Perspectives for the mechanical manipulation of hybrid hydrogels
Renate
Messing
a and
Annette M.
Schmidt
*ab
aInstitut für Makromolekulare Chemie und Organische Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstr. 1, D-40225 Düsseldorf, Germany. Fax: +49-211-81 15840; Tel: +49-211-81 14820
bDepartment für Chemie, Universität zu Köln, Luxemburger Str. 116, 50939 Köln, Germany. E-mail: annette.schmidt@uni-koeln.de; Fax: +49-221-470 5410; Tel: +49-221-470 5482
First published on 3rd August 2010
Abstract
Significant advances in the field of responsive hydrogels have been achieved by the combination of soft, gel-based matrices with the unique functions of inorganic or biological nanostructures. Like in many biological tissues, the components of such hybrid materials often have converse, yet complementary properties. The possibility of forming self-assembled and supramolecular morphologies from organic polymers in combination with inorganic nanoparticles or biological motifs is of interest for gels with new response properties. A variety of complex gel structures with unique chemical, physical, and biological properties have been engineered or discovered at the nanoscale. In this review, we highlight recent accomplishments and trends in the field of hybrid polymer hydrogels with a focus on approaches towards soft, yet tough shape-changing and actuating materials. We conclude with an outline on future directions and challenges that have to be faced in the design and application of hybrid hydrogels.
 Renate Messing | Renate Messing studied chemistry at the Heinrich-Heine-University (HHU) of Düsseldorf, Germany. She received her Diploma in 2006 at the HHU in the Department of Macromolcular and Organic Chemistry, in the research group “Smart Nanosized Systems”. In 2007, she started her PhD concerning the field of magnetoresponsive hydrogels, under the guidance of A. M. Schmidt at the HHU and will finish her thesis in 2010. |
 Annette M. Schmidt | Annette M. Schmidt received her Diploma and PhD (2001) from RWTH Aachen, with visits at the RUG Groningen and at MIT, Cambridge. Within the Emmy-Noether program of the DFG, she headed a research group on “Smart Nanosized Systems” at the Heinrich-Heine-University of Düsseldorf, before she was appointed a Professor of Physical Chemistry at the University of Cologne in 2010. Her research interests include organic–inorganic hybrid materials, functional polymers and colloids, and stimuli-responsive systems. |
Introduction
Cross-linked networks of hydrophilic polymers swollen with water, known as hydrogels, are generally soft and wet materials. They are highly swollen in aqueous media, and their swelling behaviour is influenced by a number of environmental factors. As a consequence, controllable changes in shape, volume, pore size, their mechanical or optical properties can be achieved by the tailored design of gel composition and architecture and by the introduction of functional moieties.Although hydrogels find applications in surgery and drug delivery, they are hardly considered for mechanical design owed to the generally weak nature of synthetic hydrogels. In contrast, many biological gel composites such as cartilage show amazingly strong mechanical properties.
The recent development of hybrid hydrogels by the incorporation of inorganic nanoparticles or biological moieties into the gel matrix currently results in new families of mechanically strong gels with novel response properties. It is expected that such materials would allow the design of soft biomimetic valves and actuators, which have previously not been possible.1
Stimuli-sensitive polymer-based hydrogels and their response to external triggers
A number of relevant reviews has been published on the synthesis, design and performance of polymer-based stimuli-responsive hydrogels.1–8 We give a brief overview on the most important aspects of hydrogel characteristics, and focus in this review on the recent developments concerning the hybridization of hydrogels with other types of materials in order to introduce enhanced, more complex or even fully new properties.Stimuli-sensitive polymers have the common characteristic to undergo a reversible change with respect to their conformation or phase behaviour in response to a variation in one of the environmental parameters. The reversibility of the involved processes is in many cases based on a rather delicate free-energy balance involving cooperative attractive and repulsive interactions between the polymer segments or parts of them, therefore showing the characteristics of a critical phenomenon.
The key parameter is a nonlinear response to the external signal, in contrast to a rather continuous process as observed e.g. with (enthalpic) thermal expansion. Polymeric systems can be designed to show a wide range of critical phenomena, as these high molecular, flexible macromolecules are strongly governed by entropic effects. For the same reason, temperature is an important trigger for many reversible, nonlinear effects in polymers.
Thus polymer-based hydrogel materials can respond to a variety of changes in their physicochemical environment, including thermoresponsive, pH- or ionic strength-responsive, chemosensitive and electric-field responsive gels. light or. A large number of respective references and more background information can be found in refs. 2 and 9 and other reviews.
Thermoresponsiveness
Temperature-responsive hydrogels are generally based on water-soluble polymers that show a reversible sudden change in solubility on the temperature scale, often accompanied by a sol-to-gel phase transition at a characteristic temperature. Typically, these gels are comprised of polymers with hydrophobic and hydrophilic groups that are capable to form hydrogen bonds. Due to the high number of hydrogen bonds per macromolecule, and accordingly the high degree of solvation, the polymer chains are in a swollen state. Beyond a certain temperature, commonly referred to as the cloud point temperature Tc, the interpolymer hydrophobic interactions dominate the hydrogen bond formation. Due to the release of the hydrogen bonds, water is expelled by the polymer as it undergoes a coil-to-globule collapse. As a consequence, the polymer eventually phase-separates and the gel collapses macroscopically.Solvation-related phase transitions in water are generally of the lower critical solution temperature (LCST)-type, meaning that the hydration is high in the low-temperature regime, and phase separation occures upon heating.
The most widely known LCST hydrogels are composed of poly(N-isopropylacrylamide) (PNiPAAm) (Fig. 1). The side chains of PNiPAAm exhibit amide groups that are responsible for hydrogen bond formation, while the main chain is hydrophobic.10 It is now well accepted that the solution properties of PNiPAAm in water are the result of the rather complex polarity of the polymer functionalities.11
![Examples of polymers that show thermoresponsive behaviour in water: 1poly(N-isopropylacrylamide) (PNiPAAm); 2 poly(N-vinylcaprolactam) (PVCL); 3 poly(2-isopropyl-2-oxazoline) (PiPOx); 4 poly(propylene oxide) (PPO); 5 poly(ethylene oxide) (PEO); 6poly(vinyl methyl ether) (PVME); 7 poly(2-(2-methoxyethoxy)ethyl methacrylat-co-oligo(ethyleneglycol)methacylate) (P(MEO2MA-co-OEGMA)); 8 poly(3-[N-(3-methacrylamidopropyl)-N,N-dimethyl]ammonio propiosulfonate) (PSPP).](/image/article/2011/PY/c0py00129e/c0py00129e-f1.gif) | ||
| Fig. 1 Examples of polymers that show thermoresponsive behaviour in water: 1poly(N-isopropylacrylamide) (PNiPAAm); 2 poly(N-vinylcaprolactam) (PVCL); 3 poly(2-isopropyl-2-oxazoline) (PiPOx); 4 poly(propylene oxide) (PPO); 5 poly(ethylene oxide) (PEO); 6poly(vinyl methyl ether) (PVME); 7 poly(2-(2-methoxyethoxy)ethyl methacrylat-co-oligo(ethyleneglycol)methacylate) (P(MEO2MA-co-OEGMA)); 8 poly(3-[N-(3-methacrylamidopropyl)-N,N-dimethyl]ammonio propiosulfonate) (PSPP). | ||
After the first report on the phase transition of PNiPAAm in water by Heskins and Guillet in 1968,12 the subject has fascinated chemists, physicists and engineers. One interest in this polymer originates from potential applications in biotechnological devices and drug delivery vehicles,3,4,13 as well as for soft actuators.1
The characteristics of PNiPAAm, including the cloud point temperature Tc of 32 °C, a high chemical stability, reasonable mechanical properties and modest biocompatibility, are helpful to evaluate the application potential in the lab. However, for the applicability, concepts are needed to optimize the hydrogel properties and transition characteristics.
As an elegant and versatile method to tailor the transition temperature of PNiPAAm polymers and hydrogels, a variation of the alkyl side chain has been proposed, as well as copolymerization with other more hydrophilic or more hydrophobic co-monomers. By replacing for example the monomer with a N,N-disubstituted alkyl amide, two effects can be observed. The polymer gets more hydrophobic as compared to PNiPAAm, with lower cloud point temperatures14 and lower enthalpy of dissolution values. The later is explained by the way of hydrogen bond formation: in contrast to PNiPAAm that can act both as a hydrogen bond donor and as an acceptor, the polymers of N,N-disubstituted acrylamides can only act as an acceptor.15
Another amide-based LCST polymer is poly(N-vinylcaprolactam) (PVCL). It comprises a cyclic amide in the repeating units, with the amide nitrogen attached directly to the hydrophobic polymer backbone. It comprises good biocompatibility, high complexing ability and good film-forming properties, with a cloud point temperature Tc at around 31 °C.16,17
Similarly, the temperature-responsive nature of polyoxazolines is based on the strong tendency of amide groups to form hydrogen bonds. For example, poly(2-isopropyl-2-oxazoline) (PiPOx), being isomeric to PNiPAAm, exhibits a cloud point temperature Tc between 36 °C and 39 °C, weakly depending on the polymer concentration.18
An LCST-like behaviour in water can also be found for ether-based polymers. The most common is poly(propylene oxide) (PPO) with a quite broad phase transtion temperature range that strongly depends on the molecular weight. Already at a molecular weight of about 4000 g·mol−1, the cloud point temperature Tc is below 20 °C.19 For this reason, PPO is often used in block copolymers with poly(ethylene oxide) (PEO). PEO has a much higher cloud point temperature Tc and serves as swollen segments in gels with thermoreversible PPO physical cross-links.20
Thermoresponsive polymers with ether side chains are realized e.g. in poly(vinyl methyl ether) (PVME). This all-carbon main chain polymer is known for a strong complex formation with water. The complex is further hydrated in systems with excess water and shows a rather complex phase diagram.21,22
A highly adaptive system that is of increasing interest is based on homo- and copolymers of methacrylates with oligoether side chains. By varying the composition and the side chain length, the phase transition temperature can be altered between 10 °C and 90 °C. In short to medium term applications, it furthermore comprises an excellent biocompatibility.23,24
While most thermosensitive hydrogels are based on LCST-type polymers, there are also examples for polymers exhibiting the reverse behaviour; thus possessing an upper critical solution temperature (UCST) in water.2 The most important examples are based on zwitterionic polymers, such as polysulfobetaines. For example, poly(3-[N-(3-methacrylamidopropyl)-N,N-dimethyl]ammonio propiosulfonate) (PSPP) shows an inverse cloud point temperature Tc that increases with molecular mass, attributed to the strong mutual intermolecular attraction of the zwitterionic betaine groups.25 As an additional feature, aqueous solutions and gels of polysufobetaines have a selective (preferential) binding to “soft” cations and anions, and react with an increase in viscosity on increasing salt concentration (antipolyelectrolyte effect). Other UCST type hydrogels comprise proteinogenic sequences that are responsible for the thermoresponsitivity.27 These materials will be adressed in more detail together with other biohybrid gels below.
pH- and ionsensitivity
The ability of hydrogels to possess pH-responsive properties is generally achieved by the introduction of pendant acidic or basic groups that are capable to accept or release protons in response to the environmental pH. As a consequence, the charge density along the polymer chains is changed reversibly, resulting in a dramatic change of the swelling state due to electrostatic inter- and intrachain interactions.28 Prominent example is poly(acrylic acid) (PAA) that is soluble in water at a pH of 5 and higher,29 depending on the molecular weight and the presence of co-monomers, i.e. on the charge density along the chain.30,31 In addition, the solubility behaviour is generally depending on the ionic strength. By exploiting the complex formation ability of the pending carboxylates, the volume phase transition of polyacrylate gels can be induced by calcium-sodium cation exchange.32Light responsiveness
From an application point of view, the use of electromagnetic radiation as a trigger in the switching of responsive systems has several advantages. The high degree of control, the possibility of non-invasive application, and speed of switching can not be achieved in temperature- or pH-responsive systems in a comparable way.The use of light to study and control chemical and biological systems is therefore a subject of intense research. As well the photoisomerization of organic pendant groups, as photocleavable protecting groups have been applied to add sensitivity towards light to hydrogels. The latter approach is useful on gel surfaces for patterning and/or release of biomolecules. Recently some progress has been made through the combined use of chromophores with thermoresponsive hydrogels.2,33 Such approaches may offer photo-tunable drug delivery from responsive particles34 or light-driven polymeric actuators.
Photoisomerisable constituents have been combined with stimuli-responsive polymers like PNiPAAm through copolymerisation of the respective chromophoric monomers, e.g. based on spyrobenzopyran.35–37 Although the introduction of these co-monomers often causes considerable broadening of the LCST, photo-thermal switching materials are of interest for applications such as chromatography38 or optical cell sorting.36 Recently, new pathways towards the light actuation of gel-like systems are opened by incorporation of metallic nanoparticles to hydrogels,39 as will be discussed in the further course of the review.
Response to electric fields
The employment of electric fields as an external stimulus has advantages, as it allows a precise control with regard to the magnitude of current, duration of electric pulses, intervals between pulses, etc. Under the influence of an electric field, electroresponsive hydrogels generally deswell or bend, depending on the shape of the gel and its position relative to the electrodes.40Bending occurs when the main axis of the gel lays parallel to (but does not touch) the electrodes (Fig. 2), whereas deswelling occurs when the hydrogel lays perpendicular to the electrodes.41 For polyelectrolyte gels, the effect is found to rely on the development of local pH gradients by ionic movement in modelling studies42 and in experiments.43,44 For uncharged gels, the proposed molecular mechanism of bending in response to a non-contact, static field involves the formation of a pH gradient within the gel.45
 | ||
| Fig. 2 Demonstration of polyelectrolyte hydrogel bending under electrical stimulation. Reproduced with permission from ref. 26. Copyright (2003) Society of Photo Optical Instrumentation Engineers. | ||
Hydrogel bending has mainly been studied for the production of mechanical devices such as valves, artificial muscles, switches, soft acutators and molecular machines.46,47
Nature of response effects observed in hydrogels
The response of polymer gels to external stimuli is generally based on a change in conformation or chain interaction in the presence of water as the diluent. The physicochemical processes involved can finally result in a broad range of transition effects, making them useful for many applications. However, as a consequence of the complex mechanisms involved, not all of them are fully understood or exploited in detail.One of the simpler effects is based on the reversible gelation of a macromolecular solution.48Polymer systems that undergo a reversible gelation under the influence of external stimuli are highly searched for in biomedical applications, where injectable hydrogels are proposed for soft tissue replacement and cavity filling.49,50
In general, reversible gelling in hydrogels can occur by two main mechanisms; that is, by the formation of a macrolattice due to volume occupance under reversible globule-to-coil transition, or by reversible formation of physical or chemical crosslinks.50
Hydrogels that are capable of a reversible sol-gel transition due to physical gelling are often designed by double-hydrophilic block- or graft-type copolymers or star-shaped polymer molecules with a reversibly soluble corona. While the corona induces a physical crosslinking in the temperature range of bad solubility, the more hydrophilic inner block is responsible for the high water uptake of the evolving gel.
Such double-hydrophilic architectures often show a complex phase behaviour, and the gelation can be controlled by adjustment of the molecular weight of both the more and the less hydrophilic block, and by the concentration in solution.2,51 By a proper design of the blocks, systems that respond to several stimuli with a complex phase behaviour can be obtained.52
The architecture and nature of the involved polymer segments is of high importance. For example the rheometric behaviour of doubly hydrophilic block and graft copolymers often differs significantly from that of a mixture of the pure polymers. While it is a well-known and widely investigated fact that PAAm and PNiPAAm homopolymer mixtures form molecular complexes by hydrogen bonding at low pH,53,54 the combination of both in a graft-like architecture results in a system that shows gelling due to microphase separation.55–57 The resulting increase in viscosity can be directly observed and quantified by dynamic rheology (Fig. 3).57,58
 | ||
| Fig. 3 a) Viscosity as a function of temperature at constant shear rate for a star-block copolymer sPEG-b-P(MEO2MA-co-OEGMA) in PBS solution at concentrations of 16 wt% (empty symbols) and 23 wt% (full symbols). b) Viscosity as a function of time during six heating/cooling cycles between 25 °C and 38 °C for the same polymer in PBS buffer solution at concentrations of 16 wt% (dashed line) and 23 wt% (full line). c) Photographs of the polymer solutions below (top) and above (bottom) LCST (16 wt% in PBS). Reproduced with permission from ref. 51. Copyright (2009) American Chemical Society. | ||
Another type of transition that is commonly observed in chemically cross-linked hydrogels is manifested in a discontinuous volume change.7 Here, a sudden change in the interchain interactions causes a deswelling of the chemical network. As the process of gel swelling or collapsing is rate-limited by the transport of diluent through the gel, kinetic effects of heat and mass transport play a substantial role here (see below).7 Even if the involved forces are rather weak due to the softness of hydrogels, and although the effect is limited generally to the isotropic case, the effect is of considerable interest for sensor applications, and for switchable valves for microfluidic devices.59–61 In addition, several devices have been developed that allow more complex shape changes62 or shape memory properties.63
Often closely related to the volume change is a response in permeability of the gel for hydrophilic or hydrophobic (small) molecules.1 Sumaru et al. reported the development of a membrane based on porous poly(tetrafluoroethylene) filled with spyrobenzopyran-modified PNiPAAm in an aqueous environment.38 The permeability of the membranes towards HCl solution was shown to be switched reversibly by the use of temperature or light.
Although, or just because hydrogels are soft materials, the change in mechanical stiffness or modulus can be significant upon phase transition.1 For sensor applications, this may be a high benefit. However, detailed information on the evolution of the mechanical properties during deformation is rarely found, although significant effects have occasionally been reported for PVA/PAA hydrogels64 and for PNiPAAm/PHEMA hydrogels.65
Important physical aspects of hydrogel responses
The macroscopic effects observed in phase-change hydrogel systems are controlled by parameters such as the extent of dimensional change (strain) and reversibility, the broadness of the transition and the speed of the response, the exerting force and the degree of viscoelastic loss due to dissipated heat.66Similar to the liquid-gas transition of a fluid system, the volume of a gel can change either continuously or discontinuously upon a change of the environmental conditions such as temperature and solvent composition.7 The volume transition is directly related to the coil-to-globule transition of a diluted polymer system.34,67,68 It is universal in a sense that it is observed many of polymer-solvent systems, yet for practical reasons, the transition is most easily observed in water-based systems.69
The phase transition is a result of a competitive balance between a repulsive force acting to expand the polymer network, and an attractive force acting to shrink it. The most effective repulsive force is the electrostatic interaction between the polymer charges of the same kind, as present in ionisable or polyelectrolytic hydrogels. The osmotic pressure by counter ions adds to the expanding pressure. The attractive forces can be van-der-Waals, hydrophobic or, ion-ion interactions between opposite kinds of charges, and hydrogen bonding. Today, volume phase transitions based on each of the fundamental forces were readily discovered in gels.
The gel volume phase transition was first predicted by Dusek and Patterson, using the Flory–Huggins (FH) theory for polymer solution.70 The theory predicts the volume phase transition qualitatively70,71 and has been extended, e.g. by Li and Tanaka7 and Wu et al.72 Yet, many of the observed critical phenomena are still not well understood.
A comparison of the coil-to-globule and the globule-to-coil transition in highly diluted PNiPAAm solutions reveals that there exists a hysteresis in the transition, indicating an at least partly irreversible process, as a consequence of kinetic aspects of the sticking probability.72,73 In comparison, the copolymer P(MEO2MA-co-OEGMA) exhibits a much more uniform thermal profile (i.e., heating and cooling cycles are roughly comparable).23
A drawback of macroscopic hydrogels is their slow response, since the volume transition is associated with the diffusion of molecular and/or ionic species. While the impact of the stimulus can be rapid, the actual changes in the chemical environment of a gel are slowed down by the diffusional processes.7 As a consequence, the size and the form of hydrogels are of high impact on the kinetics of swelling and shrinking. For spherical gels, the rate is proportional to the radius square, as a consequence of the diffusional nature of the rate limiting step.74 The finding resulted in the development of micro- and even nanogels, that show a greatly improved response time in swelling and deswelling processes as compared to the respective macrogels.75,76 Based on a similar idea to reduce diffusion lengths, porous macroscopic hydrogels show improved swelling response times. A clear dependence of the swelling rate on the pore size is observed in PDEAAm and PNiPAAm macroscopic hydrogels.77
More detailed studies on the swelling dynamics of hydrogels suppose that a rapidly shrinking gel tends to form a dense skin that inhibits water loss from the interior, slowing down the volume change. The effect of such skin formation and/or micro-phase separation on deswelling kinetics has been studied by Hirose and Shibayama,78 who showed that pure PNiPAAm gels shrink much slower than weakly charged copolymers of NiPAAm and acrylic acid, which retain more mobility for water in the collapsed state. Similarly, a copolymerization of NiPAAm with a carboxylated analogon (CiPAAm) has been proposed in order to improve the swelling kinetics of macrogels.79
In general, hydrogels are weak, and therefore are mostly not considered for applications where mechanical properties play a major role. As a consequence, there is not much quantitative information on the mechanical properties and the forces exerted during transition in the literature. To give a few numbers, simple hydrogels show an elastic modulus in the range of or below 1 MPa and a decrease in modulus as the water volume fraction increases. Typically, the tensile strength is below 0.5 MPa with elongations at break between 5 and 100%.1 Additionally physical gels have to deal with viscoelastic and thus time-depending effects.
There are several attempts to improve the mechanical strength and toughness of hydrogels. By modification of the network architecture, progress has been made towards elastic materials with increased load capacity and durability. For example, gels based on a double network architecture1 and interpenetrating networks (IPN)80 show a considerable improvement in the mechanical behaviour.
For actuator applications it is of importance that the material is not too soft in order to be capable to perform work against an external force, such as gravity. The energy necessary for mechanical work performance is provided by the respective stimulus – thus, from heat in temperature-responsive gels, chemical energy in pH- and ionic strength responsive gels and field energy in field-responsive gels. The effectiveness of this energy conversion is yet to be optimized.60
One mechanism of energetic loss is heat dissipation due to viscous/irreversible processes. In this respect, it is an important goal to avoid hysteretic effects, to decrease response times and to maximize the energy efficiency of hydrogel materials for actuation.
Methods of investigation
The discontinuous property change of hydrogels caused by the phase transition involves different attributes and can be investigated by partly convergent methods. Depending on the system and the type of transition to be observed, in particular optical and calorimetric setups, or combination of the both, can be employed.Among these, Cloud Point Photometry (CPP) is a widely used and simple method to monitor transitions that are accompanied with a change in optical transparency, as often observed in thermal or pH induced phase transitions and agglomeration phenomena. Here the transmittance of a liquid or gel-like sample is monitored, e.g. against temperature. As many turbidity curves show hysteresis to a more or less pronounced extent, it is essential to monitor correctly the reversibility and broadness of the transition by regarding both directions of the response. Additionally, time-related issues, such as heating rate dependence, have to be carefully considered.
For transitions that involve a first or second order enthalpic change, calorimetric methods can be applied, including differential scanning calorimetry (DSC) or microcalorimetry (MC). By the right choice of conditions, kinetic aspects can also be addressed, and a wide range of compositions can be examined. It is of interest to note that many thermoresponsive polymers show an endothermic transition enthalpy that corresponds to the loss of roughly one hydrogen bond per repeating unit, thus in the range of 1–4 kJ mol−1.2
In hydrogel micelles and in microgels, the volume transition can readily be monitored by scattering methods. Among these, dynamic light scattering (DLS) setups provide information on the diffusion properties of transparent gels and microgel dispersions. Here it is of positive impact that in consequence of their highly swollen character, gels up to the micrometer scale do not show precipitation tendency, but are subject to thermally activated Brownian diffusion. Thus, their hydrodynamic radius can be deduced from the pair correlation function obtained from scattering noise by using Stokes-like analysis. By performing temperature-dependent experiments, information on the size and diffusion properties can be obtained, and the transition characteristics can be extracted in a quantitative way.81–86
For the determination of the mechanical properties of gels, some important factors need to be considered that can often be ignored in dense materials. Apart from that gels often fracture at much higher strains than conventional engineering materials, their properties can be strongly time dependent, and depending on the environmental conditions, liquid may be taken up or lost during the test. Likewise, the properties of immersed gel samples differ from samples tested in air, as water is normally taken up in tension and excluded in compression. The degree of confinement and timescale of testing is also important for the same reasons.
Accordingly, conventional testing methods have to be customized to the mechanical properties of gels, e.g. by dynamic rheology and in tensile and compression experiments. While the results are of importance for the performance of hydrogels in the application case, they also give a high degree of information on the dynamic processes in the mechanical performance of the gels. By online measurement during stimulus application, e.g.T- or pH-change, the impact of the transition on sample shape and toughness can be monitored.1
In order to elegantly deal with the neccessity to explore the properties in the immersed state of the gel, Constantinides et al. have developed and demonstrated an approach to enable nano- to micro-scale indentation of fully immersed, hydrated polymers and tissues, enabling quantitative studies of gel and tissue mechanics as a function of temperature, pH or light irradiation (Fig. 4).87
 | ||
| Fig. 4 Schematic diagram (A) and photograph (B) of an indenter apparatus for extended nanomechanical experiments on immersed hydrated gels and tissues. Reproduced with permission from ref. 87. Copyright (2008) Elsevier. | ||
Hybrid hydrogels
Although conventional environmental-sensitive hydrogels have demonstrated tremendous potential in medicine, biotechnology and microfluidic or microactuator applications,4,5,88,89 the range of stimuli to which they can respond is rather limited. Temperature- and pH-responsiveness may be useful in drug-delivery applications, in which there is a locally distinct temperature or pH in a target region. Yet, under homeostatic conditions, in which pH and temperature are tightly regulated, these materials are less useful. In addition, changes in temperature or pH may not be desirable in the presence of cells, or are energetically unfavorable in microactuating applications. Present approaches have therefore begun to address the limitations of traditional responsive systems by developing materials reacting to more specific, different stimuli. Promising approaches involve the introduction of hybrid components that allow the application of definite external triggers to address specific responses, for example by using the electric or magnetic properties of inorganic nanoparticles, or by mimicking natural phenomena by the introduction of bioinspired functionalities.Such hybrid hydrogels can be developed as new functional materials by tailored multi-disciplinary protocols bringing together approaches from polymer chemistry, biochemistry, and colloid physics.
In hybrid hydrogels, the nature of interaction between the components can cover a broad range of forces. A useful classification introduced by Kickelbick90 distinguishes between the possible interactions connecting the inorganic, organic and or biological species. Class I hybrid materials are those that show weak interactions between the two phases, such as van-der-Waals, hydrogen bonding or weak electrostatic interactions. In contrast, Class II hybrid materials show strong chemical interations between the components. Obviously there is a steady transition between weak and strong interactions, e. g in the case of rather strong (multivalent) hydrogen bonds.
Hydrogels comprising inorganic nanoparticles
One important type of hybrid hydrogels is obtained when inorganic particles, whiskers, fibers, or lamellae are incorporated into a polymeric matrix that is capable to form a hydrogel by water swelling.Generally, the inorganic structural building blocks show a size range from about 1 nm to the micrometer range. For the very small particles of a few nanometre, the size of the inorganic units is on the same level as the organic building blocks, resulting in a variety of possible interaction pathways between organic, inorganic, and aqueous phase. At the same time, it is possible to maintain the characteristics of hydrogels, such as softness, high water uptake, and the possibility to respond to stimuli like temperature and pH.90
In general, the inorganic particle components can be divided into two major groups according to their function. There are those that reinforce the polymer gel and improve its mechanical performance, and those who add additional properties to the gels, such as response to electric and magnetic fields.
The incorporation of rigid, high density inorganic particles into hydrogels is of strong impact on the mechanical properties of the gels. Providing that the particles do not hinder the polymerization process or gel formation, and presuming a positive, or at least, not negative interaction between particles and polymer chains, the resulting materials show enhanced elastic moduli and tensile strengths. The hybrid materials combine the elastic properties of the gels with the respective physical properties of the inorganic fillers.
The interaction between particle and gel matrix, i. e. the polymer chains, and the water phase, can be of different nature. Most reported hybrid hydrogels are of the Class I type, characterized by the absence of strong chemical interactions between the inorganic and organic building blocks. They can be obtained by simply cross-linking a water-soluble polymer in the presence of the respective particles, or by generation of the particles within the gel. In both cases, hybrid gels are accessible with a broad range of gel composition and choice of particle component.90
In case there are strong physical interactions between the network chains and the surface of the solid particles, this provides a coupling between forces acting on the solid particles, and the conformational change of the surrounding macromolecules. This way, a mechanical force acting on one particle can be transmitted directly to the polymer chains. Due to cross-linking bridges in the network, changes in molecular conformation can accumulate. This explains the strong reinforcing properties of interacting particles in polymer gels.90 In addition, the interaction of the two components can have an influence on other properties, such as the magnetic or dielectric response, or on electron transfer processes between particles and matrix.
A covalent coupling of inorganic particles to the polymeric matrix is also possible, e.g. by the immobilization of a reactive functional group (anchor group) to the particle surface.91–93 This leads to materials in which the inorganic group is finally an integral part of the hybrid network. From these Class II hybrid materials, a promising development is expected towards new types of soft actuators, sensors, microengines, biomimetic energy-transducing devices, and controlled delivery systems.
Synthetic strategies towards hybrid materials
In priciple, two approaches are employed in the preparation of hybrid materials. On the one hand, one or both structural units are formed in situ from molecular precursors, and transformed into a novel (network) structure. Typically this is the case if organic polymers are formed but also if, e.g. a sol–gel process is applied within a gel to produce the inorganic component. In these cases well-defined discrete molecules are transformed to multidimensional structures. The internal structure of the final material is determined by the composition of the precursors and the reaction conditions.For the production of hybrid materials by in situ nanoparticle formation, reactions are needed that can be performed in mild, wet conditions. Commonly used processes are sol-gel processes,94,95 metal salts reduction methods96,97 and the decomposition of organometallic precursors.98
On the other hand, the preparation is based on the assembly of well-defined preformed building blocks, that are brought to reaction to form a material in which the precursors keep their original integrity at least partially. While by the building block approach, typical properties of the initial components usually survive the matrix formation and therefore enabling structure-property-predictions, this is not necessarily the case if material precursors are transferred into novel materials.
When hybrid hydrogels are obtained by the polymerization of monomeric precursors in the presence of inorganic material building blocks, often precautions have to be considered to overcome a prospective incompatibilty of the two species. Therefore, the inorganic material can be surface-modified either with non-reactive organic groups (e.g. alkyl chains) or by the immobilization of reactive surface groups, such as polymerizable functionalities.101–105
In the absence of reactive surface groups, the material can be considered to be a Class I hybrid gel (Fig. 5). In this case, the interaction between the inorganic component and the gel matrix is dominated by physical forces, that may comprise electrostatic interactions, van-der-Waals forces, and surface complex or hydrogen bond formation.106 Examples of such hybrid hydrogels are realized by the introduction of poly(vinyl pyridine) segments107 that attach to many metal nanoparticles,108,109 or the use of hydroxyethyl methacrylates co-monomers in combination with metal oxide nanoparticles.101 In the latter example, hydrogen bridges are formed between the polymer matrix and the particle surface.
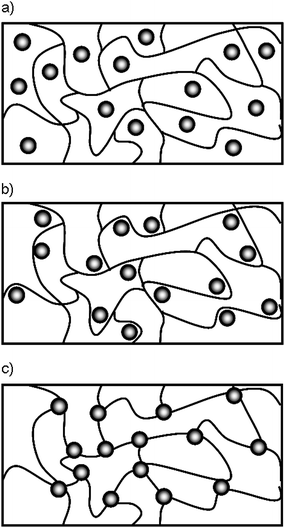 | ||
| Fig. 5 Schemes of different network architectures in hybrid hydrogels involving inorganic particles; a) chemical cross-linked polymer network with entrapped particles; b) chemical cross-linked polymer network with physical interaction between the polymer and the embedded particles, c) particle cross-linked hybrid network. | ||
Class II particle-containing hybrid hydrogels involve even stronger, generally covalent interactions between particle surface and gel phase. Recently, several methods have been reported on the modification of inorganic components with polymerizable groups, that subsequently can be copolymerized with organic monomers.103–105,110
Among the most frequently employed inorganic materials in hybrid hydrogels, silica (SiO2) nanoparticles and porous matrices play a prominent role due to the easy, size-selective synthesis, and the ease of organic functionalization. They mostly aim at the mechanical improvement of the gels, with architectures ranging from Class I type composite gels, via covalently linked silica, to IPN-like, co-continuous structures.1 By the combination with functional or templated polymer gels, they can be useful for applications like column chromatography.111
After the initial works of Haraguchi et al,99 a variety of works can be found on the incorporation of partly or fully exfoliated clay nanosheets into hydrogels. The hybrid hydrogels are prepared by in situradical polymerization using a specific solution system. Highly stable, structurally homogeneous materials with extraordinary mechanical properties are achieved (Fig. 6). Static Light Scattering (SLS) and rheology experiments on exfoliated laponite-containing gels indicate that the cross-linking proceeds mainly through the nanosheets, even if the exact nature is still subject of discussions.86
 | ||
| Fig. 6 a) Network structure model for a hybrid hydrogel consisting of uniformly dispersed hectrite clay nanosheets and poly(N-isopropylacrylamide) chains. b) Elongation model for NC gel. Reproduced with permission from ref. 99. Copyright (2002) Wiley-VCH. | ||
In the meantime, similar particles have been employed analogously in the combination with thermoresponsive PNiPAAm hydrogels (Fig. 7).112 The gels have been tensile-tested under aqueous conditions at various temperatures. The response time of the materials could further be improved by preparing porous materials by the salt-leaching technique,113 or by the incorporation of carboxymethyldextran.114 Further functionalization result in additional incorporation of pH-responsive components.115,116
 | ||
| Fig. 7 Force profiles (b, c) in response to a temperature profile (a) of clay nanosheet/PNiPAAm hydrogels with different polymer content (M1 < M2), in comparison to the force profiles of chemically cross-linked (d) poly(N,N′-dimethylacrylamide) gel, and (e) poly(N-isopropylacrylamide) gel. Reproduced with permission from ref. 100. Copyright (2005) Wiley-VCH. | ||
Other particulate materials that are introduced to improve the mechanical stability in hydrogels include carbon nanotubes117 and some examples of biological structural units118 (see below).
When particles with dipolar nature or polarizability are combined with mechanically soft materials like gels, new possibilities are opened for the manipulations of materials by the application of external fields.
For example, the incorporation of titania (TiO2) into poly(dimethyl siloxane) gels in organic solvents119 opens a new driving mechanism to induce a deformation of neutral polymer gels in a non-conductive medium. Under electric field these gels undergo a significant and quick bending.
As well temperature-depending conductivity120 as catalytic activity121 could be realized by the incorpartion of metallic gold or silver nanoparticles into PNiPAAm matrices (Fig. 8).
 | ||
| Fig. 8 Electric conductivity vs. temperature for Au/P(NiPAAm-co-ethylene glycol dimethacylate) hybrid hydrogels with different Au nanoparticle contents. Reproduced with permission from ref. 122. Copyright (2005) Wiley-VCH. | ||
A promising approach towards photo-switching is based on the optical properties of gold nanorods. Laser irradiation of the hybrid gel results in the conversion of light energy to heat through non-radiative relaxation processes within the nanorods.39 It was found that the photo-thermal heat localized within thermosensitive, NiPAAm-containing copolymer microgels resulted in a volume reduction of nearly 80%.
The combination of thermosensitive polymer with magnetic, nanoscopic particles offers an even broader variety of interesting effects. At least three different particle-field interactions are of interest for mechanical manipulation in combination with polymeric gels. First, the particles' magnetic susceptibility (or rather its deviation from the susceptibility of the surrounding medium) is responsible for the possibility to orient and guide superparamagnetic particles (and their attachments) in field gradients by particle-field interactions. By using homogeneous magnetic fields, the particles are rather oriented than moved, and interparticulate interactions may become significant. Third, the particles' energy dissipation observed in AC magnetic fields can be used to heat up the matrix and may be used to activate a thermal effect selectively at the particles location.123–129
Magnetostrictive polymer gels are a sub-class of soft materials containing magnetizable components, which have been embedded in the gel during the cross-linking process.130–132 If such a magnetic gel is exposed to an external static field, two distinct types of interactions can be identified: field-particle interaction and particle-particle interaction.133 If the field is non-uniform, the field-particle interactions are dominant, and the particles experience a magnetophoretic force. As a result, the particles are attracted to regions of stronger field intensities. Because of the cross-linking bridges in the network, changes in molecular conformation due to force impact can accumulate and lead to macroscopic shape changes and/or motion.
In uniform fields the situation is different. Due to the lack of a field gradient, there are no attractive or repulsive field-particle interactions. However, the imposed field induces magnetic dipoles, and the particle-particle interactions become dominant. As a result, mutual particle interactions occur if the particles are so closely spaced that the local field can influence their neighbours.136 This mutual interaction can be very strong, leading to a significant change in the structure of particle ensembles. The particles attract each other when aligned end to end, and repel each other in the side-by-side situation. When the particles experience enough mobility within the polymer network, a chain-like structure may be developed in response to the attractive forces. The field-induced chain formation has major implications for a number of technologies, such as magnetorheological gels.137
Zrínyi et al. have demonstrated that it is possible to use the susceptibility of superparamagnetic particles, and the induction of particle interaction in static magnetic fields to achieve considerable gel bending or contraction effects. Meanwhile, magnetostrictive gels based on different matrices like PVA,138Pluronics,139 and PHEMA have been realized.
If the synthesis of magnetically loaded gels is performed under the impact on an external magnetic field, magnetically and mechanically anisotropic materials are obtained with “frozen” anisotropy, in the sense that anisotropy is maintained also in field absence. The anisotropy manifests itself both as direction dependent elastic modulus as well as direction dependent swelling.132,140 In addition, the mechanical properties are clearly depending on the field strength and are characterized by a fast response.131,132 It is assumed that next to particle interactions, chain formation plays a significant role for the stiffening effect.126,127,130,140
The ability of magnetic nanoparticles to dissipate heat in response to a high-frequency magnetic field can be used to conveniently trigger the induction of an originally thermal response by applying an external alternating magnetic field. Accordingly, a thermosensitive hydrogel in which superparamagnetic nanoparticles are entrapped, swells or shrinks in response to magnetic heating. This behaviour may be exploited for drug release, as has recently been reported for nanoparticulate systems based on brush-coated magnetic nanoparticles,126,141 micellar solutions123 (Fig. 9) and microgel beads.142,143
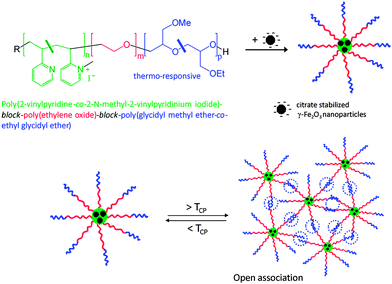 | ||
| Fig. 9 Hybrid micelles formed via complexation of citrate stabilized γ-Fe2O3 nanoparticles with a quarternized Pq2VP-b-PEO-b-P(GME-co-EGE) triblock terpolymer, and their reversible gelationvia open association by LCST behaviour of the P(GME-co-EGE) block. Reproduced with permission from ref. 123. Copyright (2010) Wiley-VCH. | ||
In core-shell-like structures, the shell solubility can be used to reversibly induce a transition between flocculation and dispersion.125 While for functional groups in the polymeric corona, good binding conditions and quasi-homogeneous behaviour are observed in the well-dispersed state, a shell collapse under temperature influence causes a severe reduction of the accessibility of these groups, and the thermo-flocculated particles can easily be separated in magnetic fields (Fig. 10). This behaviour is useful for activation control of immobilized enzymes in trypsin- and β-galactosidase containing microspheres.134,144 A lately published review summarizes recent developments in the field of thermoresponsive magnetic colloids.145
 | ||
| Fig. 10 Capture of benzylamine using FeOx-P(OEGMA-co-SIMA) particles (1) by thermoflocculation and (2) staining and UV-quantification. Reproduced with permission from ref. 134. Copyright (2010) American Chemical Society. | ||
In macroscopic gels, magnetic heating has some distinct advantages over external heating. Since the heating occurs inside the gel, a dehydrated dense “skin layer” or diffusion-limiting phase separation is not observed upon heating, as the temperature development is rather isotropic inside the gel. Recently, several works have reported on the acceleration of enzymatic reactions within thermoresponsive magnetic hydrogels. Similar to conventional heated gels, where an acceleration of as enzymatic reaction is possible by up-and-down temperature change due to swelling/deswelling (“hydraulic pump” behaviour),148 the effect is reported to be advantageously heated from incorporated magnetic nanoparticles, as the alternating frequency is less limited by thermodiffusional processes.149,150
The magnetic heatability of thermoresponsive gels can furthermore be used for the construction of chemo-mechanical or magneto-mechanical devices.123,124,127 Using different field modes, including electrical fields, Kato et al.151 demonstrated the potential of magnetic-particle containing gels that perform mechanical work with a load of up to 20 kPa.
An example for magnetic-field controlled actuation is recently reported.135 Here, the magnetic heatability of magnetic nanoparticles embedded in a PNiPAAm matrix is exploited to regulate the flow in microchannels by achieving up to 80% of gel volume shrinkage under field influence (Fig. 11).
 | ||
| Fig. 11 Demonstration of the ON-OFF flow control of a ceramic microdevice with PNiPAAm ferrohydrogel as valve by high frequency magnetic field. Reproduced with permission from ref. 135. Copyright (2005) Royal Society of Chemistry. | ||
Just recently, an elegant method has been reported to minimize particle migration/agglomeration or loss in ferrogels by chemical cross-linking Co/C nanomagnets into organic gel matrices (Fig. 12). The approach results in highly flexible gels with particle contents up to 60 wt%. Such ferrogels with modified microstructure and covalent particle-matrix interaction are expected to open perspectives for the development of soft actuators in medical implants or micro-pumps.152
 | ||
| Fig. 12 Magnetic force driven elongation of a Co-particle-linked poly(2-hydroxy-ethyl-methacrylate-co-etylene glycol dimethacrylate) based ferrohydrogel sticking to a permanent magnet (top) before (a) and after (b) switching on an additional electromagnetic field through a solenoid (bottom). Reproduced with permission from ref. 146. Copyright (2009) Wiley-VCH. | ||
Hybrid hydrogels with bioconjugates
The introduction of biological and bioinspired components into hydrogels allows the exploitation of mechanisms that mimic the large variety of highly specific environmental-responsive effects found in biological systems. These effects include as well non-covalent as covalent interactions.9The ubiquitous non-covalent interactions in biological systems are now being used to generate materials with unique, dynamic functions. They can further be divided into systems that employ bioinspired interactions between polymer segments, and such systems that make use of interactions between biologically active units immobilized in the gel and species in solution. Prominent examples for highly specific interactions include protein-protein, host-guest or antibody–antigen interaction pairs.153 The hydrogels are mainly synthesized by covalent incorporation of biochemical compounds into the polymer matrix.153
The well-known thermoresponsive effect of LCST-type polymers strongly resembles the transition of a protein from unfolded to folded structures at its cold renaturation temperature. Consequently, a protein can be thought of as a naturally thermosensitive network segment, and an increasing number of works is concerned with the introduction of repeating motifs with specific intra- or interchain interactions into polymer hydrogels. For example, elastin-like polypeptides with various “guest residues” display a temperature-depending phase behaviour in aqueous solution, which makes them attractive for drug delivery and tissue engineering.154,155 The transition temperature can be varied by employing different peptide sequences, molecular weight, and motif concentration.156,157 A rheological study on physical cross-linked protein based tri-block polymers with temperature response is reported by Wu et al (Fig. 13).158
 | ||
| Fig. 13 Rheological behaviour of a protein based triblock polymer with temperature induced sol-gel transition in water. A) Dynamic shear storageG′, loss modulus G″ and tanδ plotted as a function of temperature (γ = 2%; ω = 1 Hz). B) Dynamic shear modulus G′ and complex viscosity η* plotted as a function of frequency (γ = 2%; T = 37 °C). Reproduced with permission from ref. 147. Copyright (2009) American Chemical Society. | ||
Thermoresponsive matrices are also obtained when elastic muscle proteins27 or chitosan are introduced as physical cross-linkers into PAAm.159
The light responsiveness of certain chlorophyll-metal complexes can be used to induce gel shrinkage upon visible light irradiation of chlorophyll-modified PNiPAAm hydrogels, opening one pathway towards photo-responsive artificial muscles, switches, and memory devices.3
A powerful advantage of biological species is the highly specific ability to adsorb molecules from solution. Depending on the type of interaction and the concentration of the guest molecule, different responses can be achieved.
For example, the incorporation of alginate results in gel systems that show a reversible cross-linking by divalent metal ions (ionotropic gelation)161 or pH change.162 By combining PVA-based hydrogels with ferritin, pH-responsive materials are obtained that show a exceptional fast response time in the tens of seconds range.118
The incorporation of cyclodextrins into polymer hydrogels opens a variety of host-guest interactions between the cyclic oligoglucose rings and hydrophobic molecules or polymer main or side chains. Physical hydrogels were constructed by the formation of inclusion complexes between adamantyl-containing PAAm-copolymers, and β-CD-functional dimers and oligomers (Fig. 14),160 and the involved interactions have been studied below the LCST by viscosimetry.163 The formation of host-guest complexes between CD and PNiPAAm164 or poly(ε-lysine)165 has been shown to have a significant impact on the transition temperature.
 | ||
| Fig. 14 a) Chemical structure of thermosensitive, adamatyl-functionalized copolymers (5–7) and β-CD dimer and b) scheme of hydrogels obtained by host-guest interaction. c) Comparison of the zero-shear viscosity of three copolymer solutions and their mixtures with β-CD and CD dimer, respectively. Reproduced with permission from ref. 160. Copyright (2006). Wiley-VCH. | ||
A reversible sol-gel-sol-transition under irradiation with UV-light has been observed for α-CD-modified hydrogels with a photoresponsive guest (4,4-azodibenzoic acid).166
More specific interactions are achieved when proteins are involved. A glucose-sensitive hydrogel was prepared by introducing concanavalin A into a poly(2-glucosyloxyethyl methacrylate) (poly(GEMA)) hydrogel.167
Gels responding to specific solutes are obtained based on non-covalent protein calmodulin (CaM)-phenothiazine cross-links. CaM undergoes a conformational shift from an extended dumbbell shape to a collapsed globular conformation upon binding of specific biochemical ligands, such as the drugs trifluoperazine168 or chlorpromazine (Fig. 15).169 As a result, CaM cross-linked PEG-based hydrogel networks undergo a significant decrease in volume, demonstrating that a nanometer-scale protein conformational change can be translated into macroscopic changes in material properties. The effect can be used for the development of spatially patterned actuators170 and tunable optical biosensors.171
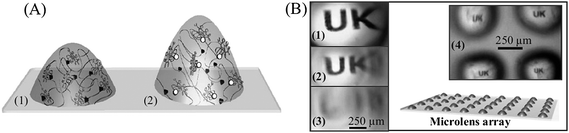 | ||
| Fig. 15 A) Chloropromazine (CPZ)-tunable hydrogel microlenses in the absence (1) and the presence (2) of CPZ, where calmodulin (CaM) releases phenothiazine (black ligand) and binds to free CPZ (white ligand). B) Patterns visualized through hydrogel microdomes utilizing an optical microscope. The “UK” pattern is best focused in the swollen state of the microdome (1) and, as the microlens becomes less swollen over time (1 min intervals) (2,3), the image becomes blurred. A collectively addressable hydrogel microdome array showing multiple patterns under different extent of focusing is exhibited in (4). Reproduced with permission from ref. 169. Copyright (2007) Wiley-VCH. | ||
Other examples of chemically responsive hydrogels are based on antigen/antibody interactions. The materials make use of specific reversible non-covalent cross-links between biological molecules linked to the polymer background with the respective binding partners. When an antigen is immobilized to the backbone of polyacrylamide (PAAm) hydrogels,172 the hydrogels have a compact structure in the presence of a soluble antibody due to the antigen-antibody interactions, while when soluble antigen is added, the hydrogel increases its swelling due to the dissociation of the (immobilized antigen)-antibody complex.
When both antigen and antibody are immobilized,173 the response behaviour could even be enhanced (Fig. 16). Such gels may be useful in applications that require antigen sensing or antigen-responsive drug release, or in membranes with antigen-responsive permeability.174
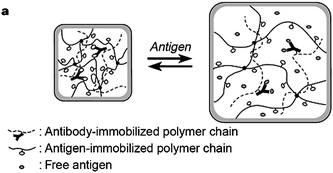 | ||
| Fig. 16 Diagram of a suggested mechanism for the swelling of an antigen-antibody semi-interpenetrating hydrogel in response to a free antigen. Reproduced with permission from ref. 173. Copyright (1999) Nature Publishing Group. | ||
Applications of mechanically manipulable hydrogels
Although responsive hydrogel materials have shown promise in many areas, they are limited to application where mechanical strength and mechanical work performance is not a critical issue. However, the combination with inorganic or biological components may open additional potential in functions where mechanical strength is of crucial importance for a proper operation. In addition, the incorporation of biologically active units gives rise to biological activity, hierarchical organization, and structural integrity that are necessary to facilitate cell interaction and to mimick biological actuation. Even if the respective concepts have been reported before in the general context of responsive soft matter, as promising breakthrough may be achieved by hybridizing hydrogels with inorganic or biological components.Artificial muscles
From an engineering point of view, muscles are soft and wet mechanical transducers, capable of performing their functions by quick and reversible shortening (unidirectional contraction). Internal forces are derived from a mechanism designed to transform other forms of energy, like thermal, chemical or field energy, into mechanical work or locomotion.Under no load conditions, the stimulation of an artificial muscle will cause it to contract to its smallest length without development of any active tension.175 In contrast, when contraction takes place against a resistance, a force is generated and mechanical work is done (Fig. 17).176 For soft gels, it is therefore important to find a highly extensile material that is able to develop considerable forces at the same time, i. e. persisting a sufficient high elastic modulus. As discussed above, the combination of both can best be found in hybrid hydrogels.
 | ||
| Fig. 17 Transport of a glass bead by a gel with anisotropic shrinking upon heating. A propagation of the phase transition from left to right is observed. Reproduced with permission from ref. 182. Copyright (2005) Wiley-VCH. | ||
It is also important to have a reliable control system. In this respect the incorporation of dipolar nanoparticles into stimulable hydrogels is of high perspective. The ability of magnetic or electric field-sensitive gels to undergo a quick controllable change of shape may be used to mimic muscular contraction, and to create a wide range of actuation, motion, and shape changes.136
Cell stimulation
The texture of hybrid hydrogel materials resembles natural living tissue due to their high water contents, soft consistency and their activation mode which is similar to natural tissue.45The mechano-electrochemical properties of cartillage give rise to a whole host of extracellular events when the tissue is subject to dynamic mechanical loads, which ultimately influence chondrocyte function and metabolism.177–179). To date, the exact mechanisms are not well understood, although several experiments indicate that combined mechano-electrochemical signals are involved.180 In the investigation and in mimicking these mechanisms, hybrid hydrogels may play an important role.
It has been shown that by using thermoresponsive gels, an equibiaxial stretching of cells can be accomplished by gel swelling with slight changes in temperature.181 It was indicated that mechanical signals were transduced into biochemical signals in cells, and the mechanical forces facilitated actin polymerization at the peripheral region, resulting in filopodia–like structures.
Microvalves
The ability to actively manipulate fluid-flow patterns through microfluidic devices is important for several current applications. Typically microvalves are controlled mechanically or electrically and rely on their intrinsic thermal, chemical or electro-optical properties. Indeed, hybrid hydrogels show promising ways of flow control for microfluidic devices based on tunable stimuli-responsive properties. An option that is gaining increasing attention is the possibility of field-generated actuation. The use of dipolar particles as antennas for electromagnetic stimulation is particularly attractive due to wide applicability and the directional specificity within a channel network. A wide range of field-material interactions is available, depending on the field nature and dynamics, and the properties of the involved particles, including direct dipolar interactions, electro- or magneto-mechanical, or –optical coupling, and the interaction with oscillating fields. The advantages of this approach towards materials with switchable flow permitivity is in the fast response time and the external tunability of the stimulating field, being of high interest for microfluidic systems in drug delivery or chemical analysis.143Conclusions
Responsive hydrogels that change properties and function in response to external stimuli are often inspired by natural systems, resulting in hybrid polymer hydrogels as new generation materials useful for a wide variety of applications. Desirable properties such as mechanical toughness and successibility to external fields are achieved with hydrogels in combination with inorganic nanostructures.The course of development suggests that the discovery of synthetic routes and fabrication technologies, and the fundamental understanding of hybrid hydrogels will continue with an emphasis on sophisticated multicomponent and complex materials. New techniques for the systematic characterization of such complex materials will, in return, impact the rational structure design.
Future directions certainly include the rational design of biomedical hybrid hydrogels that require not only control of chemical and physical properties but also the consideration of biological variables in order to simulate biological tissues. The potential impact for hybrid hydrogels on daily life ranges from stimuli-responsive sensors and actuators to microfluidics, pharmaceutical, and biomedical devices.
Acknowledgements
This work was in part supported by the Deutsche Forschungsgesellschaft (DFG) within the Emmy Noether program (SCHM1747-4) and priority program 1259 “Intelligent Hydrogels”.References
- P. Calvert, Adv. Mater., 2009, 21, 743 CrossRef CAS.
- I. Dimitrov, B. Trzebicka, A. H. E. Muller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2007, 32, 1275–1343 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- C. D. H. Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- B. Jeong and A. Gutowska, Trends Biotechnol., 2002, 20, 305–311 CrossRef CAS.
- S. K. Ahn, R. M. Kasi, S. C. Kim, N. Sharma and Y. X. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- Y. Li and T. Tanaka, Annu. Rev. Mater. Sci., 1992, 22, 243–277 CrossRef CAS.
- J. Kopecek, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5929–5946 CrossRef CAS.
- J. S. Mohammed and W. L. Murphy, Adv. Mater., 2009, 21, 2361–2374 CrossRef CAS.
- J. F. Mano, Adv. Eng. Mater., 2008, 10, 515–527 CrossRef CAS.
- P. Kujawa and F. M. Winnik, Macromolecules, 2001, 34, 4130–4135 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl. Chem., 1968, 2, 1441–1455 Search PubMed.
- S. Chaterji, I. K. Kwon and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS.
- Y. Bae and Y. H.a. n. Bae, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 923 CrossRef CAS.
- I. Idziak, D. Avoce, D. Lessard, D. Gravel and X. X. Zhu, Macromolecules, 1999, 32, 1260–1263 CrossRef CAS.
- F. Meeussen, E. Nies, H. Berghmans, S. Verbrugghe, E. Goethals and F. Du Prez, Polymer, 2000, 41, 8597–8602 CrossRef CAS.
- A. Laukkanen, L. Valtola, F. M. Winnik and H. Tenhu, Macromolecules, 2004, 37, 2268–2274 CrossRef CAS.
- H. Uyama and S. Kobayashi, Chem. Lett., 1992, 1643–1646 CAS.
- S. Saito and T. Otsuka, J. Colloid Interface Sci., 1967, 25, 531 CrossRef CAS.
- P. Alexandridis, J. F. Holzwarth and T. A. Hatton, Macromolecules, 1994, 27, 2414–2425 CrossRef CAS.
- F. Meeussen, Y. Bauwens, R. Moerkerke, E. Nies and H. Berghmans, Polymer, 2000, 41, 3737–3743 CrossRef CAS.
- H. Schäfer-Soenen, R. Moerkerke, H. Berghmans, R. Koningsveld, K. Dusek and K. Solc, Macromolecules, 1997, 30, 410–416 CrossRef.
- J. F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- S. Sugihara, S. Kanaoka and S. Aoshima, Macromolecules, 2005, 38, 1919–1927 CrossRef CAS.
- M. B. Huglin and M. A. Radwan, Polym. Int., 1991, 26, 97–104 CrossRef CAS.
- R. W. Guelch, J. Holdenried, A. Weible, T. Wallmersperger and B. Kroeplin, Proc. SPIE, 2001, 4329, 328 CAS.
- L. Chen, J. Kopecek and R. J. Stewart, Bioconjugate Chem., 2000, 11, 734–740 CrossRef CAS.
- K. Podual, F. J. Doyle and N. A. Peppas, Polymer, 2000, 41, 3975–3983 CrossRef CAS.
- H. Park and J. R. Robinson, Pharm. Res., 1987, 4, 457–464 CrossRef CAS.
- S. De and S. K. De, J. Microelectromech. Syst., 2002, 11, 544 CrossRef CAS.
- H. Li, Y. K. Yew, T. Y. Ng and K. Y. Lam, J. Electroanal. Chem., 2005, 580, 161–172 CrossRef CAS.
- F. Horkay and F. Horkay, Macromol. Biosci., 2002, 2, 207 CrossRef CAS.
- M. A. Cole, N. H. Voelcker, H. Thissen and H. J. Griesser, Biomaterials, 2009, 30, 1827–1850 CrossRef CAS.
- T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592–16596 CrossRef CAS.
- J. Edahiro, K. Sumaru, Y. Tada, K. Ohi, T. Takagi, M. Kameda, T. Shinbo, T. Kanamori and Y. Yoshimi, Biomacromolecules, 2005, 6, 970–974 CrossRef CAS.
- A. E. Ivanov, N. L. Eremeev, P. O. Wahlund, I. Y. Galaev and B. Mattiasson, Polymer, 2002, 43, 3819–3823 CrossRef CAS.
- K. Sumaru, M. Kameda, T. Kanamori and T. Shinbo, Macromolecules, 2004, 37, 4949–4955 CrossRef CAS.
- K. Sumaru and S. Kimio, Langmuir, 2006, 22, 4353 CrossRef CAS.
- M. Das, N. Sanson, D. Fava and E. Kumacheva, Langmuir, 2007, 23, 196–201 CrossRef CAS.
- S. Murdan and S. Murdan, J. Controlled Release, 2003, 92, 1 CrossRef CAS.
- C. J. Whiting, A. M. Voice, P. D. Olmsted and T. C. B. McLeish, J. Phys.: Condens. Matter, 2001, 13, 1381–1393 CrossRef CAS.
- T. Wallmersperger and D. Ballhause, Smart Mater. Struct., 2008, 17, 045012 CrossRef.
- J. R. Saunders, S. Abu-Salih and W. Mousse, J. Comput. Theor. Nanosci., 2008, 5, 1961–1975 CrossRef CAS.
- R. W. Guelch, J. Holdenried, A. Weible, T. Wallmersperger and B. Kroeplin, Proc. SPIE, 2001, 4329, 328 CAS.
- M. Bassil, J. Davenas and M. El Tahchi, Sens. Actuators, B, 2008, 134, 496–501 CrossRef.
- Y. Osada and J. P. Gong, Adv. Mater., 1998, 10, 827–837 CrossRef CAS.
- Y. Osada and M. Hasebe, Chem. Lett., 1985, 1285–1288 CAS.
- B. Jeong and B. Jeong, Adv. Drug Delivery Rev., 2002, 54, 37 CrossRef CAS.
- C. L. He, S. W. Kim and D. S. Lee, J. Controlled Release, 2008, 127, 189–207 CrossRef CAS.
- L. S. Wang, J. E. Chung, P. Pui-Yik Chan and M. Kurisawa, Biomaterials, 2010, 31, 1148–1157 CrossRef CAS.
- N. Fechler, N. Badi, K. Schade, S. Pfeifer and J. F. Lutz, Macromolecules, 2009, 42, 33–36 CrossRef CAS.
- S. Reinicke, J. Schmelz, A. Lapp, M. Karg, T. Hellweg and H. Schmalz, Soft Matter, 2009, 5, 2648–2657 RSC.
- Y. C. Wang and H. Morawetz, Macromolecules, 1989, 22, 164–167 CrossRef CAS.
- G. Staikos, K. Karayanni and Y. Mylonas, Macromol. Chem. Phys., 1997, 198, 2905–2915 CrossRef CAS.
- A. Durand and D. Hourdet, Polymer, 1999, 40, 4941–4951 CrossRef CAS.
- A. Durand and D. Hourdet, Polymer, 2000, 41, 545–557 CrossRef CAS.
- A. Durand and D. Hourdet, Macromol. Chem. Phys., 2000, 201, 858–868 CrossRef CAS.
- T. Shibanuma, T. Aoki, K. Sanui, N. Ogata, A. Kikuchi, Y. Sakurai and T. Okano, Macromolecules, 2000, 33, 444–450 CrossRef CAS.
- I. Tokarev and I. Tokarev, Soft Matter, 2009, 5, 511 RSC.
- C. Wei, Materials Research Society Symposium Proceedings, 2006, 897.
- A. Suzuki and G. Kondo, Materials Research Society Symposium Proceedings, 2007, 947.
- C. Liu, H. Qin and P. T. Mather, J. Mater. Chem., 2007, 17, 1543–1558 RSC.
- Y. Osada and A. Matsuda, Nature, 1995, 376, 219 CrossRef CAS.
- K.-F. Arndt and K. Arndt, Acta Polym., 1999, 50, 383 CrossRef CAS.
- K. K. Westbrook, J. Intell. Mater. Syst. Struct., 2007, 19, 597 CrossRef.
- P. T. Mather, Nat. Mater., 2007, 6, 93–94 CrossRef CAS.
- I. Nishio, G. Swislow, S. T. Sun and T. Tanaka, Nature, 1982, 300, 243–244 CrossRef CAS.
- G. Swislow, S. T. Sun, I. Nishio and T. Tanaka, Phys. Rev. Lett., 1980, 44, 796–798 CrossRef CAS.
- T. Amiya and T. Tanaka, Macromolecules, 1987, 20, 1162–1164 CrossRef CAS.
- K. Dušek and D. Patterso, J. Polym. Sci., Part A-2, 1968, 6, 1209 CrossRef CAS.
- S. Hirotsu, Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1987, 87, 1392–1395 CrossRef CAS.
- X. Wang, Macromolecules, 1998, 31, 2972 CrossRef CAS.
- J. E. Martin and D. Adolf, Annu. Rev. Phys. Chem., 1991, 42, 311–339 CrossRef CAS.
- T. Tanaka and D. J. Fillmore, J. Chem. Phys., 1979, 70, 1214–1218 CrossRef CAS.
- X. Jin and Y. L. Hsieh, Polymer, 2005, 46, 5149–5160 CrossRef CAS.
- T. Sakai and R. Yoshida, Langmuir, 2004, 20, 1036–1038 CrossRef CAS.
- T. Gotoh, Y. Nakatani and S. Sakohara, J. Appl. Polym. Sci., 1998, 69, 895–906 CrossRef CAS.
- H. Hirose and M. Shibayama, Macromolecules, 1998, 31, 5336–5342 CrossRef CAS.
- M. Ebara and M. Ebara, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 335 CrossRef CAS.
- X. Z. Zhang, D. Q. Wu and C. C. Chu, Biomaterials, 2004, 25, 3793–3805 CrossRef CAS.
- P. Dimitrov, S. Rangelov, A. Dworak and C. B. Tsvetanov, Macromolecules, 2004, 37, 1000–1008 CrossRef CAS.
- T. Tanaka, L. O. Hocker and G. B. Benedek, J. Chem. Phys., 1973, 59, 5151–5159 CrossRef CAS.
- P. G. de Gennes, Scaling concepts in polymer physics, Cornell University Press, Ithaca NY, 1979 Search PubMed.
- T. Tanaka, S. Ishiwata and C. Ishimoto, Phys. Rev. Lett., 1977, 38, 771–774 CrossRef CAS.
- T. Tanaka, Phys. Rev. Lett., 1978, 40, 820–823 CrossRef CAS.
- O. Okay and W. Oppermann, Macromolecules, 2007, 40, 3378–3387 CrossRef CAS.
- G. Constantinides, Z. I. Kalcioglu, M. McFarland, J. F. Smith and K. J. Van Vliet, J. Biomech., 2008, 41, 3285–3289 CrossRef.
- H. Becker and C. Gartner, Electrophoresis, 2000, 21, 12–26 CrossRef CAS.
- D. Mark, S. Haeberle, G. Roth, F. von Stetten and R. Zengerle, Chem. Soc. Rev., 2010, 39, 1153–1182 RSC.
- C. Kickelbick, in Hybrid materials: synthesis, characterization, and applications, Wiley-VCH, Weinheim, Germany, 2007, vol. 1, p. 1 Search PubMed.
- T. Gelbrich, M. Feyen and A. M. Schmidt, Z. Phys. Chem., 2006, 220, 41–49 CrossRef CAS.
- C. Gürler, M. Feyen, S. Behrens, N. Matoussevitch and A. M. Schmidt, Polymer, 2008, 49, 2211–2216 CrossRef.
- A. Kaiser, S. Dutz and A. M. Schmidt, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 7012–7020 CrossRef CAS.
- J. Li, J. P. Suo and L. T. Jia, Polymer Engineering and Science, 2010, 50, 689–696 CAS.
- Y. S. Jeon, J. Lei, D. J. Chung and J. H. Kim, J. Ind. Eng. Chem., 2009, 15, 544–549 Search PubMed.
- I. Tarnavchyk, A. Voronov, A. Kohut, N. Nosova, S. Varvarenko, V. Samaryk and S. Voronov, Macromol. Rapid Commun., 2009, 30, 1564–1569 CrossRef CAS.
- O. Ayyad, D. Munoz-Rojas, J. Oro-Sole and P. Gomez-Romero, J. Nanopart. Res., 2010, 12, 337–345 CrossRef CAS.
- P. Schexnailder and G. Schmidt, Colloid Polym. Sci., 2009, 287, 1–11 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- K. Haraguchi, S. Taniguchi and T. Takehisa, ChemPhysChem, 2005, 6, 238 CrossRef CAS.
- E. Bourgeat-Lami, M. Insulaire, S. Reculusa, A. Perro, S. Ravaine and E. Duguet, J. Nanosci. Nanotechnol., 2006, 6, 432–444 CAS.
- A. M. Alkilany and C. J. Murphy, Langmuir, 2009, 25, 13874–13879 CrossRef CAS.
- V. G. Ngo, C. Bressy, C. Leroux and A. Margaillan, Polymer, 2009, 50, 3095–3102 CrossRef CAS.
- N. Frickel, R. Messing, T. Gelbrich and A. M. Schmidt, Langmuir, 2010, 26, 2839–2846 CrossRef CAS.
- H. B. Sunkara, J. M. Jethmalani and W. T. Ford, Chem. Mater., 1994, 6, 362–364 CrossRef CAS.
- W. Loos and F. Du Prez, Macromol. Symp., 2004, 210, 483–491 CrossRef CAS.
- T. K. Tam, M. Pita, O. Trotsenko, M. Motornov, I. Tokarev, J. Halamek, S. Minko and E. Katz, Langmuir, 2010, 26, 4506–4513 CrossRef CAS.
- Y. H. Jang, S. T. Kochuveedu, M. A. Cha, Y. J. Jang, J. Y. Lee, J. Lee, J. Kim, D. Y. Ryu and D. H. Kim, J. Colloid Interface Sci., 2010, 345, 125–130 CrossRef CAS.
- Y. J. Liu, L. B. He, C. H. Xu and M. Han, Chem. Commun., 2009, 6566–6568 RSC.
- E. Bourgeat-Lami, M. Insulaire, S. Reculusa, A. Perro, S. Ravaine and E. Duguet, J. Nanosci. Nanotechnol., 2006, 6, 432–444 CAS.
- Z. D. Hua, S. Zhou and M. P. Zhao, Biosens. Bioelectron., 2009, 25, 615–622 CrossRef CAS.
- K. Haraguchi and K. Haraguchi, ChemPhysChem, 2005, 6, 238 CrossRef CAS.
- J. H. Ma, L. Zhang, Z. Li and B. R. Liang, Polym. Bull., 2008, 61, 593–602 CrossRef CAS.
- J. H. Ma, L. Zhang, B. Fan, Y. J. Xu and B. R. Liang, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1546–1555 CrossRef CAS.
- J. H. Ma, Y. J. Xu, Q. S. Zhang, L. S. Zha and B. R. Liang, Colloid Polym. Sci., 2006, 285, 479–484 CrossRef.
- X. B. Hu, L. J. Xiong, T. Wang, Z. M. Lin, X. X. Liu and Z. Tong, Polymer, 2009, 50, 1933–1938 CrossRef CAS.
- S. Ozarkar, M. Jassal and A. K. Agrawal, Smart Mater. Struct., 2008, 17, 055016 CrossRef.
- M. K. Shin, G. M. Spinks, S. R. Shin, S. I. Kim and S. J. Kim, Adv. Mater., 2009, 21, 1712 CrossRef CAS.
- J. Fehér, G. Filipcsei, J. Szalma and M. Zrínyi, Colloids Surf., A, 2001, 183–185, 505–515 CrossRef CAS.
- X. Zhao and X. Zhao, Macromol. Rapid Commun., 2005, 26, 1784 CrossRef CAS.
- Y. Lu, Y. Mei, M. Drechsler and M. Ballauff, Angew. Chem., Int. Ed., 2006, 45, 813–816 CrossRef CAS.
- X. Zhao, X. Ding, Z. Deng, Z. Zheng, Y. Peng and X. Long, Macromol. Rapid Commun., 2005, 26, 1784 CrossRef CAS.
- S. Reinicke, S. Döhler, S. Tea, M. Krekhova, R. Messing, A. M. Schmidt and H. Schmalz, Soft Matter, 2010, 6, 2760 RSC.
- A. Kaiser, M. Winkler, S. Krause, H. Finkelmann and A. M. Schmidt, J. Mater. Chem., 2009, 19, 538–543 RSC.
- A. Kaiser, T. Gelbrich and A. M. Schmidt, J. Phys.: Condens. Matter, 2006, 18, S2563–S2580 CrossRef CAS.
- A. M. Schmidt, J. Magn. Magn. Mater., 2005, 289, 5–8 CrossRef CAS.
- A. M. Schmidt, Macromol. Rapid Commun., 2006, 27, 1168–1172 CrossRef CAS.
- M. Feyen, E. Heim, F. Ludwig and A. M. Schmidt, Chem. Mater., 2008, 20, 2942–2948 CrossRef CAS.
- S. Sinnwell, A. M. Schmidt and H. Ritter, J. Macromol. Sci., Part A: Pure Appl. Chem., 2006, 43, 469–476 Search PubMed.
- G. Y. Zhou and J. R. Li, Smart Mater. Struct., 2003, 12, 859–872 CrossRef CAS.
- M. Farshad and M. Le Roux, Polym. Test., 2005, 24, 163–168 CrossRef CAS.
- M. Zrínyi, D. Szabo and L. Barsi, J. Intell. Mater. Syst. Struct., 1998, 9, 667–671 CrossRef CAS.
- R. E. Rosensweig, J. Appl. Phys., 1985, 57, 4259–4264 CrossRef.
- T. Gelbrich, M. Reinartz and A. M. Schmidt, Biomacromolecules, 2010, 11, 635–642 CrossRef CAS.
- N. S. Satarkar, W. Zhang, R. E. Eitel and J. Zach Hilt, Lab Chip, 2009, 9, 1773 RSC.
- M. Zrínyi, Colloid Polym. Sci., 2000, 278, 98–103 CrossRef CAS.
- J. M. Ginder, M. E. Nichols, L. D. Elie and J. L. Tardiff, Proc. SPIE, 1999, 3675, 131 CAS.
- R. V. Ramanujan and L. L. Lao, 2006, p. 952.
- J. Qin and J. Qin, Adv. Mater., 2009, 21, 1354 CrossRef CAS.
- G. Filipcsei, I. Csetneki, A. Szilagyi and M. Zrínyi, Adv. Polym. Sci., 2007, 206, 137–189 CAS.
- T. Gelbrich, M. Feyen and A. M. Schmidt, Macromolecules, 2006, 39, 3469–3472 CrossRef CAS.
- D. Muller-Schulte and T. Schmitz-Rode, J. Magn. Magn. Mater., 2006, 302, 267–271 CrossRef.
- S. Ghosh, S. GhoshMitra, T. Cai, D. R. Diercks, N. C. Mills and D. L. Hynds, Nanoscale Res. Lett., 2010, 5, 195–204 Search PubMed.
- A. Kondo and H. Fukuda, J. Ferment. Bioeng., 1997, 84, 337–341 CrossRef CAS.
- A. M. Schmidt, Colloid Polym. Sci., 2007, 285, 953–966 CrossRef CAS.
- R. Fuhrer, E. K. Athanassiou, N. A. Luechinger and W. J. Stark, Small, 2009, 5, 383 CrossRef CAS.
- X. Wu, R. E. Sallach, J. M. Caves, V. P. Conticello and E. L. Chaikof, Biomacromolecules, 2008, 9, 1787 CrossRef CAS.
- T. G. Park and A. S. Hoffman, Enzyme Microb. Technol., 1993, 15, 476–482 CrossRef CAS.
- N. Kato, A. Oishi and F. Takahashi, Mater. Sci. Eng., C, 1998, 6, 291–296 CrossRef.
- F. Takahashi, Y. Sakai and Y. Mizutani, J. Ferment. Bioeng., 1997, 83, 152–156 CrossRef CAS.
- N. Kato, S. Yamanobe and F. Takahashi, Mater. Sci. Eng., C, 1997, 5, 141–147 CrossRef.
- R. Fuhrer and R. Fuhrer, Small, 2009, 5, 383 CrossRef CAS.
- T. Miyata and T. Miyata, Adv. Drug Delivery Rev., 2002, 54, 79 CrossRef CAS.
- D. W. Urry, C. H. Luan, T. M. Parker, D. C. Gowda, K. U. Prasad, M. C. Reid and A. Safavy, J. Am. Chem. Soc., 1991, 113, 4346–4348 CrossRef CAS.
- D. E. Meyer and A. Chilkoti, Biomacromolecules, 2004, 5, 846–851 CrossRef CAS.
- L. Ayres, M. R. J. Vos, P. Adams, I. O. Shklyarevskiy and J. C. M. van Hest, Macromolecules, 2003, 36, 5967–5973 CrossRef CAS.
- L. Ayres, K. Koch, P. Adams and J. C. M. van Hest, Macromolecules, 2005, 38, 1699–1704 CrossRef CAS.
- X. Wu and X. Wu, Biomacromolecules, 2008, 9, 1787 CrossRef CAS.
- D. H. Fan, W. Y. Zhang, Q. S. Gu and Z. Xu, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2006, 21, 109–112 Search PubMed.
- O. Kretschmann, S. W. Choi, M. Miyauchi, I. Tomatsu, A. Harada and H. Ritter, Angew. Chem., Int. Ed., 2006, 45, 4361–4365 CrossRef.
- H. Omidian, J. G. Rocca and K. Park, Macromol. Biosci., 2006, 6, 703–710 CrossRef CAS.
- J. H. Kim, S. B. Lee, S. J. Kim and Y. M. Lee, Polymer, 2002, 43, 7549–7558 CrossRef CAS.
- C. Koopmans and H. Ritter, Macromolecules, 2008, 41, 7418–7422 CrossRef CAS.
- S. Schmitz and H. Ritter, Angew. Chem., Int. Ed., 2005, 44, 5658–5661 CrossRef CAS.
- H. S. Choi, K. M. Huh, T. Ooya and N. Yui, J. Am. Chem. Soc., 2003, 125, 6350–6351 CrossRef CAS.
- I. Tomatsu and I. Tomatsu, Macromolecules, 2005, 38, 5223 CrossRef CAS.
- T. Miyata and T. Miyata, Macromol. Chem. Phys., 1996, 197, 1135 CrossRef CAS.
- W. L. Murphy, W. S. Dillmore, J. Modica and M. Mrksich, Angew. Chem., Int. Ed., 2007, 46, 3066–3069 CrossRef CAS.
- J. D. Ehrick, S. Stokes, S. Bachas-Daunert, E. A. Moschou, S. K. Deo, L. G. Bachas and S. Daunert, Adv. Mater., 2007, 19, 4024 CrossRef CAS.
- Z. J. Sui, W. J. King and W. L. Murphy, Adv. Mater., 2007, 19, 3377 CrossRef CAS.
- Z. J. Sui, W. J. King and W. L. Murphy, Adv. Funct. Mater., 2008, 18, 1824–1831 CrossRef CAS.
- T. Miyata, N. Asami and T. Uragami, Macromolecules, 1999, 32, 2082–2084 CrossRef CAS.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS.
- R. Zhang, A. Bowyer, R. Eisenthal and J. Hubble, Biotechnol. Bioeng., 2007, 97, 976–984 CrossRef CAS.
- C. R. Bagshow, ed., Muscle contraction, Chapman & Hall, London, 1994 Search PubMed.
- L. Yeghiazarian and L. Yeghiazarian, Adv. Mater., 2005, 17, 1869 CrossRef CAS.
- R. L. Y. Sah, Y. J. Kim, J. Y. H. Doong, A. J. Grodzinsky, A. H. K. Plaas and J. D. Sandy, J. Orthop. Res., 1989, 7, 619–636 CrossRef CAS.
- Y. J. Kim, L. J. Bonassar and A. J. Grodzinsky, J. Biomech., 1995, 28, 1055–1066 CrossRef CAS.
- U. Hansen, M. Schunke, C. Domm, N. Ioannidis, J. Hassenpflug, T. Gehrke and B. Kurz, J. Biomech., 2001, 34, 941–949 CrossRef CAS.
- O. O. Akanji, D. A. Lee and D. A. Bader, Biorheology, 2008, 45, 229–243 Search PubMed.
- K. Yamaki, I. Harada, M. Goto, C. S. Cho and T. Akaike, Biomaterials, 2009, 30, 1421–1427 CrossRef CAS.
- L. Yeghiazarian, S. Mahanjan, C. Montemagno, C. Cohen and U. Wiesner, Adv. Mater., 2005, 17, 1869 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |