Cyclooligomerisation of azido-alkyne-functionalised sugars: synthesis of 1,6-linked cyclic pseudo-galactooligosaccharides and assessment of their sialylation by Trypanosoma cruzi trans-sialidase†
Vanessa L. Campoab, Ivone Carvalhob, Carlos H. T. P. Da Silvab, Sergio Schenkmanc, Lionel Hilld, Sergey A. Nepogodieva and Robert A. Field*a
aDepartment of Biological Chemistry, John Innes Centre, Colney Lane, Norwich, UK NR4 7 UH. E-mail: rob.field@bbsrc.ac.uk
bFaculdade de Ciências Farmacêuticas de Ribeirão Preto, University of São Paulo, Av. Café S/N, CEP 14040-903, Ribeirão Preto, SP, Brazil
cDepartament of Microbiology, Immunology and Parasitology, Universidade Federal de São Paulo, Rua Botucatu 862 8, Andar 04023-062, São Paulo, SP, Brazil
dDepartment of Metabolic Biology, John Innes Centre, Colney Lane, Norwich, UK NR4 7 UH
First published on 2nd July 2010
Abstract
Cyclic pseudo-galactooligosaccharides were synthesized by cyclooligomerisation of isomeric azido-alkyne derivatives of β-D-galactopyranose under Cu(I)-catalysed azide-alkyne 1,3-dipolar cycloaddition reaction conditions. The principal products isolated were cyclic dimers and trimers, with lower amounts of cyclic tetramer and pentamer also evident in some cases. Molecular mechanics calculations suggest very compact but flexible structures for the cyclic trimers, with secondary OH groups exposed outside the macrocycle and available for enzymatic glycosylation. The cyclic dimers and trimers represent a new type of acceptor substrate for Trypanosoma cruzi trans-sialidase, giving rise to doubly and triply sialylated glycomacrocycles, respectively.
Introduction
It is now widely accepted that carbohydrates play a crucial role in a diverse array of biological recognition events.1–3 The combination of many new protein targets,4,5 and a range of new tools with which to analyse them,6,7 offers numerous avenues for therapeutic intervention. In many cases individual protein–carbohydrate interactions are relatively weak, relying on the multivalent display of ligand and or receptor for enhanced affinity.8–10 This notion has found broad application in the preparation of synthetic multivalent glycoconjugates with enhanced affinities,11–14 a classic example being the work from the Bundle lab on the generation of oligovalent, water-soluble carbohydrate ligands (named STARFISH) with sub-nanomolar inhibitory activity against Escherichia coli O157:H7 Shiga-like toxin 1.15The CuI-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC),16–18 which is increasingly used in carbohydrate chemistry,19–21 provides a flexible and highly efficient route to multivalent neoglycoconjugates. Numerous examples of carbohydrate-containing linear oligomers and macrocycles prepared using this approach have been reported,18–27 as have cyclic peptides and peptidomimetics,28–32 mixed carbohydrate-amino acid macrocycles,33 crown ethers,34 glycophanes,27,35,36 cavitands,37 catenanes38,39 and other types of macrocyclic compounds.18 A particular interest of ours lies in the development of tools with which to understand the recognition of sialylated glycans,40–43 many of which occur naturally in multivalent format. This presents the opportunity for exploiting multivalent interactions for the design of more potent ligands, as demonstrated by the Wong lab in connection with flu virus adhesion and infection, for instance.44
In the current study, our goal was to devise a route to oligomeric neoglycoconjugate templates that would be compatible with enzymatic sialylation by Trypanosoma cruzi trans-sialidase (TcTS), which we45–48 and others49–52 have shown to be a versatile catalyst for enzymatic synthesis. In other studies we have demonstrated and exploited53,54 the fact that TcTS will successfully α-2,3-sialylate 1,6-disubstituted β-galactosides. We therefore targeted cyclic 1,6-linked pseudo-galactooligosaccharides, with glycosidic bonds replaced with 1,4-disubstituted 1,2,3-triazole linkages, through CuAAC-mediated oligomerisation. Examples of highly efficient syntheses of triazole-bridged cyclic pseudo-oligosaccharides assembled using this type of approach have been described in the literature.23–25,36 In these examples azido-alkyne-functionalised monomers undergo cyclooligomerisation, with preferential formation of cyclic dimers in 20 to 95% isolated yields. Such outcomes may be a result of torsional or geometric factors favouring conformations predisposed to intramolecular reactions. The high yielding cyclisation of chemoenzymatically synthesised azido-sialic acid containing compounds to give a range of macrocycles, containing up to 4 triazoles and 10 sugar units, has also been reported.26 The conformational and supramolecular properties of main chain and cyclic click oligotriazoles and polytriazoles has recently been reviewed.55 By analogy with literature examples,23–26 we anticipated that under CuAAC conditions galactose derivatives 1 and 2 would likely afford small cyclic oligomers (Fig. 1).56 In addition, given that we have recently reported on the impact of steric and stereoelectronic factors on the efficiency of carbohydrate CuAAC reactions,57–60 we expected that the site of sugar substitution by azide (e.g. anomeric centre vs. elsewhere) might impact on the efficiency of CuAAC oligomerisation and also on the flexibility of the resulting adducts. Herein we report the cyclooligomerisation of galactose derivatives 1 and 2, molecular mechanics studies on representative examples of the resulting glycomacrocycles and investigation of the TcTS-catalysed sialylation thereof.
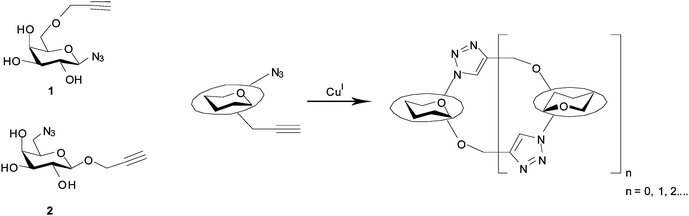 | ||
| Fig. 1 Schematic representation of the cyclooligomerisation approach to the synthesis of cyclic pseudo-oligosaccharides. | ||
Results and discussion
Synthesis of azido-alkyne-functionalised galactopyranose-based monomers
Galactopyranosyl azide building block 1 incorporating a propargyl ether at C-6 was synthesized in six steps (Scheme 1) from readily available diacetone galactose, 3. The latter was prepared by iodine-catalyzed di-O-isopropylidenation61 of the unprotected sugar in 89% yield. Alkylation of 3 with propargyl bromide in the presence of NaH in DMF gave the intermediate 4,62 which was deprotected with 80% CF3CO2H and per-O-acetylated to give protected 6-O-propargyl ether 6. Without purification, the latter was employed in the HBr/AcOH-mediated preparation of glycosyl bromide 7. Conversion of the crude bromide 7 to the corresponding β-azide 8 was carried out using the procedure described by Roy et al.,63 which involves reaction with NaN3 under phase transfer conditions. In this way β-linked azide 8 was obtained in 52% yield after purification by column chromatography. The presence of propargyl and azide functional groups in compound 8 was evident from characteristic signals in its 1H NMR [δ 2.46 (CH) and 4.19 (CH2) ppm] and IR [3278 cm−1 (propargyl) and 2104 cm−1 (N3)] spectra. The β-configuration of 8 followed from the characteristic anomeric proton signal (δ 4.61, J1,2 = 8.6 Hz) in the 1H NMR spectrum. Finally, deacetylation of protected glycoside 8 afforded target building block 1. Following the synthetic route outlined in Scheme 1, protected glycoside 8 was prepared in 39% overall yield.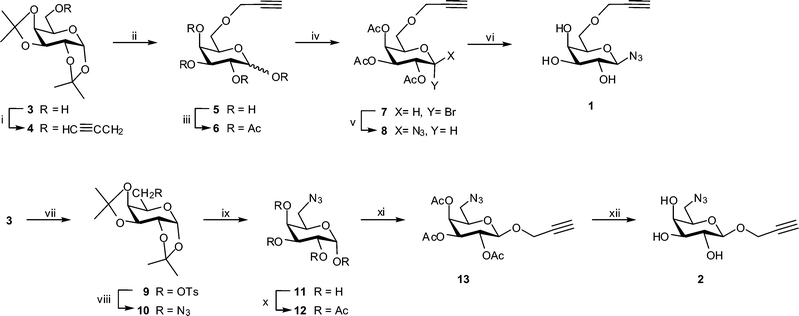 | ||
Scheme 1 Synthesis of building blocks 1 and 2. Reagents and conditions: (i) CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2Br, NaH, DMF, 89%; (ii) 80% aq. CF3CO2H, 93%; (iii) Ac2O, pyridine, 100%; (iv) 33% HBr/AcOH, 89%; (v) NaN3, aq. NaHCO3, Bu4NHSO3, CH2Cl2, 52%; (vi) NaOMe, MeOH, 90%; (vii) p-TsCl, C5H5N, 62%; (viii) NaN3, DMF, 100%; (ix) 80% aq. CF3CO2H, 90%; (x) Ac2O, C5H5N, 94%; (xi) HCCCH2OH, BF3·Et2O, 38%; (xii) NaOMe, MeOH, 86%. CCH2Br, NaH, DMF, 89%; (ii) 80% aq. CF3CO2H, 93%; (iii) Ac2O, pyridine, 100%; (iv) 33% HBr/AcOH, 89%; (v) NaN3, aq. NaHCO3, Bu4NHSO3, CH2Cl2, 52%; (vi) NaOMe, MeOH, 90%; (vii) p-TsCl, C5H5N, 62%; (viii) NaN3, DMF, 100%; (ix) 80% aq. CF3CO2H, 90%; (x) Ac2O, C5H5N, 94%; (xi) HCCCH2OH, BF3·Et2O, 38%; (xii) NaOMe, MeOH, 86%. | ||
Synthesis of the second suitable target building block, 2, was accomplished following the reaction sequence outlined in Scheme 1. Crystalline tosylate 9, prepared in 62% yield from 3, was treated with NaN3 in DMF at 120 °C in order to effect nucleophilic substitution giving known64 6-azido-6-deoxy sugar 10 in near quantitative yield. De-O-isopropylidenation of 10 in 80% CF3CO2H followed by per-O-acetylation afforded α-isomer 12, which was isolated in 94% yield. BF3·Et2O-catalysed glycosidation of 12 with propargyl alcohol62 gave the desired β-glycoside 13 in 38% yield, along with recovered starting material (18%) and trace amounts of the α-isomer. In this case, the presence of propargyl and azide functional groups in compound 13 was evident from characteristic signals in its 1H NMR [δ 2.49 (CH) and 4.42 (CH2) ppm] and IR [3278 cm−1 (propargyl) and 2104 cm−1 (N3)] spectra. The β-configuration of 13 followed from the characteristic anomeric proton signal (δ 4.80, J1,2 = 7.8 Hz) in the 1H NMR spectrum. De-O-acetylation of 13 concluded the six step synthesis, affording target compound 225 in 20% overall yield.
Cyclooligomerisation reactions
Several variations of the CuAAC reaction conditions were investigated in an attempt to optimise the formation of cyclooligomerisation products from monomers 1 and 2. In particular, the reactivity of these building blocks was tested at 0.18 M concentration in DMF with CuSO4/Cu turnings60 as a catalytic system. Our initial attempts to carry out these reactions under ambient conditions were not successful; even when elevated temperature and prolonged time (60 °C for 48 h) were used no reaction was evident. To effect these CuAAC reactions we subsequently employed microwave irradiation, which has been shown to have a profound influence on the outcome of various cycloaddition processes.65 Thus, cyclooligomerisations of 1 and 2 were carried out in a microwave reactor and the progress of the reactions was followed by TLC analysis, which revealed complete consumption of starting material and no further changes after 15 min at 100 °C. The products of the reaction of building block 1 were separated using reverse phase HPLC (see Fig. S5 in ESI†) to give cyclic dimer 14 (29%), trimer 15 (13%) and tetramer 16 (4%) (Fig. 2). Under the same reaction conditions, cyclooligomerisation of 2 led to cyclic dimer 18 (26%) and trimer 19 (6%) as the only products that could be isolated by HPLC (See Fig. S14 in ESI†). In both cases some slow moving (HPLC) products were also formed but their purification was difficult and these products were not further analysed. The overall yields of glycomacrocycles obtained were broadly similar from compounds 1 (46%) and 2 (32%), indicating only a modest difference in chemical reactivity of these isomeric azido-alkynes. The remaining material formed in both cases is likely to be intractable polymer, which was not pursued in this study. At low levels, monomer 1 gave rise to cyclic tetramer 16 and cyclic pentamer 17 (Fig. 2); the corresponding macrocycles could not be obtained from monomer 2, which may reflect a geometric preference for polymerisation rather than cyclisation in this case.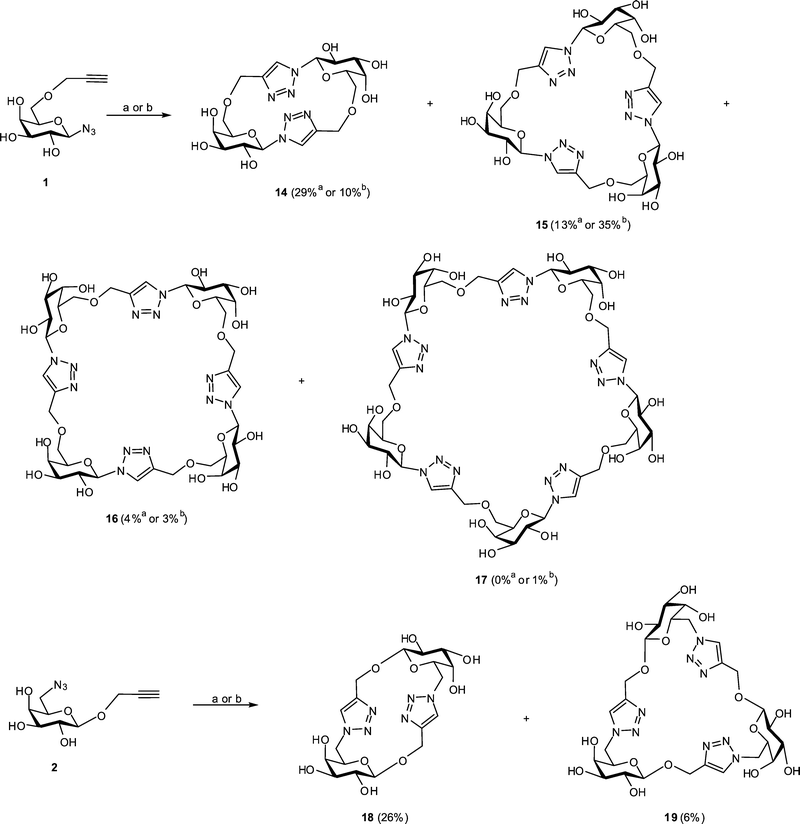 | ||
| Fig. 2 Synthesis of glycomacrocycles from building blocks 1 and 2. Reaction conditions: (a) monomer 1 or 2 (0.18 M), CuSO4 (0.25 eq.), Cu turnings (15 mg), DMF (0.5 mL), microwave irradiation (100 °C), 15 min; (b) same as (a) but monomer 1 or 2 (1.0 M). | ||
Cyclooligomerisation reactions performed at higher temperatures (120–150 °C), longer microwave irradiation times (up to 2 h) or on changing the catalytic system to CuSO4/sodium ascorbate did not significantly change the yield or ratio of cyclooligomerisation products (see Table S1 in ESI†). At high temperatures (120–150 °C) it was possible to achieve similar cyclooligomerisation results with the addition of Cu wire or even in the absence of a catalyst. In these experiments purification of products was simplified since it did not require removal of Cu salts prior to separation of macrocycles. Surprisingly these latter reactions appeared to be regiospecific, which may be due to over-riding steric hindrance/geometrical constraints.
Spectroscopic analysis of products 14–16 and 18–19 indicated the macrocyclic structures for these compounds since no signals which might correspond to a terminal azido group (ν 2100 cm−1 in IR spectra) or a propargyl CH group (δ 2.76 in 1H NMR spectra) were detected. Instead, 1H NMR and 13C NMR spectra of each product 14–16 and 18–19 contained only one set of resonances corresponding to the galactopyranose residue, triazole CH fragment and CH2 group which constitute the repeating unit of these macrocycles. Each set of signals in these spectra had distinctive chemical shift values, though the variations of these values within both series of macrocycles were relatively small. Changes were observed in the positions of certain resonances in the NMR spectra of the macrocycles compared to starting azido-alkyne derivatives as illustrated by selected data in Table 1. The macrocycle ring size for these compounds followed unequivocally from ESI MS data, which clearly show [M + Na]+ adducts (Table 1, for individual spectra see ESI†).
Carrying out cyclooligomerisation under the same microwave-assisted CuAAC conditions as described above (CuSO4/Cu turnings, 100 °C, 15 min) but employing more concentrated solutions of the building blocks (1 M), for compound 1 we observed the predominant formation of trimer 15 (35%), accompanied by dimer 14 (10%) and tetramer 16 (3%). Trace amounts (<1%) of two other products were also isolated and analyzed by the ESI-MS, revealing m/z peaks at 1216.3 and 1481.5, which were attributed to [M + Na]+ adducts of cyclic pentamer 17 and cyclic hexamer, respectively (for individual mass spectra see Fig. S12 and S13 in ESI†). In the case of building block 2, CuAAC reactions performed at 1 M substrate concentration afforded essentially the same result as the reaction with more dilute substrate, leading to dimer 18 and trimer 19 in the same proportion (Fig. 2), though traces of cyclic tetramer were also detected (see HPLC and mass spectrum in Fig. S14 and S19 in ESI†).
In most of the previously reported syntheses of triazole-linked glycomacrocycles the starting materials for CuAAC reaction were used as protected saccharides.23–25,36 In order to test the potential of acetylated precursors of monomers 1 and 2, azido-alkyne derivatives 8 and 13, were subjected to cyclooligomerisation under microwave assisted CuAAC conditions (0.16 M in DMF, CuSO4/Cu turnings as catalyst, 100 °C, 15 min). The mixtures of products from these reactions were deacetylated by treatment with 0.1 M NaOMe in MeOH and the resulting materials were analysed using reverse-phase HPLC. The same sets of glycomacrocycles, 14–16 and 18–19, as those synthesized from monomers 1 and 2 were obtained in about the same ratios, albeit with lower yields as was evident from the comparison of HPLC results.
The decreased overall efficiency of the route based on protected precursors was probably a result of the additional work up and the deprotection steps, rather than the reduced reactivities of the protected monomers in the cyclooligomerisation reaction. Overall, only reactant concentration appeared to affect the distribution of cyclic oligomers formed. Higher concentrations of reactant 1 resulted in a greater proportion of the cyclic trimer 15 and the formation of cyclic pentamer 17. The efficiency of the cyclooligomerisation reactions with both building blocks 1 and 2 proved to be essentially independent of the nature of the CuI catalyst used.
Molecular modelling
In order to investigate the 3D shape and flexibility of the triazole-linked glycomacrocycles synthesised in this study, a conformational search in the gas phase and subsequent geometry optimisation were performed for the isomeric cyclic trimers 15 and 19. The MONTE CARLO method with the MMFF molecular mechanics model implemented in the Spartan '06 software, which has been the model and method of choice for other conformational surveys,66,67 was employed in this study. Both macrocycles have relatively open structures with small cavities but they possess quite different degrees of flexibility.Calculations revealed that the set of lowest energy conformers of cyclic trimer 15 (Fig. 3 and Fig. S20 in ESI†) have notably different geometries, which presumably reflects the inherent torsional flexibility of this macrocycle that arises from the CH2–O–CH2 bridges. The relative orientation of pyranose and triazole rings in each fragment connected through the anomeric carbon atom remained approximately the same for all calculated conformers of 15. For two of these fragments the triazole and pyranose rings are present in nearly perpendicular planes, similar to their orientation in the solid sate structure of β-D-glucopyranosyl triazoles.60,68 In contrast, the third pair of triazole and pyranose rings in 15 are nearly coplanar. The overall shapes of the lowest energy conformers of 15 are rather irregular compared with those calculated for cyclic trimer 19, which have a rather flat, C3 symmetric structure (Fig. 3). Compound 19 appears to have a less flexible structure than 15, as follows from the higher degree of similarity of the lowest energy conformers calculated for the former (Fig. S20 in ESI†).
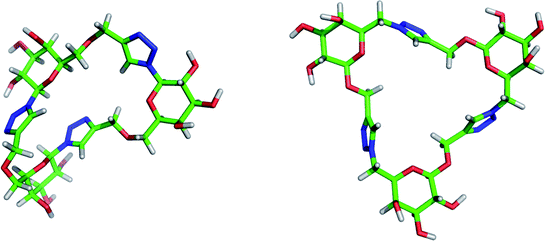 | ||
| Fig. 3 Lowest energy conformers for cyclic trimers 15 and 19 (left and right, respectively). | ||
In both cyclic trimers 15 and 19 the secondary OH groups of the sugar units are exposed – presented on the outside of the macrocycle – and available for enzymatic glycosylation (vide infra).
Enzymatic sialylation reactions
The glycomacrocycles 14–16 and 18–19 were subjected to the action of recombinant Trypanosoma cruzi trans-sialidase69 (TcTS), an α-2,3-specific enzyme capable of the transfer of sialic acid from a 2′-(4-methylumbelliferyl) α-D-N-acetylneuraminic acid (MUNANA) donor substrate to the 3-position of β-linked galactopyranose (Fig. 4). Trial reactions were carried out on a 100 μg–1 mg scale and reaction mixtures were analyzed by reverse-phase HPLC; analytical samples were purified by semi-preparative reverse-phase HPLC and initial characterisation was performed by mass spectrometry (Table S2 and Fig. S21–S24 in ESI†). Attempted sialylation of cyclic dimers 14 and 18 gave products 20 and 22 in 65 and 60% isolated yields, respectively. These doubly sialylated compounds were identified from the appearance of [M − H]− and [M − 2H]2− adducts in electrospray ionisation MS analysis following HPLC purification of the sialylated glycomacrocycles. For cyclic trimers 15 and 19, triply sialylated products 21 (56% isolated yield) and 23 (52% isolated yield), respectively, were identified from [M − 2H]2− adducts in MS analyses. In contrast, both HPLC and MS analyses of reactions failed to identify any sialylated products upon incubation of cyclic tetramer 16 with TcTS and MUNANA. Overall, this suggests that while the geometries adopted by cyclic dimers (14 and 18) and trimers (15 and 19) are compatible with access to the TcTS active site and subsequent enzymatic transformation, presumably cyclic tetramer 16 is unable to present its galactose residues in a manner consistent with TcTS binding or turnover. Further studies are ongoing to address this point.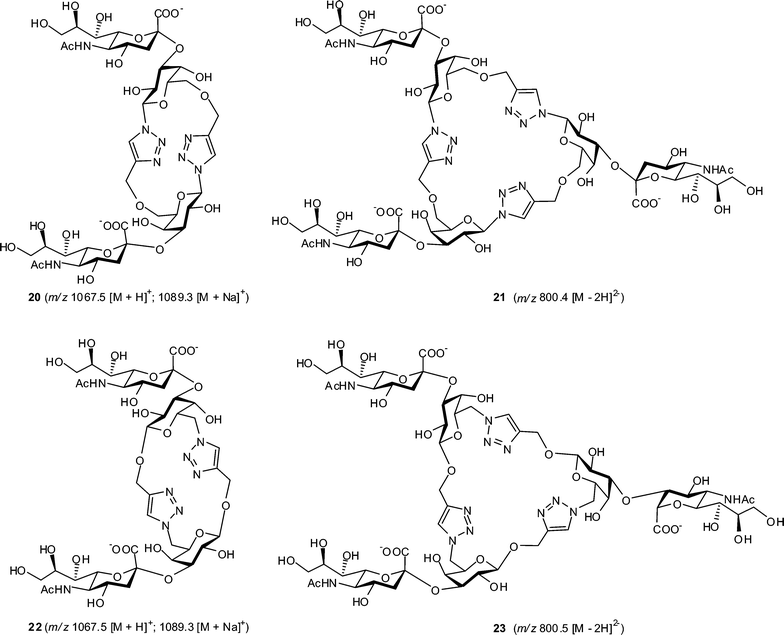 | ||
| Fig. 4 Proposed structures for products of the Trypanosoma cruzi trans-sialidase-catalyzed sialylation of cyclic pseudo-oligosaccharides 14, 15, 18 and 19 with MUNANA. | ||
Conclusion
Bifunctional building blocks incorporating an azido group either at an anomeric position or at C-6 and an alkyne functionality in the form of either a 6-O- or 1-O-propargyl ether of β-D-galactopyranose have been synthesised. These isomeric azido-alkynes have been subjected to cyclooligomerisation using a combination of microwave irradiation and Cu(I)-catalysed 1,3-dipolar cycloaddition conditions to give a set of novel glycomacrocycles with sugar units bridged by 1,4-disubstituted triazole residues. In moderately dilute solution, reaction of monomers 1 or 2 afforded only small macrocycles with a strong preference for cyclic dimers and trimers, which were obtained in reasonable yield (10–35% isolated yield of individual macrocycles). Under more concentrated conditions, formation of cyclic tetramer and pentamer from the building block 1 were also observed, albeit in low yield (a few %). In general, galactopyranosyl azide 1 proved to be slightly more efficient than its 6-azido isomer 2 in cyclooligomerisation experiments and also giving rise to higher yields and access to larger glycomacrocycles than monomer 2.In summary, the cyclooligomerisation approach employed in this study provides a practical alternative to glycosidically-linked cyclic oligosaccharides which with few exceptions (most notably the cyclodextrins) can only be obtained via multi-step chemical synthesis.70 While the synthetic routes reported give only modest yields of smaller glycomacrocycles (dimer to pentamer), clear opportunities for improvement are evident (e.g. through the introduction of linkers with different lengths and rigidities). Beyond chemical synthesis, the isomeric cyclic dimers and trimers formed by CuAAC-mediated cyclooligomerisation serve as substrates for enzyme-catalysed trans-sialylation, giving rise to multivalent sialylated macrocycles 20–23 for biomolecular recognition studies. Coupled with the inherent differences in geometric and dynamic preferences of cyclic trimers 15 and 19 identified in computational studies, these novel isomeric sialylated cyclic pseudo-galactooligosaccharides present new opportunities for dissecting the fine detail of sialic acid recognition in nature.
Experimental
General procedures, synthesis of building blocks 1 and 2 and precursors 3–13 are described in the ESI.†Typical procedure for cyclooligomerisation reaction
A solution of the azido-alkyne building block (0.09 mmol) and CuSO4 (0.042 mmol, 0.25 eq.) in DMF (0.5 mL) was placed into a microwave tube containing Cu turnings (∼15 mg), the tube was sealed and submitted to microwave irradiation in 15 min bursts (100 °C, 50 W). The reaction was followed by TLC (CH3CN–EtOAc–iPrOH–H2O 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:50:50) taking samples at 15 min intervals. After completion of the reaction the solution was carefully transferred to another flask in order to separate it from the Cu turnings and the solvent was removed under reduced pressure. The residue was re-dissolved in water (1 mL) and purified using a C18 cartridge (0.5 mL, Phenomenex) eluting first Cu salts and then carbohydrate-containing products (H2O). Individual products were obtained by HPLC purification on a semi-preparative C18 column (10 × 250 mm, Jupiter, Phenomenex) using gradient elution with 0.1% aq. CF3CO2H (A)–CH3CN (B), 0% to 35% B in 40 min, peaks were detected at 230 nm with a UV detector (see ESI†).
:20:50:50) taking samples at 15 min intervals. After completion of the reaction the solution was carefully transferred to another flask in order to separate it from the Cu turnings and the solvent was removed under reduced pressure. The residue was re-dissolved in water (1 mL) and purified using a C18 cartridge (0.5 mL, Phenomenex) eluting first Cu salts and then carbohydrate-containing products (H2O). Individual products were obtained by HPLC purification on a semi-preparative C18 column (10 × 250 mm, Jupiter, Phenomenex) using gradient elution with 0.1% aq. CF3CO2H (A)–CH3CN (B), 0% to 35% B in 40 min, peaks were detected at 230 nm with a UV detector (see ESI†).Cyclic pseudo-oligosaccharides obtained from building blocks 1 and 2
Sialylation reactions
Trial sialylation reactions were conducted with ca. 100 μg–1 mg of substrates 14–15 and 18–19 in 1 mL Eppendorf tubes. To a solution of the substrate (1 eq.) and MUNANA (5 eq.), in phosphate buffer (0.1 M, pH 7.0, 200 μL) was added the crude enzyme TcTS (50 μL), and the mixtures were incubated for 2 h at 25 °C. The mixtures were heated at 100 °C for 1 min, the denatured enzyme was removed by centrifugation (1500 rpm, 15 min), the supernatant was concentrated and the residues were purified by HPLC using the same conditions as described for purification of 14–15 and 18–19. Products were detected by UV absorption at 230 nm and collected with peak maximum at 12.35 min (20, 65%), 12.62 min (21, 56%), 12.65 min (22, 60%) and 12.6 min (23, 52%). Data for MS analysis of compounds 20–23 are shown in Fig. 4 and Table S2 in ESI.†Acknowledgements
This work was supported in the UK by the BBSRC and in Brazil by FAPESP and CNPq.Notes and references
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS.
- A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS.
- Plant Polysaccharides, ed. P. Ulskov, Blackwell, Oxford, 2010 Search PubMed.
- M. von Itzstein, Curr. Opin. Struct. Biol., 2008, 18, 558–566 CrossRef CAS.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677 CrossRef CAS.
- J. E. Turnbull and R. A. Field, Nat. Chem. Biol., 2007, 3, 74–77 CrossRef CAS.
- M. Fais, R. Karamanska, D. A. Russell and R. A. Field, J. Cereal Sci., 2009, 50, 306–311 CrossRef CAS.
- R. T. Lee and Y. C. Lee, in Neoglycoconjugates: Preparations and Applications, ed. Y. C. Lee and R. T. Lee, Academic Press, San Diego, 1994, pp. 23–52 Search PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- Y. M. Chabre and R. Roy, Adv. Carbohydr. Chem. Biochem., 2010, 63, 165–393 CAS.
- T. K. Lindhorst, Top. Curr. Chem., 2002, 218, 201–235 CAS.
- B. E. Collins and J. C. Paulson, Curr. Opin. Chem. Biol., 2004, 8, 617–625 CrossRef CAS.
- R. J. Pieters, Org. Biomol. Chem., 2009, 7, 2013–2025 RSC.
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669–672 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS.
- S. Dedola, S. A. Nepogodiev and R. A. Field, Org. Biomol. Chem., 2007, 5, 1006–1017 RSC.
- A. Dondoni, Chem.–Asian J., 2007, 2, 700–708 CrossRef CAS.
- V. Aragão-Leoneti, V. L. Campo, A. S. Gomes, R. A. Field and I. Carvalho, Tetrahedron, 2010, 66 Search PubMed , accepted.
- B. Hoffmann, B. Bernet and A. Vasella, Helv. Chim. Acta, 2002, 85, 265–287 CrossRef CAS.
- K. D. Bodine, D. Y. Gin and M. S. Gin, J. Am. Chem. Soc., 2004, 126, 1638–1639 CrossRef CAS.
- K. D. Bodine, D. Y. Gin and M. S. Gin, Org. Lett., 2005, 7, 4479–4482 CrossRef CAS.
- N. Pietrzik, D. Schmollinger and T. Ziegler, Belstein J. Org. Chem., 2008, 4 DOI:10.3762/bjoc.4.30 , No. 30.
- S. Muthana, H. Yu, H. Z. Cao, J. S. Cheng and X. Chen, J. Org. Chem., 2009, 74, 2928–2936 CrossRef CAS.
- R. Leyden and P. V. Murphy, Synlett, 2009, 1949–1950 CAS.
- R. Jagasia, J. M. Holub, M. Bollinger, K. Kirshenbaum and M. G. Finn, J. Org. Chem., 2009, 74, 2964–2974 CrossRef CAS.
- R. A. Turner, A. G. Oliver and R. S. Lokey, Org. Lett., 2007, 9, 5011–5014 CrossRef CAS.
- S. S. van Berkel, B. van der Lee, F. L. van Delft and F. P. J. T. Rutjes, Chem. Commun., 2009, 4272–4274 RSC.
- R. C. Elgersma, M. van Dijk, A. C. Dechesne, C. F. van Nostrum, W. E. Hennink, D. T. S. Rijkersa and R. M. J. Liskamp, Org. Biomol. Chem., 2009, 7, 4517–4525 RSC.
- J. M. Beierle, W. S. Horne, J. H. van Maarseveen, B. Waser, J. C. Reubi and M. R. Ghadiri, Angew. Chem., Int. Ed., 2009, 48, 4725–4729 CrossRef CAS.
- J. F. Billing and U. J. Nilsson, J. Org. Chem., 2005, 70, 4847–4850 CrossRef CAS.
- S. Binauld, C. J. Hawker, E. Fleury and E. Drockenmuller, Angew. Chem., Int. Ed., 2009, 48, 6654–6658 CrossRef CAS.
- G. R. Balou, J. P. Joly, L. Vernex-Loset and Y. Chapleur, Lett. Org. Chem., 2009, 6, 106–109 Search PubMed.
- S. Jarosz, B. Lewandowski and A. Listkowski, Synthesis, 2008, 913–916 CrossRef CAS.
- J. Morales-Sanfrutos, M. Ortega-Munoz, J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, J. Org. Chem., 2008, 73, 7768–7771 CrossRef CAS.
- A. Coskun, S. Saha, I. Aprahamian and J. F. Stoddart, Org. Lett., 2008, 10, 3187–3190 CrossRef CAS.
- S. M. Goldup, D. A. Leigh, T. Long, P. R. McGonigal, M. D. Symes and J. Wu, J. Am. Chem. Soc., 2009, 131, 15924–15929 CrossRef CAS.
- M. J. Milton, R. Harris, M. A. Probert, R. A. Field and S. W. Homans, Glycobiology, 1998, 8, 147–153 CrossRef CAS.
- R. Harris, G. R. Kiddle, R. A. Field, M. J. Milton, B. Ernst, J. L. Magnani and S. W. Homans, J. Am. Chem. Soc., 1999, 121, 2546–2551 CrossRef CAS.
- J. A. Harrison, K. P. R. Kartha, W. B. Turnbull, S. L. Scheuerl, J. H. Naismith, S. Schenkman and R. A. Field, Bioorg. Med. Chem. Lett., 2001, 11, 141–144 CrossRef CAS.
- R. Karamanska, J. Clarke, O. Blixt, J. I. Macrae, J. Q. Q. Zhang, P. R. Crocker, N. Laurent, A. Wright, S. L. Flitsch, D. A. Russell and R. A. Field, Glycoconjugate J., 2008, 25, 69–74 CrossRef CAS.
- H. Kamitakahara, T. Suzuki, N. Nishigori, Y. Suzuki, O. Kanie and C. H. Wong, Angew. Chem., Int. Ed., 1998, 37, 1524–1528 CrossRef CAS.
- M. A. Probert, M. J. Milton, R. Harris, S. Schenkman, J. M. Brown, S. W. Homans and R. A. Field, Tetrahedron Lett., 1997, 38, 5861–5864 CrossRef CAS.
- W. B. Turnbull, J. A. Harrison, K. P. R. Kartha, S. Schenkman and R. A. Field, Tetrahedron, 2002, 58, 3207–3216 CrossRef CAS.
- V. L. Campo, I. Carvalho, S. Allman, B. G. Davis and R. A. Field, Org. Biomol. Chem., 2007, 5, 2645–2657 RSC.
- R. M. van Well, B. Y. M. Collet and R. A. Field, Synlett, 2008, 2175–2177 CAS.
- V. M. Mendoza, R. Agusti, C. Gallo-Rodriguez and R. M. de Lederkremer, Carbohydr. Res., 2006, 341, 1488–1497 CrossRef.
- B. Neubacher, S. Svenja, S. Kelm, A. C. Frasch, B. Meyer and J. Thiem, ChemBioChem, 2006, 7, 896–899 CrossRef CAS.
- R. Agusti, M. E. Giorgi, V. M. Mendoza, C. Gallo-Rodriguez and R. M. de Lederkremer, Bioorg. Med. Chem., 2007, 15, 2611–2616 CrossRef CAS.
- S. A. Allman, H. H. Jensen, B. Vijayakrishnan, J. A. Garnett, E. Leon, Y. Liu, D. C. Anthony, N. R. Sibson, T. Feizi, S. Matthews and B. G. Davis, ChemBioChem, 2009, 10, 2522–2529 CrossRef CAS.
- I. Carvalho, P. Andrade, V. L. Campo, P. M. M. Guedes, R. Sesti-Costa, J. S. Silva, S. Schenkman, S. Dedola, L. Hill, M. Rejzek, S. A. Nepogodiev and R. A. Field, Bioorg. Med. Chem., 2010, 18, 2412–2427 CrossRef CAS.
- J. A. Harrison, K. P. R. Kartha, J. H. Naismith, S. Schenkman, E. J. L. Fournier, T. L. Lowary, C. Malet, U. J. Nilsson, O. Hindsgaul and R. A. Field, 2010, in preparation.
- H. F. Chow, K. N. Lau, Z. H. Ke, Y. T. Liang and C. M. Lo, Chem. Commun., 2010, 46, 3437–3453 RSC.
- O-Linked cyclic oligosaccharides have also been prepared using cyclooligomerisation approaches. However, the overall efficiency of such approaches is limited since they are based on repetitive formation of challenging O-glycosidic linkages: (a) N. K. Kochetkov, S. A. Nepogod'ev and L. V. Backinowsky, Tetrahedron, 1990, 46, 139–150 CrossRef CAS; (b) P. R. Ashton, C. L. Brown, S. Menzer, S. A. Nepogodiev, J. F. Stoddart and D. J. Williams, Chem.–Eur. J., 1996, 2, 580–591 CrossRef CAS.
- L. Marmuse, S. A. Nepogodiev and R. A. Field, Org. Biomol. Chem., 2005, 3, 2225–2227 RSC.
- S. A. Nepogodiev, S. Dedola, L. Marmuse, M. T. de Oliveira and R. A. Field, Carbohydr. Res., 2007, 342, 529–540 CrossRef CAS.
- S. Dedola, D. L. Hughes and R. A. Field, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o445–o446 CrossRef.
- S. Dedola, D. L. Hughes, S. A. Nepogodiev, M. Rejzek and R. A. Field, Carbohydr. Res., 2010, 345, 1123–1134 CrossRef CAS.
- K. P. R. Kartha, Tetrahedron Lett., 1986, 27, 3415–3416 CrossRef CAS.
- H. B. Mereyala, S. R. Gurrala and S. K. Mohan, Tetrahedron, 1999, 55, 11331–11342 CrossRef CAS.
- F. D. Tropper, F. O. Andersson, S. Braun and R. Roy, Synthesis, 1992, 618–620 CrossRef CAS.
- J. Yang, X. Fu, Q. Jia, J. Shen, J. B. Biggins, J. Q. Jiang, J. J. Zhao, J. J. Schmidt, P. G. Wang and J. S. Thorson, Org. Lett., 2003, 5, 2223–2226 CrossRef CAS.
- P. Appukkuttan, V. P. Mehta and E. V. Van der Eycken, Chem. Soc. Rev., 2010, 39, 1467–1477 RSC.
- W. J. Hehre, A Guide to Molecular Mechanics and Quantum Chemical Calculations, Wavefunction Inc., Irvine, USA, 2003 Search PubMed.
- A. R. Leach, Molecular Modelling: Principles and Applications, 2nd edn, Pearson Education, Harlow, UK, 2001 Search PubMed.
- B. L. Wilkinson, L. F. Bornaghi, T. A. Houston, S. A. Poulsen and A. R. White, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o5065–o5067 CrossRef.
- S. Schenkman, L. B. Chaves, L. C. Pontes de Carvalho and D. Eichinger, J. Biol. Chem., 1994, 269, 7970–7975 CAS.
- G. Gattuso, S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1919–1958 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, molecular modelling results, NMR and MS spectra. See DOI: 10.1039/c0sc00301h |
| This journal is © The Royal Society of Chemistry 2010 |