Electrostatic assembly of functional polymer combs onto gold nanoparticle surfaces: combining RAFT, click and LbL to generate new hybrid nanomaterials†
Antoine
Bousquet
,
Cyrille
Boyer
*,
Thomas P.
Davis
and
Martina H.
Stenzel
*
Centre for Advanced Macromolecular Design (CAMD), University of New South Wales, Sydney, Australia. E-mail: M.Stenzel@unsw.edu.au; C.Boyer@unsw.edu.au; Fax: 2-93856250; Tel: 2-93854344
First published on 2nd June 2010
Abstract
Comb polymers have been synthesized using a “grafting-onto” method via a combination of Reversible Addition–Fragmentation Chain Transfer (RAFT) polymerization and hetero-Diels–Alder (HDA) cycloaddition. A random copolymer comprised of tert-butyl acrylate (tBA) and hydroxyethyl acrylate (HEA) was post-modified via a two step process to introduce diene functionality. Polymers synthesized using benzyl pyridin-2-yldithioformate as a RAFT agent were used as the dienophile in a click reaction with polymer bearing diene pendant groups thus forming comb polymers. Polystyrene (PS), poly(n-butyl acrylate) (PnBA), poly(tert-butyl acrylate) (PtBA) and poly(oligoethyleneglycol methyl ether acrylate) (POEGMEA) were then grafted onto P(tBA-r-HEAdiene) backbones using a reaction time of 24 h. The grafting densities of the comb polymers ranged from 100% to 50%, depending on the chemical structure and the molecular weight of the linear side chain, as well as the initial molar ratio of dienophile to diene groups. Hydrolysis of the poly(tert-butyl acrylate) (PtBA) yielded hydrophilic or amphiphilic polymers with a poly(acrylic acid) (PAA) backbone. Finally a layer-by-layer approach was utilized to build up an initial layer of poly(ethylene amine) followed by a layer of PAA-g-POEGMEA onto gold nanoparticles (GNPs) yielding hybrid organic/inorganic nanoparticles. UV-vis spectroscopy, zeta-potential measurement and X-ray photoelectron spectroscopy (XPS) were used to characterize the hybrid nanoparticles showing that the POEGMA surface layer can shield the charges of the outer layer effectively.
Introduction
Comb polymers, macromolecules containing side chains (or branches) connected to a linear backbone, have a range of applications depending on the nature of the backbone and branches, including membrane preparation,1,2 drug delivery3,4 or adhesives.5 While comb polymers are attractive for many applications, they have rarely been exploited in the formation of multilayer (or ‘LbL’) assemblies. Block copolymers in contrast are frequently used to introduce specific surface functionality as the outermost layer. By coating the surface of an LbL assembly with diblock copolymers, the interaction of the surface with its environment can be mediated while the second (typically charged) block ensures good anchorage onto the LbL surface. This block copolymer approach allows the introduction of surfaces with specific functionality to improve biocompatibility, bioactivity, stimuli-responsiveness or other properties (for example).6,7 Similarly, and perhaps more efficiently, comb polymers may enhance the functionality on the outermost layer of a LbL assembly, by exploiting an ionic backbone as the anchor and a dense brush-like structure as a carrier for the functionality, screening any charge on the outermost surface. Comb polymers such as polylysine with PEG side chains or poly(2-aminoethyl methacrylate) copolymerized with poly(ethylene glycol) methyl ether methacrylate have been shown to block the surface binding of globular proteins.8 In this previous study8 the PEG chains did not exceed 2000 g mol−1 in molecular weight and we surmise that longer branches might be beneficial in the formation of denser brushes.Comb polymers are typically synthesized by living polymerization techniques, such as living ionic polymerization,9 or living radical polymerization (LRP) including Atom Transfer Radical Polymerization (ATRP),10,11 Reversible Addition–Fragmentation Chain Transfer (RAFT)4,12–14 polymerization, Nitroxide Mediated Polymerization (NMP)15,16 and Iodine Transfer Polymerization (ITP).17 There are three mains methods used to generate comb polymers: (i) the macromonomer method,18 (ii) the “grafting-from” method,19 involving the generation of a backbone bearing initiators as pendant groups for the subsequent polymerization from the branch, (iii) the “grafting-onto” method, requiring the presence of functional groups on the backbone that can react with pre-formed chains. A highly efficient reaction is desirable for the “grafting-onto” technique, involving the reaction between two polymer chains. Polymer–polymer coupling is well-suited to click chemistry.20–26 Copper(I) catalyzed azide–alkyne cycloaddition (CuAAC) fulfils click criteria20,27 and is the most popular approach, even if the elimination of the catalyst can be problematical. This CuAAC technique has already been employed to synthesize comb polymer using either azide28–30 or alkyne31–33 reactive groups along the backbone. Other types of click chemistry, such as thiol–ene chemistry,34,35 Diels–Alder cycloaddition between anthracene PS main chains and maleimide functionalized PEG or PMMA,36 or the substitution reaction of amine functionalized PEG branches on a poly(pentafluorostyrene) backbone,37 have also been used to build up comb polymers.
In recent papers, we have described the idea of using RAFT agents to both control living polymerization and as functionality to access ‘click’ type reactions, such as RAFT–HDA,38 RAFT–thiol–ene34,39 and RAFT–PDS (pyridyl disulfide).40 The RAFT–HDA approach has previously been used to prepare comb polymer PS-g-PnBA.38 Pyridinedithioesters, which are efficient RAFT agents for styrene41 and acrylates,42 can simultaneously act as heterodienophile in [4 + 2] cycloadditions. Certain dienes react with pyridinedithioesters to completion within several hours if activated by Lewis or Bronsted acids43 (Scheme 1), while cyclopentadienyl groups promote the complete reaction with the RAFT agent in less than 10 min at ambient temperatures.42 The RAFT–HDA approach has been applied to the synthesis of block copolymers,42,44,45 stars,46,47 and to surface functionalize microspheres.48 Previously, comb polymers have been obtained by the copolymerization of styrene and a diene functionalized acrylate monomer followed by efficient coupling reaction of PnBA side chains,38 or by the thiol–ene modification of poly(2-vinyloxyethyl methacrylate), both accompanied by an unwanted side-reaction involving crosslinking at higher conversions.39
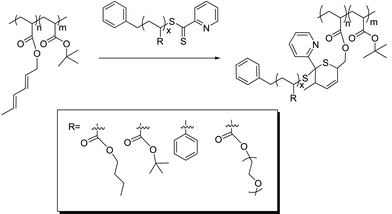 | ||
| Scheme 1 Preparation of different comb copolymers using the ‘click’ RAFT–HDA approach. | ||
In the present work we explore an improved route to graft polymers using the RAFT–HDA approach (Scheme 1). We utilize well-defined polymers prepared via RAFT polymerization and modify them in a post-functionalization step, avoiding crosslinking and any broadening of the molecular weight distribution. Different coupling reactions are described yielding hydrophobic graft copolymers, such as PtBA-g-PS, PtBA-g-PnBA, and PtBA-g-PtBA, amphiphilic graft copolymers, such as PtBA-g-POEGMEA, PAA-g-PS, PAA-g-PnBA, and hydrophilic graft copolymers, i.e. PAA-g-PAA or PAA-g-POEGMEA.
PAA-g-POEGMEA was then employed to surface modify gold nanoparticles (GNPs). Exploiting the negative charges on the PAA backbone, the graft copolymers could be self-assembled on GNP surfaces. Poly(ethylene amine) (PEI) was deposited on GNPs via electrostatic interactions to yield positively charged GNP surfaces. Subsequently, graft copolymers were self-assembled using electrostatic interactions between the PEI (cationic) and PAA (anionic) on the graft copolymers yielding gold nanoparticles surface functionalized with brush polymers.
Experimental part
1. Chemicals
Benzyl pyridin-2-yldithioformate41 and 3-benzylsulfanylthiocarbonylsulfanyl propionic acid (BPATT)49 were synthesized according to established procedures. n-Butyl acrylate (nBA, Aldrich, >99%), tert-butyl acrylate (tBA, Aldrich, 98%), hydroxyethyl acrylate (HEA, Aldrich, 96%), oligo(ethyleneglycol) methylether acrylate (OEGMEA, Aldrich, 99%, Mn = 480 g mol−1), and styrene (S, >99% Aldrich) were passed through a column of basic alumina (Ajax Finechem) and stored at −19 °C. 2,2′-Azobis(isobutyronitrile) (AIBN, Dupont) was recrystallized twice from methanol prior to use and was then stored at 4 °C. 2,2′-Dithiodipyridine (Sigma), p-toluenesulfonyl chloride (Aldrich, >98%), pyridine (AJAX, 99%), sodium hydride (Aldrich, 60% dispersion in mineral oil), anhydrous tetrahydrofurane (THF, Sigma-Aldrich, 99.9%), trans,trans-2,4-hexadien-1-ol (Aldrich, 97%) and trifluoroacetic acid (TFA, Aldrich) were used as received.2. Synthesis
1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) = 4.4–4 (4n1H, COOCH2CH2OH), 3.9–3.6 (4n1H, COOCH2CH2OH), 2.4–2.1 (1n1H + 1n2H, CH of the main chain), 2–1 (2n1H + 11n2H, CH2 of the main chain + COOC(CH3)3). n1 and n2 are the degrees of polymerization (DPn) of HEA and tBA respectively. In acetone-d6, there is an additional peak: δ (ppm) = 4.9–4.8 (1n1H, COOCH2CH2OH).
1H NMR (300 MHz, acetone-d6, 25 °C): δ (ppm) = 7.9–7.7 (2n1H, O3SCCH2CH2CCH3), 7.5–7.3 (2n1H, O3SCCH2CH2CCH3), 4.4–4.1 (4n1H, COOCH2CH2O3S), 2.5–2.1 (4n1H + 1n2H, CH of the main chain + O3SCCH2CH2CCH3), 2–1 (2n1H + 11n2H, CH2 of the main chain + COOC(CH3)3). n1 and n2 are the degrees of polymerization (DPn) of HEATS and tBA respectively.
1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) = 6.4–5.5 (4n1H, diene), 4.6–4.4 (2n1H, OCH2(CH)4CH3), 2.5–2.1 (1n1H + 1n2H, CH of the main chain), 2–1 (5n1H + 11n2H, CH2 of the main chain + OCH2(CH)4CH3 + COOC(CH3)3). n1 and n2 are the degrees of polymerization (DPn) of HEAdiene and tBA respectively.
1H NMR PnBA (300 MHz, CDCl3, 25 °C): δ (ppm) = 8.7–7.4 (4H, pyridyl protons), 7.3–7 (5H, aromatics), 4.2–3.9 (2nH, OCH2), 2.4–2.1 (1nH, CH main chain), 2–1.3 (6nH, CH2), 1–0.8 (3nH, CH3). n is the DPn of nBA.
1H NMR PS (300 MHz, CDCl3, 25 °C): δ (ppm) = 8.7–7.4 (4H, pyridyl protons), 7.4–6.2 (s, 5H + 2nH, aromatics protons), 2.1–1.7 (1nH, CH main chain), 1.7–1.2 (2nH, CH2 of the main chain). n is the DPn of S.
1H NMR PtBA (300 MHz, CDCl3, 25 °C): δ (ppm) = 8.7–7.4 (4H, pyridyl protons), 7.3–7 (5H, aromatics), 2.4–2.1 (1nH, CH main chain), 2–1 (8nH, CH2 of the main chain + CH3). n is the DPn of tBA.
1H NMR POEGMEA (300 MHz, CDCl3, 25 °C): δ (ppm) = 8.7–7.4 (m, 4H, pyridyl protons), 7.3–7 (5H, aromatics), 4.4–3.4 (s, 36nH, OCH2), 4.4–4.2 (3nH, CH3), 2.4–1.3 (3nH, CH and CH2 main chain). n is the DPn of PEGMEA.
3. Layer by layer formation
All glasswares were washed with aqua regia before use.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 40 minutes and redispersed in water (the process was repeated 3 times). The particles were characterized via UV-vis spectroscopy, TEM, DLS and zeta-potential analysis.
000 rpm for 40 minutes and redispersed in water (the process was repeated 3 times). The particles were characterized via UV-vis spectroscopy, TEM, DLS and zeta-potential analysis.
A similar process was used for PAA-g-POEGMEA copolymers
4. Analyses
The chromophoric RAFT end group (λmax = 517 nm) was employed to follow the reaction to the colorless ring that is formed during the HDA cycloaddition. A calibration curve was first established by recording the absorbance of benzyl pyridin-2-yldithioformate solution in chloroform at different concentrations.
A solution of P(tBA-r-HEAdiene) (10 mg, 1 eq, nchain = 1.05 × 10−6 moles) in chloroform was mixed with 10 equivalents of benzyl pyridin-2-yldithioformate. The solution's absorbance was recorded before the addition of TFA (A0), and again after 24 h of reaction (Af). The concentrations co and cf of the RAFT agent were then subtracted from the calibration curve and the following formula was applied to calculate the number of diene moieties per backbone chain:
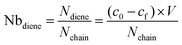
The titration was done three times yielding reproducible results indicating around 6 diene functions per backbone chain (Table S1, see ESI†). The polymer (post-titration) was precipitated in MeOH/water (1/1) and then analyzed by GPC and 1H NMR (Fig. S5 and S6, see ESI†).
Results and discussion
Synthesis of the P(tBA-r-HEA) backbone
A random copolymer P(tBA-r-HEA) (Scheme 2) was synthesized by RAFT polymerization of tBA and HEA at 70 °C in toluene, using BPATT as a chain transfer agent (CTA) and AIBN as an initiator (I) at a tBA/HEA/CTA/I ratio of 100/10/1/0.2. Conversion of both monomers, tBA and HEA, was monitored by 1H NMR (Fig. S1 in ESI†). After an inhibition period of 5 min, the polymerization proceeded according to a linear first-order kinetic relationship indicating a constant radical concentration (Fig. 1A). A linear correlation between the molecular weight and monomer conversion is consistent with the known traits of living radical polymerization (Fig. 1B). Moreover, the molecular weight distributions, obtained using GPC, show mono-modality (Fig. 1C) and the polydispersity index remained narrow during the course of the polymerization. Both monomers were consumed at a similar rate as observed using NMR, implying minimal compositional drift with the final HEA/tert-butyl acrylate molar composition of 10/90 mol% matching the feed ratio at any given time. The copolymerization was stopped at 65% conversion yielding a backbone with a molecular weight of 9300 g mol−1 and a polydispersity of 1.14 (Table 1, entry 1).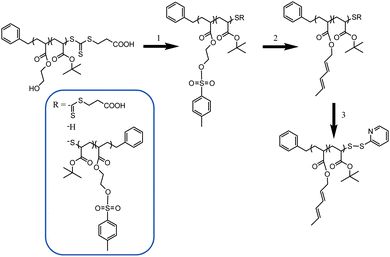 | ||
| Scheme 2 Post-functionalization of the PtBA-r-HEA backbone. (1) Tosylation step. (2) Diene introduction. (3) End chain modification. | ||
| Backbone | Branch | [M]/[R]/[I] | [M]/mol L−1 | Conv. | M n,theo | M n,NMR | M n,GPC | M w/Mn |
|---|---|---|---|---|---|---|---|---|
| a M n is calculated from monomer conversion determined by NMR. b M n is calculated from the purified polymer by comparison of the RAFT agent protons with the protons associated with the monomer. c Determined by GPC (PS standards) in THF or DMac. | ||||||||
| P(tBA-r-HEA) | 500/50/5/1 | 3 | 65% | 9200 | 9300 | 9800 | 1.14 | |
| PnBA 1 | 900/3/1 | 3 | 4.5% | 1900 | 1700 | 1800 | 1.27 | |
| PnBA 2 | 900/3/1 | 3 | 9% | 3600 | 3400 | 3300 | 1.18 | |
| PS | 900/3/1 | 3 | 15% | 4900 | 4900 | 5000 | 1.23 | |
| PtBA | 900/3/1 | 3 | 15.5% | 6200 | 6000 | 6500 | 1.11 | |
| POEGMEA | 300/3/1 | Bulk | 8% | 11500 |
11000 |
10000 |
1.17 |
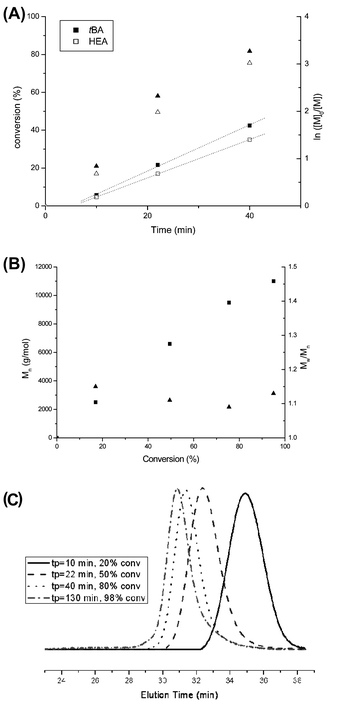 | ||
| Fig. 1 Copolymerization of tBA and HEA. (A) Conversion of tBA (full triangle) and HEA (empty triangle) versus time and a first-order kinetic plot for tBA (full square) and HEA (empty square). (B) Molecular weight and polydispersity index versus overall monomer conversion. (C) Development of molecular weight (shown as a GPC trace) with conversion. | ||
Diene functionalities were introduced to the polymer in two steps (Scheme 2, steps 1 and 2). The hydroxyl groups were modified in the presence of p-toluenesulfonyl chloride followed by a transesterification between trans-2,4-hexadien-1-ol and HEA modified tosylate. Tosylation was performed at room temperature for 15 h with P(tBA-r-HEA) and p-toluenesulfonyl chloride in pyridine. After purification the modified polymer P(tBA-r-HEATS) was characterized by 1H NMR (Fig. 2) and GPC (Fig. 3). 1H NMR analysis indicated new signals at 7.8 and 7.5 ppm, while a shift of the CH2OH signal from 3.8 to 4.2 ppm was also observed. In addition, ATR-FTIR analysis confirmed the disappearance of the OH signal (not shown here). The transesterification was performed in dry THF in the presence of sodium hydride to deprotonate the hydroxyl group. After purification, the polymer P(tBA-r-HEAdiene) was characterized by 1H NMR (Fig. 2) and GPC (Fig. 3). 1H NMR confirmed a successful modification as evidenced by the disappearance of the signals in the aromatic region (from the tosylate, at 7.8 and 7.5 ppm) and by the emergence of new signals attributed to the diene protons at 6.4 to 5.5 ppm.
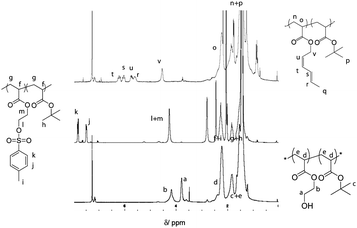 | ||
| Fig. 2 1H NMR spectra of P(tBA-r-HEA), P(tBA-r-HEATS) and P(tBA-r-HEAdiene). | ||
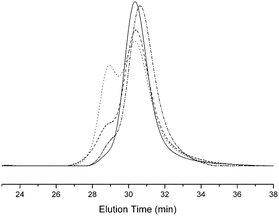 | ||
| Fig. 3 GPC traces of P(tBA-r-HEA) (full line), P(tBA-r-HEATS) (dot) and P(tBA-r-HEAdiene) (dash), P(tBA-r-HEAdiene) before end chain modification (dash), P(tBA-r-HEAdiene) after end chain modification (dashed dot) (THF used as elution solvent). | ||
Fig. 3 shows the superposition of the P(tBA-r-HEA) GPC traces at each step of the modification. The tosylation, forming P(tBA-r-HEATS), resulted in a bimodal molecular weight distribution with a first shoulder at Mn of 20000 g mol−1 and a second signal centered at 10000 g mol−1 with a relative area ratio of 40% and 60%, respectively. The GPC curve of the final polymer P(tBA-r-HEAdiene) displayed a similar molecular weight distribution with a relative area ratio changing from 40/60 to 20/80. Many previous investigations have demonstrated the degradation of the RAFT agent into thiols under basic conditions.51–53 The oxidation of free thiol end-groups by traces of oxygen can result in the formation of disulfide inter-chain coupling, and double the product molecular weights.54,55 To prove our assumption, P(tBA-r-HEAdiene) polymer was reacted in the presence of disulfide pyridine (Scheme 2) to cap the free thiol yielding a pyridyl disulfide, as previously published by CAMD.54 Pyridyl disulfide end-groups give access to a “protected thiol”54,56 able to react with another thiol at a later designated time. GPC traces (Fig. 3) showed that, after modification, the shoulder at low retention disappeared preventing the dimer formation. 1H NMR spectra (Fig. S2 in ESI†) of the purified copolymers confirmed the presence of pyridyl disulfide end-groups by the aromatic signal at 8.4, 7.7 and 7.3 ppm.
Polymerization of branched poly(n-butyl acrylate) (PnBA), poly(tert-butyl acrylate) (PtBA), poly(polyethylene glycol methylether acrylate) (PPEGMEA), and polystyrene (PS)
A prerequisite for the successful formation of comb polymers via “grafting-onto” is the synthesis of branches with pyridin-2-yldithioformate as the reactive end group. For that purpose, different monomers, styrene, nBA, tBA, OEGMEA, were polymerized using AIBN as initiator and benzyl pyridin-2-yldithioformate as the CTA (Table 1, entries 2–6). This choice of monomers was made to investigate the influence of the chemical structure of the monomer on the grafting reaction. All the polymerizations showed controlled behavior and gave narrow polydispersities (the polymers were characterized by 1H NMR and GPC, see ESI†). The chain end CTA functionality of the polymers proved to be quantitative (95 to 100%) by 1H NMR. A wide range of molecular weights were targeted to optimize the yield of the coupling reaction.Synthesis of comb polymer by HDA cycloaddition
As the RAFT–HDA reaction proceeded the reddish thiocarbonylthio functionality turned into a colorless ring structure as depicted in Scheme 1. The decline of the UV-vis absorption band at 515 nm can not only be employed to monitor the completeness and the rate of the RAFT–HDA reaction, but also to determine the amount of diene functionality along the polymer backbone. For this purpose, a known amount of polymer was reacted with excess RAFT agent, benzyl pyridin-2-yldithioformate. The difference between initial absorption and absorption after 24 hours was used to determine the exact amount of diene functionalities (complementing the results obtained from 1H NMR (Fig. S6, see ESI†)). The 1H NMR spectrum indicated a complete disappearance of the signals of the diene protons, confirming 100% conversion of the RAFT–HDA cycloaddition (Fig. S7, see ESI†). Both techniques verify the presence of six diene units per P(tBA-r-HEAdiene) backbone (Table S1, see ESI†).Five different polymer branches were conjugated to the backbone P(tBA-r-HEAdiene) using the RAFT–HDA click reaction (Scheme 1, Table 2). The coupling reactions were performed in chloroform solution at room temperature in the presence of TFA (2 eq per RAFT agent). TFA increases the reactivity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond toward Diels–Alder cycloaddition by protonating the pyridyl Z-group of the RAFT agent.43 The reactions were carried out using both an equimolar amount of diene groups and RAFT functionalities (X = 1, Table 2) and also a 50% excess of branches (X = 1.5).
S double bond toward Diels–Alder cycloaddition by protonating the pyridyl Z-group of the RAFT agent.43 The reactions were carried out using both an equimolar amount of diene groups and RAFT functionalities (X = 1, Table 2) and also a 50% excess of branches (X = 1.5).
| Entry | Branch | M n | X | C yield | M n,theo | M n,GPC | M w/Mn |
|---|---|---|---|---|---|---|---|
| a Molecular weight of the side chains. b Yield of the coupling reaction. c Molecular weight calculated by addition of the molecular weight of the backbone and the side chains coupled. d Determined by GPC (PS standards) in THF or DMAc. | |||||||
| 1 | PnBA 1 | 1800 | 1 | 100% | 19800 |
19000 |
1.21 |
| 2 | PnBA 2 | 3300 | 1 | 83% | 25500 |
24000 |
1.18 |
| 3 | PnBA 2 | 3300 | 1.5 | 100% | 28800 |
27000 |
1.17 |
| 4 | PS | 5000 | 1 | 67% | 29000 |
27000 |
1.16 |
| 5 | PS | 5000 | 1.5 | 83% | 34000 |
35000 |
1.18 |
| 6 | PtBA | 6500 | 1 | 83% | 41500 |
38000 |
1.2 |
| 7 | PtBA | 6500 | 1.5 | 100% | 48000 |
45000 |
1.18 |
| 8 | POEGMEA | 10000 |
1 | 50% | 39000 |
40000 |
1.3 |
| 9 | POEGMEA | 10000 |
1.5 | 67% | 49000 |
48500 |
1.32 |
Two PnBAs (entries 1, 2 and 3) with different molecular weights (PnBA 1, Mn = 1800 g mol−1, PnBA 2, Mn = 3300 g mol−1) were allowed to react with P(tBA-r-HEAdiene). After 24 h, the mixture of P(tBA-r-HEAdiene)/PnBA 1 (1/6) (Table 2, entry 1) was analyzed by 1H NMR and GPC. A superposition of the GPC curves (Fig. 4) of the starting materials and the final product reveals a significant shift in molecular weight. In addition, no traces of the initial macromolecules were detected. The absence of signals attributable to diene protons (δ = 6.3–5.5 ppm) and the emergence of a new 1H NMR signal, corresponding to the alkene protons of the 3,6-dihydro-2H-thiopyran ring (formed by the HDA cycloaddition, δ = 5.9–5.6 ppm) (Fig. 5) proved that the functionalization was quantitative. The GPC gave an apparent molecular weight of 19000 g mol−1, close to the theoretical molecular weight of 19800 g mol−1 (Table 2).
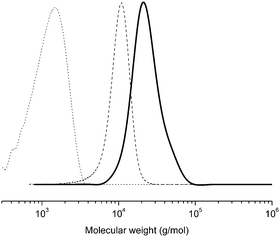 | ||
| Fig. 4 GPC traces (in THF) of the PtBA-g-PnBA 1 (full line), P(tBA-r-HEAdiene) (dash) and PnBA (dot). | ||
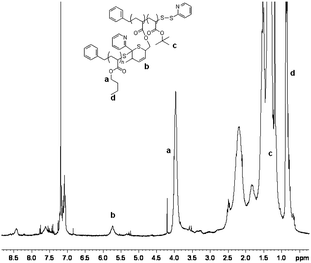 | ||
| Fig. 5 1H NMR spectra of the PtBA-g-PnBA 1 recorded in CDCl3. | ||
When the PnBA 2, which has a higher molecular weight, was reacted with P(tBA-r-HEAdiene) using a molar ratio of 6/1, the yield of the click reaction was incomplete after 48 h at room temperature, as evidenced by a biomodal GPC distribution (see Fig. S8, ESI†) with an apparent molecular weight of 24000 g mol−1 and the other with a molecular weight of 3300 g mol−1, assigned to unreacted branches. In addition, the 1H NMR of the mixture (Fig. 6(2) and Fig. S9, ESI† (full spectrum)) indicated unreacted diene protons (δ = 6.3–5.5 ppm). The grafting density was calculated using the 1H NMR integral of the unreacted diene proton δ = 6.3–5.9 ppm (X, reactant) and the integral of the cyclic alkene between δ = 5.5–5.9 ppm (Y, product). Therefore the coupling yield (Cyield, Table 2) was calculated as:
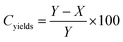
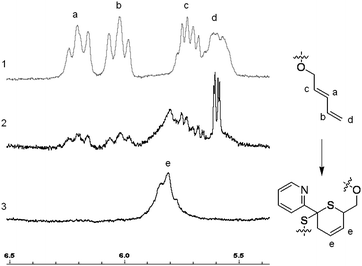 | ||
| Fig. 6 Zoom in δ = 6.5–5.5 ppm of the 1H NMR spectra of P(tBA-r-HEAdiene) (1), PtBA-g-PnBA 2 X = 1 (2) and PtBA-g-PnBA 2 X = 1.5 (3). | ||
This value was also confirmed by the apparent molecular weights found by GPC analysis:
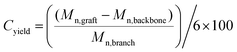
Using both methods, we calculated that an equimolar reaction (i.e., X = 1) between PnBA 2 and the backbone yielded a HDA coupling efficiency of 83%, corresponding to 5 PnBA chains per PtBA backbone. This slight decrease of reaction efficiency, compared to the coupling involving PnBA 1, can be attributed to an increase of the molecular weight, resulting in steric hindrance.
In order to increase the coupling yield, PnBA 2 was introduced in an excess of 1.5 equivalents compared to the number of diene functionalities (entry 3, Table 2). After 24 hours at room temperature, the reaction was stopped and analyzed by both 1H NMR and GPC. The 1H NMR (Fig. 6(3) and Fig. S10, see ESI† for full spectrum) proved that the coupling reaction was quantitative with a complete disappearance of signal at δ = 5.9–5.5 ppm and the appearance of a signal of the HDA cycle CHCH (δ = 5.9–5.7 ppm). Moreover, the GPC (Fig. S11, see ESI†) indicated an increase of hydrodynamic radius with an apparent molecular weight of 27000 g mol−1 (for a theoretical Mn of 28800 g mol−1). The addition of an excess on PnBA 2 branches resulted in an increase of the coupling yield to 100%.
P(tBA-r-HEAdiene) was subsequently reacted with three different polymers in equimolar amounts (X = 1) and in slight excess (X = 1.5). The reaction involving the PtBA (entries 6 and 7, Table 2) gave similar results with a grafting yield of 83% when X = 1 and 100% when X = 1.5. The GPC (Fig. S11, see ESI†) of the two reaction products indicated a clear shift from the backbone to the comb polymer and a second signal of the residual PtBA branches. 1H NMR of the fully functionalized comb polymer (Fig. S12, see ESI†) showed a complete disappearance of signal at δ = 5.9–5.5 ppm and a developing signal of the HDA cycle CHCH (δ = 5.9–5.7 ppm).
When pyridin-2-yldithio terminated PS (entries 4 and 5, Table 2) was employed as a graft, the coupling yield was lower than PnBA or PtBA, with a 67% yield for X = 1 and 83% for X = 1.5 (Fig. S13 and S14, see ESI† for GPC traces and 1H NMR spectra). This decrease may be explained by an increased stiffness of the polystyrene chains compared to poly(acrylate) chains.
Finally, POEGMEA was coupled with the P(tBA-r-HEAdiene) backbone. The relatively high molecular weight compared to the other polymers used can explain a poor click reaction efficiency of 50% when the feed ratio between POEGMEA chains' diene was 1 and 67% when the ratio was 1.5 (Fig. S15 and S16, see ESI† for GPCs and 1H NMR). In addition, the reaction may be sterically hindered by the comb-like structure of POEGMA.
Purification of the grafted product via dialysis (acetone against acetone/water (95/5)) to remove excess branch polymer results in mono-modal molecular weight distributions with PDIs below 1.3 (Fig. 7).
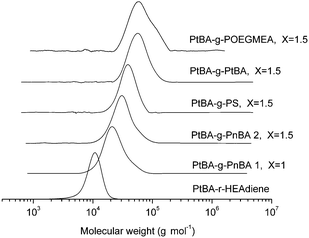 | ||
| Fig. 7 GPC traces of the backbones and the different comb polymers. | ||
The tert-butyl groups could be cleaved by the addition of TFA to yield PAA-g-PAA, PAA-g-PS and PAA-g-POEGMEA. The absence of any signals at 1.4 ppm (tert-butyl group) (Fig. S17, S18 and S19, see ESI†), as recorded by 1H NMR, confirmed the successful deprotection.
Functionalization of gold nanoparticles by layer-by-layer process
Highly uniform gold nanoparticles (GNP) were synthesized via an established citrate reduction method50 (reduction of HAuCl4 by boiling with sodium citrate) to yield spherical gold nanoparticles with a highly uniform diameter close to 20 nm as observed by transmission electron microscopy (TEM) (average diameter size assessed by TEM, 18 nm) and by dynamic light scattering (DLS) (Fig. S20, see ESI†).57 The presence of sodium citrate stabilizes GNPs in water via electrostatic repulsion forces (surface charged around −26 mV). These charges present at the surface were exploited to deposit a cationic polymer (PEI) at the surface of the GNPs. Self-assembly of different positive and negative layers was used to synthesize hybrid organic–inorganic nanoparticles, with different morphology to brush polymer nanoparticles.58–60Commercially available poly(ethylene imine) (PEI), which is positively charged at pH = 6.5 due to the protonation of the amine functionality, was employed to build up a bilayer structure. A solution of gold nanoparticles was added dropwise to an aqueous solution of PEI (pH = 6.5), and allowed to stir during two hours. The rate at which the GNPs are added proved critical to the LbL formation with rapid addition of GNPs resulting in aggregation.58,61 The mixture was then purified by centrifugation and the particles were analyzed by UV-vis spectroscopy, zeta potential (Fig. 8) and XPS (Fig. 9). UV-vis spectroscopy showed a shift of the Plasmon resonance band from 518 nm for the naked gold nanoparticles to 524 nm after the deposition of the first layer. A change of the surface charge was observed by zeta-potential analysis, shifting from −26 mV for naked nanoparticles to +28.5 mV for the PEI coated particles. XPS (Fig. 9) confirmed the presence of N (1s) at the surface of gold nanoparticles as evidenced by a signal at 399.0 eV attributable to a cationic tertiary amine.62 The characteristic signals of gold (Au 4f) at 83.5 and 87.0 eV were also observed.
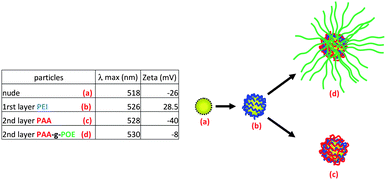 | ||
| Fig. 8 Scheme and table summarizing the LbL methodology, zeta potential and UV-vis analysis (POE = POEGMA). | ||
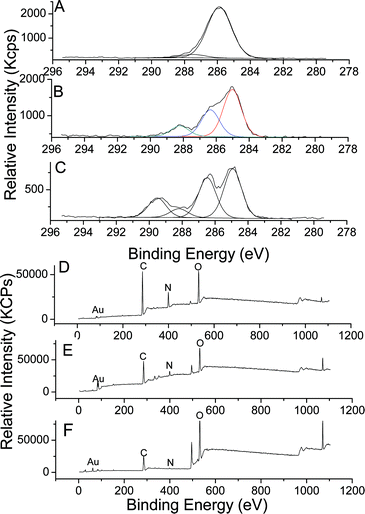 | ||
| Fig. 9 XPS spectra of GNPs modified polymers by the LbL process; C (1s) spectra of: A—GNPs/PEI; B—GNPs/PEI/PAA; C—GNPs/PEI/PAA-g-POEGMA; and wide spectra of D—GNPs/PEI; E—GNPs/PEI/PAA; F—GNPs/PEI/PAA-g-POEGMA. | ||
The positively charged GNPs/PEI were then used for two reactions: (a) to coat a layer of linear PAA homopolymer, or (b) to alternatively coat with a graft polymer such as PAA-g-POEGMEA. The positively charged GNPs were added dropwise to a solution of PAA or PAA-g-POEGMEA at a suitable concentration to prevent aggregation of the GNPs. Polymers and GNPs were mixed for two hours to reach self-limiting behavior. Finally, GNPs were purified by centrifugation and analyzed by UV-visible spectroscopy. Their plasmon resonance bands were shifted from 524 to 528–530 nm after modification. The zeta potential of the PAA modified GNP dropped to −40 mV. In contrast, the zeta potential of the PAA-g-POEGMEA grafted GNPs dropped to −8 mV, i.e. an almost neutral value. This neutral value can be explained by both a decreased acid concentration and a screening effect of the POEGMEA branches (Fig. 8).63
XPS confirmed for both particles the presence of anionic polymers by a decreased intensity of the amine peak (N (1s)) (Fig. S21, see the ESI†). In addition, a slight shift for N (1s) from 399.0 eV to 399.9 eV was observed attributable to the formation of hydrogen bonding between amine groups of PEI and carboxylic acid groups of PAA. In the case of PAA-g-POEGMEA modified GNPs, C (1s) signals at 285.0, 286.0, 286.5, 288.2 and 289.5 eV can be attributed to CH2 and CH of the backbone of PAA, –CH2–N of PEI, –CH2–O– of POEGMA and PAA, and the carbonyl of POEGMEA, respectively.62 In the case of PAA modified GNPs, a signal at 289.5 eV was not observed indicating the absence of POEGMEA.
Stability study in phosphate buffer and in acid solution
Finally, the stability of these GNPs was studied at different pH values, i.e. 6.5 (neutral pH), and at 3.0 (acid). At low pH, GNPs/PEI/PAA changed the colour from red-pink to purple blue indicating the aggregation of GNPs (in agreement with UV-visible experiments, see Fig. S23†). DLS results indicate the formation of aggregates (with a size greater than 100 nm). After 14 hours, all the particles have precipitated in the bottom of the tube. Indeed, at low pH, the electrostatic repulsion provided by PAA disappeared due to the neutralization of carboxylic acid anion. However, in the case of GNPs/PEI/PAA-g-POEGMA, the particles remain stable and no significant shift was observed in their surface plasmon resonance as measured by UV-visible and by DLS analysis after 1 hour of treatment (Fig. S23†). This proves that the stability of GNPs is conferred by steric repulsion due to the presence of POEGMA on the surface of these particles. In addition, the particles were studied in phosphate buffer (pH 7.2, 0.1 M). GNPs/PEI/PAA-g-POEGMEA nanoparticles are stable for 14 hours without precipitation, while the presence of precipitate was observed for GNPs/PEI/PAA.Conclusions
Comb copolymers were successfully synthesized by the “grafting-onto” method via a RAFT–HDA approach. A P(tBA-r-HEA) copolymer was post-functionalized in two step to introduce diene moieties. A titration by UV-visible spectroscopy showed that 6 diene functions were available on the backbone in accord with NMR results. Subsequently, different polymers synthesized by RAFT polymerization using benzyl pyridin-2-yldithioformate, PnBA, PtBA, PS and POEGMA, were used to react with P(tBA-r-HEAdiene). The coupling reaction between the polymers yielded comb copolymers in 24 hours at room temperature. The yield of the click reaction varied from 50% to 100% depending on reaction conditions. Deprotection of the comb polymers yielded a PAA backbone. As a result, hydrophobic (e.g. PtBA-g-PS), amphiphilic (PAA-r-PnBA) and hydrophilic (e.g. PAA-g-PAA) comb polymers were obtained. One of these polymers, PAA-g-POEGMEA, was self-assembled onto PEI modified gold nanoparticles via a layer by layer methodology. UV spectroscopy, zeta potential measurement and XPS proved the presence of comb copolymers on the surface of the gold nanoparticles.Acknowledgements
The authors acknowledge the Australian Research Council (ARC) for Discovery grant funding (DP0769981). In addition we would like to acknowledge significant research fellowship funding to MHS (Future Fellowship), TPD (Federation Fellowship) and CB (Australian postdoctoral Fellowship). We thank Dr M. R. Whittaker for helpful advice.Notes and references
- J. K. Koh, Y. W. Kim, S. H. Ahn, B. R. Min and J. H. Kim, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 183–189 CrossRef CAS.
- J. F. Hester, P. Banerjee and A. M. Mayes, Macromolecules, 1999, 32, 1643–1650 CrossRef CAS.
- C. J. Thompson, L. Tetley and W. P. Cheng, Int. J. Pharm., 2010, 383, 216–227 CrossRef CAS.
- C. Boyer, V. Bulmus, P. Davis Thomas, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- A. Baron, J. Rodriguez-Hernandez, E. Ibarboure, C. Derail and E. Papon, Int. J. Adhes. Adhes., 2009, 29, 1–8 CrossRef CAS.
- H. P. Yap, X. J. Hao, E. Tjipto, C. Gudipati, J. F. Quinn, T. P. Davis, C. Barner-Kowollik, M. H. Stenzel and F. Caruso, Langmuir, 2008, 24, 8981–8990 CrossRef CAS.
- C. Cortez, J. F. Quinn, X. Hao, C. S. Gudipati, M. H. Stenzel, T. P. Davis and F. Caruso, Langmuir, 2010 DOI:10.1021/la100430g.
- L. Ionov, A. Synytska, E. Kaul and S. Diez, Biomacromolecules, 2010, 11, 233–237 CrossRef CAS.
- D. Lanson, F. Ariura, M. Schappacher, R. Borsali and A. Deffieux, Macromolecules, 2009, 42, 3942–3950 CrossRef CAS.
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858–875 CrossRef CAS.
- A. Muehlebach and F. Rime, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3425–3439 CrossRef CAS.
- A. M. Granville, D. Quemener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromol. Symp., 2007, 255, 81–89 CrossRef CAS.
- M. Hernández-Guerrero, E. Min, C. Barner-Kowollik, A. H. E. Müller and M. H. Stenzel, J. Mater. Chem., 2008, 18, 4718–4730 RSC.
- J. F. Quinn, R. P. Chaplin and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2956–2966 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- D. Gromadzki, R. Makuška, M. Netopilík, P. Holler, J. Lokaj, M. Janata and P. Štěpánek, Eur. Polym. J., 2008, 44, 59–71 CrossRef CAS.
- G. David, C. Boyer, J. Tonnar, B. Ameduri, P. Lacroix-Desmazes and B. Boutevin, Chem. Rev., 2006, 106, 3936–3962 CrossRef CAS.
- B. Boutevin, G. David and C. Boyer, Adv. Polym. Sci., 2007, 206, 31–136 CAS.
- M. Hernández-Guerrero, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Eur. Polym. J., 2005, 41, 2264–2277 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, , Angew. Chem., Int. Ed., 2001, 40, 2005–2021.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987–992 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653–5661 CrossRef CAS.
- D. Quemener, C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Chem. Commun., 2006, 5051–5053 RSC.
- K. Takizawa, H. Nulwala, R. J. Thibault, P. Lowenhielm, K. Yoshinaga, K. L. Wooley and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2897–2912 CrossRef CAS.
- W. Van Camp, V. Germonpre, L. Mespouille, P. Dubois, E. J. Goethals and F. E. Du Prez, React. Funct. Polym., 2007, 67, 1168–1180 CrossRef CAS.
- J. Zhang, Y. Zhou, Z. Zhu, Z. Ge and S. Liu, Macromolecules, 2008, 41, 1444–1454 CrossRef CAS.
- R. Riva, S. Schmeits, F. Stoffelbach, C. Jerome, R. Jerome and P. Lecomte, Chem. Commun., 2005, 5334–5336 RSC.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS.
- X. Jiang, E. B. Vogel, M. R. Smith III and G. L. Baker, Macromolecules, 2008, 41, 1937–1944 CrossRef CAS.
- D. Quemener, M. Le Hellaye, C. Bissett, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 155–173 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- D. Valade, C. Boyer, T. P. Davis and V. Bulmus, Aust. J. Chem., 2009, 62, 1344–1350 CrossRef CAS.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- C. Ott, R. Hoogenboom and U. S. Schubert, Chem. Commun., 2008, 3516–3518 RSC.
- A. Bousquet, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1773–1781 CrossRef CAS.
- Z. Jia, J. Liu, T. P. Davis and V. Bulmus, Polymer, 2009, 50, 5928–5932 CrossRef CAS.
- C. Boyer, J. Liu, V. Bulmus and T. P. Davis, Aust. J. Chem., 2009, 62, 830–847 CrossRef CAS.
- A. Alberti, M. Benaglia, M. Guerra, M. Gulea, P. Hapiot, M. Laus, D. Macciantelli, S. Masson, A. Postma and K. Sparnacci, Macromolecules, 2005, 38, 7610–7618 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CAS.
- R. Bastin, H. Albadri, A. C. Gaumont and M. Gulea, Org. Lett., 2006, 8, 1033–1036 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054 RSC.
- L. Nebhani, S. Sinnwell, C. Y. Lin, M. L. Coote, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6053–6071 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120–4126 CrossRef CAS.
- S. Sinnwell, M. Lammens, M. H. Stenzel, F. E. D. Prez and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2207–2213 CrossRef CAS.
- L. Nebhani, S. Sinnwell, A. J. Inglis, M. H. Stenzel, C. Barner-Kowollik and L. Barner, Macromol. Rapid Commun., 2008, 29, 1431–1437 CrossRef CAS.
- M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4498–4512 CrossRef CAS.
- G. Frens, Nature (London), Phys. Sci., 1973, 241, 20–22.
- A. B. Lowe, C. L. McCormick, Handbook of RAFT Polymerization, Wiley Interscience, 2008, pp. 235–284 Search PubMed.
- X.-P. Qiu and F. M. Winnik, Macromol. Rapid Commun., 2006, 27, 1648–1653 CrossRef CAS.
- X.-P. Qiu and F. M. Winnik, Macromolecules, 2007, 40, 872–878 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef.
- A. W. York, C. W. Scales, F. Huang and C. L. McCormick, Biomacromolecules, 2007, 8, 2337–2341 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- C. Boyer, M. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917–6926 CrossRef CAS.
- G. Schneider and G. Decher, Nano Lett., 2004, 4, 1833–1839 CrossRef CAS.
- G. F. Schneider and G. Decher, Nano Lett., 2008, 8, 3598–3604 CrossRef CAS.
- C. Boyer, M. R. Whittaker, K. Chuah, J. Liu and T. P. Davis, Langmuir, 2010, 26, 2721–2730 CrossRef CAS.
- G. Schneider and G. Decher, Langmuir, 2008, 24, 1778–1789 CrossRef CAS.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers the Scienta ESCA300 Database, Wiley, New York, 1992 Search PubMed.
- C. Boyer, P. Priyanto, T. P. Davis, D. Pissuwan, V. Bulmus, M. Kavallaris, W. Y. Teoh, R. Amal, M. Carroll, R. Woodward and T. S. Pierre, J. Mater. Chem., 2010, 20, 255–265 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of oligomers and copolymers, GPC traces of oligomers and copolymers; TEM and DLS of naked gold nanoparticles; XPS spectra; ATR-IRFT spectra before and after deprotection of t-BuA; UV-visible spectra. See DOI: 10.1039/c0py00075b |
| This journal is © The Royal Society of Chemistry 2010 |