Differential adhesion of fibroblast and neuroblastoma cells on size- and geometry-controlled nanofibrils of the extracellular matrix†
Peter
Kaiser
ab and
Joachim P.
Spatz
*ab
aDepartment of New Materials and Biosystems, Max-Planck-Institute for Metals Research, Heisenbergstrasse 3, 70569, Stuttgart, Germany. E-mail: spatz@mf.mpg.de
bDepartment of Biophysical Chemistry, University of Heidelberg, Germany, INF 253, D - 69 120, Heidelberg, Germany. E-mail: Joachim.Spatz@urz.uni-heidelberg.de
First published on 12th October 2009
Abstract
We present a universal method to produce highly regular arrays of nanofibrils constituted by the extracellular matrix (ECM) proteins fibronectin (FN), collagen I (COL I) and EHS laminin (EHS LN). Adjusting the de-wetting parameters of a protein solution on a hydrophobic silicon micropillar structure, we control the mean nanofibril diameter between 20 nm and 160 nm. Array geometry predetermines nanofibril diameter, while protein concentration and de-wetting speed have to be adapted to yield high regularity. ECM nanofibrils span the space between adjacent pillars and immunofluorescence labelling of LN and FN nanofibrils allows us to discriminate both species in mixed networks. The biological function of ECM nanofibrils was verified in cell adhesion experiments after transfer of the nanofibrils onto non-adhesive polyethylene glycol (PEG) hydrogels. Co-cultures of fibroblasts and neuroblastoma cells on vertically arranged nanofibrils of FN and LN showed differential adhesion of the distinct cell types.
Introduction
While there is growing interest in the mechanical, structural and functional properties of fibrillar extracellular matrix (ECM),1–3 current research is hampered by the lack of suitable model systems. One of the major proteinaceous ECM constituents is fibronectin (FN), which is essential for embryonic development.4 Cells secrete FN and assemble it into nanoscale fibrils via a force-dependent mechanism.5,6The ability of FN fibrils to elastically stretch to several times their length in vivo7 has been suggested to include molecular unfolding of FN,8 which renders this protein an interesting target to study mechanochemical signalling events.
Despite the fact that cells in vivo adhere to FN fibrils, only a few investigations have used substrates based on artificial FN fibrils,9,10 which would enable the study of cellular responses entirely due to the interaction of cells with FN fibrils.
Reported methods to produce FN fibrils are:
(1) the manual pulling of single fibrils out of a concentrated drop of FN,9–11 yielding fibrils with diameters between 0.5 µm and 5 µm;11
(2) the self-assembly of FN networks underneath a lipid monolayer followed by gradual expansion of the film,12 yielding fibrils with several microns in diameter;
(3) surface-induced fibrillogenesis on hydrophobic substrates,13 yielding nanofibrils with diameters between 20 nm and 100 nm;
(4) the spontaneous aggregation of FN into fibrils via re-folding of FN during dialysis,14 yielding nanofibrils between 5 nm and 20 nm;
(5) the addition of anastellin, an aggregation-inducing fragment of FN.15
The drawbacks of these approaches are that method (1) can only yield few individual fibrils and the control of fibril align-ment and diameter is not possible for the remaining methods.
Other fibrillar ECM components that have been focus of research are: (i) collagen I (COL I), a major component of cartilage, and (ii) laminin-111, which can be isolated from Engelbreth–Holm–Swarm (EHS) tumors16 and is essential for embryonic development.
Extensive studies in recent decades have shown that fibrillogenesis of COL I is a spontaneous, entropy-driven self-assembly process.2,17 In recent years, this knowledge has been used to fabricate highly ordered collagen matrices18 and to investigate the structural19,20 and mechanical21 properties of collagen fibrils.
In contrast to collagen, the function and assembly of laminin (LN) fibrils is poorly understood. Tsiper et al.22 revealed that Schwann cells, which myelinate axons in the peripheral nerve system (PNS),23 form fibrils when supplied with EHS LN. While a role for LN fibrils in Schwann cell guidance during development was suggested by the authors, neither the mechanism of LN fibrillogenesis nor the physiological relevance of these structures has been elucidated.
Photholithography allows the production of three-dimensional microstructures, which have been successfully used as sensors of cellular traction forces,24,25 as stamps for microcontact printing,24 or as novel substrates to study motor proteins.26 Their superhydrophobic properties are of general interest in science and technology27,28 and are essential for the method presented here.
Previous work in our laboratory demonstrated the possibility to produce free-hanging FN fibrils between silicon micropillars.29 Fibrillogenesis was induced by forces generated when wetting a micropillar array after the spontaneous formation of a thin FN layer at the air–buffer interface. This method enabled limited control over fibril diameter and directionality. Since, similar to methods 2 and 3 above, FN fibrillogenesis was induced by surface forces, we hypothesized that precise control of the wetting geometry via dip-coating should allow control over FN fibril alignment.
In this work, we describe a method to produce highly regular FN fibrils of controlled mean diameters between 20 nm and 160 nm. Extending this method to other fibril-forming ECM components, like collagen I (COL I) and EHS laminin (EHS LN), we were able to produce nanofibrils with similar diameter ranges. Transfer and covalent linkage of ECM nanofibrils onto non-adhesive polyethylene glycol (PEG) substrates allowed cell adhesion and alignment of human foreskin fibroblast (HFF) and SH-SY5Y neuroblastoma cells on FN and LN, respectively.
Results and discussion
Fabrication of fibronectin nanofibrils
In our initial experiment, a 10 µl drop of FN in phosphate-buffered saline (PBS) was manually pulled over a superhydrophobic micropillar surface. While the resulting FN fibrils aligned with the pulling direction in the central region of the meniscus, they were randomly oriented at lateral areas (Fig. S1†), where the receding meniscus was highly curved. Fig. 1c shows how this problem was solved by moving the substrate relative to a droplet that is spread along a glass rod. This resulted in a meniscus that is perpendicular to the pulling direction along the micropillar array. Having developed this method on silicon micropillar arrays (Fig. 1a–c), we were able to apply it on poly(dimethylsiloxane) (PDMS) microstructures, which have the advantage of being both transparent and elastomeric and can thus be used in force-sensing experiments (Fig. 1d).30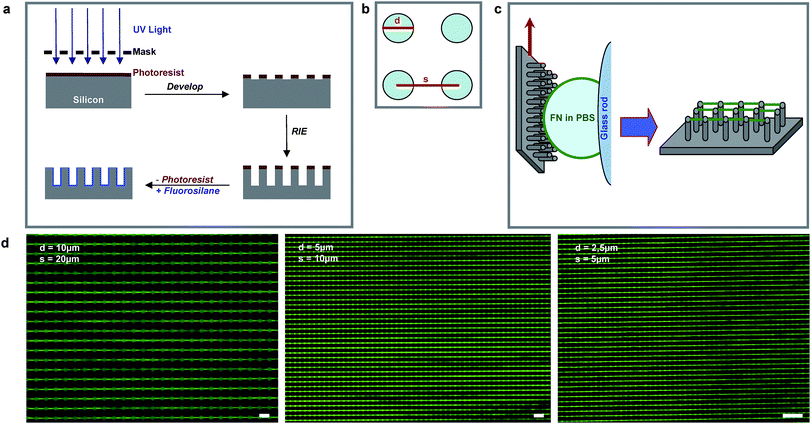 | ||
Fig. 1 (a) Fabrication of silicon microstructures was done via photolithography followed by reactive ion etching (RIE) and photoresist lift-off. The hydrophilic silicone surface was rendered hydrophobic by fluorosilane coating. (b) The pillar diameter, d, and the center-to-center distance, s, defined the microarray. (c) The microstructures were brought in contact with a drop of FN diluted in PBS. Fibrils formed during de-wetting while the substrate moved upwards. (d) Fluorescence micrographs of ATTO![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 488-labelled FN fibrils produced on PDMS microarrays with different geometries. Scale bar: 20 µm. 488-labelled FN fibrils produced on PDMS microarrays with different geometries. Scale bar: 20 µm. | ||
Bovine serum albumin (BSA) did not form nanofibrils at high protein concentrations of up to 1 mg ml−1, indicating that proteins which do not form fibrils in vivo will not form fibrils using the described method (Fig. S2†).
Substrate geometry determines nanofibril diameter
Using hydrophobic silicon micropillar arrays, we determined the optimum of both the solution concentration of FN and the pulling speed for each substrate geometry (Fig. 1b).Scanning electron microscopy (SEM) analysis provided the distribution of fibril diameters for each substrate. The parameters depicted in Fig. 2 yielded optimal regularity and narrowest distributions of the FN nanofibril diameter over substrate areas as large as 2 cm2.
 | ||
| Fig. 2 Histograms and fitted normal distributions of FN fibril diameter for the microarray geometries depicted in Fig. 1d. The inset text indicates microarray geometry with parameters s and d, as detailed in Fig. 1b, protein concentration, c, and pulling speed, v. For each histogram, 350 fibrils over three independent experiments were measured. | ||
Similar to findings by Guan et al.,31 who used an analogous method to fabricate DNA31,32 and polymer33 nanowires, the diameter of FN nanofibrils is mainly dependent on the diameter of the underlying pillars. In contrast to the fabrication of DNA nanowires by Guan et al., the range of FN concentrations and pulling speeds at which regular FN nanofibril arrays could be accomplished was specific for each substrate geometry. Applying too-high concentrations of protein or too-low pulling speeds resulted in the formation of thick and randomly oriented fibrils, while low FN concentrations or higher pulling speeds led to defects in the nanofibrillar array (Fig. S3†) and to a widening in the distribution of nanofibril diameters (Fig. S4†). This could be due to the tendency of fibronectin to accumulate at the air–buffer interface, which introduces the dynamics of protein diffusion and exchange at the buffer–substrate interface to the fabrication process. Different from DNA molecules, which are negatively charged and thus self-repellent, FN has several self-binding sites, which favors the formation of cross-connected nanofibrils, as shown in Fig. S3. The requirement for accessible self-binding sites is further supported by recent work by Pabba et al.,34 where fibrinogen formed nanofibrils only in the presence of its activator thrombin.
The observation that lower protein concentrations are needed to produce nanofibrils on small micropillars (Fig. 2) proved useful in the investigation of other protein systems. Thus, all further experiments employing ECM protein components were first performed on arrays with the smallest micropillar diameters available.
Fabrication of laminin nanofibrils
We tested the hypothesis that our method can be used to produce nanofibrils of other fibrillar ECM components.Laminin purified from Engelbreth–Holm–Swarm tumors (EHS LN) yielded regular arrays of nanofibrils at 4-fold higher protein concentrations and 10-fold lower pulling speeds (Fig. 3) compared to FN. Given the tendency of LN to quickly assemble at the air–buffer interface (Fig. 4), the reduced possibilities of LN to self-associate compared to FN might be responsible for this effect.
 | ||
| Fig. 3 SEM images of nanofibrils formed by different ECM proteins. Both EHS LN and COL I (B) formed nanofibrils with increasing thickness upon increasing the micropillar diameter. Collagen I (S) consistently yielded nanofibrils with diameters below 30 nm. Scale bar: 200 nm. | ||
 | ||
| Fig. 4 Surface activity of ECM proteins and BSA. Already after 120 seconds, the surface pressure rises to more than 75% of the value reached after 1 h (Fig. S6†). BSA (dash-dotted line, black) results in a rapid increase in surface pressure. Unexpectedly, the surface pressure resulting from LN (dashed line, red) was the highest of all three ECM proteins. | ||
We carried out a western blot analysis to rule out potential contamination of EHS LN preparations with FN, as shown in Fig. S5†. Supporting this evidence, immunofluorescence staining of the produced fibrils was positive for EHS LN and negative for FN, as shown in Fig. 5.
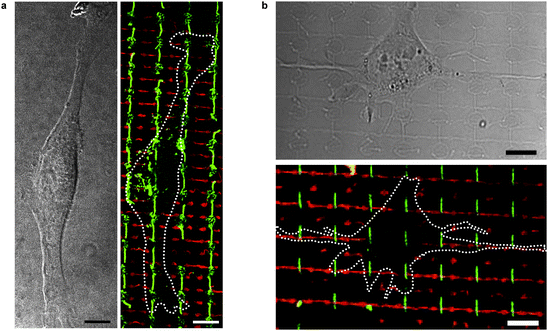 | ||
| Fig. 5 (a) HFF cell adhering to FN nanofibrils (green) on a combined LN/FN nanofibril substrate. LN is shown in red. For reasons of visibility, the panel on the right displays the circumference of the cell seen in differential interference contrast (DIC) mode on the left panel. (b) SH-SY5Y neuroblastoma cell adhering to FN (green) and LN (red) nanofibrils. The cell produces outgrowths along the LN fibril in the centre. In addition to differential adhesive behaviour, distinction between both cell types in co-culture was possible by morphological differences in size and shape. Scale bar: 10 µm. | ||
Fabrication of collagen nanofibrils
Initial experiments using COL I purified from calf skin produced highly regular arrays of nanofibrils with diameters of approximately 20 nm independently of pillar diameter, protein concentration and pulling speed (Fig. 3).Three collagen preparations from two different providers were subsequently tested for their ability to form nanofibrils:
(1) COL I purified from calf skin, referred to as COL I (C);
(2) COL I purified from rat tendon, referred to as COL I (S);
(3) COL I purified from rat tendon, referred to as COL I (B).
In summary, both COL I (S) and COL (C) yielded fibrils with diameters below 30 nm at concentrations up to 0.5 mg ml−1. COL I (B) yielded fibrils with diameters above 50 nm at concentrations below 0.2 mg ml−1.
Based on SDS PAGE and western blot analysis, we conclude that the concentration of intact COL I α-chains was lower in the protein preparations that yielded thinner compared to those that yielded thicker nanofibrils (Fig. S5). However, it cannot be ruled out that differences in protein integrity or tropocollagen helix content might be responsible for the observed effects.
Effect of ECM proteins on surface pressure
The ability of proteins to accumulate at interfaces35 is relevant for the fabrication process described here. Hence, the change in surface tension upon protein addition onto a layer of PBS was investigated (Fig. 4). While BSA, FN and LN produce quick and significant changes in surface tension of the air–buffer interface, COL I shows a less pronounced effect. Combined with the observation that LN forms nanofibrils at increased protein concentrations and BSA fails to produce nanofibrils, our results suggest that the formation of nanofibrils depends on an interplay between (i) the surface activity and (ii) the self-association capabilities of proteins located at the air–buffer interface.Cell co-culture and adhesion on crossed fibronectin and laminin nanofibril arrays
In order to demonstrate the functionality of ECM nanofibrils, we developed a model system for cell experiments, which allows us to immobilize ECM nanofibrils in an arbitrary orientation relative to each other. Since the ECM nanofibrils were formed by de-wetting of a superhydrophobic surface, the nanofibril arrays were in a dry state.Wetting of the micropillar surface followed by covalent attachment of nanofibrils onto PEG hydrogels reconstituted the nanofibrils in buffer and simultaneously eliminated substrate microtopography (Scheme 1).
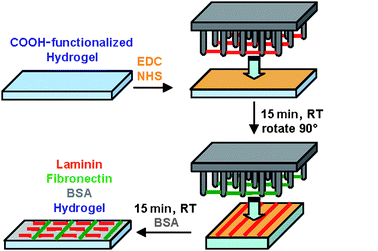 | ||
| Scheme 1 Transfer and covalent attachment of LN (red) and FN (green) nanofibrils onto PEG hydrogels. Hydrogels were copolymerized with 2-carboxyethylacrylate to introduce –COOH functional groups. Standard EDC/NHS coupling was used to couple amine functional groups on the ECM nanofibril surface to carboxyl moieties on the hydrogel surface. | ||
After sequential transfer of LN and FN nanofibrils, we investigated the differential binding of SH-SY5Y and HFF cells to LN and FN nanofibrils, respectively (Fig. 5). Cells adhere to the crossed ECM nanofibril network via paxillin-containing adhesion sites (Fig. 6), which demonstrates the biofunctionality of the fibrils after transfer. While HFF cells line perfectly with FN nanofibrils and are not influenced by the directionality of LN nanofibrils, SH-SY5Y cells interact with both LN and FN nanofibrils, but show distinct outgrowths along the LN fibrils (Figs. 5 and 6, Fig. S7†).
 | ||
| Fig. 6 Colocalization of focal adhesion protein, paxillin (blue), with FN (green) and LN (red) nanofibrils. (a) An HFF cell adhering exclusively to FN nanofibrils. The white color in the central image indicates colocalization of FN and paxillin. (b) An SH SY5Y cell adhering to both FN and LN nanofibrils. While the LN nanofibril direction dictates orientation, FN nanofibrils support cell adhesion, as indicated by white arrows in the center image. Scale bar: 5 µm. | ||
This enables the distinction of fibroblast-like cells from neuroblastoma cells in co-culture assays due to their differential adhesion to different ECM nanofibrils.
Experimental
Production of silicon and PDMS microarrays
Photoresist (#S1818G2, Rohm & Haas Deutschland GmbH) was used according to the manufacturer's protocol to yield a resist thickness of 1.7 µm. The microstructure was hard-baked for 15 min at 115 °C before transfer into a Plasmalab 80 Plus (Oxford Instruments) reactive ion etcher. The structures were etched at −10 °C to a depth ranging between 3 µm and 20 µm at an etch rate of 40 nm per cycle. Each cycle consisted of (i) a 8 s passivation step at 70 mTorr chamber pressure using a flow of 50 sccm CHF3 gas at an RF power of 30 W and an ICP power of 100 W and (ii) an etch step at 40 mTorr using 15 sccm SF6 gas together with 18 sccm CHF3 gas at an RF power of 30 W and an ICP power of 300 W.The remaining photoresist was dissolved in S1818G2 remover (Rohm&Haas Germany) for 5 min and organic residues were etched away in a 1:3 mixture of 30% v/v H2O2 and H2SO4. After copious rinsing with UltraPure water, the structure was dried and activated in an oxygen plasma etcher. Vapor deposition of 50 µl tridecafluoro-1,1,2,2-tetrahydrooctyl-1-triethoxysilane (#T2494, UCT) was performed in a desiccator at 1 mbar for 30 min.
To produce poly(dimethylsiloxane) PDMS pillar arrays, a negative PDMS mask was created using silicon micropillar structures as a casting mould. SYLGARD 184 (#608284, Sasco Holz GmbH) was cured at 140 °C for 60 min and rendered non-adhesive analogous to the silanization protocol described above. This negative was then coated with freshly degassed PDMS, pressed face-down onto a coverslip (#H878.1, Carl Roth) and cured as above.
Isolation and labelling of fibronectin
Fibronectin was isolated using a previously described procedure.11 Labelling of FN with ATTO488 maleimide (#28562, Fluka) was performed according to the manufacturer's protocol.
Production of ECM fibrils on micropillar arrays
A drop of 100 µl protein solution in PBS was pipetted onto a glass rod and the structure to be coated was immediately brought into contact with the droplet. As soon as the meniscus line covered the width of the microarray, the structure was pulled upwards at constant speed. Fibronectin concentration and pulling speed varied between 12.5–50 µg ml−1 and 5–20 mm min−1, depending on the microarray geometry as detailed in Fig. 2.EHS LN (#08-125, Millipore, Schwalbach, Germany) was used at concentrations between 25 µg ml−1 and 100 µg ml−1.
COL I from rat tail (COL (S), #C3867, SIGMA-Aldrich) and COL I from calf skin (COL (C), #C8919, SIGMA-Aldrich) were used at concentrations of 500 µg ml−1. COL I from rat tail (COL (B), #BT-274, Biomedical Technologies, Inc.) was employed at concentrations between 50 µg ml−1 and 200 µg ml−1. All LN and COL I preparations were used at a pulling speed of 2 mm min−1.
Scanning electron microscopy analysis
For each parameter set, three independent samples were produced. Three areas within the 1 × 2 cm microarray were randomly picked and fibrils were imaged in a Zeiss Ultra 55 Electron Microscope using the InLens detector at an acceleration voltage of 1 kV. Fibril diameters were manually extracted using the software provided with the instrument.Surface pressure measurement
Experiments were performed at room temperature using a NIMA 112D Langmuir–Blodgett trough. After cleaning in 2% Hellmanex (#634-0442, VWR International), the trough was copiously rinsed with UltraPure water. For all proteins, 140 µg of protein were added dropwise onto a 70 ml layer of PBS. Surface pressure was recorded over time spans of at least one hour.Transfer of fibronectin fibrils onto hydrogels
Polyethylene glycol diacrylate with a mean molecular weight of 10 kDa (1 g) was mixed with UltraPure water (1655 µl), 2-carboxyethyl acrylate (132.5 µl, #552348, Sigma-Aldrich) and 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (150 µl, Sigma-Aldrich, #410896). After UV curing (20 mW cm−2), swelling of the hydrogels in UltraPure water resulted in detachment from the casting chamber.For covalent attachment of ECM nanofibrils, hydrogels were activated for 60 minutes face-down in a drop of solution containing 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (78 mg, #153-0990, BioRad) and N-hydroxysuccinimide (19.5 mg, #56480, Fluka) in MES buffer (1000 µl, pH 5.5). Silicon microstructures containing nanofibril arrays were wetted in PBS containing MOWIOL 4–88 (0.1% w/v, #475904, Calbiochem), rinsed with PBS, and the array was put onto the activated surface of the hydrogel. This setup was enclosed between two objective slides and fixed with paper clamps. After 15 min, the hydrogel was brought into contact with the second array. The reaction was quenched by incubating the hydrogel in PBS containing bovine serum albumin (BSA, 1% w/v, #11931, SERVA) for 1 h. After rinsing in PBS, the hydrogel was transferred into a 35 mm Petri dish.
Cell culture and adhesion experiments
Human foreskin fibroblast (HFF) cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Invitrogen, #41966) containing fetal bovine serum (FBS, 10% v/v, #10050, Invitrogen). SH-SY5Y Neuroblastoma cells were cultured in DMEM containing FBS (20% v/v) and penicillin/streptomycin (1% v/v, #15140, Invitrogen). Cells were kept at 37 °C and 5% CO2 and used for experiments below passage number 10.Following trypsinization, 105 cells were plated into a 35 mm Petri dish containing the hydrogel.
After 4 h, cells were fixed in pre-warmed PBS containing paraformaldehyde (PFA, 3.5% w/v, #30525-89-4, Polysciences Inc.). Samples were incubated in blocking buffer: PBS containing BSA (1%) and Triton-X (0.1% v/v, #3051, Carl Roth). Primary antibodies against LN (#L9393, SIGMA-Aldrich), COL I (#C2456, Sigma-Aldrich) and FN (#F0791, Sigma-Aldrich and #AB2033, Chemicon) were diluted 1:2000 in blocking buffer and incubated overnight at room temperature. Samples were subsequently rinsed with PBS and incubated in blocking buffer. Secondary antibodies (#A11001 and #A21245, Invitrogen) were used as described above. After 1 h, samples were rinsed with PBS and fixed in PBS containing PFA (3.5%). Fluorescence images were recorded in PBS buffer on an upright Leica SP5 confocal microscope using a 20 × 1.0 NA water dipping objective.
Staining of focal adhesions on LN and FN nanofibrils
Substrate preparation and cell adhesion to PEG hydrogels was performed as described above, but with a modified fixation step: after 2 min pre-fixation in 3.5% PFA solution containing 0.5% v/v Triton-X-100, cells were fixed for further 45 min in 3.5% PFA solution. During the second immunostaining incubation, a mouse anti-paxillin antibody was used (#610051, BD Biosciences), which was pre-labelled with a zenon anti-mouse labelling kit (#Z25060, Invitrogen).Conclusions
We have devised a new method to produce highly regular arrays of ECM nanofibrils made of fibronectin, collagen I and EHS laminin. Our results are congruent with the model proposed by Guan et al.,31 indicating a common mechanism of polymer fibrillogenesis induced by de-wetting. The necessity of self-association sites is suggested by the observation that BSA, even at high concentrations of 1 mg ml−1, did not yield nanofibrils (Fig. S2), despite its ability to concentrate at the air–buffer interface (Fig. 4). This leads us to suggest that other proteins containing self-association sites can be used to produce protein nanofibrils. Confirming our suggestion, very recent results from Pabba et al.34 demonstrate the possibility to produce nanofibrils from highly concentrated solutions of fibrin and actin monomers. In line with this, preliminary experiments show that actin nanofibrils are also formed using our approach (Fig. S8†).It is not clear at this point how far the surface forces acting during nanofibril formation (as well as dehydration of the samples) affect protein structure. Nevertheless, the ECM nanofibrils produced using our method enabled cell adhesion. Arrays of crossed LN and FN nanofibrils allowed us to test differential adhesion of various cell lines, connective-tissue-derived HFF and neuronal-like SH-SY5Y, to these nanofibrils in co-culture assays. This is of special interest since it enables one to test differential adhesion and associated cell signalling processes entirely due to the interaction with the given ECM nanofibrils. Other ECM components as synthesised by cells are repelled by the PEG matrix.
Further investigations on the conformation of FN within nanofibrils36 are needed in order to determine the extent to which FN molecule extension and unfolding occur during the assembly process. The fact that the nanofibrils are freely suspended over distances of up to 10 µm renders them a suitable model system for mechanical studies using atomic force microscopy.37–40
Thus, precise control over ECM nanofibril geometry and alignment promises to fuel new applications and findings with relevance in nanofabrication and tissue engineering.
Acknowledgements
We thank Christine Mollenhauer and Irina Slizskaia for supplying human fibronectin, Dr. Roberto Fiammengo for providing PEG 10 kDa diacrylate and Alexandra Goldyn for critical review of the manuscript. Members of the labs of Viola Vogel, Benny Geiger and Michael Sheetz are acknowledged for helpful discussions. P.K. is a scholar of the Fonds der Chemischen Industrie. This work was mainly supported by the Volkswagen- Stiftung. This publication and the project described herein were also partly supported by the National Institutes of Health, through the NIH Roadmap for Medical Research (PN2 EY 016586). The work was also supported by the Excellence Cluster “CellNetwork” of the University of Heidelberg. JPS holds a Weston Visiting Professorship at the Weizmann Institute, Department of Molecular Cell Biology. The support of the Max Planck Society is highly acknowledged.References
- V. Vogel and M. P. Sheetz, Curr. Opin. Cell Biol., 2009, 21, 38–46 CrossRef CAS.
- K. E. Kadler, A. Hill and E. G. Canty-Laird, Curr. Opin. Cell Biol., 2008, 20, 495–501 CrossRef CAS.
- I. Vakonakis and I. D. Campbell, Curr. Opin. Cell Biol., 2007, 19, 578–583 CrossRef CAS.
- E. L. George, E. N. Georges-Labouesse, R. S. Patel-King, H. Rayburn and R. O. Hynes, Development, 1993, 119, 1079–1091 CAS.
- Y. Mao and J. E. Schwarzbauer, Matrix Biol., 2005, 24, 389–399 CrossRef CAS.
- C. Zhong, M. Chrzanowska-Wodnicka, J. Brown, A. Shaub, A. M. Belkin and K. Burridge, J. Cell Biol., 1998, 141, 539–551 CrossRef CAS.
- T. Ohashi, D. P. Kiehart and H. P. Erickson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2153–2158 CrossRef CAS.
- M. L. Smith, D. Gourdon, W. C. Little, K. E. Kubow, R. A. Eguiluz, S. Luna-Morris and V. Vogel, PLoS Biol., 2007, 5, e268 CrossRef.
- B. Wòjciak-Stothard, M. Denyer, M. Mishra and R. A. Brown, In Vitro Cell. Dev. Biol.: Anim., 1997, 33, 110–117 Search PubMed.
- Z. Ahmed and R. A. Brown, Cell Motil. Cytoskeleton, 1999, 42, 331–343 CrossRef CAS.
- W. C. Little, M. L. Smith, U. Ebneter and V. Vogel, Matrix Biol., 2008, 27, 451–461 CrossRef CAS.
- G. Baneyx and V. Vogel, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12518–12523 CrossRef CAS.
- P. Rico, J. C. R. Hernandez, D. Moratal, G. Altankov, M. M. Pradas and M. Salmeron, Tissue Eng. Part A, 2009 DOI:10.1089/ten.tea.2009.0141.
- Y. Chen, L. Zardi and D. M. Peters, Scanning, 1997, 19, 349–355 CAS.
- K. Briknarová, M. E. Akerman, D. W. Hoyt, E. Ruoslahti and K. R. Ely, J. Mol. Biol., 2003, 332, 205–215 CrossRef CAS.
- L. Schuger, Exp. Lung Res., 1997, 23, 119–129 CrossRef CAS.
- K. E. Kadler, D. F. Holmes, J. A. Trotter and J. A. Chapman, Biochem. J., 1996, 316(1), 1–11 CAS.
- D. A. Cisneros, J. Friedrichs, A. Taubenberger, C. M. Franz and D. J. Muller, Small, 2007, 3, 956–963 CrossRef CAS.
- D. F. Holmes, H. K. Graham, J. A. Trotter and K. E. Kadler, Micron, 2001, 32, 273–285 CrossRef CAS.
- D. F. Holmes and K. E. Kadler, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17249–17254 CrossRef CAS.
- A. J. Heim, T. J. Koob and W. G. Matthews, Biomacromolecules, 2007, 8, 3298–3301 CrossRef CAS.
- M. V. Tsiper and P. D. Yurchenco, J. Cell. Sci., 2002, 115, 1005–1015 CAS.
- H. Colognato, C. ffrench Constant and M. L. Feltri, Trends Neurosci., 2005, 28, 480–486 CrossRef CAS.
- J. L. Tan, J. Tien, D. M. Pirone, D. S. Gray, K. Bhadriraju and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1484–1489 CrossRef CAS.
- A. Ganz, M. Lambert, A. Saez, P. Silberzan, A. Buguin, R. M. Mège and B. Ladoux, Biol. Cell, 2006, 98, 721–730 CrossRef CAS.
- W. Roos, J. Ulmer, S. Gräter, T. Surrey and J. P. Spatz, Nano Lett., 2005, 5, 2630–2634 CrossRef CAS.
- B. Cortese, S. D'Amone, M. Manca, I. Viola, R. Cingolani and G. Gigli, Langmuir, 2008, 24, 2712–2718 CrossRef CAS.
- X.-M. Li, D. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 1350–1368 RSC.
- J. Ulmer, B. Geiger and J. P. Spatz, Soft Matter, 2008, 4, 1998–2007 RSC.
- C. A. Lemmon, C. S. Chen and L. H. Romer, Biophys. J., 2009, 96, 729–738 CrossRef CAS.
- J. Guan, N. Ferrell, B. Yu, D. J. Hansford and L. J. Lee, Soft Matter, 2007, 3, 1369–1371 RSC.
- J. Guan and L. J. Lee, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18321–18325 CrossRef CAS.
- J. Guan, B. Yu and L. Â. J. Lee, Adv. Mater., 2007, 19, 1212–1217 CrossRef CAS.
- S. Pabba, M. M. Yazdanpanah, B. H. F. Totten, V. V. Dobrokhotov, J. M. Rathfon, G. N. Tew and R. W. Cohn, Soft Matter, 2009, 5, 1378–1385 RSC.
- A. Tronin, T. Dubrovsky, S. Dubrovskaya, G. Radicchi and C. Nicolini, Langmuir, 1996, 12, 3272–3275 CrossRef CAS.
- G. Baneyx, L. Baugh and V. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14464–14468 CrossRef CAS.
- E. P. S. Tan and C. T. Lim, Appl. Phys. Lett., 2005, 87, 123106 CrossRef.
- B. Wu, A. Heidelberg and J. J. Boland, Nat. Mater., 2005, 4, 525–529 CrossRef CAS.
- L. Yang, C. F. C. Fitié, K. O. van der Werf, M. L. Bennink, P. J. Dijkstra and J. Feijen, Biomaterials, 2008, 29, 955–962 CrossRef CAS.
- L. Yang, K. O. van der Werf, C. F. C. Fitié, M. L. Bennink, P. J. Dijkstra and J. Feijen, Biophys. J., 2008, 94, 2204–2211 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary methods and Fig. S1–S8. See DOI: 10.1039/b911461k |
| This journal is © The Royal Society of Chemistry 2010 |