Adding stimuli-responsive extensions to antifouling hairy particles†
Alexandra
Muñoz-Bonilla
ab,
Alex M.
van Herk
a and
Johan P. A.
Heuts
*a
aLaboratory of Polymer Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: j.p.a.heuts@tue.nl
bInstituto de Ciencia y Tecnología de Polímeros (CSIC), C/Juan de la Cierva 3, 28006, Madrid, Spain
First published on 7th April 2010
Abstract
The use of living block copolymers as stabilisers in emulsion polymerisation allowed preparation of multilayer functional hairy particles via surface-initiated ATRP. Polymer films prepared from the obtained particles present antifouling properties along with stimuli-responsive behaviour.
Controlling surface properties, in terms of morphology and chemical functionality, by attaching polymer chains to the surface has generated a significant interest in many fields.1–6 Although several techniques have been used for grafting polymers to surfaces, such as “grafting onto” techniques, surface-initiated “grafting from” polymerisation (SIP) is probably one of the most powerful and versatile methodologies. In particular, atom transfer radical polymerization (ATRP) has been successfully used for a number of surfaces, owing to good control of molecular weight, narrow polydispersities and tolerance to various reaction conditions and monomers. In recent years, the development of aqueous ATRP has provided the possibility to grow well-defined high-density hydrophilic polymer brushes at room temperature from both flat surfaces and particles. More specifically, the surface-initiated ATRP of water-soluble monomers from functionalised particles such as polymer latex, inorganic or metallic particles has been widely used to synthesize core–shell or hairy particles with special properties and applications.2,7–10
Recently we demonstrated the functionalisation of a latex film by the migration of block copolymer surfactants used in the emulsion polymerisation to the film surface.11 In this communication we now report a new approach to prepare multi-layer polymeric particles by surface-initiated ATRP, wherein the aqueous ATRP reaction is initiated from block copolymer stabilizers, which upon film formation lead to modified surface properties. Previously we published the synthesis of hairy particles with core–shell structures by emulsion polymerisation using as stabilizers brush-type amphiphilic block copolymers of polystyrene-b-poly[poly(ethylene glycol) methyl ether methacrylate] (PS-b-P(PEGMA1100)).11 These PS-b-P(PEGMA1100) block copolymer surfactants were prepared via ATRP from a PS–Br macroinitiator, resulting in a halogen functionality at the end of the hydrophilic block of PEGMA1100. These block copolymers were used as stabilizers in the emulsion polymerization of styrene, and the bromide-functionality remains on the outside of the hairy particles, thus providing an initiating group to further polymerise by ATRP a second polymeric “shell” (see Scheme 1).
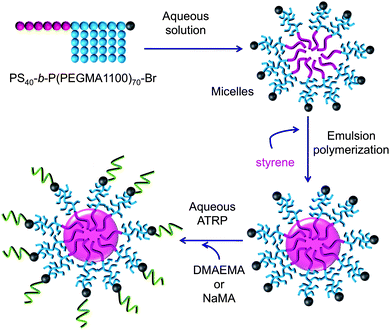 | ||
| Scheme 1 Preparation of multi-layer hairy particles by emulsion polymerisation and surface-initiated ATRP. | ||
The use of block copolymers as stabilizers and macroinitiators in emulsion or miniemulsion polymerisation under ARGET-ATRP conditions in itself is not new and has been reported previously.12 In those cases, however, the bromide end functionalities were located at the end of the hydrophobic block (i.e., on the inside of the particles), and consequently the controlled radical polymerisation was carried out to form the core of the particles. The here reported approach allows the chain extension reactions from well-defined hairy particles, without any further incorporation of ATRP initiator groups or chemical modification of the particle surface.
In the present study surface-initiated ATRP of two water-soluble monomers, sodium methacrylate (NaMA) and 2-(dimethylamino) ethyl methacrylate (DMAEMA), was carried out from polystyrene hairy particles stabilised with PS40-b-P(PEGMA1100)70 block copolymer. We previously reported that the synthesised hairy particles containing brushes of PEGMA1100 can be used for the preparation of films with antifouling properties because the PEGMA segments can successfully suppress non-specific protein adsorption.11 The addition of the second “shell” based on stimuli-responsive polymers, such as PMAA or PDMAEMA, could provide particles, and potentially latex films, with unique properties. In this study we carried out the chain extension of the PS-b-P(PEGMA1100) blocks by polymerising 1 g of monomer in 2 g of a 16.6 wt% seed latex (block copolymer content 3.2 wt% with respect to overall polymer particle content). Detailed information on the experimental conditions can be found in Table 1 and in the ESI†. The surface-initiated ATRP of DMAEMA was carried out in water using a CuBr/bpy catalyst system at 45 °C at pH = 7 from the polystyrene seed latex particles stabilised by PS40-b-P(PEGMA1100)70. The chain extension reaction with sodium methacrylate was performed at room temperature and pH = 9 using CuBr/bpy. A control experiment, carried out under the same experimental conditions but without the ATRP catalyst did not lead to any chain extension (see ESI†), proving that the ATRP system is indeed responsible for the polymerization.
| Monomer | Time/h | Conversion (%) | D DLS/nm | PDIDLS | D TEM/nm | PDITEM |
|---|---|---|---|---|---|---|
| a Polystyrene hairy particles with a diameter determined by DLS, DDLS = 367 nm and polydispersity, PDI = 0.08 and determined by TEM, DTEM = 286 nm, PDI= 1.1. b Experimental conditions: 2 g of 16.6 wt% suspension of seed particles, 1 g of monomer and [monomer] : [2,2′-bipyridine] : [Cu(I)Br] = 100 : 2 : 1. | ||||||
| DMAEMA | 7 | 60 | 523 | 0.04 | 361 | 1.21 |
| NaMA | 5 | 4 | 480 | 0.09 | 306 | 1.04 |
The experiments were carried out at high monomer concentrations to reduce chain transfer reactions to the catalyst system or solvent and also to minimize the monomer depletion effect.13,14 The polymerization of NaMA was stopped at a monomer conversion of 4%, whereas the ATRP reaction of DMAEMA reached a conversion of 60%. The kinetic investigation of the SI-ATRP of DMAEMA (see Fig. S1 in the ESI†) shows a linear first-order kinetic plot throughout the reaction, suggesting a constant number of growing radicals. Furthermore we tested the possible presence of free polymer in the aqueous phase by ultracentrifugation of the latex and analyzing the supernatant; no detectable amount of polymer by NMR in D2O or DMSO-d6 was found.
The “shell”-growth of the PDMAEMA and PNaMA from the hairy particles was studied by FTIR analysis (see Fig. S2 and S3 in the ESI†). After the chain extension of DMAEMA or NaMA, new absorption bands corresponding to the additional functionalities are observed, such as tertiary amine groups and carboxylate groups of PDMAEMA and PNaMA, respectively. The observed increase in the particle size as measured by DLS and TEM (Table 1) for both PDMAEMA and PNaMA confirms the growth of the hairy layer from the pre-existing particles. Although the DLS results in Table 1 suggest a narrow particle size distribution for both chain extension experiments, the TEM images (Fig. 1) show a non-uniform growth of the polymer layer of PDMAEMA. This result suggests a rate of propagation higher than the initiation rate.
 | ||
| Fig. 1 TEM images of (a) seed hairy particles having PS40-b-P(PEGMA1100)70 as surfactant, (b) particles after extension with PNaMA and (c) particles after extension with PDMAEMA. | ||
In contrast, the chain extension of NaMA did not markedly increase the particle size as determined by TEM, which is probably caused by the lower conversion of NaMA. However as shown in Table 1, the increase in the hydrodynamic diameter determined by DLS is appreciable and can be explained by the fact that very high molecular weight extensions are formed.
The average molecular weight of the PDMAEMA and PNaMA from the hairy particles can be estimated assuming that all the brush-type amphiphilic copolymers of PS40-b-P(PEGMA1100)70 at the surface can initiate the ATRP polymerisation. From the used recipes (see ESI†): [NaMA] : [block copolymer] = (68 × 103) : 1 and [DMAEMA] : [block copolymer] = (47 × 103) : 1, and monomer conversions of 4% and 60%, respectively, the corresponding average molecular weights were 3 × 105 and 4 × 106 g mol−1, respectively, which seem to be significantly high.
Previously, Brooks and co-workers13–15 have demonstrated that the presence of sulfate groups on the surface of the seed latex particles can significantly improve the surface-initiated polymerisation. They explained this behavior through an electrostatic model for the enrichment of the ATRP catalyst system near the surface. The Cu(I)/ligand and Cu(II)/ligand complexes could be enriched on the surface via electrostatic interactions with the anionic sulfate groups (derived from the ammonium persulfate initiator used in the emulsion polymerization), facilitating the initiation and the chain extension polymerisation, and hence the graft density. They also reported that the molecular weights of the grown polymers were much higher than those for non-sulfated surface, having a molar mass in the range of millions of Da. However, no explanation was found for the effect of surface charges on molecular weight.
Since we cannot think of any other obvious explanation, we here speculate that hydroxyl surface groups (originating in the seed latex particle preparation)16–18 could be responsible for the high molecular weights. It has been previously reported that these groups on latex particles can cause the reduction of metal ions, forming metal particles.16–18 During the chain extension reaction from the seed hairy particles, a similar effect might occur in the ATRP reaction, reducing the Cu(II) or the Cu(I). Thus, Cu(0) could be formed and also operate as reducing agent. Additionally, the possible formation of Cu(0) along with the improved disproportionation of Cu(I)Br in aqueous media could give rise to a single-electron transfer living radical polymerization (SET-LRP),19 which would explain the obtained high molecular weights.
Furthermore the stimuli-responsive behaviour of the synthesised chain-extended hairy particles was studied by zeta potential experiments as a function of pH (Fig. 2). The hairy polystyrene seed particles stabilised by PS40-b-P(PEGMA1100)70 do not exhibit an isoelectric point; negative zeta potentials were observed over the entire investigated pH range, due to the anionic sulfate groups derived from the ammonium persulfate initiator. In contrast, the introduction of the PDMAEMA blocks provides pH-responsive properties to the latex particles. The zeta potential changes from −20 mV to +52 mV with decreasing pH from basic to acidic, presenting an isoelectric point at 8.5.
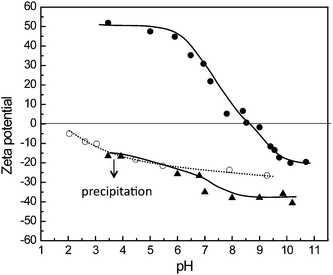 | ||
| Fig. 2 pH dependence of the zeta potential for (○) hairy seed particles stabilized by PS40-b-P(PEGMA1100)70, (▲) particles with PNaMA extensions and (●) particles with PDMAEMA extensions. | ||
The latex particles containing a poly(sodium methacrylate) “shell” present a lower zeta potential, −40 mV, than the seed polystyrene latex. The zeta potential rises as the pH decreases and below pH = 3 the particles precipitate, probably due to the protonated poly(methacrylic acid) which can form hydrogen-bonded hydrophobic complexes with the PEGMA chains20 (see Fig. 3). This characteristic can be very useful for the application of these materials, with the possibility to separate and recover the latex particles at low pH. After increasing the pH of the solution, precipitated particles redisperse well by simple mixing and sonication, recovering the hydrodynamic diameters as before precipitation, DDLS = 456 nm and PDI = 0.09.
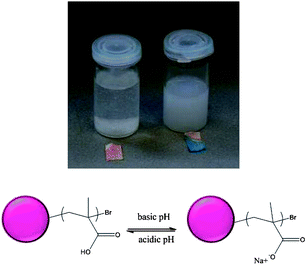 | ||
| Fig. 3 pH-responsive behaviour of the particles containing PNaMA chains at the surface. Vial depicted on the left corresponds to precipitated particles at acidic pH. Vial on the right contains stable particles at basic pH. | ||
After having demonstrated the growth of the second polymer “shell” of PDMAEMA or PNaMA on the particles, polymer films were prepared in order to study their interactions with proteins as compared with films prepared from the initial hairy seed latex. Firstly, from contact angle measurements with distilled water it was observed that the presence of the third block significantly decreases the static contact angle from 77.7° to 58.7° in the case of PDMAEMA and to 73.7° in the case of PNaMA. Table S1 of the ESI† provides the numerical data of the static, advancing and receding contact angles. At pH = 7 the carboxylic acid groups in the PMAA are ionized and the tertiary amines in the PDMAEMA are partially charged. Thus, both polymer surfaces become more hydrophilic than films containing just the diblock copolymer PS40-b-P(PEGMA1100)70.
Concerning the films' antifouling properties, the additional block of PDMAEMA or PNaMA changes the capability of the hairy particles or the coalesced films to prevent non-specific adsorption of proteins, a property associated with the P(PEGMA) chains. Non-specific adsorption of proteins (i.e. bovine serum albumin, BSA-FITC) on functionalised polystyrene films was studied by steady-state fluorescence spectroscopy. The relative fluorescence intensity was taken as a measure for the amount of adsorbed protein. Fig. 4 shows the fluorescence emission spectra of the polymer films. For comparison, the spectrum of a control experiment in which the film was prepared from polystyrene latex with a conventional surfactant, 4-dodecylbenzenesulfonic acid (SDBS),11 was also included.
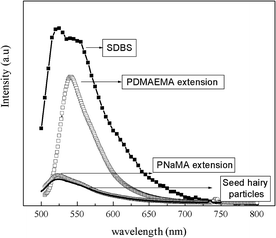 | ||
| Fig. 4 Emission spectra at 490 nm excitation wavelength of polystyrene films formed from latexes with: (–■–) SDBS,11 (□) PS40-b-P(PEGMA1100)70-b-PDMAEMA, (○) PS40-b-P(PEGMA1100)70-b-PNaMA, and (—) PS40-b-P(PEGMA1100)70.11 | ||
The interaction studies were carried out at pH = 7.4, above the isoelectric point of BSA (pH = 5.4);21 as a result, the overall charge of the protein was negative. As we published previously11 and as shown in Fig. 4, the intensity of the maximum emission is much higher in films containing SDBS, meaning that the presence of P(PEGMA) on the film surface suppresses the adsorption of BSA. Films prepared after the chain extension with NaMA show also a small maximum peak, almost with similar intensity as that observed for films prepared directly from the hairy seed particles. However, in films containing PDMAEMA the intensity of the peak is much higher (but still lower than films containing the conventional surfactant). All these results can be explained by the presence of charges on the surface along with hydrophobic interactions. In the case of the film with SDBS, two peak maxima at 521 and 540 nm are observed in the spectrum, probably caused by hydrophobic and electrostatic interactions, respectively. It was reported that the hydrophobic interactions between the alkyl chain of the surfactants and the hydrophobic packets of globular BSA are reinforced by electrostatic attraction between the anionic moieties of the surfactant and the cationic amino acids.22–24 Consequently, even though the total charge of the BSA is negative at pH = 7.4, the cationic amino acid residues adjacent to the hydrophobic patches of the BSA can interact with the anionic groups of the SDBS surfactant. The similar suppression of protein adhesion of the PNaMA–P(PEGMA) containing surfaces as compared with the original P(PEGMA)-functionalised is due to a reduction of the hydrophobic interactions caused by the presence of P(PEGMA) along with electrostatic repulsions between PNaMA and the overall negatively charged BSA.
The relatively high protein adsorption on films containing PDMAEMA–P(PEGMA) on the surface, at pH = 7.4, can be explained by the electrostatic interactions between the negatively charged BSA and the protonated amine groups of the PDMAEMA. In this case the hydrophobic interactions are less important and the electrostatic interactions become the main binding force. The electrostatic interactions are evidenced by an emission peak at 540 nm.
In summary, the use of living brush-type amphiphilic block copolymers stabilising latex particles, as macroinitiator in the surface-initiated ATRP was described. This new approach allowed preparation of functionalised multi-layer particles in a simple way, without any chemical modification reaction. The extensions by stimuli-responsive polymers, PDMAEMA and PNaMA, add new properties to the original hairy particles based on PEGMA, such as the possibility to perform further chemical reactions from the new functional groups (carboxylic acid and amine residues) or tuning the surface properties of dried latex films.
Acknowledgements
The authors acknowledge the financial support from the Foundation Emulsion Polymerization (SEP). A. Muñoz-Bonilla gratefully thanks MICINN-CSIC for her postdoctoral grant.Notes and references
- S. T. Milner, Science, 1991, 251, 905 CrossRef CAS.
- J. N. Kizhakkedathu, J. Janzen, Y. Le, R. K. Kainthan and D. E. Brooks, Langmuir, 2009, 25, 3794 CrossRef CAS.
- A. W. Bosman, R. Vestberg, A. Heumann, J. M. J. Frechet and C. J. Hawker, J. Am. Chem. Soc., 2003, 125, 715 CrossRef CAS.
- M. Q. Zhu, L. Q. Wang, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2004, 126, 2656 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062 CrossRef CAS.
- Q. L. Fan, K. G. Neoh, E. T. Kang, B. Shuter and S. C. Wang, Biomaterials, 2007, 28, 5426 CrossRef CAS.
- M. M. Guerrini, B. Charleux and J. P. Vairon, Macromol. Rapid Commun., 2000, 21, 669 CrossRef CAS.
- T. Taniguchi, M. Kasuya, Y. Kunisada, T. Miyai, H. Nagasawa and T. Nakahira, Colloids Surf., B, 2009, 71, 194 CrossRef CAS.
- V. Mittal, N. B. Matsko, A. Butte and M. Morbidelli, Eur. Polym. J., 2007, 43, 4868 CrossRef CAS.
- D. Li, J. R. Dunlap and B. Zhao, Langmuir, 2008, 24, 5911 CrossRef CAS.
- A. Muñoz-Bonilla, A. M. van Herk and J. P. A. Heuts, Macromolecules, 2010, 43, 2721 CrossRef CAS.
- F. Stoffelbach, B. Belardi, J. M. R. C. A. Santos, L. Tessier, K. Matyjaszewski and B. Charleux, Macromolecules, 2007, 40, 8813 CrossRef CAS.
- K. N. Jayachandran, A. Takacs-Cox and D. E. Brooks, Macromolecules, 2002, 35, 4247 CrossRef CAS.
- J. N. Nizhakkedathu and D. E. Brooks, Macromolecules, 2003, 36, 591 CrossRef.
- Y. Zou, K. N. Jayachandran and D. E. Brooks, Macromolecules, 2009, 42, 3258 CrossRef CAS.
- J. L. Ou, C. P. Chang, Y. Sung, K. L. Ou, C. C. Tseng, H. W. Ling and M. D. Ger, Colloids Surf., A, 2007, 305, 36 CrossRef CAS.
- C. C. Tseng, C. P. Chang, Y. Sung, J. L. Ou and M. D. Ger, Colloids Surf., A, 2009, 333, 138 CrossRef CAS.
- C. P. Chang, C. C. Tseng, J. L. Ou, W. H. Hwu and M. D. Ger, Colloid Polym. Sci., 2010, 288, 395 CrossRef CAS.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940 CrossRef CAS.
- A. M. Mathur, B. Drescher, A. B. Scranton and J. Klier, Nature, 1998, 392, 367 CrossRef CAS.
- T. Peters, in Advances in Protein Chemistry, ed. C. B. Anfinsen, J. T. Edsall and F. M. Richards, Academic Press, New York, 1985, vol. 37, p. 161 Search PubMed.
- J. M. Park, B. B. Muhoberac, P. L. Dubin and J. Xia, Macromolecules, 1992, 25, 290 CrossRef CAS.
- D. Kelley and D. J. McClements, Food Hydrocolloids, 2003, 17, 73 CrossRef CAS.
- T. Chakraborty, I. Chakraborty, S. P. Moulik and S. Ghosh, Langmuir, 2009, 25, 3062 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Text giving the experimental part including the preparation and purification of all the materials and measurement techniques. Figures showing the linear dependence of ln([M]0/[M]t) against time for the SI-ATRP of DMAEMA, FTIR result of the modified polystyrene latex. Table summarizing the contact angle data. See DOI: 10.1039/c0py00054j |
| This journal is © The Royal Society of Chemistry 2010 |