Drug-grafted seven-arm amphiphilic star poly(ε-caprolactone-co-carbonate)-b-poly(ethylene glycol)s based on a cyclodextrin core: synthesis and self-assembly behavior in water
Peng-Fei
Gou
,
Wei-Pu
Zhu
* and
Zhi-Quan
Shen
MOE Key Laboratory of Macromolecule Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310027, China. E-mail: carrols@163.com; Fax: +86 571 87953727
First published on 8th June 2010
Abstract
Novel well-defined drug-grafted seven-arm amphiphilic star poly(ε-caprolactone-co-carbonate)-b-poly(ethylene glycol)s based on a β-CD core [CDS-P(CL-co-DTC)-D-b-PEG] have been synthesized by the combination of controlled ring-opening polymerization (CROP), esterification coupling reactions and “click” reactions. First, 5,5-dibromomethyl-trimethylene carbonate (DBTC) bearing two bromide pendent groups was synthesized and used to copolymerize with CL to prepare seven-arm star random copolymers [CDS-P(CL-co-DBTC)] in the presence of per-2,3-acetyl-β-cyclodextrin and Sn(Oct)2. Second, esterification coupling reaction between CDS-P(CL-co-DBTC) and carboxyl-terminated mPEG led to amphiphilic seven-arm star copolymers [CDS-P(CL-co-DBTC)-b-PEG]. Subsequently, the bromide pendent groups on CDS-P(CL-co-DBTC)-b-PEG had been converted into azide groups by treating with NaN3. Finally, alkyne functionalized ibuprofen had been grafted onto the hydrophobic block of the star copolymers by copper(I)-catalyzed “click” reaction. 1H NMR, FT-IR and SEC analyses confirmed the well-defined drug grafted star architecture. These copolymers could self-assemble into multi-morphological aggregates in water, which were characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM).
Introduction
The synthesis of amphiphilic biodegradable and biocompatible polymers has attracted considerable interest because they can self-assemble into micelles which have great potential applications in drug delivery systems.1 Poly(ε-caprolactone) (PCL) is one of the FDA approved biodegradable and biocompatible aliphatic polyesters which can be used as synthetic biomaterial or controlled drug release matrix due to its good drug permeability, biocompatibility and nontoxicity.2 Poly(ethylene glycol) (PEG) is also a FDA approved hydrophilic, non-charged, and nonimmunogenic polymer that exhibits excellent biocompatibility.3 It is reported that PEG shelled polymeric micelles can prevent the adsorption of proteins and enzymes, lower the phagocytosis into macrophage cells and thus increase their residence time in systemic circulation.4,5Due to these advantages, polymeric micelles self-assembled from PCL and PEG based copolymers have been extensively studied as nano-drug carriers in delivery system during the past few years.6–10 However, because of the lack of functional groups on the PCL chains, biologically active moieties (such as biotin, folic acid, or drugs) cannot be easily incorporated onto the micelles in tunable amounts, while this may provide new insights for fabricating multifunctional drug carriers for special delivery system. In order to achieve this goal, ε-caprolactone (CL) bearing carboxyl,11 hydroxyl,12 bromide13 or chloride14,15 groups has been designed and used to copolymerize with other monomers to prepare biodegradable copolymers with functional pendent groups. Nevertheless, these functionalized CL monomers are difficult to synthesize and always exhibit low activities in the polymerization process. Polycarbonates are other kinds of biocompatible, biodegradable and nontoxic polymers which are usually synthesized by the ring-opening polymerization of cyclic carbonates.16 Compared with functionalized CL monomers, cyclic carbonate monomers with pendent functional groups are much easier to synthesize and polymerize. Therefore, employing the ring-opening copolymerization of functionalized cyclic carbonates with other cyclic monomers should be a promising way to synthesize biocompatible and biodegradable polymers with functional pendent groups. Ray and Grinstaff17 reported the synthesis of cyclic carbonate with benzyloxy pendent group and its copolymerization with lactide. This benzylated copolymer could be hydrogenolyzed to recover the hydroxyl groups on the main-chain for reacting with several active moieties. Lee and coworkers18 described the synthesis of biodegradable and functional poly(ester carbonate)s by the copolymerization of trans-4-hydroxy-L-proline with the cyclic carbonate bearing a carboxylic pendent group. Jing's group prepared a series of poly(ester carbonate)s with carboxyl,19 hydroxyl,20 allyl ester,21 amino,22 alkyne23 and cinnamate24 functional pendent groups by copolymerization of lactide and corresponding cyclic carbonates for developing unique biocompatible and biodegradable polymers.
Meanwhile, It has been proven that the architecture of amphiphilic copolymer may influence the morphology, stability and dimension of the self-assembled micelles,25–27 which are important parameters to determine the drug loading capacity, release behavior and circulation in vivo.28,29 Several recent publications indicate that micelles self-assembled from amphiphilic biocompatible copolymers with hyperbranched structure are of some advantages to drug delivery system. For example, Dong et al. described the synthesis of linear-dendron-like PCL-b-PEG copolymers composed of linear PEG blocks and dendron-like PCL blocks.30 These new types of copolymers provided an approach to fabricate worm-like DOX-loaded nanoparticles which showed a higher drug loading efficiency and a longer drug-release time than their linear counterparts.31 Wang and Roovers et al. also demonstrated that micelles self-assembled from PCL and PEG based multi-arm star block copolymers exhibited high drug loading capacity as well as facile release kinetics.32,33
Cyclodextrins (CDs) are naturally occurring commercial available macrocyclic oligosaccharides containing six, seven, or eight α-1,4-D-glucopyranosyl rings, and named α-,β-, or γ-CD, respectively. CDs are biocompatible, biodegradable, and do not elicit immune responses in animal and human bodies, which can be used to form inclusion complexes with many hydrophobic compounds such as toxins and drugs.34 Considering CDs' well-defined cyclic structure with large fixed numbers of hydroxyl groups, several research groups have focused on the synthesis of CD-centered star polymers in relation to potential biomedical materials.35 Li and coworkers described the synthesis of cationic star polymers based on α-CD as nonviral gene delivery vectors.36 Xiao and coworkers reported the synthesis of water-soluble cationic star polymers with a functionalized β-CD core for the sake of biomedical applications.37
In addition, because of the burst drug release behaviors and insufficient intracellular delivery caused by low drug loading capacity, conjugation of drug molecules onto the polymer chains by covalent bonds should be a better way to overcome these shortcomings.38 To date, several drug-conjugated polymeric micelles have been developed, showing that they may not only increase the drug loading capacity but also enhance the controllability of the drug delivery system.39–46
In this paper, well-defined drug-grafted seven-arm amphiphilic star poly(ε-caprolactone-co-carbonate)-b-poly(ethylene glycol)s based on a β-CD core [CDS-P(CL-co-DTC)-D-b-PEG] have been synthesized by the controlled ring-opening copolymerization of CL and 5,5-dibromomethyl-trimethylene carbonate (DBTC), esterification coupling reaction and “click” reaction. Their self-assembly behavior in water was investigated by dynamic light scattering (DLS) and transmission electron microscopy (TEM).
Experimental section
Materials
Per-2,3-acetyl-β-CD (CD-core) was synthesized according to the procedures reported previously.35 Toluene, triethylamine (TEA) and N,N-dimethylformamide (DMF) were dried over CaH2 and distilled just before use. ε-Caprolactone (99%; Acros) and 2-propyn-1-ol (99%; Aladdin, China) were distilled under reduced pressure prior to use. THF was distilled from the ketyl prepared from sodium and benzophenone. Methoxy poly(ethylene glycol)s (Fluka; Mn = 1 K, 2 K) denoted as mPEG23, mPEG45 were dried by azeotropic distillation in the presence of dry toluene. N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine (PMDETA; 99%; Aldrich), dicyclohexylcarbodiimide (DCC; 99%; Fluka), dibromoneopentyl glycol (99%; Aladdin; China) 4-dimethylaminopyridine (DMAP; 99%; Fluka), and ethyl chloroformate (99%; Aladdin; China) were used as received. Succinic anhydride (99%; Fluka) was crystallized from acetic anhydride. CuBr (99%; Acros) was purified by stirring overnight in acetic acid. After filtration, it was washed with ethanol and then dried in vacuum. Ibuprofen was obtained from Juhua Group Corporation (China). Stannous octoate [Sn(Oct)2; 97%] and other reagents were purchased from Sinopharm Chemical Reagent Co., Ltd. (China) and used as received.Synthesis of 5,5-dibromomethyl-trimethylene carbonate (Scheme 1; DBTC)
Dibromoneopentyl glycol (7.0 g, 30 mmol) and ethyl chloroformate (5.7 mL, 60 mmol) were dissolved in 50 mL anhydrous THF. The mixture was stirred at 0 °C for 30 min, and then TEA (8.3 mL, 60 mmol) was added dropwise within 20 min. Finally, the solution was stirred overnight at room temperature. After filtering the precipitate, the filtrate was evaporated under reduced pressure; and the concentrated oil-like liquid was poured into excessive cold ethyl ether to precipitate the crude product, which was recrystallized from THF/ethyl ether. Yield: 4.35 g (50.3%). 1H NMR (500 MHz, CDCl3), δ, ppm: 4.41 (s, 4H, –O–CH2–C–), 3.59 (s, 4H, –C–CH2–Br). 13C NMR in CDCl3, δ, ppm: 32.6 (2C, –CH2Br), 36.8 (1C, (–OCH2)2–C–(CH2Br)2), 72.0 (2C, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O–C–), 147.5 (1C, CO). MS for C6H8Br2O3: m/z = 311 (M + Na)+.
C–O–C–), 147.5 (1C, CO). MS for C6H8Br2O3: m/z = 311 (M + Na)+.
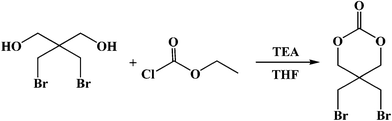 | ||
| Scheme 1 Synthesis of 5,5-dibromomethyl-trimethylene carbonate (DBTC). | ||
Synthesis of cyclodextrin-centered seven-arm star poly(ε-caprolactone-co-5,5-dibromomethyl-trimethylene carbonate)s [CDS-P(CL-co-DBTC)] by controlled ring-opening copolymerization (Scheme 2)
A typical polymerization procedure was as follows: 86 mg per-2,3-acetyl-β-CD (CD-core; 0.35 mmol of OH group) was transferred into a flame-dried ampoule. Then, 1.08 g (9.45 mmol) of CL, 300 mg (1.05 mmol) of DBTC and a magnetic stirring bar were added to the ampoule. After the CD-core and DBTC were dissolved in CL completely, the ampoule was then connected to a schlenkline, where an exhausting–refilling process was repeated three times. 5 mg Sn(Oct)2 in 1 mL dry toluene was added to the mixture, and the exhausting-refilling process was carried out again to remove the toluene. The ampoule was put into an oil bath at 120 °C for 12 h bulk polymerization. The crude polymer was dissolved in THF and poured into excess methanol to precipitate the product, which was dried in vacuum to constant weight. Yield: 1.39 g (94.5%).![Synthesis of cyclodextrin-centered drug-grafted seven-arm amphiphilic star copolymers [CDS-P(CL-co-DTC)-D-b-PEG].](/image/article/2010/PY/c0py00043d/c0py00043d-s2.gif) | ||
| Scheme 2 Synthesis of cyclodextrin-centered drug-grafted seven-arm amphiphilic star copolymers [CDS-P(CL-co-DTC)-D-b-PEG]. | ||
Synthesis of carboxyl-terminated mPEGs (mPEG-COOH)
A typical example is given below: mPEG23 (10.0 g, 10 mmol), succinic anhydride (1.56 g, 15.6 mmol), DMAP (1.28 g, 10.5 mmol), and TEA (1.06 g, 1.05 mmol) were dissolved in 50 mL of anhydrous THF, and the reaction was carried out at room temperature for 24 h under vigorous stirring. After the solvent had been evaporated completely using a rotary evaporator, the residue was dissolved in methylene chloride and then precipitated in cold diethyl ether. The purified product was dried in vacuum to constant weight. Yield: 9.90 g (90.1%). 1H NMR (CDCl3, 500 MHz): 4.27 (t, COOCH2), 3.63 (m, OCH2CH2O), 3.35 (s, CH3O), 2.60 (m, OCOCH2CH2OCO).Synthesis of cyclodextrin-centered seven-arm amphiphilic star poly(ε-caprolactone-co-5,5-dibromomethyl-trimethylene carbonate)-b-poly(ethylene glycol)s [Scheme 2; CDS-P(CL-co-DBTC)-b-PEG]
A typical example is given below: CDS-P(CL27-co-DBTC3) (Mn = 29300, 1.0 g, 0.239 mmol OH), mPEG23-COOH (316 mg, 0.287 mmol), DCC (130 mg, 0.631 mmol), and DMAP (14 mg, 0.115 mmol) were dissolved in 40 mL of anhydrous THF, and the reaction was performed at room temperature for 48 h under argon atmosphere. The reaction byproduct dicyclohexylcarbodiurea was removed by filtration. After evaporating most of the solvent, the mixture of the star copolymer and the excess unreacted mPEG23-COOH was precipitated by addition of excess cold diethyl ether.Pure CDS-P(CL27-co-DBTC3)-b-PEG23 was obtained by dialysis (dialysis membrane, MWCO 14,000) and lyophilization. 1.0 g above-mentioned mixture was first dissolved in 25 mL THF, and the distilled water (ca. 20 mL) was added dropwise to the solution with vigorous stirring. Then the solution was stirred overnight and dialyzed against distilled water over 7 days to remove THF and the excess unreacted mPEG23-COOH. The final solid-state product was recovered by lyophilization (yield > 85%).
Synthesis of azide functionalized cyclodextrin-centered seven-arm amphiphilic star poly(ε-caprolactone-co-5,5-diazidomethy-trimethylene carbonate)-b-poly(ethylene glycol)s [Scheme 2; CDS-P(CL-co-DATC)-b-PEG]
A typical procedure was as follows: CDS-P(CL27-co-DBTC3)-b-PEG23 (500 mg, 0.57 mmol of Br group) was dissolved in DMF (20 mL), and sodium azide (370 mg, 5.7 mmol) was added. The reaction mixture was stirred for 7 days at 75 °C, then filtered and evaporated to remove DMF. Methylene chloride (50 mL) was added, and the mixture was washed three times with distilled water. The organic layer was dried with anhydrous MgSO4, and the solvent was evaporated to obtain a white solid. Yield: 450 mg (94%).Synthesis of alkyne-functionalized ibuprofen (A-drug)
Ibuprofen (2.06 g, 10 mmol), 2-propyn-1-ol (1.12 g, 20 mmol) DCC (9.10 g, 44 mmol), and DMAP (1.22 g, 10 mmol) were dissolved in 100 mL anhydrous THF, and the reaction was performed at room temperature for 48 h under argon atmosphere. The byproduct dicyclohexylcarbodiurea was removed by filtration. After the solvent and unreacted 2-propyn-1-ol were removed under reduced pressure, the oil like mixture composed of alkyne-functionalized ibuprofen (A-drug), residual DCC and DMAP were dried in vacuum to constant weight and used in the following “click” reaction without further purification. However, for analysis, pure A-drug was separated by silica gel chromatography using CH2Cl2–THF (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 v/v) as mobile phase. 1H NMR (500 MHz, CDCl3), δ, ppm: 0.93 (d, 6H, –CH(CH3)2), 1.53 (d, 3H, –Ar–CH–CH3), 1.88 (h, 1H, –CH(CH3)2), 2.45 (t, 1H, –C
:20 v/v) as mobile phase. 1H NMR (500 MHz, CDCl3), δ, ppm: 0.93 (d, 6H, –CH(CH3)2), 1.53 (d, 3H, –Ar–CH–CH3), 1.88 (h, 1H, –CH(CH3)2), 2.45 (t, 1H, –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 2.48 (d, 2H, –Ar–CH2–CH(CH3)2), 3.77 (q, 1H, –Ar–CH–CH3), 4.60–4.76 (q, 2H, –O–CH2–CCH), 7.11–7.25 (q, 4H, Ar). 13C NMR in CDCl3, δ, ppm: 18.7 (1C, –Ar–CHCH3), 22.6 (2C, –CH(CH3)2), 30.3 (1C, –CH(CH3)2), 45.2 (1C, –Ar–CH2CH(CH3)2), 45.3 (1C, –Ar–CHCH3), 52.4 (1C, –O–CH2–CCH), 63.9 (1C, –O–CH2–CCH), 75.0 (1C, –O–CH2–CCH), 127.3, 129.5, 137.4, 140.8 (4C, Ar), 174.0 (1C, C = O). MS for C16H20O2: m/z = 262 (M + NH4)+, m/z = 245 (M + H)+.
CH), 2.48 (d, 2H, –Ar–CH2–CH(CH3)2), 3.77 (q, 1H, –Ar–CH–CH3), 4.60–4.76 (q, 2H, –O–CH2–CCH), 7.11–7.25 (q, 4H, Ar). 13C NMR in CDCl3, δ, ppm: 18.7 (1C, –Ar–CHCH3), 22.6 (2C, –CH(CH3)2), 30.3 (1C, –CH(CH3)2), 45.2 (1C, –Ar–CH2CH(CH3)2), 45.3 (1C, –Ar–CHCH3), 52.4 (1C, –O–CH2–CCH), 63.9 (1C, –O–CH2–CCH), 75.0 (1C, –O–CH2–CCH), 127.3, 129.5, 137.4, 140.8 (4C, Ar), 174.0 (1C, C = O). MS for C16H20O2: m/z = 262 (M + NH4)+, m/z = 245 (M + H)+.
Synthesis of cyclodextrin-centered drug-grafted seven-arm amphiphilic star poly(ε-caprolactone-co- carbonate)-b-poly(ethylene glycol)s [CDS-P(CL-co-DTC)-D-b-PEG] by “click” reaction (Scheme 2)
Drug-grafted amphiphilic star copolymer [CDS-P(CL-co-DTC)-D-b-PEG] was synthesized by “click” reaction between alkyne-functionalized ibuprofen (A-drug) and azide functionalized star copolymer precursor [CDS-P(CL-co-DATC)-b-PEG]. A typical example is given below. CDS-P(CL27-co-DATC3)-b-PEG23 (300 mg, 0.36 mmol of azide group) and crude A-drug mixture (240 mg, about 132 mg of A-drug, 0.54 mmol of alkyne group) were dissolved in argon-purged DMF (15 mL) in a flame-dried ampoule. CuBr (52 mg, 0.36 mmol) and PMDETA (62 mg, 0.36 mmol) were added in order, and the reaction mixture was degassed by three freeze-pump-thaw cycles, left in argon, and stirred at 30 °C for 48 h. The solution was passed through an alumina column to remove the copper salt. The pure copolymer was recovered by precipitating with diethyl ether and dried in vacuum. Yield: 320 mg (about 74% for polymer weight).Micelle formation
CDS-P(CL-co-DTC)-D-b-PEG (25 mg) was dissolved in 5 mL THF, and then distilled water (ca. 5 mL) was added dropwise to the solution with vigorous stirring. The micelle solution was stirred overnight and dialyzed against distilled water over 48 h to remove THF. The final concentration of the micelle solution was 103 mg L−1.Characterization
Results and discussion
Synthesis of 5,5-dibromomethyl-trimethylene carbonate (DBTC)
As shown in Scheme 1, DBTC was synthesized by a similar procedure to that described by Al-Azemi and Bisht.47 The two hydroxyl groups of dibromoneopentyl glycol were converted into cyclic carbonate esters with bromide methylene pendent groups when reacting with ethyl chloroformate in the presence of triethylamine. Compared with other kinds of cyclic carbonate monomers, DBTC only involves one-step reaction under mild conditions, which avoids tedious protection and deprotection of functional groups. The structure of DBTC was confirmed by 1H NMR, 13C NMR and MS as shown in the Experimental section.Synthesis of cyclodextrin-centered seven-arm star poly(ε-caprolactone-co-5,5-dibromomethyl-trimethylene carbonate)s [CDS-P(CL-co-DBTC)] by controlled ring-opening copolymerization
CDS-P(CL-co-DBTC) was synthesized by controlled ring-opening copolymerization (CROP) of CL and DBTC monomers using per-2,3-acetyl-β-CD (CD-core) as a multifunctional initiator catalyzed by Sn(Oct)2 at 120 °C (Scheme 2). In our previous work, we have demonstrated that for the preparing of well-defined star polymers with “core-first” method, multifunctional initiators must have good solubility to ensure that all the initiating sites could simultaneously contribute to the polymerization.48,49 For CD-core, both the intra and inter molecular hydrogen bonds become weaker due to the extinction of the 14 secondary hydroxyl groups, and its structure may be more flexible, which could provide sufficient space to permit simultaneous initiating of all the 7 primary hydroxyl groups in the copolymerization of CL and DBTC. Three different co-monomer/initiator ratios were utilized to achieve CDS-P(CL-co-DBTC) with various polymerization degrees, and the molar ratio of monomers in feed (CL/DBTC) was fixed to be 90:10 in order to reduce the functional density of bromide methylene pendent groups. Detailed characterization results are summarized in Table 1.
| Sampleb | [CL + DBTC]0 /[CD-core] (molar ratio) | Conv (%) | DBTC Content (%)c | Mn,Cal/g mol−1d | Mn,NMR/g mol−1e | Mn,SEC/g mol−1f | PDIf |
|---|---|---|---|---|---|---|---|
a Polymerization conditions: bulk, 120 °C, 12 h, CL/DBTC = 90/10.
b CDS-P(CL27-co-DBTC3), 27 and 3 represent the average repeated units of CL and DBTC in each arm calculated from 1H NMR spectra.
c Molar ratio of repeat unit (DBTC) to (CL + DBTC) in the star copolymers calculated by 1H NMR spectra.
d Calculated from the equation: 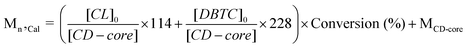 .
e Calculated from the equation: .
e Calculated from the equation: 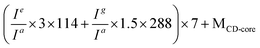 (see Fig. 1).
f Determined by SEC analysis with polystyrene standards. (see Fig. 1).
f Determined by SEC analysis with polystyrene standards.
|
|||||||
| CDS-P(CL13.5-co-DBTC1.5) | 15 | 94.2 | 9.7 | 14,700 | 13,600 | 93,00 | 1.30 |
| CDS-P(CL27-co-DBTC3) | 30 | 95.5 | 9.5 | 28,000 | 29,800 | 23,200 | 1.27 |
| CDS-P(CL36-co-DBTC4) | 40 | 93.1 | 9.4 | 36,000 | 33,200 | 32,300 | 1.42 |
Fig. 1 exhibits a representative 1H NMR spectrum of CDS-P(CL13.5-co-DBTC1.5). It clearly shows that besides the major proton signals of CL unit (Hb,c,d,e) and DBTC unit (Hg,f,f′), there are additional signals of the CD-core moiety: signals assigned to the protons on the acetyl group (Ha) and the protons on the sugar residue (4.50 ppm–5.50 ppm). A triplet (Hh) partially overlaps with a singlet (Hi) at 3.66 ppm, which are assigned to the methylene protons conjoint with the hydroxyl end groups of CL unit and DBTC unit, respectively. These results imply that in the prepared star copolymer, CL and DBTC are randomly copolymerized because both CL unit and DBTC unit appear at the end of the star copolymer chains. Meanwhile, the integral ratio of Ha and Hi+h (Ia/Ii+h) is 2.89, which is close to the theoretical value (Ia/Ii+h = 3.0). This observation confirms the well-defined seven-arm architecture. In addition, the actual molecular weights of these CDS-P(CL-co-DBTC) star copolymers determined by 1H NMR spectroscopy (Mn,NMR) are close to the values calculated from observed conversion and [CL + DBTC]0/[CD-core] (Mn,Cal). Moreover, the molar ratios of DBTC to (CL + DBTC) in the star copolymers are consistent with that in feed (Table 1). All these results verify that CDS-P(CL-co-DBTC) with designed structure and molecular weight have been successfully synthesized.
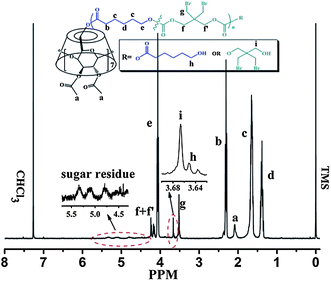 | ||
| Fig. 1 1H NMR spectrum (500 MHz) of CDS-P(CL13.5-co-DBTC1.5) in CDCl3. | ||
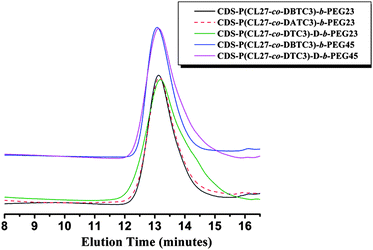
Fig. 2 shows the SEC traces of the CDS-P(CL-co-DBTC) star copolymers. For CDS-P(CL13.5-co-DBTC1.5) and CDS-P(CL27-co-DBTC3), symmetrical unimodal elution peaks with reasonably narrow molecular weight distributions (≤ 1.30) are observed, indicating the successful synthesis of CDS-P(CL-co-DBTC) with well-defined seven-arm star structure. However, as for CDS-P(CL36-co-DBTC4), when the [CL + DBTC]0/[CD-core] ratio increases to 40, the PDI value of the obtained star copolymer increases to 1.42, and tailing is detected in the low-molecular-weight region, which is attributed to unavoidable chain transfer side reactions during the CROP process. Thereby, CDS-P(CL-co-DBTC) star copolymers with higher molecular weight had not been prepared in this work.
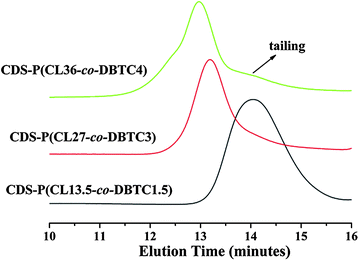 | ||
| Fig. 2 SEC traces of CDS-P(CL-co-DBTC) star copolymers. | ||
Synthesis of cyclodextrin-centered seven-arm amphiphilic star poly(ε-caprolactone-co-5,5-dibromomethyl-trimethylene carbonate)-b-poly(ethylene glycol)s [CDS-P(CL-co-DBTC)-b-PEG] by esterification coupling reaction
CDS-P(CL-co-DBTC)-b-PEG were synthesized as shown in Scheme 2. First, carboxyl-terminated mPEGs (mPEG-COOH) were prepared from the reaction of mPEG-OH with succinic anhydride. Then, the pre-synthesized CDS-P(CL-co-DBTC) bearing hydroxyl groups at the chain ends were coupled with excess amount of mPEG-COOH at room temperature in the presence of DCC and DMAP. Finally, the pure CDS-P(CL-co-DBTC)-b-PEG star copolymers were obtained by dialysis against distilled water to remove the unreacted mPEG-COOH. Fig. 3A exhibits the 1H NMR spectrum of the star copolymer CDS-P(CL27-co-DBTC3)-b-PEG23. Compared with the 1H NMR spectrum of CDS-P(CL13.5-co-DBTC1.5) in Fig. 1, new signals at δ = 3.65 and 3.38 ppm (Hj and Hk) assigned to methylene and methyl protons of PEG block could be detected. The coupling efficiency between mPEG-COOH and CDS-P(CL-co-DBTC) could be estimated from relative integral values of the methylene protons (Hj) of PEG and that of PCL (He) blocks. For our system, the coupling efficiency of all the copolymers is around 90%.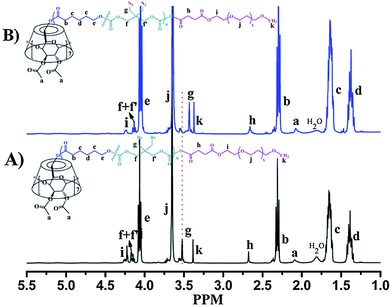 | ||
| Fig. 3 1H NMR spectra (500 MHz) of (A) CDS-P(CL27-co-DBTC3)-b-PEG23 and (B) CDS-P(CL27-co-DATC3)-b-PEG23 in CDCl3. | ||
Fig. 4 shows the representative SEC traces of the amphiphilic star copolymers [CDS-P(CL27-co-DBTC3)-b-PEG23 and CDS-P(CL27-co-DBTC3)-b-PEG45] in comparison with those of the corresponding CDS-P(CL27-co-DBTC3) and mPEG. After coupling reaction with mPEG-COOH, The SEC traces slightly shifted to high molecular mass region, indicating the incorporation of PEG macromolecules. Meanwhile, not any trace of mPEG precursors was found, which demonstrated that the excess mPEG-COOH had been completely removed by simple dialysis. However, the actual molecular weights of these CDS-P(CL-co-DBTC)-b-PEG star copolymers determined by 1H NMR spectroscopy (Mn,NMR) were higher than those determined by SEC with polystyrene standards (Mn,SEC), which could be attributed to the smaller hydrodynamic volume of the star copolymers with hyperbranched structure than the linear polymers with the same molecular weight. Detailed characterization results about the molecular weights and molecular weight distributions of these star copolymers are summarized in Table 2.
| Sample | Mn, of mPEG/g mol−1 | Mn,NMR/g mol−1a | Mn,SEC/g mol−1b | Coupling efficiency (%)c | PDIb |
|---|---|---|---|---|---|
a Mn,NMR is the molecular weight of CDS-P(CL-co-DBTC)-b-PEG which was determined by 1H NMR from the equation: 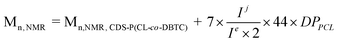 , DPPCL represents the average repeated units of CL in each arm of CDS-P(CL-co-DBTC) (calculated from Fig. 3A).
b Mn,SEC is the molecular weight of CDS-P(CL-co-DBTC)-b-PEG and was determined by SEC with polystyrene standards.
c Coupling efficiency is calculated from the relative integral values of the methylene protons (Hj) of PEG and those of PCL (He) blocks. , DPPCL represents the average repeated units of CL in each arm of CDS-P(CL-co-DBTC) (calculated from Fig. 3A).
b Mn,SEC is the molecular weight of CDS-P(CL-co-DBTC)-b-PEG and was determined by SEC with polystyrene standards.
c Coupling efficiency is calculated from the relative integral values of the methylene protons (Hj) of PEG and those of PCL (He) blocks.
|
|||||
| CDS-P(CL13.5-co-DBTC1.5)-b-PEG23 | 1,000 | 20,900 | 15,600 | 95.3 | 1.28 |
| CDS-P(CL13.5-co-DBTC1.5)-b-PEG45 | 2,000 | 27,200 | 17,300 | 92.7 | 1.26 |
| CDS-P(CL27-co-DBTC3)-b-PEG23 | 1,000 | 37,000 | 25,600 | 91.5 | 1.34 |
| CDS-P(CL27-co-DBTC3)-b-PEG45 | 2,000 | 43,200 | 28,000 | 91.1 | 1.32 |
| CDS-P(CL36-co-DBTC4)-b-PEG23 | 1,000 | 41,000 | 35,800 | 89.2 | 1.44 |
| CDS-P(CL36-co-DBTC4)-b-PEG45 | 2,000 | 47,000 | 37,500 | 87.5 | 1.46 |
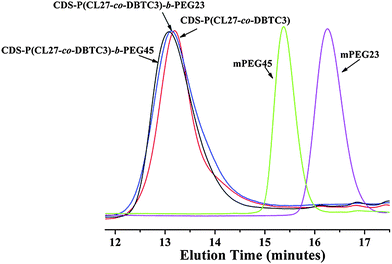 | ||
| Fig. 4 SEC traces of CDS-P(CL-co-DBTC), CDS-P(CL-co-DBTC)-b-PEG and mPEG. | ||
Synthesis of azide functionalized cyclodextrin-centered seven-arm amphiphilic star poly(ε-caprolactone-co-5,5-diazidomethy-trimethylene carbonate)-b-poly(ethylene glycol)s [CDS-P(CL-co-DATC)-b-PEG]
It is well-known in organic chemistry that bromide groups can undergo nucleophilic substitution reactions with NaN3 to form azide groups in DMF at room temperature. However, in this work, only 30% of the bromide groups had been converted to azide form in the presence of excess NaN3 in DMF at room temperature for 3 days, which was probably due to the crowded reaction sites preventing the nucleophilic substitution of the NaN3. Therefore, high degree of substitution had been achieved by increasing the temperature to 75 °C and extending the reaction time to 7 days.A representative 1H NMR spectrum of CDS-P(CL27-co-DATC3)-b-PEG23 is shown in Fig. 3B. It is noteworthy that the signal of the bromomethylene protons at 3.53 ppm [Fig. 3A (Hg)] has disappeared, whereas a new signal corresponding to methyl protons connected with azide groups [Fig. 3B (Hg)] appears at 3.43 ppm, which indicates that the bromide groups have been completely transformed to azide groups. In addition, SEC analysis proved that the molecular weight and polydispersity (Mw/Mn) of the star copolymer were not obviously changed after substitution [see Fig. 7, CDS-P(CL27-co-DATC3)-b-PEG23], which suggests that no degradation of P(CL-co-DBTC) chains occurred at such high temperature.
FT-IR measurements were performed to further confirm the azide functionalized star copolymer [CDS-P(CL27-co-DATC3)-b-PEG23]. As shown in Fig. 5, compared with the IR spectrum of CDS-P(CL27-co-DBTC3)-b-PEG23, a new absorption peak related to azide group at 2097 cm−1 has appeared, which indicates the bromide groups have been substituted by azide groups.
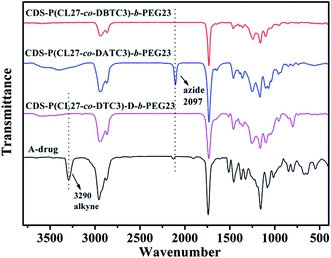 | ||
| Fig. 5 FT-IR spectra of CDS-P(CL27-co-DBTC3)-b-PEG23, CDS-P(CL27-co-DATC3)-b-PEG23, CDS-P(CL27-co-DTC3)-D-b-PEG23, and A-drug… | ||
Synthesis of alkyne-functionalized ibuprofen (A-drug)
Ibuprofen is a non-steroidal anti-inflammatory drug that is commonly used for the relief of symptoms of arthritis and fever. In order to introduce ibuprofen onto the hydrophobic block of the amphiphilic star copolymer, alkyne-functionalized ibuprofen was first synthesized from commercially available ibuprofen with 2-propyn-1-ol at room temperature in the presence of DCC and DMAP. After reaction, the crude product was oil like syrup mixed with unreacted DCC and DMAP. Considering the tolerance of most functional groups of “click” reaction,50 this mixture was directly used in the following “click” step without further purification. However, for analysis, small amount of pure A-drug was separated by silica gel chromatography and characterized by 1H NMR, 13C NMR, MS and FT-IR (see Experimental section). Fig. 6A shows the 1H NMR spectrum of A-drug. The quartet at 4.66 ppm (Hn) should be assigned to the methylene protons of propargyl group due to the coexisting of R(−) and S(+) forms of A-drug synthesized by non-enantioselective esterification.51 The resonance of the alkynyl proton (Ho) is also detected at 2.45 ppm. These results verify that alkyne group has been successfully linked to the ibuprofen molecule.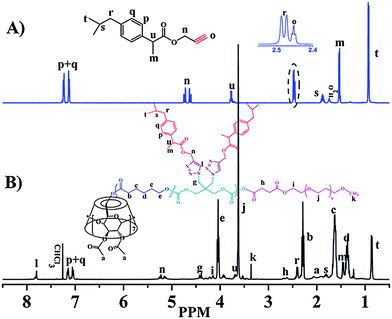 | ||
| Fig. 6 1H NMR spectra (500 MHz) of (A) A-drug in CDCl3 (B) CDS-P(CL27-co-DTC3)-D-b-PEG23. | ||
Synthesis of drug-grafted cyclodextrin-centered seven-arm amphiphilic star poly(ε-caprolactone-co-carbonate)-b-poly(ethylene glycol)s [CDS-P(CL-co-DTC)-D-b-PEG] by “click” reaction
A “click” chemistry strategy was employed to prepare drug-grafted amphiphilic star copolymer [CDS-P(CL-co-DTC)-D-b-PEG] between CDS-P(CL-co-DATC)-b-PEG and A-drug mixture using CuBr/PMDETA as catalyst in DMF at 30 °C. Considering that the exact amount of azide groups of CDS-P(CL-co-DATC)-b-PEG cannot be exactly calculated from 1H NMR and the A-drug mixture contains unreacted DCC and DMAP, about 50% excess amount of alkyne groups was used in order to prepare well-defined CDS-P(CL-co-DTC)-D-b-PEG. After “click” reaction, unreacted A-drug and impurities (DCC and DMAP) were removed by washing with ethyl ether. From the 1H NMR spectrum of CDS-P(CL27-co-DTC3)-D-b-PEG23 (Fig. 6B), the signal at 7.80 ppm (Hl) due to the proton of the triazole and signal at 5.21 ppm (Hn) due to the methylene protons of ibuprofen conjoint to the triazole ring indicate ibuprofen moieties have been successfully grafted onto the amphiphilic star copolymer. In addition, the integrals of Hl, Hn and Ht are in agreement with the theoretical results. Notably, compared with the IR spectra of CDS-P(CL27-co-DATC3)-b-PEG23 and A-drug (Fig. 5), the characteristic absorption peaks related alkyne group at 3290 cm−1 and azide group at 2097 cm−1 have completely disappeared from the IR spectrum of CDS-P(CL27-co-DTC3)-D-b-PEG23, which also confirms the successful synthesis of drug-grafted amphiphilic star copolymer. Furthermore, no corresponding signals related to DCC and DMAP in 1H NMR spectrum were detected in Fig. 6B, suggesting the impurities have been completely removed from the final products.Fig. 7 shows the SEC traces of the drug-grafted amphiphilic star copolymers [CDS-P(CL-co-DTC)-D-b-PEG] in comparison with those of the corresponding non-drug-grafted precursors. To our surprise, after “click” reaction, the SEC traces slightly shifted to low molecular mass region, which could be attributed to the much smaller hydrodynamic volume of the hyperbranched graft architecture than that of the linear PS. Some other groups also reported the similar unexpected results in their work.52,53
 | ||
| Fig. 7 SEC traces of CDS-P(CL-co-DTC)-D-b-PEG and their corresponding non-drug-grafted precursors. | ||
Self-assembly behaviors of drug-grafted amphiphilic star copolymer [CDS-P(CL-co-DTC)-D-b-PEG] in water
These synthesized drug-grafted amphiphilic star copolymers provide an opportunity to form aggregates in water. The hydrophobic P(CL-co-DTC) blocks and ibuprofen molecules constitute the core domain, which are shelled by the hydrophilic PEG chains. The critical micelle concentration (CMC) of amphiphilic copolymer was usually measured by fluorescence technique using pyrene as a probe.54 Pyrene will preferentially incorporate into hydrophobic microdomains showing strong fluorescence intensity while weak fluorescence intensity and a shift of excitation peak in a polar environment (water). In addition, the sharp rise in intensity ratio of peaks at 338 and 333 nm of pyrene in the excitation spectra indicates the on-set of micellization (CMC) for amphiphilic copolymer. Fig. 8 shows the relationship of the intensity ratios (I338/I333) as a function of copolymer concentration at room temperature. It can be obviously observed that the CMC values of these drug-grafted amphiphilic star copolymers were relatively low (ranging from 1.27 mg L−1 to 5.25 mg L−1), increasing as the weight fraction of PEG increased (Table 3), which was probably due to the hyperbranched architecture. These results indicate that the aggregates in the water are stable and the grafted hydrophobic drug molecules could be fully protected by the PEG shell.| Sample a | Weight fraction of PEG (%) | Particle size b/nm | PDI c | CMC/mg L−1 |
|---|---|---|---|---|
| a CDS-P(CL27-co-DTC3)-D-b-PEG45, 27 and 3 represent the average repeated units of CL and DBTC in each arm; 45 represents the polymerization degree of PEG in each arm. b Number-average mean diameters measured by dynamic light scattering (DLS). c PDI denotes the polydispersity of aggregates in water. | ||||
| CDS-P(CL13.5-co-DTC1.5)-D-b-PEG45 | 40.2 | 23.6 | 0.305 | 5.25 |
| CDS-P(CL13.5-co-DTC1.5)-D-b-PEG23 | 25.7 | 50.3 | 0.227 | 3.03 |
| CDS-P(CL27-co-DTC3)-D-b-PEG45 | 23.9 | 53.5 | 0.330 | 3.16 |
| CDS-P(CL27-co-DTC3)-D-b-PEG23 | 13.6 | 695.2 | 0.329 | 1.27 |
| CDS-P(CL36-co-DTC4)-D-b-PEG45 | 20.2 | 37.5 | 0.238 | 1.78 |
| CDS-P(CL36-co-DTC4)-D-b-PEG23 | 11.4 | precipitation | — | — |
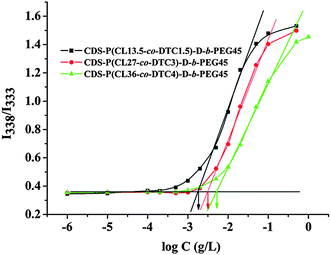 | ||
| Fig. 8 Plots of the fluorescence intensity ratio I338/I333 from pyrene excitation spectra vs. log C for CDS-P(CL-co-DTC)-D-b-PEG. | ||
The morphologies of the self-assembled aggregates from these drug-grafted amphiphilic star copolymers were investigated by DLS and TEM as shown in Fig. 9 and Table 3.
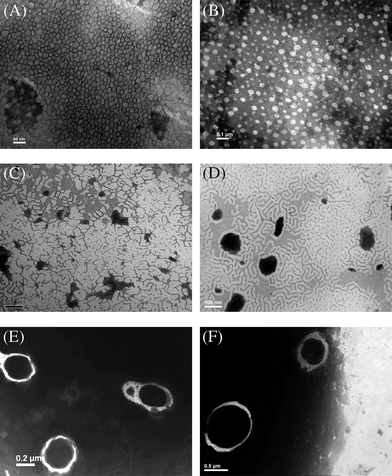 | ||
| Fig. 9 TEM micrographs of aggregates from (A) CDS-P(CL13.5-co-DTC1.5)-D-b-PEG45; (B) CDS-P(CL13.5-co-DTC1.5)-D-b-PEG23; (C) CDS-P(CL27-co-DTC3)-D-b-PEG45; (D) CDS-P(CL36-co-DTC4)-D-b-PEG45; (E, F) CDS-P(CL27-co-DTC3)-D-b-PEG23. | ||
Nearly spherical micelles were observed when the weight fraction of PEG block was 40.2% for CDS-P(CL13.5-co-DTC1.5)-D-b-PEG45 (Fig. 9A) and 25.7% for CDS-P(CL13.5-co-DTC1.5)-D-b-PEG23 (Fig. 9B). The micelle diameters, which ranged from 10 to 50 nm, increased with the decreasing of the weight fraction of hydrophilic PEG block. Nevertheless, in the case of CDS-P(CL27-co-DTC3)-D-b-PEG45 with a similar PEG weight fraction (23.9%) compared with CDS-P(CL13.5-co-DTC1.5)-D-b-PEG45, wormlike aggregates were observed (Fig. 9C). As for CDS-P(CL36-co-DTC4)-D-b-PEG45 with a PEG weight fraction of 20.2% (Fig. 9D), another kind of wormlike aggregates appeared. With a further decrease in the weight fraction of the PEG block to 13.6% within CDS-P(CL27-co-DTC3)-D-b-PEG23 sample, vesicle or polymersome aggregates with number average mean diameters ranging from 300 to 800 nm were obtained (Fig. 9E and Fig. 9F). However, CDS-P(CL36-co-DTC4)-D-b-PEG23 having the lowest PEG weight fraction (11.4%) could not form stable aggregates in water and precipitated when THF had been removed. These observations are due to the fact that the short hydrophilic PEG block could not evade the hydrophobic interactions and van der Waals interactions between the exposed hydrophobic domains, and the copolymers prefer to form large aggregates to balance these interactions. If the hydrophilic PEG block is too short to protect the hydrophobic domains, macroscopic precipitation appeared. It should be noted that the sizes of wormlike aggregates measured by DLS are not in accordance with the TEM results (Table 3), which is attributed to the fact that the basic equations to calculate the hydrodynamic diameter in DLS measurement originate from the correlation function cumulant analysis based on the assumption that the particles are noninteracting spheres and not anisotropic objects.55
Conclusions
Novel drug-grafted seven-arm amphiphilic star copolymers [CDS-P(CL-co-DTC)-D-b-PEG] based on a cyclodextrin core were successfully synthesized by the combination of CROP, esterification coupling reaction and “click” reaction. 1H NMR, FT-IR and SEC analyses confirmed the designed structures. These copolymers could self-assemble into aggregates with various morphologies in water (such as spherical micelle, wormlike aggregate and vesicle or polymersome), which can be controlled by both the macromolecular architecture and the composition of the hydrophilic PEG block. Therefore, this provides a versatile strategy not only for the synthesis of multi-morphological drug-conjugated nanoparticles as potential drug carriers in special delivery system, but also for fabricating novel hyperbranched amphiphilic copolymers with pendent functional groups.The authors are grateful for the financial support from the National Natural Science Foundation of China (20704036), special fund from the Major State Basic Research Project (2005CB623802) and the Committee of Science and Technology of Zhejiang Province.
References
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- K. E. Uhrich, S. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS.
- Y. Yamamoto, Y. Nagasaki, Y. Kato, Y. Sugiyama and K. Kataoka, J. Controlled Release, 2001, 77, 27–38 CrossRef CAS.
- P. D. Drumheller and J. A. Hubbell, J. Biomed. Mater. Res., 1995, 29, 207–215 CrossRef CAS.
- N. P. Desai and J. A. Hubbell, J. Biomed. Mater. Res., 1991, 25, 829–843 CrossRef CAS.
- H. X. Ge, Y. Hu, X. Q. Jiang, D. M. Cheng, Y. Y. Yuan, H. Bi and C. Z. Yang, J. Pharm. Sci., 2002, 91, 1463–1473 CrossRef CAS.
- L. B. Luo, J. Tam, D. Maysinger and A. Eisenberg, Bioconjugate Chem., 2002, 13, 1259–1265 CrossRef CAS.
- T. Nie, Y. Zhao, Z. W. Xie and C. Wu, Macromolecules, 2003, 36, 8825–8829 CrossRef CAS.
- W. H. Xie, W. P. Zhu and Z. Q. Shen, Polymer, 2007, 48, 6791–6798 CrossRef CAS.
- T. Noh, Y. H. Kook, C. Park, H. Youn, H. Kim, E. T. OH, E. K. Choi, H. J. Park and C. Kim, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7321–7331 CrossRef CAS.
- M. Trollsås, V. Y. Lee, D. Mecerreyes, P. Löwenhielm, M. Möller, R. D. Miller and J. L. Hedrick, Macromolecules, 2000, 33, 4619–4627 CrossRef.
- M. Trollsås, P. Löwenhielm, V. Y. Lee, M. Möller, R. D. Miller and J. L. Hedrick, Macromolecules, 1999, 32, 9062–9066 CrossRef.
- P. Lecomte, C. Detrembleur, X. Lou, M. Mazza, O. Halleux and R. Jérôme, Macromol. Symp., 2000, 157, 47–60 CrossRef CAS.
- R. Riva, S. Schmeits, F. Stoffelbach, C. Jérôme, R. Jérôme and P. Lecomte, Chem. Commun., 2005, 5334–5336 RSC.
- R. Riva, S. Schmeits, C. Jérôme, R. Jérôme and P. Lecomte, Macromolecules, 2007, 40, 796–803 CrossRef CAS.
- Y. Q. Shen, X. H. Chen and R. A. Gross, Macromolecules, 1999, 32, 3891–3897 CrossRef CAS.
- W. C. Ray and M. W. Grinstaff, Macromolecules, 2003, 36, 3557–3562 CrossRef CAS.
- R. S. Lee, J. M. Yang and T. F. Lin, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2303–2312 CrossRef CAS.
- H. L. Guan, Z. G. Xie, P. B. Zhang, X. Wang, X. S. Chen, X. H. Wang and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4771–4780 CrossRef CAS.
- X. L. Hu, S. Liu, X. S. Chen, G. J. Mo, Z. G. Xie and X. B. Jing, Biomacromolecules, 2008, 9, 553–560 CrossRef CAS.
- X. L. Hu, X. S. Chen, S. Liu, Q. Shi and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1852–1861 CrossRef CAS.
- X. L. Hu, X. S. Chen, Z. G. Xie, H. B. Cheng and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7022–7032 CrossRef CAS.
- C. H. Lu, Q. Shi, X. S. Chen, T. C. Lu, Z. G. Xie, X. L. Hu, J. Ma and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3204–3217 CrossRef CAS.
- X. L. Hu, X. S. Chen, H. B. Cheng and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 161–169 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- D. Y. Yan, Y. F. Zhou and J. Hou, Science, 2004, 303, 65–67 CrossRef CAS.
- Z. B. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS.
- L. Y. Qiu and Y. H. Bae, Pharm. Res., 2006, 23, 1–30 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS.
- C. Hua, S. M. Peng and C. M. Dong, Macromolecules, 2008, 41, 6686–6695 CrossRef CAS.
- Y. Yang, C. Hua and C. M. Dong, Biomacromolecules, 2009, 10, 2310–2318 CrossRef CAS.
- F. Wang, T. K. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2005, 16, 397–405 CrossRef CAS.
- F. Wang, T. K. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2008, 19, 1423–1429 CrossRef CAS.
- K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045–2076 CrossRef CAS.
- P. F. Gou, W. P. Zhu, N. Xu and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6455–6465 CrossRef CAS.
- C. Yang, H. Z. Li, S. H. Goh and J. Li, Biomaterials, 2007, 28, 3245–3254 CrossRef CAS.
- J. S. Li, H. N. Xiao, Y. S. Kim and T. L. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6345–6354 CrossRef CAS.
- X. B. Xiong, A. Mahmud, H. Uludag and A. Lavasanifar, Biomacromolecules, 2007, 8, 874–884 CrossRef CAS.
- P. A. Bertin, K. J. Watson and S. T. Nguyen, Macromolecules, 2004, 37, 8364–8372 CrossRef CAS.
- X. F. Zhang, Y. X. Li, X. S. Chen, X. H. Wang, X. Y. Xu, Q. Z. Liang, J. L. Hu and X. B. Jing, Biomaterials, 2005, 26, 2121–2128 CrossRef CAS.
- M. Hans, K. Shimoni, D. Danino, S. J. Siegel and A. Lowman, Biomacromolecules, 2005, 6, 2708–2717 CrossRef CAS.
- J. Khandare and T. Minko, Prog. Polym. Sci., 2006, 31, 359–397 CrossRef CAS.
- B. Parrish and T. Emrick, Bioconjugate Chem., 2007, 18, 263–267 CrossRef CAS.
- C. Giacomelli, V. Schmidt and R. Borsali, Macromolecules, 2007, 40, 2148–2157 CrossRef CAS.
- J. B. Liu, P. Zahedi, F. Q. Zeng and C. Allen, J. Pharm. Sci., 2008, 97, 3274–3290 CrossRef CAS.
- O. Perumal, J. Khandare, P. Kolhe, S. Kannan, M. Lieh-Lai and R. M. Kannan, Bioconjugate Chem., 2009, 20, 842–846 CrossRef CAS.
- T. F. Al-Azemi and K. S. Bisht, Macromolecules, 1999, 32, 6536–6540 CrossRef CAS.
- P. F. Gou, W. P. Zhu and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2108–2118 CrossRef CAS.
- P. F. Gou, W. P. Zhu, N. Zhu and Z. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2905–2916 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- M. Arroyo and J. V. Sinisterra, J. Org. Chem., 1994, 59, 4410–4417 CrossRef CAS.
- X. L. Luo, G. W. Wang, X. C. Pang and J. L. Huang, Macromolecules, 2008, 41, 2315–2317 CrossRef CAS.
- H. Li, R. Riva, R. Jérôme and P. Lecomte, Macromolecules, 2007, 40, 824–831 CrossRef CAS.
- M. Wilhelm, C. L. Zhao, Y. C. Wang, R. L. Xu, M. A. Winnik, J. L. Mura, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 1033–1040 CrossRef CAS.
- J. Zhang, L. Q. Wang, H. J. Wang and K. H. Tu, Biomacromolecules, 2006, 7, 2492–2500 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |