Synthesis and characterization of three small band gap conjugated polymers for solar cell applications†
Stefan
Hellström
a,
Lars J.
Lindgren
a,
Yi
Zhou
b,
Fengling
Zhang
b,
Olle
Inganäs
b and
Mats R.
Andersson
*a
aDepartment of Chemical and Biological Engineering, Polymer Technology, Chalmers University of Technology, SE-412 96, Göteborg, Sweden. E-mail: mats.andersson@chalmers.se; Fax: +46 31 7723418; Tel: +46 31 7723401
bDepartment of Physics, Chemistry and Biology (IFM), Linköping University, SE-581 83, Linköping, Sweden
First published on 24th June 2010
Abstract
We report on a new series of small band gap conjugated polymers utilizing donor–acceptor–donor substructures in the polymer backbone to broaden and extend the optical absorption to longer wavelengths. Three polymers were prepared by Suzuki polymerization, using the same donor–acceptor–donor segment but with different comonomers. The goal was to investigate how the optical and electronic properties of the polymers were influenced by the different comonomers. Electrochemical spectroscopy, using square-wave voltammetry, shows that increasing the electron-donating strength of the comonomer will raise the HOMO energy level of the polymer, resulting in a decreased band gap. This result is also manifested by comparing open-circuit voltages from the corresponding laboratory fabricated solar cells. The best performing photovoltaic cell, based on APFO-Green15/[60]PCBM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 w/w), reached a Jsc of 4.2 mA cm−2, a Voc of 0.73 V, and a FF of 0.54, giving a PCE of 1.7%.
:4 w/w), reached a Jsc of 4.2 mA cm−2, a Voc of 0.73 V, and a FF of 0.54, giving a PCE of 1.7%.
Introduction
Conjugated polymers are promising candidates for photoinduced charge generation and transport media in solar cells, with efficiencies of around 6% recently reported.1–3 In order to make polymer solar cells competitive, their efficiencies need to be increased, a challenge addressed to polymer design, synthesis, and device fabrication. An ideal p-type conjugated polymer has to simultaneously combine strong absorption, small band gap, high hole mobility, good film-forming properties, and suitable energy levels with respect to the n-type material. If the band gap of the polymer is decreased, the absorption spectrum will be broadened, making it possible to utilize more photons for charge generation in the photovoltaic cell. Decreasing the band gap means that either the highest occupied molecular orbital (HOMO) of the conjugated polymer is raised or the lowest unoccupied molecular orbital (LUMO) is lowered. A raised HOMO gives a lower photovoltage of the device and a lowered LUMO decreases the offset to the LUMO of the n-type material used. An offset of approximately 0.3 eV is required to maintain a sufficient driving force for exciton dissociation and prevent recombination of photogenerated charges.4,5In this report we present three small band gap polymers with alternating electron-donating (D) and electron-accepting (A) units. This design, using DAD-segments in the polymer backbone, creates an internal charge transfer, which reduces the band gap.6–9 To investigate how the choice of comonomer influences the polymer properties, one DAD-monomer was Suzuki polymerized with three different comonomers resulting in two low band gap para-phenylene (LBPP) polymers and one alternating polyfluorene (APFO) polymer. The three comonomers differ in electron-donating strength, which results in three polymers with different optical and electrochemical properties. Together with an n-type material, e.g. [6,6]-phenyl-C61-butyric acid methyl ester ([60]PCBM) or [6,6]-phenyl-C71-butyric acid methyl ester ([70]PCBM), charge generation, charge separation, and transportation of electrons and holes are possible in the active layer of the photovoltaic cell. From a solar cell efficiency point of view, this series of materials so far show quite modest performance. However, they serve as a good example of how the molecular structure influences optical and electronic performance.
Results and discussion
Polymer design and synthesis
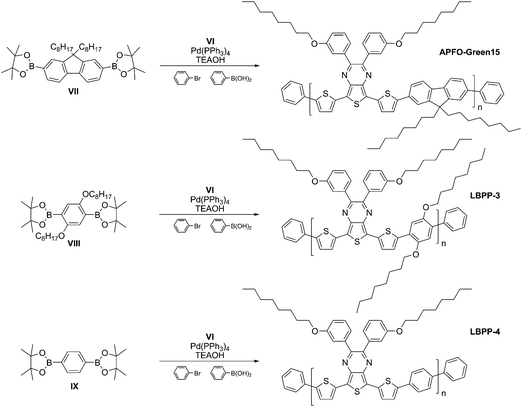
Inspired by previously prepared polymers, i.e. APFO-Greens,10–13 we wanted to make a new series of polymers to investigate the influence of different comonomers. A new monomer (VI) was synthesized (Scheme 1) and copolymerized with either 2,2′-(9,9-dioctylfluorene-2,7-diyl)bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) (VII),14 2,2′-(2,5-bis(octyloxy)-1,4-phenylene)bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolane) (VIII),15 or 1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene (IX), resulting in APFO-Green15, LBPP-3, and LBPP-4, respectively. The DAD-monomer (VI) was designed with thiophene units as electron donating groups and a thieno[3,4-b]pyrazine derivative as electron accepting group, reducing the band gap by intramolecular charge transfer. Furthermore, the chemical structure of thieno[3,4-b]pyrazine, with electronegative nitrogen atoms in combination with the relatively low aromaticity of thiophene, induces an increased preference for adopting a quinoid structure of the polymer backbone, which further decreases the band gap through reduced bond length alternation.16 Solubilizing octyloxy side-chains were attached in meta-position of the pendent phenyl rings, instead of para-position, to maintain the strong electron-accepting character of the thieno[3,4-b]pyrazine unit.17 For the polymerization reactions (Scheme 2), the Suzuki cross-coupling reaction was employed and phenylboronic acid and bromobenzene were added as end-capping agents after the polymerization time.18 The polymers were obtained using similar reaction conditions in all three cases (0.03 M total monomer concentration, equimolar amounts of monomers, 3 mol% catalyst, refluxing toluene, and nitrogen atmosphere) and reaction times were 4 hours. After end capping, precipitation, and washing with ammonia and water, the polymeric materials were extracted with diethyl ether to remove low molecular-weight oligomers, and the final products were obtained by extraction with chloroform. | ||
| Scheme 1 Synthetic route to the DAD-monomer. | ||
 | ||
| Scheme 2 Polymerization reactions. | ||
Synthetic steps are similar to the syntheses of previously published APFO polymers,12,19 with the exception of the use of protective-group chemistry prior to the alkylation. The idea with a protected monomer is that after deprotection, the resulting phenolic compound can be alkylated with several alkylating agents, yielding monomers with different lengths of the side-chains. This can be achieved without the necessity of preparing all different types of diones via a Grignard reagent.
Molecular weight
A high polymer molecular weight will guarantee an adequate conjugation length and enhance interchain interactions, yielding an increased hole mobility via strengthened intramolecular charge transport properties and intermolecular hopping. All polymer syntheses were performed using stoichiometric proportions of the comonomers to achieve the highest molecular weight possible. Molecular-weight distributions of the polymers were determined by size-exclusion chromatography (SEC). Number-average (Mn) and weight-average molecular weights (Mw) are presented in Table 1. The molecular weights of APFO-Green15 and LBPP-3 are high to very high as a result of both polymers having solubilizing side-chains on the comonomers. However, the molecular weight of LBPP-3 is most probably overestimated due to polymer aggregation, as indicated by the presence of a high-molecular weight tail of the SEC-curve. The molecular weight of LBPP-4, lacking solubilizing substituents on the para-phenylene comonomer, is somewhat low. Even after repeating the polymerization of LBPP-4, higher molecular weights were not obtained.Thermal properties
The thermal stability of the polymers was investigated by thermogravimetric analysis (TGA) in nitrogen atmosphere. All polymers display good thermal stability with a large weight loss around 400 °C, although LBPP-3 degrades partially at lower temperatures. LBPP-4 is the most stable polymer, which can be explained by good packing properties as a result of a small comonomer, free from bulky side-chains. Results from TGA are presented in the ESI†.Electrochemical properties
The positions of the HOMO and the LUMO of the polymers are of great importance when evaluating the materials from a solar cell perspective. From the energy levels one can estimate driving forces for charge-transfer processes together with an estimation of the open-circuit photovoltage. These positions can be obtained from electrochemical spectroscopy, where the oxidation peak corresponds to the HOMO and the LUMO can be related to the reduction peak. To measure the electrochemical energy levels, we employed square-wave voltammetry (SWV)20 and cyclic voltammetry (CV).21 SWV is a pulse-voltammetric technique based on chronoamperometry, where the current is sampled as a function of time after applying a potential pulse. The fast-decay nature of the capacitive charging current results in electrochemical data originating from pure Faradic current. Another advantage with SWV is efficient suppression of background currents, yielding well resolved spectra.Fig. 1 shows SWV and CV spectra of the polymers and estimated HOMO and LUMO levels from SWV together with the electrochemical band gap are presented in Table 2. SWV allows estimation of HOMO and LUMO levels from peak potentials, described in the Experimental section below. However, as the oxidation peak of LBPP-3 turned out to be relatively broad, CV spectra were recorded to improve the reliability of the estimated energy levels. The relative position of the onsets of the CV oxidation waves is in agreement with the SWV data presented in Table 2. Additionally, the results from CV in Fig. 1b clearly indicate the electrochemical reversibility of the polymers.
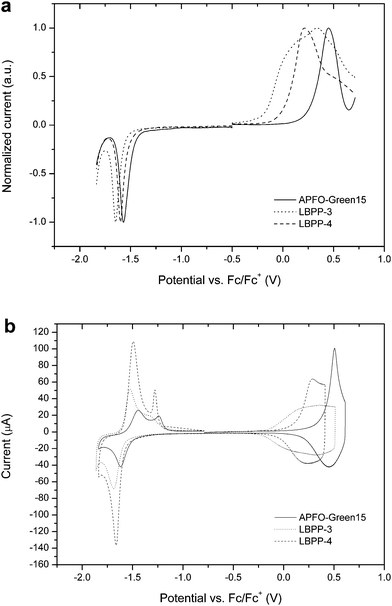 | ||
| Fig. 1 Electrochemical voltammograms of polymer films on Pt-wire from (a) square-wave voltammetry and (b) cyclic voltammetry. | ||
| Polymer | E 0 ox /V | E 0 red /V | HOMOb/eV | LUMOb/eV | E g ec /eV |
|---|---|---|---|---|---|
| a Peak potentials vs. Fc/Fc+. b Energy levels are estimated from HOMO/LUMO = −(Eox/red + 5.13) eV. c Electrochemical band gap. d Estimated due to broad oxidation peak. | |||||
| APFO-Green15 | 0.45 | −1.57 | −5.58 | −3.56 | 2.0 |
| LBPP-3 | 0.05d | −1.65 | −5.2 | −3.48 | 1.7 |
| LBPP-4 | 0.21 | −1.60 | −5.34 | −3.53 | 1.8 |
The electrochemical data show, that when going from APFO-Green15 to LBPP-4 and finally to LBPP-3, the oxidation waves are raised towards vacuum implying a smaller band gap. The reason for this is explained by the increasing electron-donating strength of the comonomers. The dioctyloxy phenylene unit present in LBPP-3 is very electron rich due to the electron-donating character of the octyloxy side-chains. This strengthens the electron-donating moieties of the DAD-segment which results in a strong intramolecular charge transfer. The smaller band gap of LBPP-3 compared to LBPP-4 is mainly a result of a raised HOMO, whereas the LUMO position is practically unaffected by the substitution of comonomer. Consequently, APFO-Green15 shows the lowest lying HOMO and largest electrochemical band gap as a result of having the weakest electron-donating comonomer of the three polymers. Looking at the estimated LUMO positions it is evident that all three polymers guarantee a downhill energy offset for charge transfer to [60]PCBM and [70]PCBM, which LUMO positions previously have been estimated to be −4.1 eV by SWV.17 The electrochemical band gaps of the three polymers range from 1.7–2.0 eV.
Optical absorption
Absorption spectroscopy, measuring transitions between electronic states, provides essential information about the spectral coverage and the magnitude of the band gap. Although information about the location of molecular orbital energies cannot be obtained, optical absorption measurements have great resemblance with the charge-generation process in a photovoltaic cell and are of utmost importance when characterizing solar cell materials. The UV-Vis-NIR absorption spectra of the polymers are given in Fig. 2. The optical band gap of the polymers were estimated from the onset of absorption (Table 3) because vibronic structures often yield broad low-energy absorption peaks, making it hard to distinguish absorption maxima. Estimation of optical band gap from absorption onset is also the preferred method used in scientific publications on conjugated polymers, facilitating comparisons of polymer band gap reported by different research groups. The optical band gaps (1.2–1.6 eV) are smaller than the electrochemical band gaps, which is almost always the case when characterizing conjugated polymers. It is important to realize that there should be a difference which can be explained by the dissimilar nature of the measurements where different energy processes contribute to band gap estimation. However, results from optical absorption are in agreement with the conclusion from electrochemistry that an electron-rich comonomer gives a smaller band gap. LBPP-3 shows an absorption spectrum furthest extended to lower energies, corresponding to an optical band gap of 1.2 eV, as a direct result of incorporation of the electron-rich dioctyloxy para-phenylene comonomer into the polymer backbone.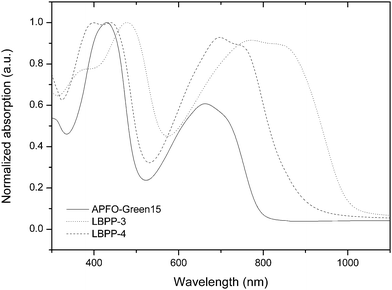 | ||
| Fig. 2 Optical absorption of polymer films, normalized by the high-energy peak. | ||
Fabrication and characterization of photovoltaic cells
A number of photovoltaic cells were fabricated in order to evaluate the polymers from a solar cell perspective. The cells had the general structure glass/ITO/PEDOT:PSS/polymer:PCBM/LiF/Al. Previously, the overall performance of polymer solar cells based on alternating polyfluorene copolymers has shown to be dependent on charge transport rather than on the generation of charges.22 Therefore, we used a 1:4 (w/w) ratio of polymer:PCBM to improve charge-carrier transport and establish an adequate balance between the hole and electron mobility in the active layer. Fig. 3 shows the EQE of the fabricated devices and all photovoltaic data are summarized in Tables 4 and 5 for [60]PCBM and [70]PCBM as n-type materials, respectively. [70]PCBM has a stronger absorption in the visible region compared to [60]PCBM, yielding photovoltaic cells with superior charge generation properties. Cells made from APFO-Green15 display the highest EQE over the whole visible range despite the relatively low absorption coefficient of the low energy peak of the neat polymer (Fig. 2).
![EQE of the best performing devices based on blends of individual polymers and (a) [60]PCBM or (b) [70]PCBM, see Tables 4 and 5 for film thickness.](/image/article/2010/PY/c0py00152j/c0py00152j-f3.gif) | ||
| Fig. 3 EQE of the best performing devices based on blends of individual polymers and (a) [60]PCBM or (b) [70]PCBM, see Tables 4 and 5 for film thickness. | ||
| Polymer | Spin speed/rpm | Active layer thickness/nm | J sc/mA cm−2 | V oc/V | FF | PCE (%) |
|---|---|---|---|---|---|---|
| APFO-Green15 | 4000 | 77 | 2.9 | 0.74 | 0.53 | 1.1 |
| 6000 | 70 | 3.2 | 0.72 | 0.56 | 1.3 | |
| 7000 | 65 | 3.1 | 0.73 | 0.54 | 1.2 | |
| LBPP-3 | 1000 | 98 | 1.5 | 0.35 | 0.51 | 0.3 |
| 3000 | 68 | 2.1 | 0.35 | 0.55 | 0.4 | |
| LBPP-4 | 2000 | 92 | 2.3 | 0.56 | 0.61 | 0.8 |
| 4000 | 80 | 2.4 | 0.54 | 0.55 | 0.7 |
The active layers were spin coated using different spin speeds to investigate how the thickness and morphology of the active layer influence device performance. Looking at the results using [60]PCBM as electron acceptor, one notices the trend that an increased spin speed, i.e. a thinner active layer, increases the performance of the cells. A thin active layer absorbs less photons but will benefit from good charge collection. Making the layer thicker will result in problems with trapping of photogenerated charges and back reactions in the form of recombination. However, if the charge-carrier mobility is raised, e.g. by improving the hole-transporting properties of the p-type polymer, the extent of charge recombination will be reduced which in turn allows a thicker active layer, enhancing the solar cell performance by increased photon absorption.
APFO-Green15 shows the highest open-circuit voltage (Voc) which is expected from the low HOMO level of the fluorene unit, and is also in agreement with the anodic peak potential results from SWV. The excellent hole-transporting properties of the fluorene segment are signified by a satisfactory short-circuit current (Jsc) using APFO-Green15 as p-type material. The same trends are seen from the photovoltaic cells with [70]PCBM as n-type material. However, these cells generally show a poorer performance compared with the cells using [60]PCBM.
Morphology
Separation of photogenerated charges takes place at the interface between the polymer and n-type material in the active layer. A large interfacial area is required due to the short lifetimes of excitons, limiting their diffusion length to approximately 10 nm.23–25 The photovoltaic cells were prepared using the bulk-heterojunction concept,26 not only to create a large interfacial area but also to provide continuous pathways for efficient charge collection. Atomic force microscopy (AFM) was used to investigate how incorporation of different comonomers into the polymer backbone influences the nano- and microstructure of the active layers. AFM height-images of the three polymers and [60]PCBM or [70]PCBM are shown in Fig. 4–6. The general tendency is that the morphology using [60]PCBM as n-type material is far superior than using [70]PCBM. AFM-images with [70]PCBM reveal domains in the active layer, which limit transport and collection of charge carriers. This explains the moderate photovoltaic performance for cells using [70]PCBM as p-type material, in spite of the strong absorption of [70]PCBM in the visible region suggesting enhanced device performance via improved charge generation. Blends of LBPP-4:[70]PCBM display smoother microstructures with smaller domains compared to LBPP-3:[70]PCBM. This indicates that an increase in side-chain density promotes phase separation of the materials.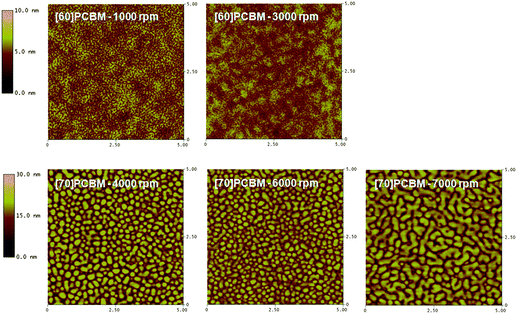 | ||
| Fig. 4 AFM height-images of 5 × 5 µm diodes using APFO-Green15 as p-type material. | ||
 | ||
| Fig. 5 AFM height-images of 5 × 5 µm diodes using LBPP-3 as p-type material. | ||
 | ||
| Fig. 6 AFM height-images of 5 × 5 µm diodes using LBPP-4 as p-type material. | ||
The AFM-images suggest that a higher spin speed improves morphology, resulting in a larger interfacial area and increased charge-carrier mobility for both holes and electrons in the active layer. To some extent, the morphology of the active layer in a bulk-heterojunction polymer solar cell can be controlled by the choice of solvent or by addition of a small amount of a solvent with low evaporation rate during the spin coating procedure.27 However, this kind of morphological optimization has not yet been done for polymers presented in this paper.
Conclusions
By copolymerizing one DAD monomer with three different comonomers, three conjugated polymers were prepared with the aim of investigating how different chemical and electronic structures influence the properties of the final polymers. The results show, that when selecting a comonomer with a high electron-donating strength, the band gap of the polymer is decreased, primarily by raising the HOMO energy level. This lowers the Voc of the photovoltaic cell, whereas the downhill driving force for exciton dissociation remains unchanged. The choice of comonomer will also partly influence the morphology of the active layer. Side-chains increase the solubility of the polymer, but will induce formation of domains in the active layer, resulting in an inefficient charge separation and charge collection at the electrodes.Experimental
General
All chemicals, except (3-bromophenoxy)(tert-butyl)dimethylsilane (I) and 5,5″-dibromo-3′,4′-dinitro-2,2′:5′,2″-terthiophene (III), were purchased from Sigma-Aldrich and used without further purification. Compounds I28 and III29 were synthesized following previously reported procedures. NMR-spectra of chloroform-d or DMSO-d6 solutions were recorded at ambient temperature on a Varian VXR-300S spectrometer. The peak values were calibrated relative to tetramethylsilane (δH = 0 ppm). J values are given in Hz.Mass spectra were obtained by MALDI-TOF on a Bruker Daltronics Autoflex, operating in reflectron mode. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was used as matrix.
Size-exclusion chromatography was performed on a Waters Alliance 2000 with refractive index detector. The working temperature was 135 °C and 1,2,4-trichlorobenzene was used as eluent at a flow rate of 1 mL min−1. Molecular weights and polydispersity indices were obtained by calibration relative to polystyrene standards.
Electrochemical spectroscopy
All electrochemical measurements were performed on a CH-Instruments 650A Potentiostat/Galvanostat. A three electrode setup, with platinum wires both as working electrode and counter electrode together with a quasi Ag/Ag+ reference electrode, was used. A solution of 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) in anhydrous acetonitrile was used as supporting electrolyte. Thin polymer films were cast from o-dichlorobenzene solution onto the working electrode. A small amount of Bu4NPF6 was added to the polymer solution prior to deposition to improve charge transport in the polymer matrix during the scans. In order to remove oxygen, the electrolyte was purged with nitrogen gas prior to each experiment. During the scans the nitrogen inlet was moved above the electrolyte surface. The third scan of the anodic and cathodic electrochemical waves was recorded and calibrated vs. the oxidative peak potential of ferrocene. HOMO and LUMO levels were estimated from peak potentials setting the oxidative peak potential of Fc/Fc+vs. the normal-hydrogen electrode (NHE) to 0.630 V30 and the NHE vs. the vacuum level to 4.5 V.21Atomic force microscopy (AFM)
The surface morphology of the active layers was imaged by AFM, using a Dimension 3100 system (Digital Instruments/Veeco) operating in tapping mode. Silicon cantilevers (NSG10) with a force constant of 5.5–22.5 N m−1, a resonance frequency of 190–355 kHz, and a tip curvature radius of 10 nm were used.Device fabrication
A conductive layer of PEDOT–PSS (Baytron P VP Al 4038) was spin coated onto an ITO-coated glass slide. The film was annealed at 120 °C for 10 minutes in order to remove water. Chloroform solutions of conjugated polymer and [60]PCBM or [70]PCBM (1:4 w/w) were spin coated on top of the PEDOT–PSS. Finally, the cathode, consisting of LiF (0.6 nm) and Al (80 nm), was deposited in vacuum. The thickness of the active layer was measured using a surface profiler, Dektak 6M. The size of the diodes was defined by a mask to be approximately 4–6 mm2 when depositing Al.
External quantum efficiencies (EQEs) were calculated from the photocurrents at short-circuit conditions. The currents were recorded by a Keithley 485 picoammeter under illumination of monochromatic light through the anodic side of the devices. Current–voltage characteristics were recorded using a Keithley 2400 Source Meter under illumination of AM 1.5. The light intensity of the solar simulator (Model SS-50A, Photo Emission Tech., Inc.) was 100 mW cm−2. All laboratory fabrication of photovoltaic cells was made in ambient environment.
Synthetic procedures
:1 as eluent) gave II (1.14 g, 2.42 mmol, 29%) as a yellow oil. νmax (film)/cm−1 2956 (CH3) 2886 (CH3), 1675 (CO), 1596 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) 1484 (CC), 1278 (COSi), 860, 840 and 782 (m-sub. benzene); δH (300 MHz, CDCl3, Me4Si) 7.48 (2H, d, J 7.8, ArH), 7.44 (2H, s, ArH), 7.35 (2H, t, J 7.6, ArH), 7.12 (2H, d, J 8.0, ArH), 0.98 (18H, s, C(CH3)3) and 0.20 (12H, s, CH3Si); δC (100 MHz, CDCl3, Me4Si) 194.7, 156.4, 134.4, 130.2, 127.0, 123.5, 120.4, 25.7, 18.3, −4.4; λmax (CHCl3)/nm 264 and 314 (ε/dm3 mol−1 cm−1 24500 and 6600); m/z (MALDI-TOF) 470.47 (M+, C26H38O4Si2 requires 470.23).
C) 1426, 1280 (COSi), 933 (COSi), 830 and 778 (m-sub. benzene); δH (300 MHz, CDCl3, Me4Si) 7.30 (2H, d, J 3.9, ArH), 7.23 (2H, s, ArH), 7.16 (2H, t, J 7.8, ArH), 7.05 (2H, d, J 3.9, ArH), 7.03 (2H, d, J 6.0, ArH), 6.88 (2H, d, J 7.2, ArH), 1.00 (18H, s, C(CH3)3) and 0.21 (12H, s, CH3Si); δC (100 MHz, CDCl3, Me4Si) 155.8, 153.0, 140.1, 137.3, 135.9, 129.7, 129.2, 124.1, 123.8, 123.3, 121.4, 121.3, 114.8, 25.8, 18.3, −4.2; λmax (CHCl3)/nm 357 and 563 (ε/dm3 mol−1 cm−1 37800 and 9000); m/z (MALDI-TOF) 867.83 (M+, C38H42Br2N2O2S3Si2 requires 868.03).
C), 1427, 1283, 782 (m-sub. benzene); δH (300 MHz, DMSO, Me4Si) 9.65 (2H, s, ArOH), 7.49 (2H, d, J 3.0, ArH), 7.30 (2H, d, J 3.6, ArH), 7.17 (2H, t, J 7.8, ArH), 7.03 (2H, s, ArH), 6.85 (2H, d, J 7.5, ArH) and 6.82 (2H, d, J 7.5, ArH); δC (100 MHz, DMSO, Me4Si) 157.6, 153.6, 140.3, 137.0, 135.4, 130.7, 129.5, 124.5, 123.9, 121.1, 116.9, 114.6; λmax (CHCl3)/nm 357 and 563 (ε/dm3 mol−1 cm−1 70900 and 17000); m/z (MALDI-TOF) 639.86 (M+, C26H14Br2N2O2S3 requires 639.86).
C), 1424, 1268 (arom. ether), 780 (m-sub. benzene); δH (300 MHz, CDCl3, Me4Si) 7.31 (2H, d, J 3.9, ArH), 7.23 (2H, s, ArH), 7.20 (2H, d, J 6.3, ArH), 7.10 (2H, d, J 6.8, ArH), 7.04 (2H, d, J 3.9, ArH), 6.93 (2H, d, J 6.8, ArH), 3.89 (4H, t, J 6.5, OCH2), 1.75 (4H, m, CH2), 1.42 (4H, m, CH2), 1.20–1.40 (16H, br, CH2) and 0.89 (6H, t, J 6.1, CH3); δC (100 MHz, CDCl3, Me4Si) 159.0, 153.2, 139.9, 137.2, 136.0, 129.9, 129.2, 124.2, 124.1, 122.5, 116.8, 115.3, 114.8, 68.2, 31.9, 29.4, 29.4, 29.2, 26.1, 22.8, 14.2; λmax (CHCl3)/nm 357 and 565 (ε/dm3 mol−1 cm−1 33200 and 8100); m/z (MALDI-TOF) 864.05 (M+, C42H46Br2N2O2S3 requires 864.11).
C) 1484 (CC), 1278 (COSi), 860, 840 and 782 (m-sub. benzene); δH (300 MHz, CDCl3, Me4Si) 7.48 (2H, d, J 7.8, ArH), 7.44 (2H, s, ArH), 7.35 (2H, t, J 7.6, ArH), 7.12 (2H, d, J 8.0, ArH), 0.98 (18H, s, C(CH3)3) and 0.20 (12H, s, CH3Si); δC (100 MHz, CDCl3, Me4Si) 194.7, 156.4, 134.4, 130.2, 127.0, 123.5, 120.4, 25.7, 18.3, −4.4; λmax (CHCl3)/nm 264 and 314 (ε/dm3 mol−1 cm−1 24500 and 6600); m/z (MALDI-TOF) 470.47 (M+, C26H38O4Si2 requires 470.23).
C) 1426, 1280 (COSi), 933 (COSi), 830 and 778 (m-sub. benzene); δH (300 MHz, CDCl3, Me4Si) 7.30 (2H, d, J 3.9, ArH), 7.23 (2H, s, ArH), 7.16 (2H, t, J 7.8, ArH), 7.05 (2H, d, J 3.9, ArH), 7.03 (2H, d, J 6.0, ArH), 6.88 (2H, d, J 7.2, ArH), 1.00 (18H, s, C(CH3)3) and 0.21 (12H, s, CH3Si); δC (100 MHz, CDCl3, Me4Si) 155.8, 153.0, 140.1, 137.3, 135.9, 129.7, 129.2, 124.1, 123.8, 123.3, 121.4, 121.3, 114.8, 25.8, 18.3, −4.2; λmax (CHCl3)/nm 357 and 563 (ε/dm3 mol−1 cm−1 37800 and 9000); m/z (MALDI-TOF) 867.83 (M+, C38H42Br2N2O2S3Si2 requires 868.03).
C), 1427, 1283, 782 (m-sub. benzene); δH (300 MHz, DMSO, Me4Si) 9.65 (2H, s, ArOH), 7.49 (2H, d, J 3.0, ArH), 7.30 (2H, d, J 3.6, ArH), 7.17 (2H, t, J 7.8, ArH), 7.03 (2H, s, ArH), 6.85 (2H, d, J 7.5, ArH) and 6.82 (2H, d, J 7.5, ArH); δC (100 MHz, DMSO, Me4Si) 157.6, 153.6, 140.3, 137.0, 135.4, 130.7, 129.5, 124.5, 123.9, 121.1, 116.9, 114.6; λmax (CHCl3)/nm 357 and 563 (ε/dm3 mol−1 cm−1 70900 and 17000); m/z (MALDI-TOF) 639.86 (M+, C26H14Br2N2O2S3 requires 639.86).
C), 1424, 1268 (arom. ether), 780 (m-sub. benzene); δH (300 MHz, CDCl3, Me4Si) 7.31 (2H, d, J 3.9, ArH), 7.23 (2H, s, ArH), 7.20 (2H, d, J 6.3, ArH), 7.10 (2H, d, J 6.8, ArH), 7.04 (2H, d, J 3.9, ArH), 6.93 (2H, d, J 6.8, ArH), 3.89 (4H, t, J 6.5, OCH2), 1.75 (4H, m, CH2), 1.42 (4H, m, CH2), 1.20–1.40 (16H, br, CH2) and 0.89 (6H, t, J 6.1, CH3); δC (100 MHz, CDCl3, Me4Si) 159.0, 153.2, 139.9, 137.2, 136.0, 129.9, 129.2, 124.2, 124.1, 122.5, 116.8, 115.3, 114.8, 68.2, 31.9, 29.4, 29.4, 29.2, 26.1, 22.8, 14.2; λmax (CHCl3)/nm 357 and 565 (ε/dm3 mol−1 cm−1 33200 and 8100); m/z (MALDI-TOF) 864.05 (M+, C42H46Br2N2O2S3 requires 864.11).
APFO-Green15 (general procedure for polymerization). The monomers, VI (140 mg, 0.16 mmol) and VII (104 mg, 0.16 mmol), were dissolved in toluene (10 mL). The solution was purged with nitrogen for 15 min and tetrakis(triphenylphosphine)palladium(0) (19 mg, 0.016 mmol) was added. The reaction mixture was heated to reflux. After 10 min, tetraethylammonium hydroxide solution (20 wt% in water, 0.6 mL, 0.81 mmol) was added and the polymerization continued at reflux for 4 h. Bromobenzene (41 mg, 0.26 mmol) was added and after 60 min phenylboronic acid (39 mg, 0.32 mmol) was added. After an additional 60 min, the mixture was cooled to room temperature and precipitated from methanol (200 mL). The polymer was collected by filtration, dissolved in boiling chloroform (75 mL), and washed with ammonia solution (25 wt% in water, 50 mL) overnight. The polymer solution was washed once more with ammonia solution (50 mL) and twice with water (100 mL) before it was concentrated and precipitated from methanol (200 mL). After filtration, the crude material was soxhlet extracted with diethyl ether for 4 h. The remaining material was soxhlet extracted with chloroform, yielding APFO-Green15 (143 mg, 81%) as a dark green powder.
LBPP-3. Synthesized following the general procedure with VI (140 mg, 0.16 mmol) and VIII (95 mg, 0.16 mmol) giving LBPP-3 (155 mg, 92%) as a green powder.
LBPP-4. Synthesized following the general procedure with VI (140 mg, 0.16 mmol) and IX (53 mg, 0.16 mmol) giving LBPP-4 (102 mg, 81%) as a green powder.
Acknowledgements
We thank the National Graduate School in Materials Science, the Swedish Research Council, and the Strategic Research Foundation (SSF) through the Center of Organic Electronics (COE) for financial support. The Wallenberg foundation is acknowledged for financial support regarding the MALDI-TOF.Notes and references
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 Search PubMed.
- J. Hou, H.-Y. Chen, S. Zhang, R. I. Chen, Y. Yang, Y. Wu and G. Li, J. Am. Chem. Soc., 2009, 131, 15586–15587 CrossRef CAS.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 Search PubMed.
- J. J. M. Halls, J. Cornil, D. A. dos Santos, R. Silbey, D. H. Hwang, A. B. Holmes, J. L. Brédas and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 5721 CrossRef CAS.
- J.-L. Bredas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef CAS.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Synth. Met., 1993, 55, 299–306 CrossRef CAS.
- C. Kitamura, S. Tanaka and Y. Yamashita, Chem. Mater., 1996, 8, 570–578 CrossRef.
- T. Yamamoto, Z.-h. Zhou, T. Kanbara, M. Shimura, K. Kizu, T. Maruyama, Y. Nakamura, T. Fukuda, B.-L. Lee, N. Ooba, S. Tomaru, T. Kurihara, T. Kaino, K. Kubota and S. Sasaki, J. Am. Chem. Soc., 1996, 118, 10389–10399 CrossRef CAS.
- H. A. M. van Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1–40 CrossRef.
- E. Perzon, X. Wang, F. Zhang, W. Mammo, J. L. Delgado, P. de la Cruz, O. Inganäs, F. Langa and M. R. Andersson, Synth. Met., 2005, 154, 53–56 CrossRef CAS.
- E. Perzon, X. Wang, S. Admassie, O. Inganäs and M. R. Andersson, Polymer, 2006, 47, 4261–4268 CrossRef CAS.
- F. Zhang, W. Mammo, L. M. Andersson, S. Admassie, M. R. Andersson and O. Inganäs, Adv. Mater., 2006, 18, 2169–2173 CrossRef CAS.
- F. Zhang, J. Bijleveld, E. Perzon, K. Tvingstedt, S. Barrau, O. Inganas and M. R. Andersson, J. Mater. Chem., 2008, 18, 5468–5474 RSC.
- M. Ranger, D. Rondeau and M. Leclerc, Macromolecules, 1997, 30, 7686–7691 CrossRef CAS.
- P.-H. Aubert, M. Knipper, L. Groenendaal, L. Lutsen, J. Manca and D. Vanderzande, Macromolecules, 2004, 37, 4087–4098 CrossRef CAS.
- J. L. Bredas, J. Chem. Phys., 1985, 82, 3808–3811 CrossRef CAS.
- S. Hellström, F. Zhang, O. Inganäs and M. R. Andersson, Dalton Trans., 2009, 10032–10039 RSC.
- M. Svensson, F. Zhang, O. Inganäs and M. R. Andersson, Synth. Met., 2003, 135–136, 137–138 CrossRef CAS.
- F. Zhang, E. Perzon, X. Wang, W. Mammo, M. R. Andersson and O. Inganäs, Adv. Funct. Mater., 2005, 15, 745–750 CrossRef CAS.
- J. G. Osteryoung and R. A. Osteryoung, Anal. Chem., 1985, 57, 101–110 CrossRef.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- K. G. Jespersen, F. Zhang, A. Gadisa, V. Sundström, A. Yartsev and O. Inganäs, Org. Electron., 2006, 7, 235–242 CrossRef CAS.
- J. J. M. Halls, K. Pichler, R. H. Friend, S. C. Moratti and A. B. Holmes, Synth. Met., 1996, 77, 277–280 CrossRef CAS.
- M. Theander, A. Yartsev, D. Zigmantas, V. Sundström, W. Mammo, M. R. Andersson and O. Inganäs, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 12957 CrossRef CAS.
- A. Haugeneder, M. Neges, C. Kallinger, W. Spirkl, U. Lemmer, J. Feldmann, U. Scherf, E. Harth, A. Gügel and K. Müllen, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15346 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- F. Zhang, K. G. Jespersen, C. Björström, M. Svensson, M. R. Andersson, V. Sundström, K. Magnusson, E. Moons, A. Yartsev and O. Inganäs, Adv. Funct. Mater., 2006, 16, 667–674 CrossRef CAS.
- L. Zhang and E. Meggers, J. Am. Chem. Soc., 2005, 127, 74–75 CrossRef CAS.
- W. Mammo, S. Admassie, A. Gadisa, F. Zhang, O. Inganäs and M. R. Andersson, Sol. Energy Mater. Sol. Cells, 2007, 91, 1010–1018 CrossRef CAS.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Thermogravimetric analyses (TGA) of polymers in nitrogen atmosphere. See DOI: 10.1039/c0py00152j |
| This journal is © The Royal Society of Chemistry 2010 |