Site-specific photocatalytic splitting of methanol on TiO2(110)†
Chuanyao
Zhou‡
a,
Zefeng
Ren‡
a,
Shijing
Tan‡
a,
Zhibo
Ma
a,
Xinchun
Mao
a,
Dongxu
Dai
a,
Hongjun
Fan
*a,
Xueming
Yang
*a,
Jerry
LaRue
b,
Russell
Cooper
b,
Alec M.
Wodtke
b,
Zhuo
Wang
c,
Zhenyu
Li
c,
Bing
Wang
*c,
Jinlong
Yang
c and
Jianguo
Hou
c
aState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, 457 Zhongshan Road, Dalian, 116023, Liaoning, P. R. China. E-mail: fanhj@dicp.ac.cn; xmyang@dicp.ac.cn
bDepartment of Chemistry and Biochemistry, University of California at Santa Barbara, CA 93106, USA
cHefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui, China. E-mail: bwang@ustc.edu
First published on 6th September 2010
Abstract
Clean hydrogen production is highly desirable for future energy needs, making the understanding of molecular-level phenomena underlying photocatalytic hydrogen production both fundamentally and practically important. Water splitting on pure TiO2 is inefficient, however, adding sacrificial methanol could significantly enhance the photocatalyzed H2 production. Therefore, understanding the photochemistry of methanol on TiO2 at the molecular level could provide important insights to its photocatalytic activity. Here, we report the first clear evidence of photocatalyzed splitting of methanol on TiO2 derived from time-dependent two-photon photoemission (TD-2PPE) results in combination with scanning tunneling microscopy (STM). STM tip induced molecular manipulation before and after UV light irradiation clearly reveals photocatalytic bond cleavage, which occurs only at Ti4+ surface sites. TD-2PPE reveals that the kinetics of methanol photodissociation is clearly not of single exponential, an important characteristic of this intrinsically heterogeneous photoreaction.
1 Introduction
Titanium dioxide is one of the most important materials for real world applications in chemical catalysis.1–8 Photocatalysis by TiO2, especially its application in water splitting,1 has attracted much attention. It has been reported that pure TiO2 is minimally active for water splitting to produce hydrogen,9 while the same catalyst is much more active for hydrogen production from water–methanol mixture.10 Clearly, the chemistry of methanol on TiO2 has played a crucial role in this process. Therefore, it is essential to understand the photocatalytic chemistry of methanol on TiO2, which could potentially provide clues for developing new efficient photocatalysts for water splitting.The TiO2(110) surface has been extensively studied both theoretically and experimentally6,11 and the physical and chemical properties of the methanol/TiO2 surface have been investigated in detail.12–15 Recent experiments using scanning tunnelling microscopy (STM) show that intact CH3OH molecules can diffuse along the Ti4+ rows and dissociate after encountering a nearby vacancy site on the bridge-bonded oxygen (BBO) row (see Fig. 1) suggesting that CH3OH is not dissociated on Ti4+ sites. Temperature programmed desorption (TPD) also suggested that the majority of adsorbed methanol molecules on the Ti4+ sites of TiO2(110) is undissociated.13,14 A recent 2PPE study has detected an excited electronic state at about 2.4 eV above the Fermi level on the CH3OH/TiO2(110) surface.15,16 This electronic excitation was attributed to a “wet electronic state”.17 DFT calculations18,19 suggest that the molecular state of methanol adsorbed on a Ti4+ site is nearly isoenergetic with the dissociated state. A very recent theoretical study shows that undissociated methanol is the most stable structure by 0.08 eV,17 forming a surface hydrogen bonded dimer; however, these calculations are not sufficiently accurate to unambiguously derive whether methanol is dissociated or not on Ti4+ sites.
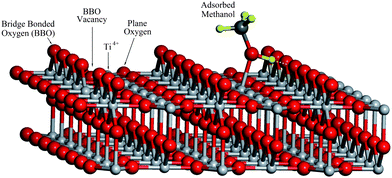 | ||
| Fig. 1 The TiO2(110) surface model with about 4% concentration of BBO vacancy. | ||
In order to understand the photochemistry of methanol on TiO2, we have performed a combined experimental and theoretical study on the methanol/TiO2(110) system. Time-dependent two-photon photoemission (TD-2PPE) experiment shows an excited resonance observed previously was photoinduced, and this was supported by a careful study using the scanning tunneling microscopy (STM) technique. Theoretical analysis allows us to attribute the photoinduced excited resonance to the photodissociated methanol on TiO2. TD-2PPE further indicates that the kinetics of methanol photodissociation is of this intrinsically heterogeneous photoreaction.
This paper is organized in the following: after a brief introduction, a description of the experimental and theoretical methods has been provided, experimental results from both 2PPE and STM studies are then provided, followed by the presentation of the theoretical calculated results. Finally, a short conclusion is provided.
2 Experimental
2.1 Femtosecond 2PPE experimental method
Two-photon photoemission technique has been developed for applications in different surface related studies. Fauster and co-workers20 employed this technique to study the structure and dynamics of image potential states on metal surfaces. This method has also been used for the study of hot electron dynamics of metals21 and semiconductors,22 and to probe the electronically excited states and the dynamics of atoms and molecules adsorbed metal surfaces.23–28 In recent years, 2PPE has been applied to investigate the surface electron dynamics of molecular adsorbed metal oxide surfaces.29 In this work, we have used a recently developed femtosecond 2PPE apparatus to study the methanol/TiO2(110) photochemistry. The apparatus used in this work has been described in great detail in another paper.30A detailed description of the experimental setup of the femtosecond two-photon photoemission (2PPE) spectrometer used in this work is also given in the electronic supplementary information (ESI†). The 2PPE experimental apparatus is comprised of an ultrahigh vacuum system, which includes a sample preparation and characterization chamber and a main probing chamber with a hemispherical electron energy analyzer, as well as a Ti:Sapphire femtosecond laser system with a frequency doubling setup and a home built Mach-Zehnder interferometer that allows for ultrafast excited electron dynamics studies. The vacuum system consists of a main chamber for two-photon photoemission measurement, a sample preparation and characterization chamber and a load-lock system. The upper chamber is designed for sample preparation and characterization. The key element of this apparatus is the hemispherical electron energy analyzer (PHOIBOS 100, SPECS) for low energy photoelectron detection from the sample surface. In the current 2PPE experiment of methanol/TiO2, the first photon is used to excite electrons in the surface defect states to the conducting band followed by another photon excitation of the excited electrons to the vacuum (Fig. 2). More detailed descriptions of the 2PPE experiments can be found in the ESI.†
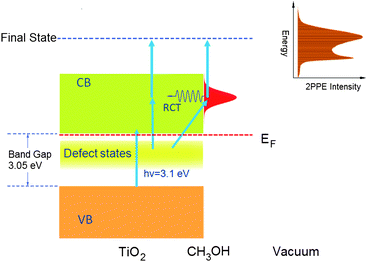 | ||
| Fig. 2 The scheme of the two photon photoemission experiment on the methanol/TiO2(110) surface. | ||
An experimental scheme is also developed using the imaging detector for time-dependent 2PPE (TD-2PPE) measurement over the energy range from the work function edge to twice of the photon energy relative to the Fermi level. On this apparatus, we have developed a data acquisition program for 2PPE imaging with a real time measurement window of 0.1 s. This means that 2PPE spectra can be obtained in less than 0.1 s. The TD-2PPE technique allows us to measure kinetics of surface photochemical and chemical reactions that induce considerable excited electronic structure changes.
The experiment in this work was carried out on methanol adlayers on TiO2(110) at a surface temperature of 105 K in an ultrahigh vacuum (UHV) chamber with a base pressure of ∼5 × 10−11 mbar, using a 400 nm femtosecond laser light source. Most of the 2PPE experiments described here were carried out on a nearly stoichiometric TiO2(110) surface, which was prepared by several cycles of Ar+ sputtering and annealing with 3 × 10−7 mbar of oxygen at 850 K for 60 min. The TiO2 sample was then cooled down with oxygen present in the system to prepare the TiO2 surface with little defects. We have also prepared the surface by cooling down the TiO2 sample in UHV. The 2PPE results are similar for both cases. The 2PPE results presented in this work were all done with the sample cooled down with oxygen present. The surface quality was confirmed by Auger electron spectroscopy (AES) and low energy electron diffraction (LEED). This procedure allows us to prepare a TiO2(110) surface with a low concentration of BBOV.
2.2 Scanning tunnelling microscopy technique
In order to understand the photochemical changes occurring on the methanol/TiO2(110) surface, we have also carried out a high resolution STM study on the methanol/TiO2(110) surface under similar conditions to the 2PPE experiments.31–33 The STM experiment on the methanol/TiO2(110) surface was conducted with a low temperature scanning tunnelling microscope (Omicron, MATRIX) in an UHV system. In the STM experiment on methanol on TiO2(110), the vacuum system was baked for about 100 h and had a base pressure of 3 × 10−11 mbar. A one-side polished rutile TiO2(110) sample was prepared by several cycles of sputtering with Ar+ ions for about 15 min, followed by annealing at 900 K for about 30 min in each cycle. After such treatments, the sample was transferred into the cryostat of the microscope, which had been pre-cooled to 80 K. During methanol dosing and light illumination, the sample was kept in the cryostat at 80 K, but the STM tip was retracted from the surface by about 10 μm. In this way, it is easy for us to compare the changes of the same areas. The chemically etched tungsten tip was cleaned by Ar+ ion sputtering and annealing before use. To study the UV photocatalytic effect, we illuminated the methanol adsorbed TiO2(110) surface using a focused UV light from a mercury-xenon lamp with 200 mW power. The UV light was focused to a spot size of ∼500 μm after passing through a band pass filter with a cutoff wavelength of 400 nm.3 Results and discussions
3.1 2PPE spectra of methanol/TiO2
Photochemistry of methanol on TiO2 has been studied by the 2PPE technique described above. Fig. 3 shows regular 2PPE spectra measured for both bare (purple line) and 1 ML methanol covered TiO2. The lower x-axis indicates the final energy of the two-photon induced emitted electron. The upper x-axis shows the energy of one photon intermediate excited electronic states. In both cases, the Fermi level is the zero of energy. The spectrum of the bare surface exhibits an exceedingly small 2PPE signal and a high work function.15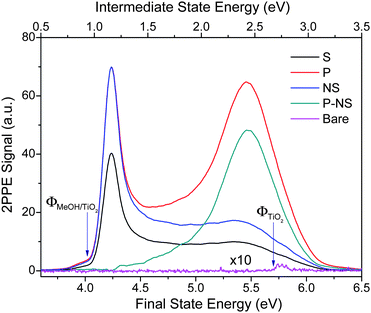 | ||
| Fig. 3 2PPE spectra for the bare (Bare) and CH3OH adsorbed stoichiometric TiO2(110) surface. For the CH3OH adsorbed surface, 2PPE spectra were measured for both p-polarization (P) and s-polarization (S) after the surface was illuminated by the femtosecond probing laser for more than 1000 s. The excited peak is obtained by subtracting the normalized s-polarization (NS) data from the p-polarization (P) data. The small bump in the s-polarization data at about 5.5 eV is likely due to the impurity of the p-polarized light in the s-polarized light beam. The spectra shown here are integrated between −5 and +5 degrees. The lower x-axis indicates the final energy of the electron emitted to the vacuum relative to the Fermi level. The upper x-axis shows the energy of any intermediate excited electronic state present after absorption of the first photon. | ||
The 2PPE spectra are shown in Fig. 3 for methanol adlayers on TiO2 after irradiating the surface for 15 min with p-polarized 400 nm light.34 Both p-polarized (red line) and s-polarized (black line) light yield 2PPE spectra, which however differ significantly from one another. Specifically, the p-polarized 2PPE spectrum exhibits an additional feature at about 5.5 eV, which is not as apparent with s-polarization. This feature has been previously assigned as an electronically excited resonance above the Fermi level and dubbed as a “wet electron state”.15
In order to gain insight into the nature of this excited resonance state, we have measured TD-2PPE spectra (Fig. 4A). Here, one clearly sees that the 5.5 eV excited resonance feature is initially absent and grows monotonically with exposure time of the probing laser light. This unambiguously proves that the observed resonance feature is photoinduced on the CH3OH/TiO2(110) surface. Furthermore, a similar photoinduced excited resonance feature is also observed on the CH3OH/TiO2(110) surface at a methanol coverage of 0.50 ML and 0.16 ML. This suggests that the surface excited resonance observed is not coverage dependent. These time-dependent and furthermore dramatic changes to the excited state electronic structure induced by irradiation at 400 nm clearly show that the methanol covered TiO2 surface is photochemically active.
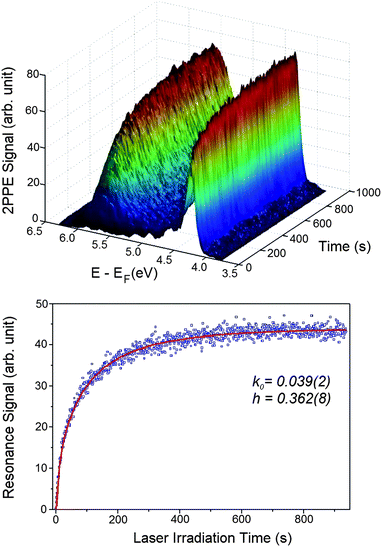 | ||
| Fig. 4 A) Time-dependent 2PPE spectra were measured for the freshly CH3OH adsorbed stoichiometric TiO2(110) surface after it had been exposed for different time durations. A) This plot shows the evolution of the 2PPE spectra after the surface was exposed for certain time durations. B) The time dependent signal of the excited resonance feature in Fig. 2 integrated between 4.9 and 6.1 eV measured with the laser power of 64 mW. | ||
3.2 STM results on methanol/TiO2
To understand the photochemical changes occurring on methanol covered TiO2, we have designed a high resolution STM study to see how STM images of the methanol molecules adsorbed on TiO2(110) are affected by UV light irradiation using a low temperature scanning tunnelling microscope. Fig. 5A shows an STM image of a bare TiO2(110) surface at 80 K. In the STM image, the bright rows are the Ti4+ sites, while the dark rows are the BBO sites. The bright spots interspersed along the dark rows are the bridge-bonded oxygen vacancy (BBOV) sites. The surface used for STM study here has a BBOv concentration of about 4%, similar to the surface used for the 2PPE study. Fig. 5B shows the same area as in Fig. 5A with the methanol coverage of 0.02 ML. In the STM image of this area, six adsorbed methanol molecules appear as uniform bright spots at Ti4+ sites. Only one methanol molecule in this area was adsorbed at BBOV (marked by “+” in Fig. 5A) and appears to be dissociated, which is consistent with the previous STM study.8 All other methanol molecules in this area were adsorbed at Ti4+ sites marked by small white circles in Fig. 5A. These molecules can be moved intact along the Ti4+ row or even desorbed by the STM tip (Fig. S3 in ESI†). This implies in a very direct way that the methanol molecules adsorbed on Ti4+ sites are mobile and undissociated, consistent with previous STM evidence of undissociated methanol on Ti4+ sites.8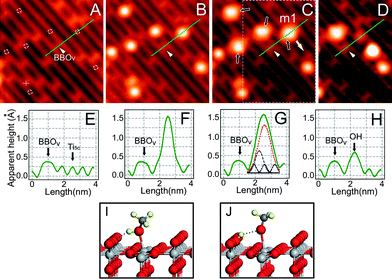 | ||
| Fig. 5 Imaging photocatalyzed dissociation of methanol. STM images (acquired at bias of 1.0 V and set point current of 10 pA, size of 7.3 × 7.3 nm2) of A) bare TiO2(110)−1 × 1 surface; B) surface with adsorbed CH3OH. C) after 10 min irradiation by 400 nm light. Dashed circles in A): sites for CH3OH adsorption on Ti4+ row; Cross: site for CH3OH on BBOV. Black arrows in C) indicate the four dissociated molecules after UV irradiation. D) STM Image after manipulation of the dissociated molecule m1 in the marked area of C). The dissociated molecule m1 can be separated into two parts, where the part left on the BBO row is due to the H atom, and the part moved to another location is attributed to the mothoxy species. Manipulation performed at 0.4 V and 700 pA by pulling m1 with the STM tip along the direction of the white arrow. E–H) Cut profiles along the green lines in A–D), respectively. The fitted peaks in G) reflect the contributions from methoxy group, OH group, and Ti4+ row, respectively. I) The calculated structure of molecular adsorbed methanol on TiO2(110). J) The calculated structure of dissociated methanol on TiO2(110). | ||
After 10 min with UV light irradiation on the methanol adsorbed surface, we observe subtle but distinct changes to the STM image (see Fig. 5C). Specifically, among the six methanol molecules adsorbed at Ti4+ sites, four methanol molecules (marked by black arrows) appear to be “stretched” in the STM image after UV light illumination. This can be more clearly seen by comparison of STM line scans before (Fig. 5F) and after (Fig. 5G) UV light illumination.
To further confirm the interpretation of the STM images, we performed STM manipulation of single methanol molecules. For example, consider the molecule labeled “m1” in Fig. 5C (i.e. after illumination), which was pulled by the STM tip in the direction indicated by the white arrow thus yielding the STM image of Fig. 5D. Remarkably, the ”stretched” (m1) molecule is separated into two spots, one dim spot left on the BBO site and one bright spot translated in the direction indicated by the white arrow. The dim spot left on the BBO site is identified as an H-atom bonded to an oxygen on the BBO row by varying the imaging conditions, while the bright spot moved along the white arrow is likely a methoxy radical. This suggests that the “stretched” methanol molecules after UV light irradiation can be separated by the STM tip, unlike the original undissociated methanol molecules adsorbed on Ti4+. We therefore conclude that the “stretched” methanol molecule is a dissociated methanol species, induced by the UV light, with its hydroxyl H-atom transferred to the nearby BBO site (see Fig. 5I and 5J).
More examples of single molecule manipulations which further support these conclusions can be found in the ESI.† We have also performed similar experiments using light at wavelengths longer than 440 nm, where the photon energy is insufficient to promote electrons from the valence band to the conduction band of TiO2. Here, no changes in the STM images were seen for methanol molecules adsorbed at the Ti4+ sites. This strongly suggests that electron-hole-pair excitation from the valence band to the conduction band is responsible for methanol photodissociation on TiO2.
3.3 Assignment of the excited resonance
Since the photoinduced excited resonance observed in the 2PPE experiment is not dependent on the methanol coverage, the 2PPE and STM results can be directly related. Therefore, the STM observation of methanol photodissociation under UV light irradiation clearly suggests that the photoinduced resonance observed in the 2PPE experiments should arise from the photodissociation of methanol on TiO2.DFT calculations on the electronic structure based on a cluster model support this assignment and provide a simple physical picture describing how the excited state arises. Theoretical study was carried out to give further insight into the 2PPE spectrum of the CH3OH on TiO2(110) surface. A computational model was shown in Fig. S4 in the ESI.† It consists of 4 layers, and 286 atoms. The model was designed by cropping the bulk rutile, then saturating all surface Ti atoms, except those in the 110 surface, by OH/OH2 to maintain the coordination environment and oxidation states of Ti in bulk. Two CH3OH molecules were put on the surface adjacently. We have used BLYP functionals in this work. The BLYP calculations were carried out using the Amsterdam Density Functional (ADF 2008) package,35 utilizing the double-basis set (DZ for hydrogens bond to TiO2 and DZP for all other atoms) and the frozen-core approximation. B3LYP calculations were carried out using LACVP** basis sets and Jaguar 7.5 package.36 This cluster model gives similar results to those in ref. 17 for the geometry and methanol dissociation energy, which shows that the cluster model is quite reasonable.
Theoretical calculations show that when the CH3O–H bond breaks, the Ti4+ puckers significantly out of the plane of surface Ti4+ atoms due to the strong chemical Ti–O bond formed with the methoxy radical. Specifically, the calculations show that the puckering induced by the newly formed Ti–O bond raises the energy of the Ti d-orbitals dramatically forming the new electronic resonance (see Figs. S7 and S8 in ESI†). The puckering produces a new excited state ∼2.5 eV above the LUMO, which serves as an approximate Fermi energy in this case,37,38 in good agreement with the energy of the photoinduced resonance feature observed in the 2PPE spectra (see Fig. 3). Other unoccupied orbitals calculated in this work are found to be inconsistent with the 2PPE observations. The calculations shown here clearly support that the photoinduced excited resonance observed in the 2PPE experiment is due to the dissociation of the methanol molecule on TiO2 under UV irradiation.
Based on our experimental results and DFT calculations, we believe that the assignment of the photoinduced excited resonance to a surface Ti d-orbitals state due to methanol dissociation is more reasonable than the “wet electron state” proposed,16,17 because of the following reasons:
1) In ref. 17, theoretical calculations have shown that the “wet electron state” has a broad distribution between 2.4 to 3.3 eV above the CBM with its centre around 2.9 eV, this is not consistent with the experimental observation which gives a peak at 2.3 eV, and then decreases quickly. This means that the calculated “wet electron state” peaks are more than 0.5 eV higher than the experimental value. While our calculated surface state that is associated with the dissociated methanol is within 0.1 eV. In addition, both the current calculations and previous calculations39 show that the “wet electrons state” is much higher in energy than the 2PPE experimental observed resonance.
2) In the current 2PPE study at both 0.50 ML and 0.16 ML methanol coverage, similar photoinduced resonances were observed. This certainly cannot be explained by the “wet electron state”, which could only exist at the methanol coverage close to a monolayer.
3) Surface Ti d-orbitals should be geometrically more close to the defect state sites where the electrons were excited from, while the methyl “wet electron state” is further away and has little geometric overlap with the defect states. From the Franck–Condon principle, such excitation is much less efficient.
3.4 Measurement of photochemical kinetics
We now consider the observed kinetics of the methanol photodissociation on TiO2. In the TD-2PPE experiment, we can measure the rising signal of the resonance feature in real time. By integrating the signal between 4.9 and 6.1 eV in Fig. 4A, the time dependent resonance signal can be acquired (Fig. 4B). Since the rise of the resonance signal is associated with the methanol dissociation, the time-dependent resonance signal illustrates essentially the kinetics of photocatalyzed methanol dissociation. The time-dependent resonance signal shown in Fig. 4B appears to be rising monotonically. However, a single exponential model cannot properly simulate the rising curve. This implies that the photocatalyzed methanol dissociation on TiO2 is not a homogenous process, in which the rate constant should be time independent. Instead, it appears that the rate constant of methanol dissociation on TiO2 is strongly time-dependent, implying that the photocatalyzed reaction is intrinsically a heterogeneous process. Indeed, the rising kinetics can be very well described by a fractal-like kinetic model for heterogeneous reactions:40,41| I = I0 (1 − exp(−k0/(1−h) t(1−h))) | (1) |
4 Conclusions
A combined experimental and theoretical study has been carried out on the methanol/TiO2(110) system. A photoinduced excited resonance state on the CH3OH/TiO2(110) surface has been observed using time-dependent two-photon photoemission method. High resolution STM experiments performed in this work show that methanol adsorbed on the Ti4+ sites can be dissociated by UV light illumination with wavelength < 400 nm. In the dissociation of methanol, its hydroxyl hydrogen was transferred to the neighboring BBO site. This suggests that the excited electronic resonance observed is likely due to the dissociated methanol on TiO2 surface. DFT calculations found a new excited surface resonance state due to the dissociated methanol on TiO2. Time-dependent 2PPE measurements show that the rising kinetics of the excited resonance is clearly of heterogeneous nature.The results obtained in this work have significant implications for photocatalysis on TiO2. Our results unambiguously show that the O–H bond in methanol is photochemically active on TiO2. This is undoubtedly an important aspect of the explanation of why hydrogen production is more efficient for methanol–water mixtures. Our results furthermore show that the Ti4+ out-of-plane puckering is an essential element of bond dissociation on this surface, suggesting that Ti4+ is the key catalytic site for photoinduced chemistry on TiO2.
Acknowledgements
We would like to thank the Chinese Academy of Sciences, National Natural Science Foundation of China and the Ministry of Science and Technology for financial support of this work. AMW, JL and RC acknowledge the support of the Partnership for International research and Education Electron Chemistry and Catalysis at Interfaces NSF Grant OISE-0530268Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243 CrossRef CAS.
- E. Wahlström, E. K. Vestergaard, R. Schaub, A. Rønnau, M. Vestergaard, E. Lægsgaard, I. Stensgaard and F. Besenbacher, Science, 2004, 303, 511 CrossRef.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341 CrossRef CAS.
- P. V. Kamat, Chem. Rev., 1993, 93, 267 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Jr, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- Z. Zhang, O. Bondarchuk, J. M. White, B. D. Kay and Z. Dohnalek, J. Am. Chem. Soc., 2006, 128, 4198 CrossRef CAS.
- S. Sato and J. M. White, Chem. Phys. Lett., 1980, 72, 83 CrossRef CAS.
- T. Kawai and T. Sakata, J. Chem. Soc., Chem. Commun., 1980, 694 RSC.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53 CrossRef CAS.
- S. P. Bates, M. J. Gillan and G. Kresse, J. Phys. Chem. B, 1998, 102, 2017 CrossRef CAS.
- M. A. Henderson, S. Otero-Tapia and M. E. Castro, Faraday Discuss., 1999, 114, 313 RSC.
- C. L. Pang, R. Lindsay and G. Thornton, Chem. Soc. Rev., 2008, 37, 2328 RSC.
- K. Onda, B. Li, J. Zhao and Petek, Surf. Sci., 2005, 593, 32 CrossRef CAS.
- B. Li, et al, Science, 2006, 311, 1436 CrossRef CAS.
- J. Zhao, J. Yang and H. Petek, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 235416 CrossRef.
- R. Sanchez de Armas, J. Oviedo, M. A. San Miguel and J. F. Sanz, J. Phys. Chem. C, 2007, 111, 10023 CrossRef CAS.
- J. Oviedo, R. Sanchez de Armas, M. A. San Miguel and J. F. Sanz, J. Phys. Chem. C, 2008, 112, 17737 CrossRef CAS.
- U. Hofer, I. L. Shumay, C. Russs, U. Thomann, W. Wallauer and T. Fauster, Science, 1997, 277, 1480 CrossRef CAS.
- S. Ogawa, H. Nagano and H. Petek, Phys. Rev. Lett., 1997, 78, 1339 CrossRef CAS.
- E. Knosel, A. Hotzel and M. Wolf, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 12812 CrossRef CAS.
- R. L. Ringle, Jr., N.-H. Ge, R. E. Jordan, J. D. McNeil and C. B. Harris, Chem. Phys., 1996, 205, 191 CrossRef CAS.
- H. Petek, M. J. Weida, H. Nagano and S. Ogawa, Science, 2000, 288, 1402 CrossRef CAS.
- L. Bartels, G. Meyer, K.-H. Rieder, D. Velic, E. Knosel, A. Hotzel, M. Wolf and G. Ertl, Phys. Rev. Lett., 1998, 80, 2004 CrossRef CAS.
- C. Gahl, K. Ishioka, Q. Zhong, A. Hotzel and M. Wolf, Faraday Discuss., 2000, 117, 191 RSC.
- T. Vondrak, H. Wang, P. Winget, C. J. Cramer and X.-Y. Zhu, J. Am. Chem. Soc., 2000, 122, 4700 CrossRef CAS.
- T. Hertel, E. Knoesel, M. Wolf and G. Ertl, Phys. Rev. Lett., 1996, 76, 535 CrossRef CAS.
- K. Onda, B. Li and H. Petek, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 045415 CrossRef.
- Z. Ren, C. Zhou, Z. Ma, C. Xiao, X. Mao, D. Dai, J. LaRue, R. Cooper, A. M. Wodtke and X. Yang, Chin. J. Chem. Phys., 2010, 23, 255 CrossRef CAS.
- Y. Zhao, et al, J. Am. Chem. Soc., 2009, 131, 7958 CrossRef CAS.
- X. F. Cui, et al, J. Phys. Chem. C, 2009, 113, 13204 CrossRef CAS.
- X. F. Cui, et al, J. Chem. Phys., 2008, 129, 044703 CrossRef.
- The 400 nm used for the 2PPE measurement was generated by frequency doubling of the 800 nm light from a Ti:Saphire femtosecond laser (Synergy PRO, Femtolasers Produktions GmbH), pumped by a Verdi V6 diode laser (Coherent Co.). The laser power used in the experiment is about 60 mW, the laser pulse width measured the surface is about 28 fs. The laser beam was focused onto an area with a radius of ∼100 μm radius on the surface.
- G. T. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- Jaguar, 7. 5 ed Schrödinger, L.L.C, Portland, OR, 1991–2008.
- R. L. Kurtz, R. Stock-Bauer, T. E. Msdey, E. Roman and J. L. de Segovia, Surf. Sci., 1989, 218, 178 CrossRef CAS.
- A. K. See and R. A. Bartynski, J. Vac. Sci. Technol., A, 1992, 10, 2591 CrossRef CAS.
- T. Koitaya, H. Nakamura and K. Yamashita, J. Phys. Chem. C, 2009, 113, 7236 CrossRef CAS.
- R. Kopelman, J. Stat. Phys., 1986, 42, 185 CrossRef.
- R. Kopelman, Science, 1988, 241, 1620 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c0sc00316f |
| ‡ These authors have made similar contributions to this work. |
| This journal is © The Royal Society of Chemistry 2010 |