanage, J. Org. Chem., 2016, 81, 4103–4111 CrossRef PubMed.
- B. Li, J. Lan, D. Wu and J. You, Angew. Chem., Int. Ed., 2015, 54, 14008–14012 CrossRef PubMed.
- G. Chen, T. Shigenari, P. Jain, Z. Zhang, Z. Jin, J. He, S. Li, C. Mapelli, M. M. Miller, M. A. Poss, P. M. Scola, K.-S. Yeung and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 3338–3351 CrossRef PubMed.
- Z. Chen, L. a. Hu, F. Zeng, R. Zhu, S. Zheng, Q. Yu and J. Huang, Chem. Commun., 2017, 53, 4258–4261 RSC.
- J.-Q. Wu, Z.-P. Qiu, S.-S. Zhang, J.-G. Liu, Y.-X. Lao, L.-Q. Gu, Z.-S. Huang, J. Li and H. Wang, Chem. Commun., 2015, 51, 77–80 RSC.
- W. Liu, Q. Yu, L. A. Hu, Z. Chen and J. Huang, Chem. Sci., 2015, 6, 5768–5772 RSC.
- R. Das and M. Kapur, Chem. – Eur. J., 2016, 22, 16986–16990 CrossRef PubMed.
- G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef PubMed.
- J. Wen, A. Wu, P. Chen and J. Zhu, Tetrahedron Lett., 2015, 56, 5282–5286 CrossRef.
- C. Yu, F. Li, J. Zhang and G. Zhong, Chem. Commun., 2017, 53, 533–536 RSC.
- X. Wang, H. Tang, H. Feng, Y. Li, Y. Yang and B. Zhou, J. Org. Chem., 2015, 80, 6238–6249 CrossRef PubMed.
- S. L. Yedage and B. M. Bhanage, Green Chem., 2016, 18, 5635–5642 RSC.
- J.-Q. Wu, S.-S. Zhang, H. Gao, Z. Qi, C.-J. Zhou, W.-W. Ji, Y. Liu, Y. Chen, Q. Li, X. Li and H. Wang, J. Am. Chem. Soc., 2017, 139, 3537–3545 CrossRef PubMed.
- J.-P. Krieger, D. Lesuisse, G. Ricci, M.-A. Perrin, C. Meyer and J. Cossy, Org. Lett., 2017, 19, 2706–2709 CrossRef PubMed.
- R. Boobalan, P. Gandeepan and C.-H. Cheng, Org. Lett., 2016, 18, 3314–3317 CrossRef PubMed.
- X. Zhou, Z. Peng, H. Zhao, Z. Zhang, P. Lu and Y. Wang, Chem. Commun., 2016, 52, 10676–10679 RSC.
- X. Wang, T. Gensch, A. Lerchen, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2017, 139, 6506–6512 CrossRef PubMed.
- S. Sharma, S. H. Han, Y. Oh, N. K. Mishra, S. Han, J. H. Kwak, S.-Y. Lee, Y. H. Jung and I. S. Kim, J. Org. Chem., 2016, 81, 2243–2251 CrossRef PubMed.
- S. Sharma, S. H. Han, H. Jo, S. Han, N. K. Mishra, M. Choi, T. Jeong, J. Park and I. S. Kim, Eur. J. Org. Chem., 2016, 3611–3618 CrossRef.
- T. K. Beng, S. Langevin, H. Braunstein and M. Khim, Org. Biomol. Chem., 2016, 14, 830–834 RSC.
- F. M. Moghaddam, G. Tavakoli, B. Saeednia, P. Langer and B. Jafari, J. Org. Chem., 2016, 81, 3868–3876 CrossRef PubMed.
- T. Morita, T. Satoh and M. Miura, Org. Lett., 2017, 19, 1800–1803 CrossRef PubMed.
- S. Kovacs, B. L. Toth, G. Borsik, T. Bihari, N. V. May, A. Stirling and Z. Novak, Adv. Synth. Catal., 2017, 359, 527–532 CrossRef.
- D. Yamauchi, T. Nishimura and H. Yorimitsu, Angew. Chem., Int. Ed., 2017, 56, 7200–7204 CrossRef PubMed.
- T. Nishikata, A. R. Abela, S. Huang and B. H. Lipshutz, Beilstein J. Org. Chem., 2016, 12, 1040–1064 CrossRef PubMed.
- H. Lv, J. Shi, J. Huang, C. Zhang and W. Yi, Org. Biomol. Chem., 2017, 15, 7088–7092 RSC.
- M. K. Manna, A. Hossian and R. Jana, Org. Lett., 2015, 17, 672–675 CrossRef PubMed.
- S.-S. Chen, M.-S. Wu and Z.-Y. Han, Angew. Chem., Int. Ed., 2017, 56, 6641–6645 CrossRef PubMed.
- Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef PubMed.
- J.-H. Liu, M. Cui, X.-Y. Lu, Z.-Q. Zhang, B. Xiao and Y. Fu, Chem. Commun., 2016, 52, 1242–1245 RSC.
- J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC.
- A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440–5453 RSC.
- R. Sharma, K. Thakur, R. Kumar, I. Kumar and U. Sharma, Catal. Rev.: Sci. Eng., 2015, 57, 345–405 CrossRef.
- N. E. Ihanainen, E. T. T. Kumpulainen and A. M. P. Koskinen, Eur. J. Org. Chem., 2015, 3226–3229 CrossRef.
- Y. Ren, C. Yan, J. Jia and H.-S. Wu, J. Organomet. Chem., 2016, 824, 88–98 CrossRef.
- S. Li, L. Cai, H. Ji, L. Yang, G. Li and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef PubMed.
- A. Modak, A. Mondal, R. Watile, S. Mukherjee and D. Maiti, Chem. Commun., 2016, 52, 13916–13919 RSC.
- L. Yang, L. Fu and G. Li, Adv. Synth. Catal., 2017, 359, 2235–2240 CrossRef.
- J. Wencel-Delord and F. Colobert, Org. Chem. Front., 2016, 3, 394–400 RSC.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef.
- Y. Deng and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 888–891 CrossRef PubMed.
- A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147–3153 RSC.
- M. Bera, S. Agasti, R. Chowdhury, R. Mondal, D. Pal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 5272–5276 CrossRef PubMed.
- A. Modak, T. Patra, R. Chowdhury, S. Raul and D. Maiti, Organometallics, 2017, 36, 2418–2423 CrossRef.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888–11891 CrossRef PubMed.
- T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef PubMed.
- S. Maity, E. Hoque, U. Dhawa and D. Maiti, Chem. Commun., 2016, 52, 14003–14006 RSC.
- F. Hu and M. Szostak, Org. Lett., 2016, 18, 4186–4189 CrossRef PubMed.
- S. J. Pastine, D. V. Gribkov and D. Sames, J. Am. Chem. Soc., 2006, 128, 14220–14221 CrossRef PubMed.
- W. Xu and N. Yoshikai, Chem. Sci., 2017, 8, 5299–5304 RSC.
- M. Y. Wong, T. Yamakawa and N. Yoshikai, Org. Lett., 2015, 17, 442–445 CrossRef PubMed.
- K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 400–402 CrossRef PubMed.
- W. Xu, J. H. Pek and N. Yoshikai, Adv. Synth. Catal., 2016, 358, 2564–2568 CrossRef.
- W. Xu and N. Yoshikai, Angew. Chem., Int. Ed., 2016, 55, 12731–12735 CrossRef PubMed.
- R. Pollice, N. Dastbaravardeh, N. Marquise, M. D. Mihovilovic and M. Schnürch, ACS Catal., 2014, 5, 587–595 CrossRef PubMed.
- H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 6339–6342 CrossRef PubMed.
- S.-S. Zhang, X.-G. Liu, S.-Y. Chen, D.-H. Tan, C.-Y. Jiang, J.-Q. Wu, Q. Li and H. Wang, Adv. Synth. Catal., 2016, 358, 1705–1710 CrossRef.
- Q. Lu, S. Gressies, S. Cembellin, F. J. R. Klauck, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12778–12782 CrossRef PubMed.
- J. H. Kim, S. Gressies and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5577–5581 CrossRef PubMed.
- J. Wang, S. Zha, K. Chen, F. Zhang, C. Song and J. Zhu, Org. Lett., 2016, 18, 2062–2065 CrossRef PubMed.
- X. Wang and N. Jiao, Org. Lett., 2016, 18, 2150–2153 CrossRef PubMed.
- X. Jin, X. Yang, Y. Yang and C. Wang, Org. Chem. Front., 2016, 3, 268–272 RSC.
- N. Lv, Z. Chen, Y. Liu, Z. Liu and Y. Zhang, Org. Lett., 2017, 19, 2588–2591 CrossRef PubMed.
- Y. Yi, H. Lee and C.-H. Jun, Chem. Commun., 2016, 52, 10171–10174 RSC.
- S. Raju, P. Annamalai, P.-L. Chen, Y.-H. Liu and S.-C. Chuang, Org. Lett., 2017, 19, 4134–4137 CrossRef PubMed.
- C.-Z. Luo, J. Jayakumar, P. Gandeepan, Y.-C. Wu and C.-H. Cheng, Org. Lett., 2015, 17, 924–927 CrossRef PubMed.
- J. Jayakumar and C.-H. Cheng, Chem. – Eur. J., 2016, 22, 1800–1804 CrossRef PubMed.
- C. Lin, Z. Chen, Z. Liu and Y. Zhang, Org. Lett., 2017, 19, 850–853 CrossRef PubMed.
- S.-J. Lou, Y.-J. Mao, D.-Q. Xu, J.-Q. He, Q. Chen and Z.-Y. Xu, ACS Catal., 2016, 6, 3890–3894 CrossRef.
- D. M. Reddy, S.-C. Wang, K. Du and C.-F. Lee, J. Org. Chem., 2017, 82, 10070–10076 CrossRef PubMed.
- J. Peng, C. Chen and C. Xi, Chem. Sci., 2016, 7, 1383–1387 RSC.
- H. Wang, S. Yu, Z. Qi and X. Li, Org. Lett., 2015, 17, 2812–2815 CrossRef PubMed.
- X. Zhou, S. Yu, L. Kong and X. Li, ACS Catal., 2016, 6, 647–651 CrossRef.
- T. Nanjo, C. Tsukano and Y. Takernoto, Chem. Pharm. Bull., 2016, 64, 1384–1392 CrossRef PubMed.
- J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863–870 CrossRef PubMed.
- K. Guo, X. Chen, M. Guan and Y. Zhao, Org. Lett., 2015, 17, 1802–1805 CrossRef PubMed.
- M. Sen, D. Kalsi and B. Sundararaju, Chem. – Eur. J., 2015, 21, 15529–15533 CrossRef PubMed.
- H. Wang, J. Koeller, W. Liu and L. Ackermann, Chem. – Eur. J., 2015, 21, 15525–15528 CrossRef PubMed.
- K. Muralirajan, R. Kuppusamy, S. Prakash and C.-H. Cheng, Adv. Synth. Catal., 2016, 358, 774–783 CrossRef.
- L. Zheng, Y. Bin, Y. Wang and R. Hua, J. Org. Chem., 2016, 81, 8911–8919 CrossRef PubMed.
- Y.-F. Liang, X. Li, X. Wang, Y. Yan, P. Feng and N. Jiao, ACS Catal., 2015, 5, 1956–1963 CrossRef.
- Y.-F. Liang, X. Wang, C. Tang, T. Shen, J. Liu and N. Jiao, Chem. Commun., 2016, 52, 1416–1419 RSC.
- J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490–498 CrossRef PubMed.
- Z. Ren, J. E. Schulz and G. Dong, Org. Lett., 2015, 17, 2696–2699 CrossRef PubMed.
- L.-Y. Shao, C. Li, Y. Guo, K.-K. Yu, F.-Y. Zhao, W.-L. Qiao, H.-W. Liu, D.-H. Liao and Y.-F. Ji, RSC Adv., 2016, 6, 78875–78880 RSC.
- Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293–3298 CrossRef PubMed.
- S. J. Thompson, D. Q. Thach and G. Dong, J. Am. Chem. Soc., 2015, 137, 11586–11589 CrossRef PubMed.
- Y.-F. Liang, X. Wang, Y. Yuan, Y. Liang, X. Li and N. Jiao, ACS Catal., 2015, 5, 6148–6152 CrossRef.
- M. Zou, J. Liu, C. Tang and N. Jiao, Org. Lett., 2016, 18, 3030–3033 CrossRef PubMed.
- H. Wang, G. Tang and X. Li, Angew. Chem., Int. Ed., 2015, 54, 13049–13052 CrossRef PubMed.
- S. Yu, B. Wan and X. Li, Org. Lett., 2015, 17, 58–61 CrossRef PubMed.
- K. Muralirajan, R. Haridharan, S. Prakash and C.-H. Cheng, Adv. Synth. Catal., 2015, 357, 761–766 CrossRef.
- S. Zhou, J. Wang, F. Zhang, C. Song and J. Zhu, Org. Lett., 2016, 18, 2427–2430 CrossRef PubMed.
- Z. Yang, X. Lin, L. Wang and X. Cui, Org. Chem. Front., 2017, 4, 2179–2183 RSC.
- B. V. S. Reddy, C. R. Reddy, M. R. Reddy, S. Yarlagadda and B. Sridhar, Org. Lett., 2015, 17, 3730–3733 CrossRef PubMed.
- P. Shi, L. Wang, S. Guo, K. Chen, J. Wang and J. Zhu, Org. Lett., 2017, 19, 4359–4362 CrossRef PubMed.
- A. Lerchen, S. Vasquez-Cespedes and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3208–3211 CrossRef PubMed.
- B. Yu, Y. Chen, M. Hong, P. Duan, S. Gan, H. Chao, Z. Zhao and J. Zhao, Chem. Commun., 2015, 51, 14365–14368 RSC.
- J. Wang, S. Zha, K. Chen and J. Zhu, Org. Chem. Front., 2016, 3, 1281–1285 RSC.
- S. Zhang, D. Huang, G. Xu, S. Cao, R. Wang, S. Peng and J. Sun, Org. Biomol. Chem., 2015, 13, 7920–7923 RSC.
- Z. Huang, C. Wang and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 5299–5303 CrossRef PubMed.
- J. Wen, A. Wu, Y. Miao and J. Zhu, Tetrahedron Lett., 2015, 56, 5512–5516 CrossRef.
- Y. Yu, C. Kuai, R. Chauvin, N. Tian, S. Ma and X. Cui, J. Org. Chem., 2017, 82, 8611–8616 CrossRef PubMed.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef PubMed.
- M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896–9898 CrossRef PubMed.
- G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef PubMed.
- H. Tang, X.-R. Huang, J. Yao and H. Chen, J. Org. Chem., 2015, 80, 4672–4682 CrossRef PubMed.
- O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef PubMed.
- R. K. Rit, M. R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450–4459 CrossRef.
- X. Yang, G. Shan, L. Wang and Y. Rao, Tetrahedron Lett., 2016, 57, 819–836 CrossRef.
- Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222–234 CrossRef PubMed.
- Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC.
- C. Zhong and X. Shi, Eur. J. Org. Chem., 2010, 2999–3025 CrossRef.
- Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef PubMed.
- D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef PubMed.
- S. Afewerki and A. Córdova, Chem. Rev., 2016, 116, 13512–13570 CrossRef PubMed.
- Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337–1378 RSC.
- H. Sun, N. Guimond and Y. Huang, Org. Biomol. Chem., 2016, 14, 8389–8397 RSC.
- P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 Search PubMed.
- Y. Dou, Z. Xie, Z. Sun, H. Fang, C. Shen, P. Zhang and Q. Zhu, ChemCatChem, 2016, 8, 3570–3574 CrossRef.
- C. J. Whiteoak, O. Planas, A. Company and X. Ribas, Adv. Synth. Catal., 2016, 358, 1679–1688 CrossRef.
- Y. Yin, J. Xie, F.-Q. Huang, L.-W. Qi and B. Zhang, Adv. Synth. Catal., 2017, 359, 1037–1042 CrossRef.
- M. D. Reddy, F. R. Fronczek and E. B. Watkins, Org. Lett., 2016, 18, 5620–5623 CrossRef PubMed.
- J.-M. Li, J. Weng, G. Lu and A. S. C. Chan, Tetrahedron Lett., 2016, 57, 2121–2124 CrossRef.
- J. Wei, J. Jiang, X. Xiao, D. Lin, Y. Deng, Z. Ke, H. Jiang and W. Zeng, J. Org. Chem., 2016, 81, 946–955 CrossRef PubMed.
- H. Chen, P. Li, M. Wang and L. Wang, Org. Lett., 2016, 18, 4794–4797 CrossRef PubMed.
- H. Qiao, S. Sun, F. Yang, Y. Zhu, W. Zhu, Y. Dong, Y. Wu, X. Kong, L. Jiang and Y. Wu, Org. Lett., 2015, 17, 6086–6089 CrossRef PubMed.
- Y. Kuninobu, M. Nishi and M. Kanai, Org. Biomol. Chem., 2016, 14, 8092–8100 RSC.
- L.-K. Jin, G.-P. Lu and C. Cai, Org. Chem. Front., 2016, 3, 1309–1313 RSC.
- R. Parella and S. A. Babu, J. Org. Chem., 2015, 80, 2339–2355 CrossRef PubMed.
- B. Gopalakrishnan, S. A. Babu and R. Padmavathi, Tetrahedron, 2015, 71, 8333–8349 CrossRef.
- R. Parella and S. A. Babu, J. Org. Chem., 2015, 80, 12379–12396 CrossRef PubMed.
- H.-Y. Xiong, T. Besset, D. Cahard and X. Pannecoucke, J. Org. Chem., 2015, 80, 4204–4212 CrossRef PubMed.
- X. Cheng, Z. Chen, Y. Gao, F. Xue and C. Jiang, Org. Biomol. Chem., 2016, 14, 3298–3306 RSC.
- L. Grigorjeva and O. Daugulis, Org. Lett., 2015, 17, 1204–1207 CrossRef PubMed.
- S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2015, 137, 531–539 CrossRef PubMed.
- B. K. Singh and R. Jana, J. Org. Chem., 2016, 81, 831–841 CrossRef PubMed.
- D. P. Affron and J. A. Bull, Eur. J. Org. Chem., 2016, 139–149 CrossRef PubMed.
- Q. Gui, X. Chen, L. Hu, D. Wang, J. Liu and Z. Tan, Adv. Synth. Catal., 2016, 358, 509–514 CrossRef.
- L. Hu, Q. Gui, X. Chen, Z. Tan and G. Zhu, Org. Biomol. Chem., 2016, 14, 11070–11075 RSC.
- L. Hu, X. Chen, Q. Gui, Z. Tan and G. Zhu, Chem. Commun., 2016, 52, 6845–6848 RSC.
- L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237–18240 CrossRef PubMed.
- C. Li, D. Zhang, W. Zhu, P. Wan and H. Liu, Org. Chem. Front., 2016, 3, 1080–1083 RSC.
- A. Chabrier, A. Bruneau, S. Benmahdjoub, B. Benmerad, S. Belaid, J.-D. Brion, M. Alami and S. Messaoudi, Chem. – Eur. J., 2016, 22, 15006–15010 CrossRef PubMed.
- Y. Liu, K. Yang and H. Ge, Chem. Sci., 2016, 7, 2804–2808 RSC.
- O. Verho, M. Pourghasemi Lati and M. Oschmann, J. Org. Chem., 2018, 83, 4464–4476 CrossRef PubMed.
- B. B. Yann, G. Corinne and D. Grégory, Chem. – Eur. J., 2017, 23, 10043–10047 CrossRef PubMed.
- M. Berger, R. Chauhan, C. A. B. Rodrigues and N. Maulide, Chem. – Eur. J., 2016, 22, 16805–16808 CrossRef PubMed.
- T. Deguchi, H.-L. Xin, H. Morimoto and T. Ohshima, ACS Catal., 2017, 7, 3157–3161 CrossRef.
- S. A. Ruider and N. Maulide, Angew. Chem., Int. Ed., 2015, 54, 13856–13858 CrossRef PubMed.
- R. Padmavathi, R. Sankar, B. Gopalakrishnan, R. Parella and S. A. Babu, Eur. J. Org. Chem., 2015, 3727–3742 CrossRef.
- N. Bisht and S. A. Babu, Tetrahedron, 2016, 72, 5886–5897 CrossRef.
- M. Sattar, Praveen, C. Durga Prasad, A. Verma, S. Kumar and S. Kumar, Adv. Synth. Catal., 2016, 358, 240–253 CrossRef.
- H. Qiao, S. Sun, F. Yang, Y. Zhu, W. Zhu, Y. Wu and Y. Wu, RSC Adv., 2016, 6, 59319–59326 RSC.
- C. Shan, X. Luo, X. Qi, S. Liu, Y. Li and Y. Lan, Organometallics, 2016, 35, 1440–1445 CrossRef.
- Y. Xie, D. Xu, W.-W. Sun, S.-J. Zhang, X.-P. Dong, B. Liu, Y. Zhou and B. Wu, Asian J. Org. Chem., 2016, 5, 961–965 CrossRef.
- C. Du, P.-X. Li, X. Zhu, J.-F. Suo, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2016, 55, 13571–13575 CrossRef PubMed.
- K. Takamatsu, K. Hirano and M. Miura, Angew. Chem., Int. Ed., 2017, 56, 5353–5357 CrossRef PubMed.
- T.-Y. Zhang, J.-B. Lin, Q.-Z. Li, J.-C. Kang, J.-L. Pan, S.-H. Hou, C. Chen and S.-Y. Zhang, Org. Lett., 2017, 19, 1764–1767 CrossRef PubMed.
- Y. Liu, Y. Zhang, M. Huang and J.-P. Wan, RSC Adv., 2015, 5, 46192–46196 RSC.
- Y. Aihara, J. Wuelbern and N. Chatani, Bull. Chem. Soc. Jpn., 2015, 88, 438–446 CrossRef.
- L. Ilies, S. Ichikawa, S. Asako, T. Matsubara and E. Nakamura, Adv. Synth. Catal., 2015, 357, 2175–2179 CrossRef.
- R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2015, 137, 7660–7663 CrossRef PubMed.
- H. Wang, S. Zhang, Z. Wang, M. He and K. Xu, Org. Lett., 2016, 18, 5628–5631 CrossRef PubMed.
- K. Xu, Z. Tan, H. Zhang and S. Zhang, Synthesis, 2017, 3931–3936 CrossRef.
- T. Kubo and N. Chatani, Org. Lett., 2016, 18, 1698–1701 CrossRef PubMed.
- J. Singh, M. Deb and A. J. Elias, Organometallics, 2015, 34, 4946–4951 CrossRef.
- D. Schmiel and H. Butenschoen, Eur. J. Org. Chem., 2017, 3041–3048 CrossRef.
- K. Shibata and N. Chatani, Chem. Sci., 2016, 7, 240–245 RSC.
- P. Gandeepan, P. Rajamalli and C.-H. Cheng, Angew. Chem., Int. Ed., 2016, 55, 4308–4311 CrossRef PubMed.
- K. Shibata, T. Yamaguchi and N. Chatani, Org. Lett., 2015, 17, 3584–3587 CrossRef PubMed.
- W. Ma and L. Ackermann, ACS Catal., 2015, 5, 2822–2825 CrossRef.
- W. Miura, K. Hirano and M. Miura, Org. Lett., 2015, 17, 4034–4037 CrossRef PubMed.
- R. Manoharan and M. Jeganmohan, Org. Lett., 2017, 19, 5884–5887 CrossRef PubMed.
- Q. He, T. Yamaguchi and N. Chatani, Org. Lett., 2017, 19, 4544–4547 CrossRef PubMed.
- N. Barsu, D. Kalsi and B. Sundararaju, Chem. – Eur. J., 2015, 21, 9364–9368 CrossRef PubMed.
- S. Maity, R. Kancherla, U. Dhawa, E. Hoque, S. Pimparkar and D. Maiti, ACS Catal., 2016, 6, 5493–5499 CrossRef.
- R. Manoharan, G. Sivakumar and M. Jeganmohan, Chem. Commun., 2016, 52, 10533–10536 RSC.
- T. Yamaguchi, Y. Kommagalla, Y. Aihara and N. Chatani, Chem. Commun., 2016, 52, 10129–10132 RSC.
- H.-W. Liang, W. Ding, K. Jiang, L. Shuai, Y. Yuan, Y. Wei and Y.-C. Chen, Org. Lett., 2015, 17, 2764–2767 CrossRef PubMed.
- R. Boobalan, R. Kuppusamy, R. Santhoshkumar, P. Gandeepan and C.-H. Cheng, ChemCatChem, 2017, 9, 273–277 CrossRef.
- A. Deb, A. Hazra, Q. Peng, R. S. Paton and D. Maiti, J. Am. Chem. Soc., 2017, 139, 763–775 CrossRef PubMed.
- M. Liu, Y. Niu, Y.-F. Wu and X.-S. Ye, Org. Lett., 2016, 18, 1836–1839 CrossRef PubMed.
- K. Shibata, S. Natsui and N. Chatani, Org. Lett., 2017, 19, 2234–2237 CrossRef PubMed.
- C. Shao, G. Shi and Y. Zhang, Eur. J. Org. Chem., 2016, 5529–5538 CrossRef.
- J. Yi, L. Yang, C. Xia and F. Li, J. Org. Chem., 2015, 80, 6213–6221 CrossRef PubMed.
- V. G. Landge, C. H. Shewale, G. Jaiswal, M. K. Sahoo, S. P. Midya and E. Balaraman, Catal. Sci. Technol., 2016, 6, 1946–1951 RSC.
- V. G. Landge, G. Jaiswal and E. Balaraman, Org. Lett., 2016, 18, 812–815 CrossRef PubMed.
- L. C. Misal Castro, A. Obata, Y. Aihara and N. Chatani, Chem. – Eur. J., 2016, 22, 1362–1367 CrossRef PubMed.
- Z. He and Y. Huang, ACS Catal., 2016, 6, 7814–7823 CrossRef.
- S. Kathiravan and I. A. Nicholls, Org. Lett., 2017, 19, 4758–4761 CrossRef PubMed.
- T. Matsubara, L. Ilies and E. Nakamura, Chem. – Asian J., 2016, 11, 380–384 CrossRef PubMed.
- K. Takamatsu, K. Hirano and M. Miura, Org. Lett., 2015, 17, 4066–4069 CrossRef PubMed.
- W. Hao, J. Tian, W. Li, R. Shi, Z. Huang and A. Lei, Chem. – Asian J., 2016, 11, 1664–1667 CrossRef PubMed.
- F. Zou, X. Chen and W. Hao, Tetrahedron, 2017, 73, 758–763 CrossRef.
- D. Kalsi, N. Barsu, P. Dahiya and B. Sundararaju, Synthesis, 2017, 3937–3944 Search PubMed.
- Z.-Y. Gu, C.-G. Liu, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2017, 82, 2223–2230 CrossRef PubMed.
- L. Yu, H. Huang, X. Chen, L. Hu, Y. Yu and Z. Tan, Chem. Commun., 2017, 53, 4597–4600 RSC.
- X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2015, 137, 4924–4927 CrossRef PubMed.
- B. Khan, A. Ali Khan, R. Kant and D. Koley, Adv. Synth. Catal., 2016, 358, 3753–3758 CrossRef.
- J. Ni, J. Li, Z. Fan and A. Zhang, Org. Lett., 2016, 18, 5960–5963 CrossRef PubMed.
- T. T. Nguyen, L. Grigorjeva and O. Daugulis, Chem. Commun., 2017, 53, 5136–5138 RSC.
- B. Khan, R. Kant and D. Koley, Adv. Synth. Catal., 2016, 358, 2352–2358 CrossRef.
- Q. Yan, Z. Chen, W. Yu, H. Yin, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 2482–2485 CrossRef PubMed.
- J. Roane and O. Daugulis, J. Am. Chem. Soc., 2016, 138, 4601–4607 CrossRef PubMed.
- B. K. Singh, A. Polley and R. Jana, J. Org. Chem., 2016, 81, 4295–4303 CrossRef PubMed.
- P. Sadhu and T. Punniyamurthy, Chem. Commun., 2016, 52, 2803–2806 RSC.
- C. Du, P.-X. Li, X. Zhu, J.-N. Han, J.-L. Niu and M.-P. Song, ACS Catal., 2017, 7, 2810–2814 CrossRef.
- Z. Li, H. Yu and C. Bolm, Angew. Chem., Int. Ed., 2017, 56, 9532–9535 CrossRef PubMed.
- J. Liu, S. Zhuang, Q. Gui, X. Chen, Z. Yang and Z. Tan, Adv. Synth. Catal., 2015, 357, 732–738 CrossRef.
- X.-S. Yin, Y.-C. Li, J. Yuan, W.-J. Gu and B.-F. Shi, Org. Chem. Front., 2015, 2, 119–123 RSC.
- C. Lin, D. Li, B. Wang, J. Yao and Y. Zhang, Org. Lett., 2015, 17, 1328–1331 CrossRef PubMed.
- V. P. Reddy, R. Qiu, T. Iwasaki and N. Kambe, Org. Biomol. Chem., 2015, 13, 6803–6813 RSC.
- J. Liu, L. Yu, S. Zhuang, Q. Gui, X. Chen, W. Wang and Z. Tan, Chem. Commun., 2015, 51, 6418–6421 RSC.
- Q. Zhao, T. Poisson, X. Pannecoucke, J.-P. Bouillon and T. Besset, Org. Lett., 2017, 19, 5106–5109 CrossRef PubMed.
- Y. Liu, M. Huang and L. Wei, Asian J. Org. Chem., 2017, 6, 41–43 CrossRef.
- A. Mandal, H. Sahoo and M. Baidya, Org. Lett., 2016, 18, 3202–3205 CrossRef PubMed.
- M. Iwasaki, N. Miki, Y. Tsuchiya, K. Nakajima and Y. Nishihara, Org. Lett., 2017, 19, 1092–1095 CrossRef PubMed.
- J.-L. Pan, C. Chen, Z.-G. Ma, J. Zhou, L.-R. Wang and S.-Y. Zhang, Org. Lett., 2017, 19, 5216–5219 CrossRef PubMed.
- X. Wu, Y. Zhao and H. Ge, Chem. Sci., 2015, 6, 5978–5983 RSC.
- Q. Gou, Z.-F. Zhang, Z.-C. Liu and J. Qin, J. Org. Chem., 2015, 80, 3176–3186 CrossRef PubMed.
- K. Chen, X. Li, S.-Q. Zhang and B.-F. Shi, Org. Chem. Front., 2016, 3, 204–208 RSC.
- S. Mohan, B. Gopalakrishnan and S. A. Babu, Tetrahedron, 2016, 72, 5853–5863 CrossRef.
- Y. Liu, Y. Zhang, X. Cao and J.-P. Wan, Beilstein J. Org. Chem., 2016, 12, 1122–1126 CrossRef PubMed.
- M. D. Reddy and E. B. Watkins, J. Org. Chem., 2015, 80, 11447–11459 CrossRef PubMed.
- P. Hu and T. Bach, Synlett, 2015, 2853–2857 Search PubMed.
- X. Wang, L. Zhu, S. Chen, X. Xu, C.-T. Au and R. Qiu, Org. Lett., 2015, 17, 5228–5231 CrossRef PubMed.
- X. Wang, P. Xie, R. Qiu, L. Zhu, T. Liu, Y. Li, T. Iwasaki, C.-T. Au, X. Xu, Y. Xia, S.-F. Yin and N. Kambe, Chem. Commun., 2017, 53, 8316–8319 RSC.
- H. M. Omer and P. Liu, J. Am. Chem. Soc., 2017, 139, 9909–9920 CrossRef PubMed.
- S. Singh, K. Surya and R. B. Sunoj, J. Org. Chem., 2017, 82, 9619–9626 CrossRef PubMed.
- A. Dey, S. Pimparkar, A. Deb, S. Guin and D. Maiti, Adv. Synth. Catal., 2017, 359, 1301–1307 CrossRef.
- R. Parella and S. A. Babu, J. Org. Chem., 2017, 82, 7123–7150 CrossRef PubMed.
- W. A. Nack, B. Wang, X. Wu, R. Jiao, G. He and G. Chen, Org. Chem. Front., 2016, 3, 561–564 RSC.
- N. Hoshiya, M. Kondo, H. Fukuda, M. Arisawa, J. I. Uenishi and S. Shuto, J. Org. Chem., 2017, 82, 2535–2544 CrossRef PubMed.
- X. Yang, G. Shan, Z. Yang, G. Huang, G. Dong, C. Sheng and Y. Rao, Chem. Commun., 2017, 53, 1534–1537 RSC.
- S.-B. Yan, S. Zhang and W.-L. Duan, Org. Lett., 2015, 17, 2458–2461 CrossRef PubMed.
- R. Feng, B. Wang, Y. Liu, Z. Liu and Y. Zhang, Eur. J. Org. Chem., 2015, 142–151 CrossRef.
- Q.-Y. Yu, H.-M. Zhong, W.-W. Sun, S.-J. Zhang, P. Cao, X.-P. Dong, H.-B. Qin, J.-K. Liu and B. Wu, Asian J. Org. Chem., 2016, 5, 608–612 CrossRef.
- B. Mondal, B. Roy and U. Kazmaier, J. Org. Chem., 2016, 81, 11646–11655 CrossRef PubMed.
- Z.-Z. Zhang, Y.-Q. Han, B.-B. Zhan, S. Wang and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 13145–13149 CrossRef PubMed.
- K. Chen, X. Li, S.-Q. Zhang and B.-F. Shi, Chem. Commun., 2016, 52, 1915–1918 RSC.
- N. Hoshiya, K. Takenaka, S. Shuto and J.-I. Uenishi, Org. Lett., 2016, 18, 48–51 CrossRef PubMed.
- Q. Yang and S.-D. Yang, ACS Catal., 2017, 7, 5220–5224 CrossRef.
- G. Liao, X.-S. Yin, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Nat. Commun., 2016, 7, 12901 CrossRef PubMed.
- G. Shan, G. Huang and Y. Rao, Org. Biomol. Chem., 2015, 13, 697–701 RSC.
- M. Li, Y. Yang, D. Zhou, D. Wan and J. You, Org. Lett., 2015, 17, 2546–2549 CrossRef PubMed.
- S. Maity, S. Agasti, A. M. Earsad, A. Hazra and D. Maiti, Chem. – Eur. J., 2015, 21, 11320–11324 CrossRef PubMed.
- L. Ilies, Y. Itabashi, R. Shang and E. Nakamura, ACS Catal., 2017, 7, 89–92 CrossRef.
- J. Zhang, H. Chen, C. Lin, Z. Liu, C. Wang and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 12990–12996 CrossRef PubMed.
- J. Zhang, D. Li, H. Chen, B. Wang, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2016, 358, 792–807 CrossRef.
- F.-X. Luo, Z.-C. Cao, H.-W. Zhao, D. Wang, Y.-F. Zhang, X. Xu and Z.-J. Shi, Organometallics, 2017, 36, 18–21 CrossRef.
- P. Williamson, A. Galvan and M. J. Gaunt, Chem. Sci., 2017, 8, 2588–2591 RSC.
- N. Barsu, S. K. Bolli and B. Sundararaju, Chem. Sci., 2017, 8, 2431–2435 RSC.
- L. Zeng, S. Tang, D. Wang, Y. Deng, J.-L. Chen, J.-F. Lee and A. Lei, Org. Lett., 2017, 19, 2170–2173 CrossRef PubMed.
- H.-Y. Xiong, D. Cahard, X. Pannecoucke and T. Besset, Eur. J. Org. Chem., 2016, 3625–3630 CrossRef.
- R.-Y. Zhu, K. Tanaka, G.-C. Li, J. He, H.-Y. Fu, S.-H. Li and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 7067–7070 CrossRef PubMed.
- Q. Zhang, X.-S. Yin, K. Chen, S.-Q. Zhang and B.-F. Shi, J. Am. Chem. Soc., 2015, 137, 8219–8226 CrossRef PubMed.
- Q. Zhu, D. Ji, T. Liang, X. Wang and Y. Xu, Org. Lett., 2015, 17, 3798–3801 CrossRef PubMed.
- H. Sun, Y. Zhang, P. Chen, Y.-D. Wu, X. Zhang and Y. Huang, Adv. Synth. Catal., 2016, 358, 1946–1957 CrossRef.
- J. Hu, T. Lan, Y. Sun, H. Chen, J. Yao and Y. Rao, Chem. Commun., 2015, 51, 14929–14932 RSC.
- K. Chen, S.-Q. Zhang, H.-Z. Jiang, J.-W. Xu and B.-F. Shi, Chem. – Eur. J., 2015, 21, 3264–3270 CrossRef PubMed.
- X. Yang, T.-Y. Sun and Y. Rao, Chem. – Eur. J., 2016, 22, 3273–3277 CrossRef PubMed.
- C. Wang, L. Zhang and J. You, Org. Lett., 2017, 19, 1690–1693 CrossRef PubMed.
- C. Wang, Y. Yang, D. Qin, Z. He and J. You, J. Org. Chem., 2015, 80, 8424–8429 CrossRef PubMed.
- S.-J. Zhang, W.-W. Sun, P. Cao, X.-P. Dong, J.-K. Liu and B. Wu, J. Org. Chem., 2016, 81, 956–968 CrossRef PubMed.
- C. Yamamoto, K. Takamatsu, K. Hirano and M. Miura, J. Org. Chem., 2016, 81, 7675–7684 CrossRef PubMed.
- Q. Gou, Y.-W. Yang, Z.-N. Liu and J. Qin, Chem. – Eur. J., 2016, 22, 16057–16061 CrossRef PubMed.
- C. Wang and Y. Yang, Tetrahedron Lett., 2017, 58, 935–940 CrossRef.
- X. Ye, J. L. Petersen and X. Shi, Chem. Commun., 2015, 51, 7863–7866 RSC.
- C. Lin, W. Yu, J. Yao, B. Wang, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 1340–1343 CrossRef PubMed.
- S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu, Z.-Z. Zhang and B.-F. Shi, Chem. Commun., 2015, 51, 7341–7344 RSC.
- X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S.-F. Yin, Org. Lett., 2015, 17, 1970–1973 CrossRef PubMed.
- W.-H. Rao, B.-B. Zhan, K. Chen, P.-X. Ling, Z.-Z. Zhang and B.-F. Shi, Org. Lett., 2015, 17, 3552–3555 CrossRef PubMed.
- J.-L. Pan, Q.-Z. Li, T.-Y. Zhang, S.-H. Hou, J.-C. Kang and S.-Y. Zhang, Chem. Commun., 2016, 52, 13151–13154 RSC.
- Y.-J. Liu, Y.-H. Liu, Z.-Z. Zhang, S.-Y. Yan, K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2016, 55, 13859–13862 CrossRef PubMed.
- A. Deb, S. Singh, K. Seth, S. Pimparkar, B. Bhaskararao, S. Guin, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 8171–8175 CrossRef.
- A. Yokota and N. Chatani, Chem. Lett., 2015, 44, 902–904 CrossRef.
- M. L. Czyz, D. W. Lupton and A. Polyzos, Chem. – Eur. J., 2017, 23, 14450–14453 CrossRef PubMed.
- T. Kubo, Y. Aihara and N. Chatani, Chem. Lett., 2015, 44, 1365–1367 CrossRef.
- X. Yang, Y. Sun, T.-Y. Sun and Y. Rao, Chem. Commun., 2016, 52, 6423–6426 RSC.
- Y. Aihara and N. Chatani, ACS Catal., 2016, 6, 4323–4329 CrossRef.
- P.-X. Ling, S.-L. Fang, X.-S. Yin, Q. Zhang, K. Chen and B.-F. Shi, Chem. Commun., 2017, 53, 6351–6354 RSC.
- H. Gang, Z. Shu-Yu, W. A. Nack, L. Qiong and C. Gong, Angew. Chem., Int. Ed., 2013, 52, 11124–11128 CrossRef PubMed.
- O. Planas, C. J. Whiteoak, A. Company and X. Ribas, Adv. Synth. Catal., 2015, 357, 4003–4012 CrossRef.
- D. Kalsi and B. Sundararaju, Org. Lett., 2015, 17, 6118–6121 CrossRef PubMed.
- Y. Ran, Y. Yang and L. Zhang, Tetrahedron Lett., 2016, 57, 3322–3325 CrossRef.
- M. D. Reddy, A. N. Blanton and E. B. Watkins, J. Org. Chem., 2017, 82, 5080–5095 CrossRef PubMed.
- F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187–4192 RSC.
- Q. Zhang, K. Chen, W. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588–13592 CrossRef PubMed.
- S. Zhao, B. Liu, B.-B. Zhan, W.-D. Zhang and B.-F. Shi, Org. Lett., 2016, 18, 4586–4589 CrossRef PubMed.
- S. Zhao, Y.-J. Liu, S.-Y. Yan, F.-J. Chen, Z.-Z. Zhang and B.-F. Shi, Org. Lett., 2015, 17, 3338–3341 CrossRef PubMed.
- Y.-J. Liu, Y.-H. Liu, X.-S. Yin, W.-J. Gu and B.-F. Shi, Chem. – Eur. J., 2015, 21, 205–209 CrossRef PubMed.
- Y.-J. Liu, Y.-H. Liu, S.-Y. Yan and B.-F. Shi, Chem. Commun., 2015, 51, 6388–6391 RSC.
- W.-H. Rao and B.-F. Shi, Org. Lett., 2015, 17, 2784–2787 CrossRef PubMed.
- H. Wang, Y. Niu, G. Zhang and X.-S. Ye, Tetrahedron Lett., 2016, 57, 4544–4548 CrossRef.
- B.-B. Zhan, Y.-H. Liu, F. Hu and B.-F. Shi, Chem. Commun., 2016, 52, 4934–4937 RSC.
- K. Yang, Y. Wang, X. Chen, A. A. Kadi, H.-K. Fun, H. Sun, Y. Zhang and H. Lu, Chem. Commun., 2015, 51, 3582–3585 RSC.
- H.-Y. Bai, Z.-G. Ma, M. Yi, J.-B. Lin and S.-Y. Zhang, ACS Catal., 2017, 7, 2042–2046 CrossRef.
- B. Liu, Z.-Z. Zhang, X. Li and B.-F. Shi, Org. Chem. Front., 2016, 3, 897–900 RSC.
- S. Zhao, J. Yuan, Y.-C. Li and B.-F. Shi, Chem. Commun., 2015, 51, 12823–12826 RSC.
- D. Dailler, G. Danoun and O. Baudoin, Angew. Chem., Int. Ed., 2015, 54, 4919–4922 CrossRef PubMed.
- Q. Li, Y. Li, W. Hu, R. Hu, G. Li and H. Lu, Chem. – Eur. J., 2016, 22, 12286–12289 CrossRef PubMed.
- Q. Li, W. Hu, R. Hu, H. Lu and G. Li, Org. Lett., 2017, 19, 4676–4679 CrossRef PubMed.
- Y.-H. Liu, Y.-J. Liu, S.-Y. Yan and B.-F. Shi, Chem. Commun., 2015, 51, 11650–11653 RSC.
- S. Zhao, F.-J. Chen, B. Liu and B.-F. Shi, Sci. China: Chem., 2015, 58, 1302–1309 CrossRef.
- M. Iwasaki, M. Iyanaga, Y. Tsuchiya, Y. Nishimura, W. Li, Z. Li and Y. Nishihara, Chem. – Eur. J., 2014, 20, 2459–2462 CrossRef PubMed.
- Y. Yang, W. Hou, L. Qin, J. Du, H. Feng, B. Zhou and Y. Li, Chem. – Eur. J., 2014, 20, 416–420 CrossRef PubMed.
- S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 4069–4072 RSC.
- Y. Li, Y.-J. Liu and B.-F. Shi, Adv. Synth. Catal., 2017, 359, 4117–4121 CrossRef.
- S.-L. Liu, X.-H. Li, S.-S. Zhang, S.-K. Hou, G.-C. Yang, J.-F. Gong and M.-P. Song, Adv. Synth. Catal., 2017, 359, 2241–2246 CrossRef.
- B. Liu and B.-F. Shi, Synlett, 2016, 2396–2400 Search PubMed.
- J. Miao, K. Yang, M. Kurek and H. Ge, Org. Lett., 2015, 17, 3738–3741 CrossRef PubMed.
- X. Zhang, G. He and G. Chen, Org. Biomol. Chem., 2016, 14, 5511–5515 RSC.
- G. He, S.-Y. Zhang, W. A. Nack, R. Pearson, J. Rabb-Lynch and G. Chen, Org. Lett., 2014, 16, 6488–6491 CrossRef PubMed.
- M. Larrosa, S. Heiles, J. Becker, B. Spengler and R. Hrdina, Adv. Synth. Catal., 2016, 358, 2163–2171 CrossRef.
- S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124–2127 CrossRef PubMed.
- R. Odani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2013, 78, 11045–11052 CrossRef PubMed.
- L.-S. Zhang, G. Chen, X. Wang, Q.-Y. Guo, X.-S. Zhang, F. Pan, K. Chen and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 3899–3903 CrossRef PubMed.
- X. Wang, S. Niu, L. Xu, C. Zhang, L. Meng, X. Zhang and D. Ma, Org. Lett., 2017, 19, 246–249 CrossRef PubMed.
- J. Xu, L. Qiao, J. Shen, K. Chai, C. Shen and P. Zhang, Org. Lett., 2017, 19, 5661–5664 CrossRef PubMed.
- A. M. Martinez, J. Echavarren, I. Alonso, N. Rodriguez, R. Gomez Arrayas and J. C. Carretero, Chem. Sci., 2015, 6, 5802–5814 RSC.
- Á. M. Martínez, N. Rodríguez, R. Gómez-Arrayás and J. C. Carretero, Chem. – Eur. J., 2017, 23, 11669–11676 CrossRef PubMed.
- Z. Li, S. Sun, H. Qiao, F. Yang, Y. Zhu, J. Kang, Y. Wu and Y. Wu, Org. Lett., 2016, 18, 4594–4597 CrossRef PubMed.
- S. Pradhan, P. B. De and T. Punniyamurthy, J. Org. Chem., 2017, 82, 4883–4890 CrossRef PubMed.
- B. F. Van Steijvoort, N. Kaval, A. A. Kulago and B. U. W. Maes, ACS Catal., 2016, 6, 4486–4490 CrossRef.
- P.-L. Wang, Y. Li, Y. Wu, C. Li, Q. Lan and X.-S. Wang, Org. Lett., 2015, 17, 3698–3701 CrossRef PubMed.
- J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, J. Am. Chem. Soc., 2016, 138, 10750–10753 CrossRef PubMed.
- V. Rajkumar, Naveen and S. A. Babu, ChemistrySelect, 2016, 1, 1207–1219 CrossRef.
- R. Parella and S. A. Babu, J. Org. Chem., 2017, 82, 6550–6567 CrossRef PubMed.
- Z.-L. Zang, S. Karnakanti, S. Zhao, P. Hu, Z. Wang, P.-L. Shao and Y. He, Org. Lett., 2017, 19, 1354–1357 CrossRef PubMed.
- V. G. Landge, S. P. Midya, J. Rana, D. R. Shinde and E. Balaraman, Org. Lett., 2016, 18, 5252–5255 CrossRef PubMed.
- S. He, L. Xiaochong, W. Chenglong, Q. Jingyi, C. Wenyi and S. Zhizhong, Asian J. Org. Chem., 2017, 6, 1693–1698 CrossRef.
- J. Lan, H. Xie, X. Lu, Y. Deng, H. Jiang and W. Zeng, Org. Lett., 2017, 19, 4279–4282 CrossRef PubMed.
- Naveen, V. Rajkumar, S. A. Babu and B. Gopalakrishnan, J. Org. Chem., 2016, 81, 12197–12211 CrossRef PubMed.
- H. Sahoo, S. Mukherjee, G. S. Grandhi, J. Selvakumar and M. Baidya, J. Org. Chem., 2017, 82, 2764–2771 CrossRef PubMed.
- K. Takamatsu, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2015, 80, 3242–3249 CrossRef PubMed.
- C. Yamamoto, K. Takamatsu, K. Hirano and M. Miura, J. Org. Chem., 2017, 82, 9112–9118 CrossRef PubMed.
- D. Li, M. Yu, J. Zhang, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 5300–5303 CrossRef PubMed.
- Q. Gu, H. H. Al Mamari, K. Graczyk, E. Diers and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 3868–3871 CrossRef PubMed.
- K. Graczyk, T. Haven and L. Ackermann, Chem. – Eur. J., 2015, 21, 8812–8815 CrossRef PubMed.
- K. Graczyk, T. Haven and L. Ackermann, Chem. – Eur. J., 2015, 21, 8812–8815 CrossRef PubMed.
- W. Liu, G. Cera, J. C. A. Oliveira, Z. Shen and L. Ackermann, Chem. – Eur. J., 2017, 23, 11524–11528 CrossRef PubMed.
- X. Ye, C. Xu, L. Wojtas, N. G. Akhmedov, H. Chen and X. Shi, Org. Lett., 2016, 18, 2970–2973 CrossRef PubMed.
- G. Cera, T. Haven and L. Ackermann, Chem. – Eur. J., 2017, 23, 3577–3582 CrossRef PubMed.
- G. Cera, T. Haven and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 1484–1488 CrossRef PubMed.
- G. Cera, T. Haven and L. Ackermann, Chem. – Eur. J., 2017, 23, 3577–3582 CrossRef PubMed.
- X. Ye, Y. Zhang, Y. He and X. Shi, Tetrahedron, 2016, 72, 2756–2762 CrossRef.
- H.-L. Wang, M. Shang, S.-Z. Sun, Z.-L. Zhou, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Org. Lett., 2015, 17, 1228–1231 CrossRef PubMed.
- B. Liu, X. Huang, X. Wang, Z. Ge and R. Li, Org. Chem. Front., 2015, 2, 797–800 RSC.
- S.-Z. Sun, M. Shang, H.-L. Wang, H.-X. Lin, H.-X. Dai and J.-Q. Yu, J. Org. Chem., 2015, 80, 8843–8848 CrossRef PubMed.
- S. Liang, N.-W. Liu and G. Manolikakes, Adv. Synth. Catal., 2016, 358, 159–163 CrossRef.
- J. Liu, Z. Xue, Z. Zeng, Y.-X. Chen and G. Chen, Adv. Synth. Catal., 2016, 358, 3694–3699 CrossRef.
- J. Selvakumar, G. S. Grandhi, H. Sahoo and M. Baidya, RSC Adv., 2016, 6, 79361–79365 RSC.
- C. Reddy, N. Bisht, R. Parella and S. A. Babu, J. Org. Chem., 2016, 81, 12143–12168 CrossRef PubMed.
- S.-L. Liu, X.-H. Li, T.-H. Shi, G.-C. Yang, H.-L. Wang, J.-F. Gong and M.-P. Song, Eur. J. Org. Chem., 2017, 2280–2289 CrossRef.
- D. Mu, F. Gao, G. Chen and G. He, ACS Catal., 2017, 7, 1880–1885 CrossRef.
- S. Jerhaoui, F. Chahdoura, C. Rose, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2016, 22, 17397–17406 CrossRef PubMed.
- S. Jerhaoui, J. Wencel-Delord, F. Colobert and J.-P. Djukic, Chem. – Eur. J., 2017, 23, 15594–15600 CrossRef PubMed.
- B. Gopalakrishnan, S. Mohan, R. Parella and S. A. Babu, J. Org. Chem., 2016, 81, 8988–9005 CrossRef PubMed.
- S. Jerhaoui, F. Chahdoura, C. Rose, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2016, 22, 17397–17406 CrossRef PubMed.
- D. Mu, F. Gao, G. Chen and G. He, ACS Catal., 2017, 7, 1880–1885 CrossRef.
- H. Miura, S. Terajima, K. Tsutsui and T. Shishido, J. Org. Chem., 2017, 82, 1231–1239 CrossRef PubMed.
- R. Mei, H. Wang, S. Warratz, S. A. Macgregor and L. Ackermann, Chem. – Eur. J., 2016, 22, 6759–6763 CrossRef PubMed.
- L.-B. Zhang, X.-Q. Hao, S.-K. Zhang, Z.-J. Liu, X.-X. Zheng, J.-F. Gong, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2015, 54, 272–275 CrossRef PubMed.
- L.-B. Zhang, S.-K. Zhang, D. Wei, X. Zhu, X.-Q. Hao, J.-H. Su, J.-L. Niu and M.-P. Song, Org. Lett., 2016, 18, 1318–1321 CrossRef PubMed.
- J. Liu, Y. Xie, W. Zeng, D. Lin, Y. Deng and X. Lu, J. Org. Chem., 2015, 80, 4618–4626 CrossRef PubMed.
- S.-K. Zhang, X.-Y. Yang, X.-M. Zhao, P.-X. Li, J.-L. Niu and M.-P. Song, Organometallics, 2015, 34, 4331–4339 CrossRef.
- G. Tan, L. Zhang, X. Liao, Y. Shi, Y. Wu, Y. Yang and J. You, Org. Lett., 2017, 19, 4830–4833 CrossRef PubMed.
- R. Mei, H. Wang, S. Warratz, S. A. MacGregor and L. Ackermann, Chem. – Eur. J., 2016, 22, 6759–6763 CrossRef PubMed.
- X.-Q. Hao, C. Du, X. Zhu, P.-X. Li, J.-H. Zhang, J.-L. Niu and M.-P. Song, Org. Lett., 2016, 18, 3610–3613 CrossRef PubMed.
- X.-X. Zheng, C. Du, X.-M. Zhao, X. Zhu, J.-F. Suo, X.-Q. Hao, J.-L. Niu and M.-P. Song, J. Org. Chem., 2016, 81, 4002–4011 CrossRef PubMed.
- L.-B. Zhang, X.-Q. Hao, Z.-J. Liu, X.-X. Zheng, S.-K. Zhang, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2015, 54, 10012–10015 CrossRef PubMed.
- C. Wang, C. Chen, J. Zhang, J. Han, Q. Wang, K. Guo, P. Liu, M. Guan, Y. Yao and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 9884–9888 CrossRef PubMed.
- J. Hu, G. Li, C. Yuan, Z.-B. Huang, D.-Q. Shi and Y. Zhao, Org. Lett., 2016, 18, 5998–6001 CrossRef PubMed.
- L. Zhang, C. Wang, J. Han, Z.-B. Huang and Y. Zhao, J. Org. Chem., 2016, 81, 5256–5262 CrossRef PubMed.
- C. Chen, M. Guan, J. Zhang, Z. Wen and Y. Zhao, Org. Lett., 2015, 17, 3646–3649 CrossRef PubMed.
- C. Wang, L. Zhang, C. Chen, J. Han, Y. Yao and Y. Zhao, Chem. Sci., 2015, 6, 4610–4614 RSC.
- J. Han, N. Wang, Z.-B. Huang, Y. Zhao and D.-Q. Shi, J. Org. Chem., 2017, 82, 6831–6839 CrossRef PubMed.
- J. Han, L. Zhang, Y. Zhu, Y. Zheng, X. Chen, Z.-B. Huang, D.-Q. Shi and Y. Zhao, Chem. Commun., 2016, 52, 6903–6906 RSC.
- J. Han, N. Wang, Z.-B. Huang, Y. Zhao and D.-Q. Shi, Org. Biomol. Chem., 2017, 15, 5112–5116 RSC.
- P. Liu, J. Han, Q. Wang, Z. Huang, D. Shi, R. Zeng and Y. S. Zhao, RSC Adv., 2015, 5, 60646–60649 RSC.
- J. Han, Y. Zheng, C. Wang, Y. Zhu, D.-Q. Shi, R. Zeng, Z.-B. Huang and Y. Zhao, J. Org. Chem., 2015, 80, 9297–9306 CrossRef PubMed.
- J. Han, Y. Zheng, C. Wang, Y. Zhu, Z.-B. Huang, D.-Q. Shi, R. Zeng and Y. Zhao, J. Org. Chem., 2016, 81, 5681–5689 CrossRef PubMed.
- M. Wang, Y. Yang, Z. Fan, Z. Cheng, W. Zhu and A. Zhang, Chem. Commun., 2015, 51, 3219–3222 RSC.
- N. Chatani, Y. Ie, F. Kakiuchi and S. Murai, J. Org. Chem., 1997, 62, 2604–2610 CrossRef PubMed.
- N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935–10941 CrossRef PubMed.
- N. Chatani, S. Yorimitsu, T. Asaumi, F. Kakiuchi and S. Murai, J. Org. Chem., 2002, 67, 7557–7560 CrossRef PubMed.
- L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542–9543 CrossRef PubMed.
- A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300–2301 CrossRef PubMed.
- D. Shabashov and O. Daugulis, Org. Lett., 2005, 7, 3657–3659 CrossRef PubMed.
- S. Schröter, C. Stock and T. Bach, Tetrahedron, 2005, 61, 2245–2267 CrossRef.
- L.-C. Campeau and K. Fagnou, Chem. Soc. Rev., 2007, 36, 1058–1068 RSC.
- J. De Houwer and B. U. W. Maes, Synthesis, 2014, 2533–2550 Search PubMed.
- K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef PubMed.
- J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC.
- F. Juliá-Hernández, M. Simonetti and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 11458–11460 CrossRef PubMed.
- G. Meng and M. Szostak, ACS Catal., 2017, 7, 7251–7256 CrossRef.
- G. M. Reddy, N. S. S. Rao, P. Satyanarayana and H. Maheswaran, RSC Adv., 2015, 5, 105347–105352 RSC.
- Y. Kita, H. Tohma, K. Hatanaka, T. Takada, S. Fujita, S. Mitoh, H. Sakurai and S. Oka, J. Am. Chem. Soc., 1994, 116, 3684–3691 CrossRef.
- R. A. Jagtap, V. Soni and B. Punji, ChemSusChem, 2017, 10, 2242–2248 CrossRef PubMed.
- L. Su, D.-D. Guo, B. Li, S.-H. Guo, G.-F. Pan, Y.-R. Gao and Y.-Q. Wang, ChemCatChem, 2017, 9, 2001–2008 CrossRef.
- P. Nareddy, F. Jordan and M. Szostak, Org. Biomol. Chem., 2017, 15, 4783–4788 RSC.
- Y.-L. Zou, Z.-Y. Wang, Y.-M. Feng, Y.-G. Li and E. A. B. Kantchev, Eur. J. Org. Chem., 2017, 6274–6282 CrossRef.
- S.-S. Zhang, C.-Y. Jiang, J.-Q. Wu, X.-G. Liu, Q. Li, Z.-S. Huang, D. Li and H. Wang, Chem. Commun., 2015, 51, 10240–10243 RSC.
- Z. Li, X. Zhou, P. Lu and Y. Wang, J. Org. Chem., 2016, 81, 9433–9437 CrossRef PubMed.
- H. Wu, T. Liu, M. Cui, Y. Li, J. Jian, H. Wang and Z. Zeng, Org. Biomol. Chem., 2017, 15, 536–540 RSC.
- V. K. Tiwari, N. Kamal and M. Kapur, Org. Lett., 2015, 17, 1766–1769 CrossRef PubMed.
- A. Schischko, H. Ren, N. Kaplaneris and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1576–1580 CrossRef PubMed.
- R. Odani, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2014, 53, 10784–10788 CrossRef PubMed.
- D. Das, P. Poddar, S. Maity and R. Samanta, J. Org. Chem., 2017, 82, 3612–3621 CrossRef PubMed.
- P. Peng, J. Wang, H. Jiang and H. Liu, Org. Lett., 2016, 18, 5376–5379 CrossRef PubMed.
- K. Anil Kumar, P. Kannaboina and P. Das, Org. Biomol. Chem., 2017, 15, 5457–5461 RSC.
- D. Zell, S. Warratz, D. Gelman, S. J. Garden and L. Ackermann, Chem. – Eur. J., 2016, 22, 1248–1252 CrossRef PubMed.
- Y. Ebe and T. Nishimura, J. Am. Chem. Soc., 2015, 137, 5899–5902 CrossRef PubMed.
- Y. Ebe, M. Onoda, T. Nishimura and H. Yorimitsu, Angew. Chem., Int. Ed., 2017, 56, 5607–5611 CrossRef PubMed.
- Y. Wu and B. Zhou, ACS Catal., 2017, 7, 2213–2217 CrossRef.
- G.-F. Zhang, Y. Li, X.-Q. Xie and C.-R. Ding, Org. Lett., 2017, 19, 1216–1219 CrossRef PubMed.
- B. Zhou, Y. Hu and C. Wang, Angew. Chem., Int. Ed., 2015, 54, 13659–13663 CrossRef PubMed.
- Y. Hara, S. Onodera, T. Kochi and F. Kakiuchi, Org. Lett., 2015, 17, 4850–4853 CrossRef PubMed.
- J. A. Boerth and J. A. Ellman, Chem. Sci., 2016, 7, 1474–1479 RSC.
- T. J. Potter and J. A. Ellman, Org. Lett., 2016, 18, 3838–3841 CrossRef PubMed.
- J. Kim, S.-W. Park, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2015, 137, 13448–13451 CrossRef PubMed.
- H. Cheng, W. Dong, C. A. Dannenberg, S. Dong, Q. Guo and C. Bolm, ACS Catal., 2015, 5, 2770–2773 CrossRef.
- Y.-F. Liang, L. Massignan, W. Liu and L. Ackermann, Chem. – Eur. J., 2016, 22, 14856–14859 CrossRef PubMed.
- S.-L. Liu, Y. Li, J.-R. Guo, G.-C. Yang, X.-H. Li, J.-F. Gong and M.-P. Song, Org. Lett., 2017, 19, 4042–4045 CrossRef PubMed.
- C. Zhu, J. L. Schwarz, S. Cembellín, S. Greßies and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 437–441 CrossRef PubMed.
- Q. Lu, S. Cembellín, S. Greßies, S. Singha, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 1399–1403 CrossRef PubMed.
- W. Miura, K. Hirano and M. Miura, J. Org. Chem., 2017, 82, 5337–5344 CrossRef PubMed.
- M. Barday, C. Janot, N. R. Halcovitch, J. Muir and C. Aïssa, Angew. Chem., Int. Ed., 2017, 56, 13117–13121 CrossRef PubMed.
- A. C. B. Burtoloso, R. M. P. Dias and I. A. Leonarczyk, Eur. J. Org. Chem., 2013, 5005–5016 CrossRef.
- H. Wang, S. Yu, Z. Qi and X. Li, Org. Lett., 2015, 17, 2812–2815 CrossRef PubMed.
- D. Das, A. Biswas, U. Karmakar, S. Chand and R. Samanta, J. Org. Chem., 2016, 81, 842–848 CrossRef PubMed.
- V. Soni, R. A. Jagtap, R. G. Gonnade and B. Punji, ACS Catal., 2016, 6, 5666–5672 CrossRef.
- T. Yamakawa, Y. W. Seto and N. Yoshikai, Synlett, 2015, 340–344 Search PubMed.
- D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508–4511 CrossRef PubMed.
- J. H. Kim, T. Gensch, D. Zhao, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 10975–10979 CrossRef PubMed.
- X. Cong, S. Zhai and X. Zeng, Org. Chem. Front., 2016, 3, 673–677 RSC.
- G. S. Kumar and M. Kapur, Org. Lett., 2016, 18, 1112–1115 CrossRef PubMed.
- Q. Lu, F. J. R. Klauck and F. Glorius, Chem. Sci., 2017, 8, 3379–3383 RSC.
- H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 6339–6342 CrossRef PubMed.
- T. H. Meyer, W. Liu, M. Feldt, A. Wuttke, R. A. Mata and L. Ackermann, Chem. – Eur. J., 2017, 23, 5443–5447 CrossRef PubMed.
- D. Zell, Q. Bu, M. Feldt and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7408–7412 CrossRef PubMed.
- D. Zell, U. Dhawa, V. Müller, M. Bursch, S. Grimme and L. Ackermann, ACS Catal., 2017, 7, 4209–4213 CrossRef.
- D. Zell, V. Müller, U. Dhawa, M. Bursch, R. R. Presa, S. Grimme and L. Ackermann, Chem. – Eur. J., 2017, 23, 12145–12148 CrossRef PubMed.
- J. Ni, H. Zhao and A. Zhang, Org. Lett., 2017, 19, 3159–3162 CrossRef PubMed.
- X. Xue, J. Xu, L. Zhang, C. Xu, Y. Pan, L. Xu, H. Li and W. Zhang, Adv. Synth. Catal., 2016, 358, 573–583 CrossRef.
- R. Qiu, L. Zhang, C. Xu, Y. Pan, H. Pang, L. Xu and H. Li, Adv. Synth. Catal., 2015, 357, 1229–1236 CrossRef.
- S. Wang, J.-T. Hou, M.-L. Feng, X.-Z. Zhang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2016, 52, 2709–2712 RSC.
- H. Wang, F. Pesciaioli, J. C. A. Oliveira, S. Warratz and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15063–15067 CrossRef PubMed.
- Q. Lu, S. Gressies, F. J. R. Klauck and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 6660–6664 CrossRef PubMed.
- X.-F. Yang, X.-H. Hu, C. Feng and T.-P. Loh, Chem. Commun., 2015, 51, 2532–2535 RSC.
- Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef PubMed.
- S. Prakash, K. Muralirajan and C.-H. Cheng, Angew. Chem., Int. Ed., 2016, 55, 1844–1848 CrossRef PubMed.
- Y.-X. Lao, S.-S. Zhang, X.-G. Liu, C.-Y. Jiang, J.-Q. Wu, Q. Li, Z.-S. Huang and H. Wang, Adv. Synth. Catal., 2016, 358, 2186–2191 CrossRef.
- Y. Yang, B. Li, W. Liu, R. Zhang, L. Yu, Q.-G. Ma, R. Lv, D. Du and T. Li, J. Org. Chem., 2016, 81, 11335–11345 CrossRef PubMed.
- J. Sanjosé-Orduna, D. Gallego, A. Garcia-Roca, E. Martin, J. Benet-Buchholz and M. H. Pérez-Temprano, Angew. Chem., Int. Ed., 2017, 56, 12137–12141 CrossRef PubMed.
- C.-Z. Luo, P. Gandeepan, Y.-C. Wu, C.-H. Tsai and C.-H. Cheng, ACS Catal., 2015, 5, 4837–4841 CrossRef.
- Y. R. Han, S.-H. Shim, D.-S. Kim and C.-H. Jun, Org. Lett., 2017, 19, 2941–2944 CrossRef PubMed.
- N. S. Upadhyay, J. Jayakumar and C.-H. Cheng, Adv. Synth. Catal., 2016, 358, 3381–3386 CrossRef.
- B. Butschke and H. Schwarz, Chem. Sci., 2012, 3, 308–326 RSC.
- D. Ghorai, C. Dutta and J. Choudhury, ACS Catal., 2016, 6, 709–713 CrossRef.
- R. Morioka, K. Nobushige, T. Satoh, K. Hirano and M. Miura, Org. Lett., 2015, 17, 3130–3133 CrossRef PubMed.
- T. Li, Z. Wang, W.-B. Qin and T.-B. Wen, ChemCatChem, 2016, 8, 2146–2154 CrossRef.
- T. Li, Z. Wang, M. Zhang, H.-J. Zhang and T.-B. Wen, Chem. Commun., 2015, 51, 6777–6780 RSC.
- Y. Li, F. Xie and X. Li, J. Org. Chem., 2016, 81, 715–722 CrossRef PubMed.
- S. M. Khake, V. Soni, R. G. Gonnade and B. Punji, Chem. – Eur. J., 2017, 23, 2907–2914 CrossRef PubMed.
- A. Funayama, T. Satoh and M. Miura, J. Am. Chem. Soc., 2005, 127, 15354–15355 CrossRef PubMed.
- S. B. Wang, Q. Gu and S. L. You, J. Org. Chem., 2017, 82, 11829–11835 CrossRef PubMed.
- T. Li, Z. Wang, K. Xu, W. Liu, X. Zhang, W. Mao, Y. Guo, X. Ge and F. Pan, Org. Lett., 2016, 18, 1064–1067 CrossRef PubMed.
- T. Li, C. Fu, Q. Ma, Z. Sang, Y. Yang, H. Yang, R. Lv and B. Li, J. Org. Chem., 2017, 82, 10263–10270 CrossRef PubMed.
- (a) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3635–3638 CrossRef PubMed; (b) A. B. Pawar and S. Chang, Org. Lett., 2015, 17, 660–663 CrossRef PubMed.
- C. Qi, X. Hu and H. Jiang, Chem. Commun., 2017, 53, 7994–7997 RSC.
- Z.-B. Chen, F.-L. Zhang, Q. Yuan, H.-F. Chen, Y.-M. Zhu and J.-K. Shen, RSC Adv., 2016, 6, 64234–64238 RSC.
- W. Liu, S. C. Richter, R. Mei, M. Feldt and L. Ackermann, Chem. – Eur. J., 2016, 22, 17958–17961 CrossRef PubMed.
- M. Chaitanya and P. Anbarasan, J. Org. Chem., 2015, 80, 3695–3700 CrossRef PubMed.
- M. Chaitanya and P. Anbarasan, Org. Lett., 2015, 17, 3766–3769 CrossRef PubMed.
- Y.-F. Liang, X. Li, X. Wang, Y. Yan, P. Feng and N. Jiao, ACS Catal., 2015, 5, 1956–1963 CrossRef.
- Y.-S. Bao, D. Zhang, M. Jia and B. Zhaorigetu, Green Chem., 2016, 18, 2072–2077 RSC.
- X. Liu, Z. Yi, J. Wang and G. Liu, RSC Adv., 2015, 5, 10641–10646 RSC.
- F. Xiao, S. Chen, H. Huang and G.-J. Deng, Eur. J. Org. Chem., 2015, 7919–7925 CrossRef.
- S. Yu, Y. Li, L. Kong, X. Zhou, G. Tang, Y. Lan and X. Li, ACS Catal., 2016, 6, 7744–7748 CrossRef.
- S. Maiti, L. Burgula, G. Chakraborti and J. Dash, Eur. J. Org. Chem., 2017, 332–340 CrossRef.
- Z. Wang, Y. Li, F. Zhu and X.-F. Wu, Adv. Synth. Catal., 2016, 358, 2855–2859 CrossRef.
- F. Zhou, D.-S. Wang, X. Guan and T. G. Driver, Angew. Chem., 2017, 129, 4601–4605 CrossRef.
- W. Liu, J. Bang, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 14137–14140 CrossRef PubMed.
- J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef PubMed.
- M. Yoshida, K. Kawai, R. Tanaka, T. Yoshino and S. Matsunaga, Chem. Commun., 2017, 53, 5974–5977 RSC.
- X.-G. Liu, Q. Li and H. Wang, Adv. Synth. Catal., 2017, 359, 1942–1946 CrossRef.
- P. Sharma, S. Rohilla and N. Jain, J. Org. Chem., 2015, 80, 4116–4122 CrossRef PubMed.
- X.-S. Zhang, G. Li, X.-G. Zhang and X.-H. Zhang, Tetrahedron, 2015, 71, 5458–5464 CrossRef.
- F. Wang, X. Yu, Z. Qi and X. Li, Chem. – Eur. J., 2016, 22, 511–516 CrossRef PubMed.
- T. Liu, W. Zhou and J. Wu, Org. Lett., 2017, 19, 6638–6641 CrossRef PubMed.
- Q. Wang, F. Xie and X. Li, J. Org. Chem., 2015, 80, 8361–8366 CrossRef PubMed.
- P. Peng, J. Wang, C. Li, W. Zhu, H. Jiang and H. Liu, RSC Adv., 2016, 6, 57441–57445 RSC.
- C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef PubMed.
- A. C. Shaikh, D. S. Ranade, S. Thorat, A. Maity, P. P. Kulkarni, R. G. Gonnade, P. Munshi and N. T. Patil, Chem. Commun., 2015, 51, 16115–16118 RSC.
- V. F. Pais, M. M. Alcaide, R. López-Rodríguez, D. Collado, F. Nájera, E. Pérez-Inestrosa, E. Álvarez, J. M. Lassaletta, R. Fernández, A. Ros and U. Pischel, Chem. – Eur. J., 2015, 21, 15369–15376 CrossRef PubMed.
- N. Kano, M. Yamamura and T. Kawashima, Dalton Trans., 2015, 44, 16256–16265 RSC.
- Y. Yoshigoe and Y. Kuninobu, Org. Lett., 2017, 19, 3450–3453 CrossRef PubMed.
- S. D. Sarkar, N. Y. P. Kumar and L. Ackermann, Chem. – Eur. J., 2017, 23, 84–87 CrossRef PubMed.
- W. Miura, K. Hirano and M. Miura, Org. Lett., 2016, 18, 3742–3745 CrossRef PubMed.
- S. Okada, T. Namikoshi, S. Watanabe and M. Murata, ChemCatChem, 2015, 7, 1531–1534 CrossRef.
- L. Rubio-Perez, M. Iglesias, J. Munarriz, V. Polo, V. Passarelli, J. J. Perez-Torrente and L. A. Oro, Chem. Sci., 2017, 8, 4811–4822 RSC.
- T. Wakaki, M. Kanai and Y. Kuninobu, Org. Lett., 2015, 17, 1758–1761 CrossRef PubMed.
- X.-H. Hu, X.-F. Yang and T.-P. Loh, ACS Catal., 2016, 6, 5930–5934 CrossRef.
- A. A. Kantak, L. Marchetti and B. DeBoef, Chem. Commun., 2015, 51, 3574–3577 RSC.
- M. A. Ali, X. Yao, H. Sun and H. Lu, Org. Lett., 2015, 17, 1513–1516 CrossRef PubMed.
- P. Patel and S. Chang, ACS Catal., 2015, 5, 853–858 CrossRef.
- G. Li, C. Jia, Q. Chen, K. Sun, F. Zhao, H. Wu, Z. Wang, Y. Lv and X. Chen, Adv. Synth. Catal., 2015, 357, 1311–1315 CrossRef.
- Y. Park, K. T. Park, J. G. Kim and S. Chang, J. Am. Chem. Soc., 2015, 137, 4534–4542 CrossRef PubMed.
- Y. Park, S. Jee, J. G. Kim and S. Chang, Org. Process Res. Dev., 2015, 19, 1024–1029 CrossRef.
- J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103–14107 CrossRef PubMed.
- Y. Liang, Y.-F. Liang, C. Tang, Y. Yuan and N. Jiao, Chem. – Eur. J., 2015, 21, 16395–16399 CrossRef PubMed.
- R. Mei, J. Loup and L. Ackermann, ACS Catal., 2016, 6, 793–797 CrossRef.
- S.-B. Wang, Q. Gu and S.-L. You, Organometallics, 2017, 36, 4359–4362 CrossRef.
- D. Das and R. Samanta, Adv. Synth. Catal., 2018, 360, 379–384 CrossRef.
- M. A. Ali, X. Yao, G. Li and H. Lu, Org. Lett., 2016, 18, 1386–1389 CrossRef PubMed.
- L. Shi and B. Wang, Org. Lett., 2016, 18, 2820–2823 CrossRef PubMed.
- S. Yu, Y. Li, X. Zhou, H. Wang, L. Kong and X. Li, Org. Lett., 2016, 18, 2812–2815 CrossRef PubMed.
- C. S. Azad and A. K. Narula, RSC Adv., 2015, 5, 100223–100227 RSC.
- Y. Wu and B. Zhou, Org. Lett., 2017, 19, 3532–3535 CrossRef PubMed.
- S. K. Santra, A. Banerjee, N. Khatun and B. K. Patel, Eur. J. Org. Chem., 2015, 350–356 CrossRef.
- S. Korwar, K. Brinkley, A. R. Siamaki, B. F. Gupton and K. C. Ellis, Org. Lett., 2015, 17, 1782–1785 CrossRef PubMed.
- Y. Zhang, Y. Zhao, Y. Luo, L. Xiao, Y. Huang, X. Li, Q. Peng, Y. Liu, B. Yang, C. Zhu, X. Zhou and J. Zhang, Org. Lett., 2017, 19, 6470–6473 CrossRef PubMed.
- J. Dong, P. Liu and P. Sun, J. Org. Chem., 2015, 80, 2925–2929 CrossRef PubMed.
- T. Yamaguchi, E. Yamaguchi, N. Tada and A. Itoh, Adv. Synth. Catal., 2015, 357, 2017–2021 CrossRef.
- W. Lu, H. Xu and Z. Shen, Org. Biomol. Chem., 2017, 15, 1261–1267 RSC.
- P. Das, D. Saha, D. Saha and J. Guin, ACS Catal., 2016, 6, 6050–6054 CrossRef.
- N. Khatun, A. Banerjee, S. K. Santra, W. Ali and B. K. Patel, RSC Adv., 2015, 5, 36461–36466 RSC.
- S. Han, S. Sharma, J. Park, M. Kim, Y. Shin, N. K. Mishra, J. J. Bae, J. H. Kwak, Y. H. Jung and I. S. Kim, J. Org. Chem., 2014, 79, 275–284 CrossRef PubMed.
- T. Okada, K. Nobushige, T. Satoh and M. Miura, Org. Lett., 2016, 18, 1150–1153 CrossRef PubMed.
- P. Zhang, L. Hong, G. Li and R. Wang, Adv. Synth. Catal., 2015, 357, 345–349 CrossRef.
- V. Pascanu, F. Carson, M. V. Solano, J. Su, X. Zou, M. J. Johansson and B. Martín-Matute, Chem. – Eur. J., 2016, 22, 3729–3737 CrossRef PubMed.
- H. Fei and S. M. Cohen, J. Am. Chem. Soc., 2015, 137, 2191–2194 CrossRef PubMed.
- G. Zhang, S. Sun, F. Yang, Q. Zhang, J. Kang, Y. Wu and Y. Wu, Adv. Synth. Catal., 2015, 357, 443–450 CrossRef.
- F. Xie, Z. Zhang, X. Yu, G. Tang and X. Li, Angew. Chem., Int. Ed., 2015, 54, 7405–7409 CrossRef PubMed.
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef PubMed.
- G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169–178 RSC.
- D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330–7331 CrossRef PubMed.
- F. Kakiuchi, K. Tsuchiya, M. Matsumoto, E. Mizushima and N. Chatani, J. Am. Chem. Soc., 2004, 126, 12792–12793 CrossRef PubMed.
- B. Liu, P. Hu, X. Zhou, D. Bai, J. Chang and X. Li, Org. Lett., 2017, 19, 2086–2089 CrossRef PubMed.
- S. Han, J. Park, S. Kim, S. H. Lee, S. Sharma, N. K. Mishra, Y. H. Jung and I. S. Kim, Org. Lett., 2016, 18, 4666–4669 CrossRef PubMed.
- S. Kim, S. Han, J. Park, S. Sharma, N. K. Mishra, H. Oh, J. H. Kwak and I. S. Kim, Chem. Commun., 2017, 53, 3006–3009 RSC.
- W. Hou, Y. Yang, Y. Wu, H. Feng, Y. Li and B. Zhou, Chem. Commun., 2016, 52, 9672–9675 RSC.
- S.-Y. Yan, P.-X. Ling and B.-F. Shi, Adv. Synth. Catal., 2017, 359, 2912–2917 CrossRef.
- X. Wang, D.-G. Yu and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 10280–10283 CrossRef PubMed.
- P. Gao, W. Guo, J. Xue, Y. Zhao, Y. Yuan, Y. Xia and Z. Shi, J. Am. Chem. Soc., 2015, 137, 12231–12240 CrossRef PubMed.
- J. H. Kim, S. Greßies, M. Boultadakis-Arapinis, C. Daniliuc and F. Glorius, ACS Catal., 2016, 6, 7652–7656 CrossRef.
- G. Tan and J. You, Org. Lett., 2017, 19, 4782–4785 CrossRef PubMed.
- X. Zhou, S. Yu, L. Kong and X. Li, ACS Catal., 2016, 6, 647–651 CrossRef.
- S. Han, W. Ma, Z. Zhang, L. Liu, M. Tang and J. Li, Asian J. Org. Chem., 2017, 6, 1014–1018 CrossRef.
- M. Sen, B. Emayavaramban, N. Barsu, J. R. Premkumar and B. Sundararaju, ACS Catal., 2016, 6, 2792–2796 CrossRef.
- L. Kong, B. Liu, X. Zhou, F. Wang and X. Li, Chem. Commun., 2017, 53, 10326–10329 RSC.
- V. G. Landge, M. K. Sahoo, S. P. Midya, G. Jaiswal and E. Balaraman, Dalton Trans., 2015, 44, 15382–15386 RSC.
- L. Kong, X. Zhou, Y. Xu and X. Li, Org. Lett., 2017, 19, 3644–3647 CrossRef PubMed.
- B. Liu, B. Li and B. Wang, Chem. Commun., 2015, 51, 16334–16337 RSC.
- H. Wang, G. Tang and X. Li, Angew. Chem., Int. Ed., 2015, 54, 13049–13052 CrossRef PubMed.
- N. Barsu, M. A. Rahman, M. Sen and B. Sundararaju, Chem. – Eur. J., 2016, 22, 9135–9138 CrossRef PubMed.
- X. Huang, Y. Wang, J. Lan and J. You, Angew. Chem., Int. Ed., 2015, 54, 9404–9408 CrossRef PubMed.
- T. Jeong, N. K. Mishra, P. Dey, H. Oh, S. Han, S. H. Lee, H. S. Kim, J. Park and I. S. Kim, Chem. Commun., 2017, 53, 11197–11200 RSC.
- X. Zhang, R. Wu, W. Liu, D.-W. Qian, J. Yang, P. Jiang and Q.-Z. Zheng, Org. Biomol. Chem., 2016, 14, 4789–4793 RSC.
- S. Yu, G. Tang, Y. Li, X. Zhou, Y. Lan and X. Li, Angew. Chem., Int. Ed., 2016, 55, 8696–8700 CrossRef PubMed.
- C. Tang, M. Zou, J. Liu, X. Wen, X. Sun, Y. Zhang and N. Jiao, Chem. – Eur. J., 2016, 22, 11165–11169 CrossRef PubMed.
- W. Zhang, S. Ren, J. Zhang and Y. Liu, J. Org. Chem., 2015, 80, 5973–5978 CrossRef PubMed.
- G. S. Kumar, J. W. Boyle, C. Tejo and P. W. H. Chan, Chem. – Asian J., 2016, 11, 385–389 CrossRef PubMed.
- J. De Houwer, K. Abbaspour Tehrani and B. U. W. Maes, Angew. Chem., Int. Ed., 2012, 51, 2745–2748 CrossRef PubMed.
- H. Sterckx, J. De Houwer, C. Mensch, I. Caretti, K. A. Tehrani, W. A. Herrebout, S. Van Doorslaer and B. U. W. Maes, Chem. Sci., 2016, 7, 346–357 RSC.
- G. Wang, L. Liu, H. Wang, Y.-S. Ding, J. Zhou, S. Mao and P. Li, J. Am. Chem. Soc., 2017, 139, 91–94 CrossRef PubMed.
- E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2012, 18, 10092–10142 CrossRef PubMed.
- W. Li, X. Huang and J. You, Org. Lett., 2016, 18, 666–668 CrossRef PubMed.
- L. Kong, S. Yu, G. Tang, H. Wang, X. Zhou and X. Li, Org. Lett., 2016, 18, 3802–3805 CrossRef PubMed.
- K. Muralirajan, S. Prakash and C.-H. Cheng, Adv. Synth. Catal., 2017, 359, 513–518 CrossRef.
- Y. Unoh, T. Satoh, K. Hirano and M. Miura, ACS Catal., 2015, 5, 6634–6639 CrossRef.
- Z. Qi and X. Li, Angew. Chem., Int. Ed., 2013, 52, 8995–9000 CrossRef PubMed.
- C. Sollert, K. Devaraj, A. Orthaber, P. J. Gates and L. T. Pilarski, Chem. – Eur. J., 2015, 21, 5380–5386 CrossRef PubMed.
- X. Zhu, J.-H. Su, C. Du, Z.-L. Wang, C.-J. Ren, J.-L. Niu and M.-P. Song, Org. Lett., 2017, 19, 596–599 CrossRef PubMed.
- X. Zhou, Y. Pan and X. Li, Angew. Chem., Int. Ed., 2017, 56, 8163–8167 CrossRef PubMed.
- M. Gao, Y. Yang, H. Chen and B. Zhou, Adv. Synth. Catal., 2018, 360, 100–105 CrossRef.
- S.-S. Zhang, J. Xia, J.-Q. Wu, X.-G. Liu, C.-J. Zhou, E. Lin, Q. Li, S.-L. Huang and H. Wang, Org. Lett., 2017, 19, 5868–5871 CrossRef PubMed.
- P. Lu, C. Feng and T.-P. Loh, Org. Lett., 2015, 17, 3210–3213 CrossRef PubMed.
- Y. Xu, X. Zhou, G. Zheng and X. Li, Org. Lett., 2017, 19, 5256–5259 CrossRef PubMed.
- Z. Zhang, S. Han, M. Tang, L. Ackermann and J. Li, Org. Lett., 2017, 19, 3315–3318 CrossRef PubMed.
- S.-Y. Yan, Z.-Z. Zhang and B.-F. Shi, Chem. Commun., 2017, 53, 10287–10290 RSC.
- J. Xia, L. Kong, X. Zhou, G. Zheng and X. Li, Org. Lett., 2017, 19, 5972–5975 CrossRef PubMed.
- Z. Qi, L. Kong and X. Li, Org. Lett., 2016, 18, 4392–4395 CrossRef PubMed.
- M. Choi, J. Park, S. Sharma, H. Jo, S. Han, M. Jeon, N. K. Mishra, S. H. Han, J. S. Lee and I. S. Kim, J. Org. Chem., 2016, 81, 4771–4778 CrossRef PubMed.
- X. Jiang, J. Chen, W. Zhu, K. Cheng, Y. Liu, W.-K. Su and C. Yu, J. Org. Chem., 2017, 82, 10665–10672 CrossRef PubMed.
- S.-Y. Chen, Q. Li and H. Wang, J. Org. Chem., 2017, 82, 11173–11181 CrossRef PubMed.
- X. Zhou, Y. Luo, L. Kong, Y. Xu, G. Zheng, Y. Lan and X. Li, ACS Catal., 2017, 7, 7296–7304 CrossRef.
- L. Shi, X. Zhong, H. She, Z. Lei and F. Li, Chem. Commun., 2015, 51, 7136–7139 RSC.
- C. Wang, A. Wang and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 9935–9938 CrossRef PubMed.
- S.-Y. Chen, X.-L. Han, J.-Q. Wu, Q. Li, Y. Chen and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 9939–9943 CrossRef PubMed.
- X. Zhou, Z. Fan, Z. Zhang, P. Lu and Y. Wang, Org. Lett., 2016, 18, 4706–4709 CrossRef PubMed.
- M. Moselage, N. Sauermann, S. C. Richter and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 6352–6355 CrossRef PubMed.
- S.-H. Cai, L. Ye, D.-X. Wang, Y.-Q. Wang, L.-J. Lai, C. Zhu, C. Feng and T.-P. Loh, Chem. Commun., 2017, 53, 8731–8734 RSC.
- L. Kong, X. Zhou and X. Li, Org. Lett., 2016, 18, 6320–6323 CrossRef PubMed.
- N. Sauermann, M. J. González and L. Ackermann, Org. Lett., 2015, 17, 5316–5319 CrossRef PubMed.
- Z. Ruan, N. Sauermann, E. Manoni and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 3172–3176 CrossRef PubMed.
- Z.-Z. Zhang, B. Liu, C.-Y. Wang and B.-F. Shi, Org. Lett., 2015, 17, 4094–4097 CrossRef PubMed.
- Y. Wu, Y. Yang, B. Zhou and Y. Li, J. Org. Chem., 2015, 80, 1946–1951 CrossRef PubMed.
- D. Kang and S. Hong, Org. Lett., 2015, 17, 1938–1941 CrossRef PubMed.
- N. K. Mishra, T. Jeong, S. Sharma, Y. Shin, S. Han, J. Park, J. S. Oh, J. H. Kwak, Y. H. Jung and I. S. Kim, Adv. Synth. Catal., 2015, 357, 1293–1298 CrossRef.
- G. Kumar and G. Sekar, RSC Adv., 2015, 5, 28292–28298 RSC.
- C. Li, W. Zhu, S. Shu, X. Wu and H. Liu, Eur. J. Org. Chem., 2015, 3743–3750 CrossRef.
- Y. Zhao, U. K. Sharma, F. Schröder, N. Sharma, G. Song and E. V. Van der Eycken, RSC Adv., 2017, 7, 32559–32563 RSC.
- U. K. Sharma, H. P. L. Gemoets, F. Schröder, T. Noël and E. V. Van der Eycken, ACS Catal., 2017, 7, 3818–3823 CrossRef.
- M. K. Manna, G. Bairy and R. Jana, Org. Biomol. Chem., 2017, 15, 5899–5903 RSC.
- T. Jeong, S. Han, N. K. Mishra, S. Sharma, S.-Y. Lee, J. S. Oh, J. H. Kwak, Y. H. Jung and I. S. Kim, J. Org. Chem., 2015, 80, 7243–7250 CrossRef PubMed.
- J. Xu, N. Sharma, U. K. Sharma, Z. Li, G. Song and E. V. Van der Eycken, Adv. Synth. Catal., 2015, 357, 2615–2621 CrossRef.
- A. S. Tsai, M. E. Tauchert, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 1248–1250 CrossRef PubMed.
- J. Shi, B. Zhou, Y. Yang and Y. Li, Org. Biomol. Chem., 2012, 10, 8953–8955 RSC.
- B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Chem. Commun., 2015, 51, 4659–4661 RSC.
- M. Jeon, N. K. Mishra, U. De, S. Sharma, Y. Oh, M. Choi, H. Jo, R. Sachan, H. S. Kim and I. S. Kim, J. Org. Chem., 2016, 81, 9878–9885 CrossRef PubMed.
- N. K. Mishra, M. Jeon, Y. Oh, H. Jo, J. Park, S. Han, S. Sharma, S. H. Han, Y. H. Jung and I. S. Kim, Org. Chem. Front., 2017, 4, 241–249 RSC.
- H. Li, J. Jie, S. Wu, X. Yang and H. Xu, Org. Chem. Front., 2017, 4, 250–254 RSC.
- L. Zhu, X. Cao, R. Qiu, T. Iwasaki, V. P. Reddy, X. Xu, S.-F. Yin and N. Kambe, RSC Adv., 2015, 5, 39358–39365 RSC.
- R. Qiu, V. P. Reddy, T. Iwasaki and N. Kambe, J. Org. Chem., 2015, 80, 367–374 CrossRef PubMed.
- W. Xie, B. Li and B. Wang, J. Org. Chem., 2016, 81, 396–403 CrossRef PubMed.
- T. Gensch, F. J. R. Klauck and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 11287–11291 CrossRef PubMed.
- P. Gandeepan, J. Koeller and L. Ackermann, ACS Catal., 2017, 7, 1030–1034 CrossRef.
- C. K. Jana, S. Grimme and A. Studer, Chem. – Eur. J., 2009, 15, 9078–9084 CrossRef PubMed.
- V. Smout, A. Peschiulli, S. Verbeeck, E. A. Mitchell, W. Herrebout, P. Bultinck, C. M. L. Vande Velde, D. Berthelot, L. Meerpoel and B. U. W. Maes, J. Org. Chem., 2013, 78, 9803–9814 CrossRef PubMed.
- S. Pan, K. Endo and T. Shibata, Org. Lett., 2011, 13, 4692–4695 CrossRef PubMed.
- S. J. Pastine, D. V. Gribkov and D. Sames, J. Am. Chem. Soc., 2006, 128, 14220–14221 CrossRef PubMed.
- J.-H. Chu, P.-S. Lin and M.-J. Wu, Organometallics, 2010, 29, 4058–4065 CrossRef.
- M. Tobisu, J. Zhao, H. Kinuta, T. Furukawa, T. Igarashi and N. Chatani, Adv. Synth. Catal., 2016, 358, 2417–2421 CrossRef.
- H. Kinuta, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 1593–1600 CrossRef PubMed.
- J. Li and Z.-X. Wang, Org. Lett., 2017, 19, 3723–3726 CrossRef PubMed.
- C. Huang, N. Chernyak, A. S. Dudnik and V. Gevorgyan, Adv. Synth. Catal., 2011, 353, 1285–1305 CrossRef PubMed.
- A. García-Rubia, M. Á. Fernández-Ibáñez, R. Gómez Arrayás and J. C. Carretero, Chem. – Eur. J., 2011, 17, 3567–3570 CrossRef PubMed.
- M. Sun, Z. Wang, J. Wang, P. Guo, X. Chen and Y.-M. Li, Org. Biomol. Chem., 2016, 14, 10585–10588 RSC.
- H. Richter, S. Beckendorf and O. G. Mancheño, Adv. Synth. Catal., 2011, 353, 295–302 CrossRef.
- N. K. Mishra, M. Choi, H. Jo, Y. Oh, S. Sharma, S. H. Han, T. Jeong, S. Han, S.-Y. Lee and I. S. Kim, Chem. Commun., 2015, 51, 17229–17232 RSC.
- H. Jiang, S. Gao, J. Xu, X. Wu, A. Lin and H. Yao, Adv. Synth. Catal., 2016, 358, 188–194 CrossRef.
- G.-D. Tang, C.-L. Pan and X. Li, Org. Chem. Front., 2016, 3, 87–90 RSC.
- Z. Ruan, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 3153–3157 CrossRef PubMed.
- Z. Ruan, S. Lackner and L. Ackermann, ACS Catal., 2016, 6, 4690–4693 CrossRef.
- T. Müller and L. Ackermann, Chem. – Eur. J., 2016, 22, 14151–14154 CrossRef PubMed.
- S. Yang, B. Feng and Y. Yang, J. Org. Chem., 2017, 82, 12430–12438 CrossRef PubMed.
- L. Wang, Z. Yang, M. Yang, R. Zhang, C. Kuai and X. Cui, Org. Biomol. Chem., 2017, 15, 8302–8307 RSC.
- G. G. Pawar, A. Brahmanandan and M. Kapur, Org. Lett., 2016, 18, 448–451 CrossRef PubMed.
- D. Nageswar Rao, S. Rasheed, G. Raina, Q. N. Ahmed, C. K. Jaladanki, P. V. Bharatam and P. Das, J. Org. Chem., 2017, 82, 7234–7244 CrossRef PubMed.
- M. Spettel, R. Pollice and M. Schnürch, Org. Lett., 2017, 19, 4287–4290 CrossRef PubMed.
- K. Yu, Y. Liang, B. Li and B. Wang, Adv. Synth. Catal., 2016, 358, 661–666 CrossRef.
- S. Bai, X. Chen, X. Hu, Y. Deng, H. Jiang and W. Zeng, Org. Biomol. Chem., 2017, 15, 3638–3647 RSC.
- P. V. Reddy, M. Annapurna, P. Srinivas, P. R. Likhar and M. Lakshmi Kantam, New J. Chem., 2015, 39, 3399–3404 RSC.
- X. Hu, X. Chen, Y. Zhu, Y. Deng, H. Zeng, H. Jiang and W. Zeng, Org. Lett., 2017, 19, 3474–3477 CrossRef PubMed.
- Z.-J. Wu, K. L. Huang and Z.-Z. Huang, Org. Biomol. Chem., 2017, 15, 4978–4983 RSC.
- J. Xia, X. Yang, Y. Li and X. Li, Org. Lett., 2017, 19, 3243–3246 CrossRef PubMed.
- M. Ravi, S. Allu and K. C. K. Swamy, J. Org. Chem., 2017, 82, 2355–2363 CrossRef PubMed.
- L. Wang, Y. Yu, M. Yang, C. Kuai, D. Cai, J. Yu and X. Cui, Adv. Synth. Catal., 2017, 359, 3818–3825 CrossRef.
- J.-H. Chu, S.-T. Chen, M.-F. Chiang and M.-J. Wu, Organometallics, 2015, 34, 953–966 CrossRef.
- D. C. McAteer, E. Javed, L. Huo and S. Huo, Org. Lett., 2017, 19, 1606–1609 CrossRef PubMed.
- L. Wang, Z. Yang, M. Yang, M. Tian, C. Kuai and X. Cui, Chem. – Asian J., 2017, 12, 2634–2643 CrossRef PubMed.
- L. Wang, L. Pan, Y. Huang, Q. Chen and M. He, Eur. J. Org. Chem., 2016, 3113–3118 CrossRef.
- K. Raghuvanshi, K. Rauch and L. Ackermann, Chem. – Eur. J., 2015, 21, 1790–1794 CrossRef PubMed.
- Y. Xu, P. Liu, S.-L. Li and P. Sun, J. Org. Chem., 2015, 80, 1269–1274 CrossRef PubMed.
- C. Gao, H. Li, M. Liu, J. Ding, X. Huang, H. Wu, W. Gao and G. Wu, RSC Adv., 2017, 7, 46636–46643 RSC.
- S.-J. Lou, Q. Chen, Y.-F. Wang, D.-Q. Xu, X.-H. Du, J.-Q. He, Y.-J. Mao and Z.-Y. Xu, ACS Catal., 2015, 5, 2846–2849 CrossRef.
- D. Sarkar and V. Gevorgyan, Chem. – Eur. J., 2016, 22, 11201–11204 CrossRef PubMed.
- Y. Zhang, H. Jiang, D. Chen and Y. Zhang, Org. Biomol. Chem., 2016, 14, 4585–4589 RSC.
- Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 3191–3195 CrossRef PubMed.
- D. Sarkar, A. V. Gulevich, F. S. Melkonyan and V. Gevorgyan, ACS Catal., 2015, 5, 6792–6801 CrossRef.
- J. Guilbaud, M. Labonde, H. Cattey, S. Contal, C. Montalbetti, N. Pirio, J. Roger and J.-C. Hierso, Adv. Synth. Catal., 2017, 359, 3792–3804 CrossRef.
- H. Andersson, R. Olsson and F. Almqvist, Org. Biomol. Chem., 2011, 9, 337–346 RSC.
- G. Yan, A. J. Borah and M. Yang, Adv. Synth. Catal., 2014, 356, 2375–2394 CrossRef.
- Y. Wang and L. Zhang, Synthesis, 2015, 289–305 Search PubMed.
- H. Sterckx, C. Sambiagio, V. Médran-Navarrete and B. U. W. Maes, Adv. Synth. Catal., 2017, 359, 3226–3236 CrossRef.
- H. Hwang, J. Kim, J. Jeong and S. Chang, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef PubMed.
- S. Yu, B. Wan and X. Li, Org. Lett., 2015, 17, 58–61 CrossRef PubMed.
- X. Chen, X. Cui and Y. Wu, Org. Lett., 2016, 18, 2411–2414 CrossRef PubMed.
- X. Chen, X. Cui and Y. Wu, Org. Lett., 2016, 18, 3722–3725 CrossRef PubMed.
- D. E. Stephens, J. Lakey-Beitia, A. C. Atesin, T. A. Ateşin, G. Chavez, H. D. Arman and O. V. Larionov, ACS Catal., 2015, 5, 167–175 CrossRef PubMed.
- D. E. Stephens, J. Lakey-Beitia, G. Chavez, C. Ilie, H. D. Arman and O. V. Larionov, Chem. Commun., 2015, 51, 9507–9510 RSC.
- J. Jeong, P. Patel, H. Hwang and S. Chang, Org. Lett., 2014, 16, 4598–4601 CrossRef PubMed.
- R. Sharma, I. Kumar, R. Kumar and U. Sharma, Adv. Synth. Catal., 2017, 359, 3022–3028 CrossRef.
- X. Zhang, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2014, 53, 10794–10798 CrossRef PubMed.
- U. Sharma, Y. Park and S. Chang, J. Org. Chem., 2014, 79, 9899–9906 CrossRef PubMed.
- N. Barsu, M. Sen, J. R. Premkumar and B. Sundararaju, Chem. Commun., 2016, 52, 1338–1341 RSC.
- D. Kalsi, R. A. Laskar, N. Barsu, J. R. Premkumar and B. Sundararaju, Org. Lett., 2016, 18, 4198–4201 CrossRef PubMed.
- B. Wang, C. Li and H. Liu, Adv. Synth. Catal., 2017, 359, 3029–3034 CrossRef.
- T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 356, 1516–1520 CrossRef.
- R. Sharma, R. Kumar, I. Kumar and U. Sharma, Eur. J. Org. Chem., 2015, 7519–7528 CrossRef.
- T. Asaumi, N. Chatani, T. Matsuo, F. Kakiuchi and S. Murai, J. Org. Chem., 2003, 68, 7538–7540 CrossRef PubMed.
- A. Armstrong, L. H. Jones, J. D. Knight and R. D. Kelsey, Org. Lett., 2005, 7, 713–716 CrossRef PubMed.
- N. Gulia and O. Daugulis, Angew. Chem., Int. Ed., 2017, 56, 3630–3634 CrossRef PubMed.
- M. Moselage, J. Li, F. Kramm and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 5341–5344 CrossRef PubMed.
- C. Yuan, G. Tu and Y. Zhao, Org. Lett., 2017, 19, 356–359 CrossRef PubMed.
- C. J. Teskey, S. M. A. Sohel, D. L. Bunting, S. G. Modha and M. F. Greaney, Angew. Chem., Int. Ed., 2017, 56, 5263–5266 CrossRef PubMed.
- N. Barsu, B. Emayavaramban and B. Sundararaju, Eur. J. Org. Chem., 2017, 4370–4374 CrossRef.
- J. A. Boerth, J. R. Hummel and J. A. Ellman, Angew. Chem., Int. Ed., 2016, 55, 12650–12654 CrossRef PubMed.
- A. Wangweerawong, J. R. Hummel, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2016, 81, 1547–1557 CrossRef PubMed.
- Y. Takahama, Y. Shibata and K. Tanaka, Org. Lett., 2016, 18, 2934–2937 CrossRef PubMed.
- S. Shome and S. P. Singh, Eur. J. Org. Chem., 2015, 6025–6032 CrossRef.
- M. Sen, P. Dahiya, J. R. Premkumar and B. Sundararaju, Org. Lett., 2017, 19, 3699–3702 CrossRef PubMed.
- J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 8551–8554 CrossRef PubMed.
- J. R. Hummel and J. A. Ellman, Org. Lett., 2015, 17, 2400–2403 CrossRef PubMed.
- S. Shu, Z. Fan, Q. Yao and A. Zhang, J. Org. Chem., 2016, 81, 5263–5269 CrossRef PubMed.
- C. Testa, J. Roger, S. Scheib, P. Fleurat-Lessard and J.-C. Hierso, Adv. Synth. Catal., 2015, 357, 2913–2923 CrossRef.
- X.-M. Fan, Y. Guo, Y.-D. Li, K.-K. Yu, H.-W. Liu, D.-H. Liao and Y.-F. Ji, Asian J. Org. Chem., 2016, 5, 499–505 CrossRef.
- J. Shi, J. Zhou, Y. Yan, J. Jia, X. Liu, H. Song, H. E. Xu and W. Yi, Chem. Commun., 2015, 51, 668–671 RSC.
- N. Umeda, K. Hirano, T. Satoh, N. Shibata, H. Sato and M. Miura, J. Org. Chem., 2011, 76, 13–24 CrossRef PubMed.
- D. L. Davies, C. E. Ellul, S. A. Macgregor, C. L. McMullin and K. Singh, J. Am. Chem. Soc., 2015, 137, 9659–9669 CrossRef PubMed.
- J. Jie, H. Li, S. Wu, Q. Chai, H. Wang and X. Yang, RSC Adv., 2017, 7, 20548–20552 RSC.
- A. G. Algarra, D. L. Davies, Q. Khamker, S. A. Macgregor, C. L. McMullin, K. Singh and B. Villa-Marcos, Chem. – Eur. J., 2015, 21, 3087–3096 CrossRef PubMed.
- W. Yang, S. Ye, D. Fanning, T. Coon, Y. Schmidt, P. Krenitsky, D. Stamos and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 2501–2504 CrossRef PubMed.
- W. Yang, S. Ye, Y. Schmidt, D. Stamos and J.-Q. Yu, Chem. – Eur. J., 2016, 22, 7059–7062 CrossRef PubMed.
- F. Zhao, Y. Liu, S. Yang, K. Xie and Y. Jiang, Org. Chem. Front., 2017, 4, 1112–1115 RSC.
- S. Zhao, F. Wu, Y. Ma, W. Chen, M. Liu and H. Wu, Org. Biomol. Chem., 2016, 14, 2550–2555 RSC.
- F. Zhao, Z. Chen, Y. Liu, K. Xie and Y. Jiang, Eur. J. Org. Chem., 2016, 5971–5979 CrossRef.
- A. Irastorza, J. M. Aizpurua and A. Correa, Org. Lett., 2016, 18, 1080–1083 CrossRef PubMed.
- A. Goitia, E. Gomez-Bengoa and A. Correa, Org. Lett., 2017, 19, 962–965 CrossRef PubMed.
- T. L. Mindt, H. Struthers, L. Brans, T. Anguelov, C. Schweinsberg, V. Maes, D. Tourwé and R. Schibli, J. Am. Chem. Soc., 2006, 128, 15096–15097 CrossRef PubMed.
- N. Belskaya, J. Subbotina and S. Lesogorova, in Chemistry of 1,2,3-triazoles, ed. W. Dehaen and V. A. Bakulev, Springer International Publishing, Cham, 2015, pp. 51–116, DOI:10.1007/7081_2014_125.
- C. Premi, S. S. Patel and N. Jain, Eur. J. Org. Chem., 2016, 3788–3795 CrossRef.
- X. Yu, Q. Ma, S. Lv, J. Li, C. Zhang, L. Hai, Q. Wang and Y. Wu, Org. Chem. Front., 2017, 4, 2184–2190 RSC.
- X. Wang, C. Zhang, J. Li, C. Jiang, F. Su, Z. Zhan, L. Hai, Z. Chen and Y. Wu, RSC Adv., 2016, 6, 68929–68933 RSC.
- B. Zhu, X. Cui, C. Pi, D. Chen and Y. Wu, Adv. Synth. Catal., 2016, 358, 326–332 CrossRef.
- V. Gayakhe, Y. S. Sanghvi, I. J. S. Fairlamb and A. R. Kapdi, Chem. Commun., 2015, 51, 11944–11960 RSC.
- L. Liang, M.-S. Xie, H.-X. Wang, H.-Y. Niu, G.-R. Qu and H.-M. Guo, J. Org. Chem., 2017, 82, 5966–5973 CrossRef PubMed.
- Y. Bunno, N. Murakami, Y. Suzuki, M. Kanai, T. Yoshino and S. Matsunaga, Org. Lett., 2016, 18, 2216–2219 CrossRef PubMed.
- A. B. Pawar and D. M. Lade, Org. Biomol. Chem., 2016, 14, 3275–3283 RSC.
- M. Yu, Z. Wang, M. Tian, C. Lu, S. Li and H. Du, J. Org. Chem., 2016, 81, 3435–3442 CrossRef PubMed.
- S.-S. Li, W.-H. Li, G.-T. Zhang, Y.-Q. Xia, C.-F. Liu, F. Su, X.-M. Zhang and L. Dong, Org. Biomol. Chem., 2016, 14, 7859–7863 RSC.
- B. Liu, R. Li, W. Zhan, X. Wang, Z. Ge and R. Li, RSC Adv., 2016, 6, 48205–48211 RSC.
- W.-H. Li, L. Wu, S.-S. Li, C.-F. Liu, G.-T. Zhang and L. Dong, Chem. – Eur. J., 2016, 22, 17926–17929 CrossRef PubMed.
- R. Morioka, T. Satoh and M. Miura, Chem. Lett., 2016, 45, 682–684 CrossRef.
- B. Liu, X. Wang, Z. Ge and R. Li, Org. Biomol. Chem., 2016, 14, 2944–2949 RSC.
- A. Mishra, T. K. Vats and I. Deb, J. Org. Chem., 2016, 81, 6525–6534 CrossRef PubMed.
- M. Jeon, J. Park, P. Dey, Y. Oh, H. Oh, S. Han, S. H. Um, H. S. Kim, N. K. Mishra and I. S. Kim, Adv. Synth. Catal., 2017, 359, 3471–3478 CrossRef.
- A. Mishra, T. K. Vats, M. P. Nair, A. Das and I. Deb, J. Org. Chem., 2017, 82, 12406–12415 CrossRef PubMed.
- C.-F. Liu, G.-T. Zhang, J.-S. Sun and L. Dong, Org. Biomol. Chem., 2017, 15, 2902–2905 RSC.
- S.-S. Li, C.-F. Liu, Y.-Q. Xia, W.-H. Li, G.-T. Zhang, X.-M. Zhang and L. Dong, Org. Biomol. Chem., 2016, 14, 5214–5218 RSC.
- S.-S. Li, C.-Q. Wang, H. Lin, X.-M. Zhang and L. Dong, Org. Lett., 2015, 17, 3018–3021 CrossRef PubMed.
- S.-S. Li, C.-F. Liu, G.-T. Zhang, Y.-Q. Xia, W.-H. Li and L. Dong, Chem. – Asian J., 2017, 12, 415–418 CrossRef PubMed.
- S.-S. Li, H. Lin, C.-F. Liu, Y.-Q. Xia, X.-M. Zhang and L. Dong, Adv. Synth. Catal., 2016, 358, 1595–1601 CrossRef.
- F. Yang, J. Yu, Y. Liu and J. Zhu, Org. Lett., 2017, 19, 2885–2888 CrossRef PubMed.
- Y. Nishii, A.-K. Bachon, S. Moon, C. Bolm and M. Miura, Chem. Lett., 2017, 46, 1347–1349 CrossRef.
- X. Yu, K. Chen, F. Yang, S. Zha and J. Zhu, Org. Lett., 2016, 18, 5412–5415 CrossRef PubMed.
- Y. Xiaolong, C. Kehao, W. Qi, G. Shan, Z. Shanke and Z. Jin, Angew. Chem., Int. Ed., 2017, 56, 5222–5226 CrossRef PubMed.
- T. Noguchi, Y. Nishii and M. Miura, Chem. Lett., 2017, 46, 1512–1514 CrossRef.
- M. Suresh Babu and K. M. Lokanatha Rai, Tetrahedron Lett., 2004, 45, 7969–7970 CrossRef.
- T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297–2360 CrossRef.
- T. M. M. Maiden, S. Swanson, P. A. Procopiou and J. P. A. Harrity, Chem. – Eur. J., 2015, 21, 14342–14346 CrossRef PubMed.
- R. Mei and L. Ackermann, Adv. Synth. Catal., 2016, 358, 2443–2448 CrossRef.
- M. Tobisu, K. Yasui, Y. Aihara and N. Chatani, Angew. Chem., Int. Ed., 2017, 56, 1877–1880 CrossRef PubMed.
- Q. Zhou, J.-F. Zhang, H. Cao, R. Zhong and X.-F. Hou, J. Org. Chem., 2016, 81, 12169–12180 CrossRef PubMed.
- T. M. M. Maiden, S. Swanson, P. A. Procopiou and J. P. A. Harrity, J. Org. Chem., 2016, 81, 10641–10650 CrossRef PubMed.
- K. Seth, M. Nautiyal, P. Purohit, N. Parikh and A. K. Chakraborti, Chem. Commun., 2015, 51, 191–194 RSC.
- K. Kon, H. Suzuki, K. Takada, Y. Kohari, T. Namikoshi, S. Watanabe and M. Murata, ChemCatChem, 2016, 8, 2202–2205 CrossRef.
- F. Babudri, G. Bartoli, F. Ciminale, S. Florio and G. Ingrosso, Tetrahedron Lett., 1984, 25, 2047–2050 CrossRef.
- J. Yan, Y. Zhou, N. Zhang, H. Chen, X. He, S. Xiao and K. Zheng, Res. Chem. Intermed., 2017, 43, 5395–5402 CrossRef.
- S. S. Ramos, L. V. Reis, R. E. F. Boto, P. F. Santos and P. Almeida, Tetrahedron Lett., 2013, 54, 5441–5444 CrossRef.
- J. Rashkova, P. Tzvetkova, S. Simova, L. Viteva and T. Gospodova, Dyes Pigm., 2009, 82, 360–364 CrossRef.
- L. Viteva, T. Gospodova, J. Rashkova, I. Abrahams, I. Timtcheva, S. Simova, M. R. Mazieres and J. G. Wolf, Eur. J. Org. Chem., 2007, 3102–3114 CrossRef.
- B. V. Pipaliya and A. K. Chakraborti, ChemCatChem, 2017, 9, 4191–4198 CrossRef.
- B. V. Pipaliya and A. K. Chakraborti, J. Org. Chem., 2017, 82, 3767–3780 CrossRef PubMed.
- P. Ranjan Mohanta, A. Banerjee, S. Kumar Santra, A. Behera and B. K. Patel, Adv. Synth. Catal., 2016, 358, 2047–2052 CrossRef.
- B. Vinayak, A. Ashok and M. Chandrasekharam, Eur. J. Org. Chem., 2017, 7127–7132 CrossRef.
- M. Yu, Y. Xie, C. Xie and Y. Zhang, Org. Lett., 2012, 14, 2164–2167 CrossRef PubMed.
- J. Yao, M. Yu and Y. Zhang, Adv. Synth. Catal., 2012, 354, 3205–3210 CrossRef.
- X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem. – Eur. J., 2013, 19, 11898–11903 CrossRef PubMed.
- X.-S. Zhang, Y.-F. Zhang, K. Chen and Z.-J. Shi, Org. Chem. Front., 2014, 1, 1096–1100 RSC.
- P. Villuendas and E. P. Urriolabeitia, Org. Lett., 2015, 17, 3178–3181 CrossRef PubMed.
- B. Wang, C. Lin, Y. Liu, Z. Fan, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 973–977 RSC.
- M. Tobisu, Y. Masuya, K. Baba and N. Chatani, Chem. Sci., 2016, 7, 2587–2591 RSC.
- R. Che, Z. Wu, Z. Li, H. Xiang and X. Zhou, Chem. – Eur. J., 2014, 20, 7258–7261 CrossRef PubMed.
- X.-S. Zhang, Y.-F. Zhang, Z.-W. Li, F.-X. Luo and Z.-J. Shi, Angew. Chem., Int. Ed., 2015, 54, 5478–5482 CrossRef PubMed.
- H. L. Li, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2017, 56, 1495–1499 CrossRef PubMed.
- H.-L. Li, M. Kanai and Y. Kuninobu, Org. Lett., 2017, 19, 5944–5947 CrossRef PubMed.
- Y. Unoh, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2015, 17, 704–707 CrossRef PubMed.
- L. Liu, G. Wang, J. Jiao and P. Li, Org. Lett., 2017, 19, 6132–6135 CrossRef PubMed.
- D. Wang, X. Yu, W. Yao, W. Hu, C. Ge and X. Shi, Chem. – Eur. J., 2016, 22, 5543–5546 CrossRef PubMed.
- L. Wang, Y.-B. Xie, N.-Y. Huang, N.-N. Zhang, D.-J. Li, Y.-L. Hu, M.-G. Liu and D.-S. Li, Adv. Synth. Catal., 2017, 359, 779–785 CrossRef.
- R. Samanta and A. P. Antonchick, Angew. Chem., Int. Ed., 2011, 50, 5217–5220 CrossRef PubMed.
- B. Wang, Y. Liu, C. Lin, Y. Xu, Z. Liu and Y. Zhang, Org. Lett., 2014, 16, 4574–4577 CrossRef PubMed.
- B. Wang, C. Shen, J. Yao, H. Yin and Y. Zhang, Org. Lett., 2014, 16, 46–49 CrossRef PubMed.
- K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188–1191 CrossRef PubMed.
- K. Padala and M. Jeganmohan, Chem. Commun., 2014, 50, 14573–14576 RSC.
- T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139–2144 CrossRef.
- G. Solladié, J. Hutt and A. Girardin, Synthesis, 1987, 173 CrossRef.
- Q. Dherbassy, G. Schwertz, M. Chesse, C. K. Hazra, J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2016, 22, 1735–1743 CrossRef PubMed.
- C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871–13875 CrossRef PubMed.
- Q. Dherbassy, J. Wencel-Delord and F. Colobert, Tetrahedron, 2016, 72, 5238–5245 CrossRef.
- Q. Dherbassy, J. P. Djukic, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2018, 57, 4668–4672 CrossRef PubMed.
- T. Wesch, A. Berthelot-Bréhier, F. R. Leroux and F. Colobert, Org. Lett., 2013, 15, 2490–2493 CrossRef PubMed.
- Q. Huang, S. Fu, S. Ke, H. Xiao, X. Zhang and S. Lin, Eur. J. Org. Chem., 2015, 6602–6605 CrossRef.
- B. Li, D. D. Guo, S. H. Guo, G. F. Pan, Y. R. Gao and Y. Q. Wang, Chem. – Asian J., 2017, 12, 130–144 CrossRef PubMed.
- K. Nobushige, K. Hirano, T. Satoh and M. Miura, Tetrahedron, 2015, 71, 6506–6512 CrossRef.
- D. Chen, G. Xing and H. Zhou, Org. Chem. Front., 2015, 2, 947–950 RSC.
- H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222–7228 CrossRef PubMed.
- R. Vanjari, T. Guntreddi and K. N. Singh, Chem. – Asian J., 2016, 11, 696–699 CrossRef PubMed.
- W. J. Kerr, M. Reid and T. Tuttle, ACS Catal., 2015, 5, 402–410 CrossRef.
- A. Burhop, R. Weck, J. Atzrodt and V. Derdau, Eur. J. Org. Chem., 2017, 1418–1424 CrossRef.
- G. Cheng, P. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 8183–8186 CrossRef PubMed.
- W. Dong, L. Wang, K. Parthasarathy, F. Pan and C. Bolm, Angew. Chem., Int. Ed., 2013, 52, 11573–11576 CrossRef PubMed.
- Y. Cheng and C. Bolm, Angew. Chem., Int. Ed., 2015, 54, 12349–12352 CrossRef PubMed.
- W. H. Jeon, J.-Y. Son, J. E. Kim and P. H. Lee, Org. Lett., 2016, 18, 3498–3501 CrossRef PubMed.
- G. H. Ko, J.-Y. Son, H. Kim, C. Maeng, Y. Baek, B. Seo, K. Um and P. H. Lee, Adv. Synth. Catal., 2017, 359, 3362–3370 CrossRef.
- J. Huang, Y. Huang, T. Wang, Q. Huang, Z. Wang and Z. Chen, Org. Lett., 2017, 19, 1128–1131 CrossRef PubMed.
- Y. Li and L. Dong, Org. Biomol. Chem., 2017, 15, 9983–9986 RSC.
- J. Wen, H. Cheng, G. Raabe and C. Bolm, Org. Lett., 2017, 19, 6020–6023 CrossRef PubMed.
- J. Wen, D. P. Tiwari and C. Bolm, Org. Lett., 2017, 19, 1706–1709 CrossRef PubMed.
- G. Zheng, M. Tian, Y. Xu, X. Chen and X. Li, Org. Chem. Front., 2018, 5, 998–1002 RSC.
- Y. Cheng, W. Dong, H. Wang and C. Bolm, Chem. – Eur. J., 2016, 22, 10821–10824 CrossRef PubMed.
- Y. Cheng, W. Dong, K. Parthasarathy and C. Bolm, Org. Lett., 2017, 19, 726–729 CrossRef PubMed.
- M. R. Yadav, R. K. Rit and A. K. Sahoo, Chem. – Eur. J., 2012, 18, 5541–5545 CrossRef PubMed.
- R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2014, 16, 968–971 CrossRef PubMed.
- M. R. Yadav, R. K. Rit and A. K. Sahoo, Org. Lett., 2013, 15, 1638–1641 CrossRef PubMed.
- M. R. Yadav, R. K. Rit, M. Shankar and A. K. Sahoo, J. Org. Chem., 2014, 79, 6123–6134 CrossRef PubMed.
- M. R. Yadav, M. Shankar, E. Ramesh, K. Ghosh and A. K. Sahoo, Org. Lett., 2015, 17, 1886–1889 CrossRef PubMed.
- K. Ghosh, R. K. Rit, E. Ramesh and A. K. Sahoo, Angew. Chem., Int. Ed., 2016, 55, 7821–7825 CrossRef PubMed.
- M. Shankar, T. Guntreddi, E. Ramesh and A. K. Sahoo, Org. Lett., 2017, 19, 5665–5668 CrossRef PubMed.
- M. Shankar, K. Ghosh, K. Mukherjee, R. K. Rit and A. K. Sahoo, Org. Lett., 2016, 18, 6416–6419 CrossRef PubMed.
- K. Raghuvanshi, D. Zell and L. Ackermann, Org. Lett., 2017, 19, 1278–1281 CrossRef PubMed.
- Y. Yokoyama, Y. Unoh, R. A. Bohmann, T. Satoh, K. Hirano, C. Bolm and M. Miura, Chem. Lett., 2015, 44, 1104–1106 CrossRef.
- J. E. Spangler, Y. Kobayashi, P. Verma, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11876–11879 CrossRef PubMed.
- P. Jain, P. Verma, G. Xia and J.-Q. Yu, Nat. Chem., 2016, 9, 140–144 CrossRef PubMed.
- C. Zhong-Jian, L. Chen-Xu, G. Qing and Y. Shu-Li, Angew. Chem., Int. Ed., 2018, 57, 1296–1299 CrossRef PubMed.
- A. T. Tran and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10530–10534 CrossRef PubMed.
- Y. Zhao, F. Han, L. Yang and C. Xia, Org. Lett., 2015, 17, 1477–1480 CrossRef PubMed.
- Y.-N. Ma, S.-X. Li and S.-D. Yang, Acc. Chem. Res., 2017, 50, 1480–1492 CrossRef PubMed.
- (a) Y.-N. Ma, H.-Y. Zhang and S.-D. Yang, Org. Lett., 2015, 17, 2034–2037 CrossRef PubMed; (b) Z. S. Han, L. Zhang, Y. Xu, J. D. Sieber, M. A. Marsini, Z. Li, J. T. Reeves, K. R. Fandrick, N. D. Patel, J.-N. Desrosiers, B. Qu, A. Chen, D. M. Rudzinski, L. P. Samankumara, S. Ma, N. Grinberg, F. Roschangar, N. K. Yee, G. Wang, J. J. Song and C. H. Senanayake, Angew. Chem., Int. Ed., 2015, 54, 5474–5477 CrossRef PubMed; (c) S.-B. Yan, S. Zhang and W.-L. Duan, Org. Lett., 2015, 17, 2458–2461 CrossRef PubMed.
- S.-X. Li, Y.-N. Ma and S.-D. Yang, Org. Lett., 2017, 19, 1842–1845 CrossRef PubMed.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, ChemCatChem, 2017, 9, 3117–3120 CrossRef.
- Y. Unoh, T. Satoh, K. Hirano and M. Miura, ACS Catal., 2015, 5, 6634–6639 CrossRef.
- Z. Liu, J.-Q. Wu and S.-D. Yang, Org. Lett., 2017, 19, 5434–5437 CrossRef PubMed.
- Y. Yang, X. Qiu, Y. Zhao, Y. Mu and Z. Shi, J. Am. Chem. Soc., 2016, 138, 495–498 CrossRef PubMed.
- R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172–8174 CrossRef PubMed.
- Y. Yang, R. Li, Y. Zhao, D. Zhao and Z. Shi, J. Am. Chem. Soc., 2016, 138, 8734–8737 CrossRef PubMed.
- Y. Yokoyama, Y. Unoh, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 7649–7655 CrossRef PubMed.
- Y. Unoh, Y. Yokoyama, T. Satoh, K. Hirano and M. Miura, Org. Lett., 2016, 18, 5436–5439 CrossRef PubMed.
- W. H. Jeon, J.-Y. Son, S.-E. Kim and P. H. Lee, Adv. Synth. Catal., 2015, 357, 811–817 CrossRef.
- J.-Y. Son, H. Kim, W. H. Jeon, Y. Baek, B. Seo, K. Um, K. Lee and P. H. Lee, Adv. Synth. Catal., 2017, 359, 3194–3206 CrossRef.
- L. Y. Chan, X. Meng and S. Kim, J. Org. Chem., 2013, 78, 8826–8832 CrossRef PubMed.
- B. C. Chary, S. Kim, Y. Park, J. Kim and P. H. Lee, Org. Lett., 2013, 15, 2692–2695 CrossRef PubMed.
- L. Y. Chan, S. Kim, T. Ryu and P. H. Lee, Chem. Commun., 2013, 49, 4682 RSC.
- L. Y. Chan, L. Cheong and S. Kim, Org. Lett., 2013, 15, 2186–2189 CrossRef PubMed.
- X.-H. Hu, X.-F. Yang and T.-P. Loh, Angew. Chem., Int. Ed., 2015, 54, 15535–15539 CrossRef PubMed.
- L.-Y. Jiao, A. V. Ferreira and M. Oestreich, Chem. – Asian J., 2016, 11, 367–370 CrossRef PubMed.
- Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632–635 CrossRef PubMed.
- Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 364–367 CrossRef PubMed.
- Y. Sun and N. Cramer, Chem. Sci., 2018, 9, 2981–2985 RSC.
- K. M. Crawford, T. R. Ramseyer, C. J. A. Daley and T. B. Clark, Angew. Chem., Int. Ed., 2014, 53, 7589–7593 CrossRef PubMed.
- X. Qiu, M. Wang, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 7233–7237 CrossRef PubMed.
- Q. Ge, J. Zong, B. Li and B. Wang, Org. Lett., 2017, 19, 6670–6673 CrossRef PubMed.
- F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC.
- G. R. Jones and Y. Landais, Tetrahedron, 1996, 52, 7599–7662 CrossRef.
- S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486–1499 CrossRef PubMed.
- M. Parasram and V. Gevorgyan, Acc. Chem. Res., 2017, 50, 2038–2053 CrossRef PubMed.
- J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef PubMed.
- P. I. Djurovich, A. R. Dolich and D. H. Berry, J. Chem. Soc., Chem. Commun., 1994, 1897–1898 RSC.
- E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70–73 CrossRef PubMed.
- Y. Kuninobu, T. Nakahara, H. Takeshima and K. Takai, Org. Lett., 2013, 15, 426–428 CrossRef PubMed.
- M. Murai, H. Takeshima, H. Morita, Y. Kuninobu and K. Takai, J. Org. Chem., 2015, 80, 5407–5414 CrossRef PubMed.
- B. Su and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 12137–12140 CrossRef PubMed.
- Y. Kuninobu, K. Yamauchi, N. Tamura, T. Seiki and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 1520–1522 CrossRef PubMed.
- Q.-W. Zhang, K. An, L.-C. Liu, Q. Zhang, H. Guo and W. He, Angew. Chem., Int. Ed., 2017, 56, 1125–1129 CrossRef PubMed.
- T. Shibata, T. Shizuno and T. Sasaki, Chem. Commun., 2015, 51, 7802–7804 RSC.
- M. Murai, K. Matsumoto, Y. Takeuchi and K. Takai, Org. Lett., 2015, 17, 3102–3105 CrossRef PubMed.
- Q.-W. Zhang, K. An, L.-C. Liu, Y. Yue and W. He, Angew. Chem., Int. Ed., 2015, 54, 6918–6921 CrossRef PubMed.
- E. M. Simmons and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 17092–17095 CrossRef PubMed.
- T. Lee, T. W. Wilson, R. Berg, P. Ryberg and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 6742–6745 CrossRef PubMed.
- T. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 4879–4886 CrossRef PubMed.
- B. Su, T.-G. Zhou, X.-W. Li, X.-R. Shao, P.-L. Xu, W.-L. Wu, J. F. Hartwig and Z.-J. Shi, Angew. Chem., Int. Ed., 2017, 56, 1092–1096 CrossRef PubMed.
- B. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 6586–6589 CrossRef PubMed.
- C. Karmel, B. Li and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 1460–1470 CrossRef PubMed.
- A. Bunescu, T. W. Butcher and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 1502–1507 CrossRef PubMed.
- T. Lee and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 8723–8727 CrossRef PubMed.
- T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534–7535 CrossRef PubMed.
- S. H. Cho and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8157–8160 CrossRef PubMed.
- D. W. Robbins, T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 4068–4069 CrossRef PubMed.
- M. A. Larsen, S. H. Cho and J. Hartwig, J. Am. Chem. Soc., 2016, 138, 762–765 CrossRef PubMed.
- B. Su, T.-G. Zhou, P.-L. Xu, Z.-J. Shi and J. F. Hartwig, Angew. Chem., Int. Ed., 2017, 56, 7205–7208 CrossRef PubMed.
- C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2009, 131, 10844–10845 CrossRef PubMed.
- C. Huang and V. Gevorgyan, Org. Lett., 2010, 12, 2442–2445 CrossRef PubMed.
- M. Mewald, J. A. Schiffner and M. Oestreich, Angew. Chem., Int. Ed., 2012, 51, 1763–1765 CrossRef PubMed.
- C. Huang, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 12406–12409 CrossRef PubMed.
- C. Huang, N. Ghavtadze, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 17630–17633 CrossRef PubMed.
- C. Wang and H. Ge, Chem. – Eur. J., 2011, 17, 14371–14374 CrossRef PubMed.
- Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2015, 54, 2255–2259 CrossRef PubMed.
- Y. Lin, K.-Z. Jiang, J. Cao, Z.-J. Zheng, Z. Xu, Y.-M. Cui and L.-W. Xu, Adv. Synth. Catal., 2017, 359, 2247–2252 CrossRef.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef.
- D.-G. Yu, F. de Azambuja and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 7710–7712 CrossRef PubMed.
- R. Kuppusamy, K. Muralirajan and C.-H. Cheng, ACS Catal., 2016, 6, 3909–3913 CrossRef.
- B. Chattopadhyay, J. E. Dannatt, I. L. Andujar-De Sanctis, K. A. Gore, R. E. Maleczka, D. A. Singleton and M. R. Smith, J. Am. Chem. Soc., 2017, 139, 7864–7871 CrossRef PubMed.
- G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245–1247 CrossRef PubMed.
- S. Kawamorita, H. Ohmiya, K. Hara, A. Fukuoka and M. Sawamura, J. Am. Chem. Soc., 2009, 131, 5058–5059 CrossRef PubMed.
- E. Tan, O. Quinonero, M. E. de Orbe and A. M. Echavarren, ACS Catal., 2018, 8, 2166–2172 CrossRef PubMed.
- S. Xiaochao, N. Masayoshi and H. Zhaomin, Angew. Chem., Int. Ed., 2016, 55, 14812–14817 CrossRef PubMed.
- X. Shi, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2016, 138, 6147–6150 CrossRef PubMed.
- Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2014, 43, 2799–2823 RSC.
- D.-W. Gao, Q. Gu, C. Zheng and S.-L. You, Acc. Chem. Res., 2017, 50, 351–365 CrossRef PubMed.
- L. A. López and E. López, Dalton Trans., 2015, 44, 10128–10135 RSC.
- D.-Y. Zhu, P. Chen and J.-B. Xia, ChemCatChem, 2016, 8, 68–73 CrossRef.
- W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560–14561 CrossRef PubMed.
- W. C. P. Tsang, R. H. Munday, G. Brasche, N. Zheng and S. L. Buchwald, J. Org. Chem., 2008, 73, 7603–7610 CrossRef PubMed.
- B.-J. Li, S.-L. Tian, Z. Fang and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1115–1118 CrossRef PubMed.
- J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184–16186 CrossRef PubMed.
- M. Miura, T. Tsuda, T. Satoh, S. Pivsa-Art and M. Nomura, J. Org. Chem., 1998, 63, 5211–5215 CrossRef.
- Z. Liang, L. Ju, Y. Xie, L. Huang and Y. Zhang, Chem. – Eur. J., 2012, 18, 15816–15821 CrossRef PubMed.
- V. Rajeshkumar, T.-H. Lee and S.-C. Chuang, Org. Lett., 2013, 15, 1468–1471 CrossRef PubMed.
- Z. Liang, J. Zhang, Z. Liu, K. Wang and Y. Zhang, Tetrahedron, 2013, 69, 6519–6526 CrossRef.
- Z. Liang, R. Feng, H. Yin and Y. Zhang, Org. Lett., 2013, 15, 4544–4547 CrossRef PubMed.
- Z. Liang, J. Yao, K. Wang, H. Li and Y. Zhang, Chem. – Eur. J., 2013, 19, 16825–16831 CrossRef PubMed.
- Z.-J. Cai, C. Yang, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2015, 80, 7928–7936 CrossRef PubMed.
- K. Takamatsu, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 2892–2895 CrossRef PubMed.
- S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef PubMed.
- C. Jean-Ho, L. Pi-Shan, L. Ya-Ming, S. Wei-Ting and W. Ming-Jung, Chem. – Eur. J., 2011, 17, 13613–13620 CrossRef PubMed.
- C. Suzuki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2015, 17, 1597–1600 CrossRef PubMed.
- A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef PubMed.
- A. P. Smalley, J. D. Cuthbertson and M. J. Gaunt, J. Am. Chem. Soc., 2017, 139, 1412–1415 CrossRef PubMed.
- M. Nappi, C. He, W. G. Whitehurst, B. G. N. Chappell and M. J. Gaunt, Angew. Chem., Int. Ed., 2018, 57, 3178–3182 CrossRef PubMed.
- D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Gaunt, Science, 2016, 354, 851–857 CrossRef PubMed.
- J. R. Cabrera-Pardo, A. Trowbridge, M. Nappi, K. Ozaki and M. J. Gaunt, Angew. Chem., Int. Ed., 2017, 56, 11958–11962 CrossRef PubMed.
- K. F. Hogg, A. Trowbridge, A. Alvarez-Pérez and M. J. Gaunt, Chem. Sci., 2017, 8, 8198–8203 RSC.
- C. He and M. J. Gaunt, Chem. Sci., 2017, 8, 3586–3592 RSC.
- C. He and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 15840–15844 CrossRef PubMed.
- J. C. Fox, R. E. Gilligan, A. K. Pitts, H. R. Bennett and M. J. Gaunt, Chem. Sci., 2016, 7, 2706–2710 RSC.
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef.
- Q. Ding, S. Ye, G. Cheng, P. Wang, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 417–425 CrossRef PubMed.
- H. J. Davis, M. T. Mihai and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759–12762 CrossRef PubMed.
- H. J. Davis, G. R. Genov and R. J. Phipps, Angew. Chem., Int. Ed., 2017, 56, 13351–13355 CrossRef PubMed.
- G. Olivo, G. Farinelli, A. Barbieri, O. Lanzalunga, S. Di Stefano and M. Costas, Angew. Chem., Int. Ed., 2017, 56, 16347–16351 CrossRef PubMed.
- T. Nakamura, K. Suzuki and M. Yamashita, J. Am. Chem. Soc., 2017, 139, 17763–17766 CrossRef PubMed.
- Z. Qi, M. Wang and X. Li, Org. Lett., 2013, 15, 5440–5443 CrossRef PubMed.
- Z. Zhou, G. Liu, Y. Chen and X. Lu, Adv. Synth. Catal., 2015, 357, 2944–2950 CrossRef.
- H. Wang, M. Moselage, M. J. González and L. Ackermann, ACS Catal., 2016, 6, 2705–2709 CrossRef.
- X. Guo, J. Han, Y. Liu, M. Qin, X. Zhang and B. Chen, J. Org. Chem., 2017, 82, 11505–11511 CrossRef PubMed.
- Y. Li, F. Xie, Y. Liu, X. Yang and X. Li, Org. Lett., 2018, 20, 437–440 CrossRef PubMed.
- F. Xie, S. Yu, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2016, 55, 15351–15355 CrossRef PubMed.
- Q. Wang, Y. Xu, X. Yang, Y. Li and X. Li, Chem. Commun., 2017, 53, 9640–9643 RSC.
- D. Bai, Q. Jia, T. Xu, Q. Zhang, F. Wu, C. Ma, B. Liu, J. Chang and X. Li, J. Org. Chem., 2017, 82, 9877–9884 CrossRef PubMed.
- H. Deng, H. Li, W. Zhang and L. Wang, Chem. Commun., 2017, 53, 10322–10325 RSC.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- C. Sambiagio, R. H. Munday, A. John Blacker, S. P. Marsden and P. C. McGowan, RSC Adv., 2016, 6, 70025–70032 RSC.
- C. J. Pink, H.-T. Wong, F. C. Ferreira and A. G. Livingston, Org. Process Res. Dev., 2008, 12, 589–595 CrossRef.
- Akanksha and D. Maiti, Green Chem., 2012, 14, 2314–2320 RSC.
- A. Mouret, L. Leclercq, A. Muhlbauer and V. Nardello-Rataj, Green Chem., 2014, 16, 269–278 RSC.
- R. Savela, J. Warna, D. Y. Murzin and R. Leino, Catal. Sci. Technol., 2015, 5, 2406–2417 RSC.
- http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180284.pdf .
- S. Santoro, S. I. Kozhushkov, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 3471–3493 RSC.
- A. J. Hunt, Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, The Royal Society of Chemistry, 2016, pp. 54–62, 10.1039/9781782625940-00054.
- C. A. Hone, D. M. Roberge and C. O. Kappe, ChemSusChem, 2017, 10, 32–41 CrossRef PubMed.
- L. V. Desai, K. J. Stowers and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 13285–13293 CrossRef PubMed.
- H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 10386–10390 CrossRef PubMed.
- D. Lapointe, T. Markiewicz, C. J. Whipp, A. Toderian and K. Fagnou, J. Org. Chem., 2010, 76, 749–759 CrossRef PubMed.
- Y. J. Liu, H. Xu, W. J. Kong, M. Shang, H. X. Dai and J. Q. Yu, Nature, 2014, 515, 389–393 CrossRef PubMed.
Footnotes |
| † Dedicated to the memory of Prof. Walther Schmidt. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cs00201k |
| This journal is © The Royal Society of Chemistry 2018 |