Recent advances in the design of bioconjugates from controlled/living radical polymerization
Benjamin
Le Droumaguet
and
Julien
Nicolas
*
Laboratoire de Physico-Chimie, Pharmacotechnie et Biopharmacie, Université Paris-Sud, UMR CNRS 8612, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, F-92296, Châtenay-Malabry cedex, France. E-mail: julien.nicolas@u-psud.fr; Fax: +33 1 46 83 59 46; Tel: +33 1 46 83 58 53
First published on 29th January 2010
Abstract
Since its discovery, controlled/living radical polymerization (CLRP) has proven to be a mature technology for building tailor-made (block) copolymers, functional polymers and polymers with a wide range of biological recognition. Due to these considerable advantages over other synthetic approaches, CLRP techniques have been successfully exploited to construct novel polymer-protein/peptide bioconjugates with a high level of structural control and varied interesting, somehow unexpected, features. A comprehensive review of the recent advances in the rapidly expanding field of bioconjugation is presented and outlines work up to early 2010.
 Benjamin Le Droumaguet | Benjamin Le Droumaguet obtained his Masters degree in organic chemistry in 2004 from the University of Rennes. Then he moved to the research group of Dr Kelly Velonia at the University of Geneva to take up a PhD position in the field of polymer-protein bioconjugates. After obtaining his PhD in 2008, he came back to France for a post-doctoral fellowship in the research group of Prof. Patrick Couvreur, where he is currently working on the synthesis of functionalized poly(alkyl cyanoacrylate) nanoparticles for active targeting. His research interests are directed towards the conjugation of biological molecules with organic materials for the design of functional “smart” biohybrid systems. |
 Julien Nicolas | Julien Nicolas graduated from the Ecole Supérieure de Chimie Organique et Minérale (ESCOM), France, in 2001. He completed his PhD in 2005 under the supervision of Prof. Bernadette Charleux at the University Pierre and Marie Curie, Paris, where he studied nitroxide-mediated polymerization. He then joined the group of Prof. David M. Haddleton at the University of Warwick, UK, for a postdoctoral fellowship to design polymer-protein bioconjugates by controlled/living radical polymerization. In 2007, he was appointed as researcher at the CNRS in the group of Prof. Patrick Couvreur, University Paris-Sud, France, where his current research activities are focused on the synthesis of novel nanoparticles and biopolymers for drug delivery purposes. |
1 Introduction
The exciting world of polymer science has been in a state of almost perpetual (r)evolution since the pioneering work of Staudinger1 and has now developed into a modern and multidisciplinary research field. Due to the cross-fertilization of polymer science with diverse areas, it covers not only primarily synthetic polymers mainly devoted to structural applications such as coatings or packaging materials, but also a broad range of higher value-added functional applications in nanomaterials, opto-electronic technology as well as biomedical-related areas.Macromolecular synthesis certainly represents one of the most prolific sub-disciplines in polymer science, as attested by the discovery of controlled/living radical polymerization (CLRP) fifteen years ago,2–13 that brought about an important breakthrough in the field. Indeed, this new synthetic tool provided the polymer chemist with an efficient and easy route for achieving various macromolecular architectures of high complexity and functionality, unavailable by other polymerization methods. Among the numerous arenas where CLRP has been successfully exploited is the design of a new-class of bioconjugates.14–20 Despite the many possible definitions of the word bioconjugate,21 in this review the term refers to the coupling or the binding between a synthetic macromolecule (here a (co)polymer synthesized by CLRP) and a biomacromolecule.
Innovative drug delivery technologies are now a key component of pharmaceutical development. Various therapeutic peptides and proteins represent a rapidly growing section of marketed drugs and have an uncontested place alongside other well-established therapies. However, they suffer from severe limitations due to their inherent physicochemical properties such as a variable solubility, a low bioavailability and a limited stability. The attachment of polyethylene glycol (PEG) to protein or peptide therapeutics, termed PEGylation, is an example of a highly successful strategy that gives rise to several benefits including increased bioavailability and plasma half-lives, decreased immunogenicity, reduced proteolysis and enhanced solubility and stability.22 Recent advances in PEGylation technology are enabling even more opportunities to create novel, efficient products. Whereas the initial strategy is a random PEG attachment on lysine residues or at the N-terminus, a recent promising approach consists of site-specific bioconjugation23 at areas on a pharmaceutical drug that do not interfere with its biological activity while still making the most of the PEG shield. Due to high robustness, flexibility and mild experimental conditions as well as to the possibility to insert varied functional groups within the polymer chains, CLRP is becoming a believable technology regarding this public health research field.
The explosive number of publications within the past couple of years devoted to the design of bioconjugates from CLRP methods prompted us to provide an updated survey that includes proteins, peptides and oligonucleotides, and which covers achievements up to early 2010. An overview of the employed CLRP techniques is first presented followed by a detailed description of the various synthetic strategies to achieve well-defined bioconjugates. A distinction is established between whether the conjugation is performed via a covalent linkage or using a non-covalent approach.
2 Controlled/living radical polymerization (CLRP)
CLRP techniques have emerged as simple routes for preparing well-defined polymers of predetermined molecular weight (MW) and narrow molecular weight distribution (MWD), as well as various block copolymers and a wide range of complex macromolecular architectures.2–13 Prior to the development of CLRP, all these features were hardly achievable. Indeed, only with living ionic polymerization24 could such a degree of structural uniformity be reached but under very drastic polymerization conditions and with a limited choice of monomers.In contrast to conventional free-radical polymerization where propagating radicals exhibit a very short lifetime (∼1 s) that hampers the conception of well-defined architectures, the concept of CLRP is to increase this lifetime up to the timescale of the polymerization reaction, via the establishment of a reversible equilibrium between active species, which can propagate, and dormant/capped species, which cannot propagate. CLRP then proceeds in such a manner that all polymer chains grow at the same rate and the detrimental impact of irreversible termination events over the MW and the MWD are made almost negligible.25 An ideal living polymerization system should exhibit the following features: (i) a linear evolution of the logarithmic conversion (ln[1/(1 − conversion)]) with time, accounting for a constant propagating radicals concentration; (ii) a linear increase in the number-average molar mass, Mn, with monomer conversion, where the degree of polymerization, DPn, is predetermined by the consumed monomer to initially introduced initiator molar ratio; (iii) low polydispersity indexes, Mw/Mn, close to a Poisson distribution (Mw/Mn ≈ 1 + 1/DPn); (iv) a quantitative α- and ω-functionalization and (v) the possibility for polymer chains, after monomer consumption, to further grow when additional monomer is introduced which allows block copolymer synthesis to be performed by sequential monomer addition.25
Nowadays, a plethora of well-established CLRP methods offers the polymer chemist an impressive toolbox for making advanced macromolecular architectures of increasing complexity. Among them, nitroxide-mediated polymerization (NMP),2 atom transfer radical polymerization (ATRP)3,4,7,9,10 and reversible addition-fragmentation chain transfer (RAFT),5,6,12,13 the latter including macromolecular design via the interchange of xanthates (MADIX), represent the three most well-known CLRP techniques. In addition to these famous approaches, one can find the iniferter26–29 system and cyanoxyl-mediated polymerization30 as well as recently emerged CLRP techniques, such as iodine transfer polymerization (ITP),31 single electron transfer-living radical polymerization (SET-LRP),32 organotellurium-mediated polymerization (TERP),8 organostibine-mediated polymerization (SBRP)8 and cobalt-mediated polymerization (CMRP).33,34
The following section will briefly describe the CLRP techniques that have been used for the synthesis of bioconjugates so far. The reader who would like a more exhaustive point of view about all CLRP methods is referred to the above-mentioned references associated to each technique.
2.1 Iniferter
In the early 80s, the use of iniferters in radical polymerization, defined as agents (such as tetraethylthiuram disulfide, phenylazotriphenylmethane or benzyl dithiocarbamate) that are able to initiate, transfer and terminate, was actually the first successful step to date towards CLRP.26,27 Iniferters can be either: asymmetrical (denoted A–B), where A is a reactive radical which participates in initiation and propagation reactions whereas B is only involved in termination reaction, or symmetrical (denoted C–C), where C participates in both initiation and termination reactions. Besides this, iniferters can be activated upon heating (thermal iniferters) or by UV irradiation (photoiniferters).28,29Even though the use of iniferters led to broad MWDs and poor initiation efficiencies, this technique has allowed a broad range of monomers to be polymerized in rather mild conditions as well as several macromolecular architectures to be prepared.28,29
2.2 Cyanoxyl-mediated polymerization
In the early 90s, it was shown that cyanoxyl radicals (˙OC≡N) could be successfully used as control agents during the polymerization of methyl (meth)acrylate, n-butyl acrylate and acrylic acid to form homopolymers as well as block, statistical and grafted copolymers in relatively mild experimental conditions.30,35,36 Moreover, the polymerization can be carried out in aqueous solution and is tolerant of a broad range of functional groups. However, the use of cyanoxyl-mediated polymerization was strongly restrained by the emergence of another type of oxygen-centered persistent radicals as mediators for CLRP, namely nitroxides.2.3 Nitroxide-mediated polymerization (NMP)
NMP is based on a reversible termination reaction between a growing radical, P˙, and a free nitroxide, N˙, to form a (macro)alkoxyamine, P–N (Fig. 1). This equilibrium between active and dormant species presents the advantage of being a purely thermal process: the (macro)alkoxyamine regenerates the propagating radical and the nitroxide by homolytic cleavage at high temperature (usually > 70 °C).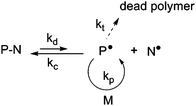 | ||
| Fig. 1 The activation–deactivation equilibrium in nitroxide-mediated polymerization (NMP). | ||
A typical NMP can be initiated following two different pathways: (i) by using a bicomponent initiating system (i.e. a conventional radical initiator and a free nitroxide) or (ii) via a monocomponent initiating system (i.e. a preformed alkoxyamine). Georges and co-workers first reported the controlled radical polymerization of styrene with (2,2,6,6-tetramethylpiperidinyl-1-oxy) (TEMPO, N1, Fig. 2) as the mediator.37 However, as TEMPO was almost exclusively limited to styrenic monomers (only a sterically hindered TEMPO derivative allowed the control of the n-butyl acrylate polymerization),38 new acyclic nitroxides have been designed to improved the range of polymerizable monomers under controlled/living conditions. More precisely, N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)] nitroxide (SG1 or DEPN, N2, Fig. 2)39–42 and N-tert-butyl-N-[1-phenyl-2-(methylpropyl)]nitroxide (TIPNO, N3, Fig. 2)43–47 are now able to control the polymerization of styrenics, alkyl acrylates, acrylic acid, acrylamides and dienes.39,43,48–50 The polymerization of methacrylic esters can be controlled either: (i) by using a particular nitroxide such as 2,2-diphenyl-3-phenylimino-2,3-dihydroindol-1-yloxyl nitroxide (DPAIO, N4, Fig. 2),51 specific to methacrylates or (ii) by a copolymerization approach under SG1 control with a small amount of comonomer such as styrene52–54 or acrylonitrile.55
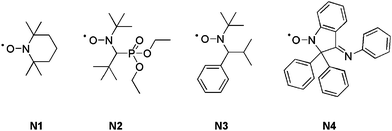 | ||
| Fig. 2 The structures of nitroxides used as mediators in NMP: TEMPO (N1), SG1 or DEPN (N2), TIPNO (N3) and DPAIO (N4). | ||
2.4 Atom transfer radical polymerization (ATRP)
ATRP was discovered independently by Sawamoto56 and Matyjaszewski.57–59 The ATRP process is based on a rapid exchange of a halide atom (especially Cl or Br) between a growing radical and a dormant species, via a redox process involving a transition metal complex (Fig. 3). To ensure a good control of the polymerization, this equilibrium is strongly shifted towards the dormant species.3,4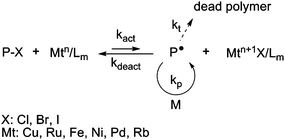 | ||
| Fig. 3 The activation–deactivation equilibrium in atom transfer radical polymerization (ATRP). | ||
Various transition metals can be employed in ATRP (Cu, Ru, Fe, Ni, etc.). In the first ATRP process, called direct ATRP, the transition metal complex in a lower oxidation state (Mtn/Lm, where Mt is the metal and L is the ligand) is directly added to the reaction as an activator and reacts reversibly with the dormant species (P-X, with X a halogen atom) to form a deactivator (Mtn+1X/Lm) and the active species P˙. In contrast, when the polymerization is initiated by a conventional initiator and a metal complex at the higher oxidation state, the process is called reverse ATRP. The simultaneous reverse and normal initiation (SR&NI) process takes advantage of both normal and reverse ATRP as Cu(II) (which is tolerant to oxygen), an alkyl halide and a radical initiator are initially present in the reaction medium.60 It provides a way to reduce the amount of copper complex and to prepare more complex macromolecular architectures.
Recently, new ATRP processes have been developed, namely activators generated by electron transfer (AGET)61 and activators regenerated by electron transfer (ARGET).62 The AGET process employs a reducing agent (e.g. ascorbic acid or tin(II) 2-ethylhexanoate) which reacts with Mtn+1X/Lm to generate the active catalyst (Mtn/Lm). The process then follows a direct ATRP process. AGET ATRP allows the preparation of pure block copolymers with no homopolymer of the second monomer. The ARGET process uses an excess of reducing agent which allows a significant reduction of the amount of metal in the media.
This powerful and versatile technique can be used under mild experimental conditions and in various polymerization media with a wide range of monomers including styrenics, alkyl (meth)acrylates, acrylonitrile, (meth)acrylamides as well as water-soluble monomers to give tailor-made polymers.3,4,7,10,11,63,64 An additional flexibility is provided by the possibility of using commercially available functionalized initiators and to functionalize chain ends. However, if ATRP can be used with a large range of monomers, the polymerization of functional monomers bearing acid or amine function remains hardly achievable.
2.5 Reversible addition fragmentation chain transfer (RAFT)
RAFT polymerization is governed by a reversible transfer reaction between a growing (macro)radical (active species) and a (macro)RAFT agent (dormant species).5,12,13,65 RAFT agents, denoted Z–C(![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) S)SR, act as transfer agents by a two-step addition-fragmentation mechanism (Fig. 4). The RAFT group is typically a thiocarbonylthio group such as dithioester (Z = alkyl), trithiocarbonate (Z = S-alkyl), xanthate (O-alkyl) or dithiocarbamate (Z = N(alkyl)2). The RAFT process using thiocarbonylthio compounds, including dithioesters and trithiocarbonates, was reported by the CSIRO laboratory in early 199866 whereas a similar process using xanthates as RAFT agents (the so-called MADIX) was reported in late 1998.67 RAFT is potentially universal and can be applied to a wide range of functional monomers (styrenics, alkyl (meth)acrylates, acrylic acid, vinyl acetate etc.), which allows polymers with precisely controlled structural parameters to be prepared such as random, block, gradient, grafted and star copolymers.5,12,13
S)SR, act as transfer agents by a two-step addition-fragmentation mechanism (Fig. 4). The RAFT group is typically a thiocarbonylthio group such as dithioester (Z = alkyl), trithiocarbonate (Z = S-alkyl), xanthate (O-alkyl) or dithiocarbamate (Z = N(alkyl)2). The RAFT process using thiocarbonylthio compounds, including dithioesters and trithiocarbonates, was reported by the CSIRO laboratory in early 199866 whereas a similar process using xanthates as RAFT agents (the so-called MADIX) was reported in late 1998.67 RAFT is potentially universal and can be applied to a wide range of functional monomers (styrenics, alkyl (meth)acrylates, acrylic acid, vinyl acetate etc.), which allows polymers with precisely controlled structural parameters to be prepared such as random, block, gradient, grafted and star copolymers.5,12,13
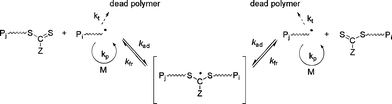 | ||
| Fig. 4 Mechanism of reversible addition fragmentation chain transfer (RAFT). | ||
Even though Moad, Rizzardo and co-workers recently succeeded in elaborating a switchable RAFT agent,68 one of the major drawback of this technique was the lack of a universal RAFT agent. In particular, dithioesters or trithiocarbonates were suitable for controlling polymerization of more activated monomers such as styrene (S) and derivatives, methacrylic esters (e.g. methyl methacrylate MMA), methacrylic acid (MA), methacrylamide (MAM), acrylic acid (AA), acrylamide (AM) or acrylonitrile (AN). However they inhibit or retard the polymerization of less activated monomers such as vinyl acetate (VAc), N-vinylpyrrolidone (NVP) or N-vinylcarbazole (NVC), for which xanthates or dithiocarbamates are more suitable.5,12,13 The choice of R and Z groups is thus crucial to achieve a good control of the polymerization.
3 Synthesis of polymer-protein/peptide bioconjugates using a covalent approach
3.1 Bioconjugation with preformed polymer: the “grafting to” method
The direct conjugation of preformed polymers to proteins/peptides is certainly the most widespread approach for creating bioconjugates, the best example being the protein PEGylation,87–90 discovered by Abuchowski et al. in the late seventies.91,92 Even though a broad range of monomers have been polymerized by CLRP for further coupling to proteins/peptides, comb-like polymers based on poly(ethylene glycol) methacrylate (PEGMA) and poly(ethylene glycol) acrylate (PEGA) were advantageously employed as an alternative to traditional PEGylation. Such branched PEG polymers indeed exhibit two main benefits over traditional linear PEGs: (i) a higher flexibility of control over the macromolecular characteristics of both the PEG chains and the polymer backbone and (ii) a better resistance against proteolysis and antibodies action, due to their “umbrella-like” shape.93,94Moreover, one of the biggest advantages of CLRP techniques (especially ATRP and RAFT) over traditional polymerization methods is that they allow a fine tuning of the functionalities present at both the α- and the ω-termini of polymer chains by using functional initiators/transfer agents (see Fig. 5 and Fig. 7) or via post-polymerization modification steps.
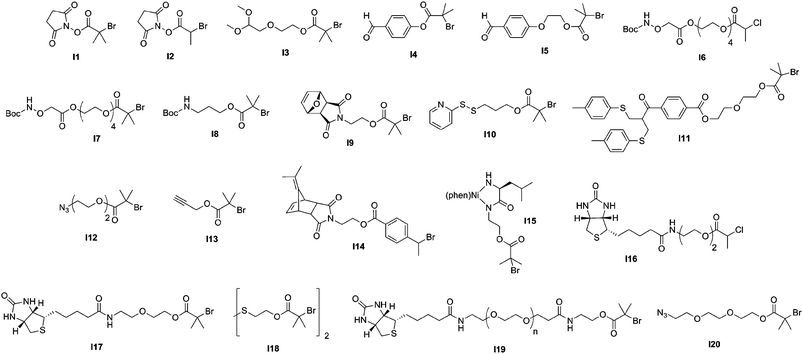 | ||
| Fig. 5 Structure of ATRP initiators employed in the synthesis of bioconjugates. | ||
| Protein | α-Functional polymer | ||||
|---|---|---|---|---|---|
| Name | Target group | α-Functional groupb | Monomer/sc | CLRP technique | Ref. |
| a via lysine residues. b NHS: N-hydroxysuccinimide; PDS: pyridyl disulfide. c PEGMA: poly(ethylene glycol) methyl ether methacrylate; NVP: N-vinyl pyrrolidone; MPC: 2-methacryloyloxyethyl phosphorylcholine; HPMA: N-(2-hydroxypropyl)methacrylamide; NIPPAm: N-isopropyl acrylamide; HEMA hydroxyethyl methacrylate; GMA: glycidyl methacrylate; PEGA: poly(ethylene glycol) methyl ether acrylate. | |||||
| lysozyme | NH2 | NHS | PEGMA | ATRP | 69 |
| lysozyme | NH2 | NHS | NVP | RAFT | 70 |
| lysozyme | NH2 | NHS | MPC | ATRP | 71 |
| papain | NH2 | NHS | MPC | iniferter | 72,73 |
| lysozyme | NH2 | aldehyde | PEGMA | ATRP | 74 |
| lysozyme | NH2 | aldehyde | MPC | ATRP | 71 |
| sCT | NH2 (N-terminus) | aldehyde | PEGMA | ATRP | 75,76 |
| erythropoietin | NH2 | aldehyde | MPC | ATRP | 71 |
| G-CSF | NH2 | aldehyde | MPC | ATRP | 71 |
| lysozyme | NH2 | thiazolidine-2-thione | HPMA, PEGMA | RAFT | 77 |
| BSA | N ε -levulinatea | aminooxy | NIPAAm, PEGMA, HEMA | ATRP | 78 |
| BSA | N ε -levulinatea | aminooxy | NIPAAm | RAFT | 79 |
| BSA | SH | maleimide | PEGMA, GMA | ATRP | 80 |
| BSA | SH | maleimide | GMA, PMA, HMA (terpolymer) | ATRP | 81 |
| T4 lysozyme | SH | maleimide | PEGA | RAFT | 82 |
| BSA | SH | PDS | HEMA | ATRP | 83 |
| BSA | SH | PDS | NIPAAm | ATRP | 84 |
| T4 lysozyme | SH | PDS | NIPAAm | ATRP | 84 |
| BSA | SH | PDS | PEGA, NIPAAm | RAFT | 85 |
| IFN | disulfide bridge | bis-sulfone | MPC | ATRP | 86 |
3.1.1.1 Conjugation via an amine reactive group. N-hydroxysuccinimide terminated polymers. The Haddleton and Stenzel groups used ATRP69 and RAFT70, respectively, to construct well-defined N-hydroxysuccinimide (NHS) α-functional polymers for protein bioconjugation purposes. For instance, NHS α-functional poly[poly(ethylene glycol) methyl ether methacrylate] (poly(PEGMA)) polymers (Mn = 2.8 and 6.4 kDa, Mw/Mn < 1.15) were synthesized from 2 different NHS-based ATRP initiators (I1 and I2, Fig. 5) in the presence of CuBr/N-(ethyl)-2-pyridylmethanimine and used for the bioconjugation to lysine residues of lysozyme.69 The coupling between lysozyme and an excess of NHS α-functional poly(PEGMA) polymers was performed in anhydrous DMSO in the presence of TEA. A quantitative coupling was obtained after 6 h and SDS-PAGE analysis of the bioconjugates indicated that approximately 6 to 7 polymer chains were anchored to each protein, in good agreement with the 7 free lysine residues available per lysozyme. More recently, α-NHS poly(N-vinylpyrrolidone) (PVP) polymers were prepared by RAFT polymerization from RA1 (Fig. 7) upon AIBN initiation (Mn = 16.9–33.4 kDa, Mw/Mn = 1.38–1.41) and coupled to lysozyme.70 Similarly to the ATRP route, an average of 7 PVP chains were tethered to lysozyme.
Ishihara’s group reported the functionalization of the papain protein with NHS α-functional water-soluble poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) polymer using a photoinduced iniferter-mediated polymerization.72,73 2-Methacryloyloxyethyl phosphorylcholine (MPC) monomer and the resulting polymers95,96 exhibit excellent biocompatibility97 as they mimic the structure of natural phospholipids found in cell membranes. The polymerization of MPC was triggered by the photo-irradiation of 4-(N,N-diethyldithiocarbamoylmethyl)-benzoic acid (BDC) at ambient temperature. It was shown that increasing the molecular weight of the PMPC-NHS polymer resulted in a lower degree of functionalization (from 42 to 19%). Besides, the resulting conjugates retained 35% of the catalytic activity of the native enzyme, whatever the molecular weight of the PMPC-NHS moiety. Recently, ATRP has been investigated by Emrick and co-workers for the preparation of NHS α-functional PMPC polymers (Mn = 2.3–8.2 kDa, Mw/Mn = 1.2–1.5) derived from initiators I1 and I2 (Fig. 5) for bioconjugation to lysozyme in quantitative yields.71
Aldehyde terminated polymers. Another strategy, based on the work of Bentley and co-workers regarding reductive amination,98 was developed by Haddleton’s group for the synthesis of aldehyde α-functional branched PEG polymers by ATRP and their subsequent conjugation to proteins.74,75 In the first report, a ketal-protected aldehyde ATRP initiator (I3, Fig. 5) was used to initiate the polymerization of PEGMA (Mn = 11 kDa, Mw/Mn = 1.14). After deprotection upon acidic catalysis, the α-aldehyde poly(PEGMA) was reacted with lysozyme at pH 5 or 6 in the presence of sodium cyanoborohydride (NaCNBH3) to give, after in situ reduction of the imine moiety into an amine bond, the corresponding lysozyme-poly(PEGMA) bioconjugate. The conjugation was quantitative and occurred faster at pH 5. The same reductive amination pathway was also applied to the functionalization of the N-terminal cysteine of salmon calcitonin (sCT), a 3432 Da peptide used for the treatment of post-menopausal osteoporosis and Paget's disease, by a 6.5 kDa α-aldehyde poly(PEGMA) in the presence of NaCNBH3 (Fig. 6).75 The poly(PEGMA)-sCT bioconjugate (9.8 kDa) was observed to be non-cytotoxic, even at a relatively high concentration and to retain approximately 90% of the initial native sCT activity. Stability studies also indicated that poly(PEGMA)-sCT displayed resistance toward proteolytic activity of 3 individual intestinal enzymes (trypsin, chymotrypsin, elastase) whereas native sCT was degraded within a few minutes. The bioconjugates also reduced the serum calcium levels. All these results indicated that the poly(PEGMA) polymers have a tremendous potential to improve the pharmacokinetics of injected peptides in therapeutic applications. Another complementary study regarding the functionalization of sCT by poly(PEGMA) with Mn varying from 6.5 up to 109 kDa showed that sCT activity was not altered by the polymer chain length.76
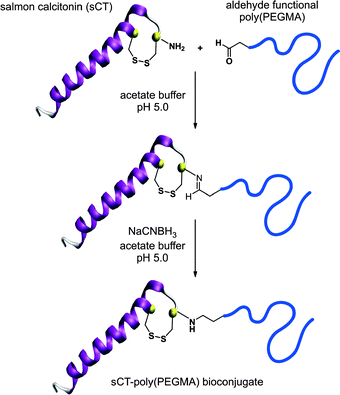 | ||
| Fig. 6 Design of sCT-poly(PEGMA) bioconjugates by a combination of ATRP and reductive amination.75 | ||
The reductive amination pathway was also applied to the synthesis of bioconjugates based on granulocyte colony stimulating factor (G-CSF) and erythropoietin (EPO) by Emrick and co-workers. Briefly, ATRP of MPC from two benzaldehyde-based ATRP initiators (I4 and I5, Fig. 5) afforded a range of well defined aldehyde α-functional PMPC polymers (Mn = 4.5–7 kDa, Mw/Mn = 1.1) that were suitable for bioconjugation with the lysine residues of the two above-mentioned proteins in the presence of NaCNBH3.71 SEC-HPLC indicated the formation of the corresponding (G-CSF)-PMPC and EPO-PMPC bioconjugates together with the presence of unreacted native protein.
Thiazolidine-2-thione terminated polymers. Recently, Davis and co-workers presented the synthesis of new lysine-reactive α-functional polymers based on the functionalization of a RAFT agent with the thiazolidine-2-thione moiety.77 In this study, the 2-cyano-5-oxo-5-(2-thioxothiazolidin-3-yl)pental-2-yl benzodithioate RAFT agent (RA2, Fig. 7) mediated the polymerization of N-(2-hydroxypropyl) methacrylamide (HPMA). Lysozyme was reacted with 40 eq. of the α-thiazolidine-2-thione PHPMA (Mn = 3.5 kDa, Mw/Mn = 1.09) and SEC analysis demonstrated the efficiency of the coupling reaction. It was also shown that pH was an important parameter as it influenced the number of polymer chains attached to the protein (a low pH protonates the amines that lose their nucleophilic character). Finally, the bioactivity of the resulting lysozyme-PHPMA bioconjugates was evaluated in the presence of a lysozyme substrate (Micrococcus lysodeikticus cells) and gave only 4.8% of the original activity of the native protein for the conjugate formed at pH 6.5. Besides this, the same group described the successful synthesis of a well-defined, biodegradable poly(PEGMA) using a thiazolidine-2-thione-based RAFT agent bearing a disulfide bridge (RA3, Fig. 7).99 After conjugation to lysozyme via amide linkages, cleavage of the polymer chains from the conjugate was triggered and the released protein displayed a marked increase in bioactivity.
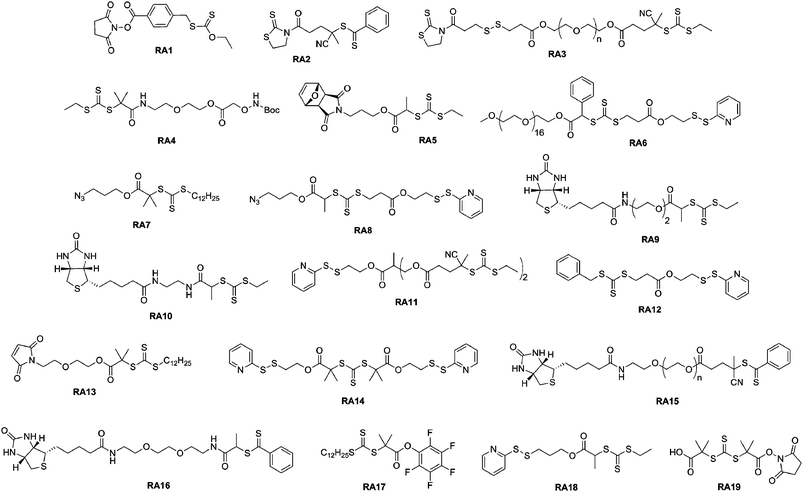 | ||
| Fig. 7 Structure of RAFT agents employed in the synthesis of bioconjugates. | ||
Aminooxy terminated polymers. Another option for selectively functionalizing lysine residues of proteins with functional polymers is the use of aminooxy terminated polymers. Maynard’s group reported the design of aminooxy α-functional poly(N-isopropylacrylamide) (PNIPAAm) polymers for the synthesis of BSA-PNIPAAm bioconjugates via ATRP78 and RAFT79 methodologies. In the first approach, Boc-protected aminooxy ATRP initiators (I6 and I7, Fig. 5) initiated the polymerization of NIPAAm in a controlled fashion, followed by Boc deprotection under acidic conditions. As the use of the aminooxy coupling requires the derivatization of the protein with ketone or aldehyde moieties, BSA was first reacted with NHS-activated levulinate (Fig. 8). The resulting Nε-levulinyl lysine-modified BSA was then reacted with α-aminooxy PNIPAAm to give the desired BSA-PNIPAAm bioconjugate via oxime bonds formation, further purified from unreacted BSA by thermal precipitation at 35 °C. SDS PAGE analysis confirmed that the coupling was achieved and UV-Vis turbidity experiments revealed the effective thermoresponsive features of the bioconjugates. This approach was also employed with different molecular weight ATRP polymers from PEGMA, NIPAAm and 2-hydroxyethyl methacrylate (HEMA)78 as well as with α-aminooxy PNIPAAm (Mn = 4.2 kDa, Mw/Mn < 1.15) from RAFT using RA4 (Fig. 7).79
 | ||
| Fig. 8 Aminooxy end-functionalized polymers from ATRP for selective conjugation to proteins.78 | ||
This study also reported the possibility of modifying the trithiocarbonate end-group of the PNIPAAm by aminolysis in the presence of butylamine and tris(2-carboxyethyl)phosphine (TCEP), to avoid disulfide coupling, and to further immobilize the resulting thiol ω-functional PNIPAAm polymer onto a gold surface. In a final step, the authors were able to immobilize heparin, a sulfated polysaccharide, previously decorated with aldehyde functions by reaction with NaIO4.
Carboxylic acid terminated polymers. Börner and co-workers used ATRP to synthesize hybrid nanotubes composed of cyclooctapeptides (stacked by intermolecular hydrogen bonds) decorated with two poly(n-butyl acrylate) (PnBA) polymer chains tethered to the two opposite lysine residues of each cyclic oligopeptide.100 Polymerization of nBA was initiated from benzyl 2-bromopropionate in ACN using CuBr/CuBr2/PMDETA at 60 °C for 2 h. The benzyl ester group was then cleaved from the PnBA polymer under reductive conditions (Mn = 2.1 kDa, Mw/Mn = 1.10). Final amidation between lysine residues and carboxy-terminated PnBA was achieved in the presence of EDC and DIPEA to yield a triblock-like biopolymer able to self-assemble in nanotubular superstructures. A similar polymer was also attached to the N-terminus of a linear dodecapeptide by classical amidation reaction followed by an O-N-acyl switch that guided the self assembly of the bioconjugate into densely twisted tapelike microstructures.101 AFM and TEM were used to characterize these helical superstructures. The stable assembly was ascribed to the formation of antiparallel β-sheets between peptide segments whereas PnBA tails constitute the shell of the superhelices.
3.1.1.2 Conjugation via a thiol reactive group. Amine targeting via lysine residues is one of the techniques most used for bioconjugates synthesis. However, it always results in the formation of a broad range of bioconjugates differing in their degree of functionalization and thus displaying different molecular weights. This explains the recent increasing interest for α-functional polymers derived from CLRP towards the targeting of free cysteine residues that are present in protein at lower percentages than amine counterparts.
Maleimide terminated polymers. The direct use of a maleimido ATRP initiator is not suitable as the maleimide moiety is polymerizable and branching is thus likely to occur upon polymerization. Two synthetic pathways were proposed by Haddleton and co-workers80 to efficiently circumvent this difficulty: (i) from a Boc-protected amino ATRP initiator (I8, Fig. 5) where the maleimide moiety was introduced during a post-polymerization step via an amidation reaction with 3-maleimidopropionyl chloride in the presence of DIPEA or (ii) from a furan-protected maleimido ATRP initiator (I9, Fig. 5), the maleimide moiety being recovered by a retro Diels–Alder reaction after polymerization. This was applied to the synthesis of well-defined and pure maleimide α-functional poly(PEGMA) and poly(glycerol methacrylate) (PGMA) polymers (Mn = 4.1–35.4 kDa, Mw/Mn = 1.06–1.27), using N-(ethyl)-2-pyridylmethanimine/CuBr as the catalytic system. Coupling experiments were successfully performed on glutathione (γ-ECG) and BSA (which contains a single free cysteine residue at position 34)102 as a model tripeptide and protein, respectively. SDS-PAGE and FPLC analyses revealed the successful formation of the bioconjugates.
The same furan-protected maleimido ATRP initiator also allowed BSA-polymer giant amphiphiles to be prepared in a controlled fashion as demonstrated by Velonia and co-workers (Fig. 9).81 Polymerization of a ketal-protected glycerol methacrylate, a very small amount of hostasol methacrylate (HMA) as a fluorescent monomer and trimethylsilyl-protected propargyl methacrylate (incorporated in the reaction medium after ∼50% conversion) yielded the corresponding terpolymer. Its subsequent deprotection afforded a fluorescent α-maleimide poly(glycerol methacrylate-co-hostasol methacrylate-co-propargyl methacrylate) (P(GMA-co-HMA-co-PMA)) copolymer containing alkyne side chains available for further click chemistry.81 After bioconjugation, hydrophilic BSA-(PGMA-co-HMA-co-PMA) biohybrids were then hydrophobized by copper catalyzed click chemistry reaction with small hydrophobic azides (1-azidodecane and 1-(azidomethyl)benzene) to afford amphiphilic protein-polymer biomacromolecules that self-assembled in aqueous solutions into well-defined spherical superstructures. Depending on the nature of the clicked azide, average diameters varied from 20 to 200 nm.
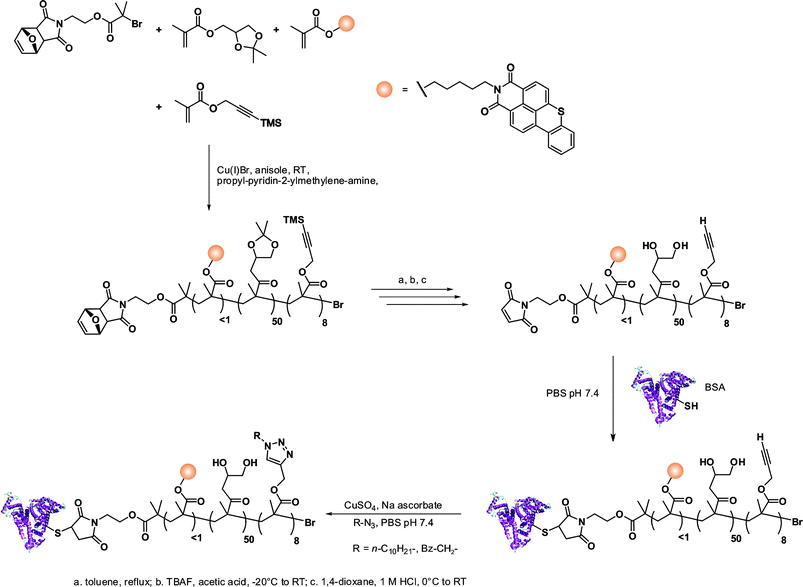 | ||
| Fig. 9 Design of giant amphiphiles by a combination of ATRP and click chemistry.81 | ||
Maynard’s group used RAFT polymerization for the formation of semitelechelic maleimide-containing poly(PEGA) polymers for bioconjugation purposes.82 A furan-protected maleimide α-functional RAFT agent (RA5, Fig. 7) was used to mediate the polymerization of PEGA at 60 °C in the presence of AIBN to afford the corresponding poly(PEGA) polymers (Mn = 20.0–39 kDa, Mw/Mn = 1.25–1.36). In order to avoid partial hydrolysis of the ester bond linking the maleimide to the polymer upon retro Diels–Alder reaction, the polymer was prepared from an amide linked RAFT agent. Maleimido poly(PEGA) was then coupled to V131C T4 lysozyme (T4L, a mutant protein genetically engineered to present a cysteine residue at position 131 instead of a valine residue) in the presence of EDTA and TCEP as confirmed by SDS-PAGE and SEC-HPLC.
Pyridyl disulfide terminated polymers. The use of a pyridyl disulfide (PDS) group to target the free cysteine residues of proteins represents a convenient alternative to the maleimide-thiol Michael-type addition approach. Importantly, it has a significantly higher stability in PBS compared to maleimide group and has the advantage of being cleavable under reductive environments, allowing for further release of the polymer/protein.
Two complementary studies published by Maynard and co-workers reported the synthesis of PDS α-functional polymers from ATRP. In a first report, HEMA was polymerized from a PDS-containing ATRP initiator (I10, Fig. 5) in the presence of CuBr/bpy as the catalyst to give the corresponding PHEMA (Mn = 3.9, 7.9, 15.7 kDa, Mw/Mn = 1.20–1.25) with 90% of PDS functionality.83 The bioconjugation was performed with BSA and assessed by SDS-PAGE in reducing or non-reducing conditions, and by Ellman's assay in order to highlight the specificity of the coupling on the single free cysteine residue. This approach was then extended to BSA-PNIPAAm and T4L-PNIPAAm “smart” bioconjugates with bioconjugation yields higher than 65% for both proteins.84 The PNIPAAm block was recovered upon cleavage of the disulfide bridge and exhibited a PDI as low as 1.34. Interestingly, in the case of the T4L-PNIPAAm bioconjugates, no significant difference in bioactivity was observed when compared to the native protein.
Recently, Davis and co-workers used two PDS-functionalized RAFT agents (RA6 and R12, Fig. 7) for the synthesis of a range of α-PDS PNIPAAm (Mn = 17.2–23.3 kDa, Mw/Mn < 1.36) and poly(PEGA) (Mn = 8–18 kDa, Mw/Mn = 1.14–1.45) in water or acetonitrile. The kinetic data indicated that the PDS moiety is largely benign in free radical polymerizations, remaining intact for subsequent reaction with thiol groups. This has been examplified by a successful conjugation to BSA, as evidenced by SEC and PAGE analysis.85 A series of block copolymers was also prepared from the PEG macro-RAFT agent (RA6, Fig. 7).
3.1.1.3 Disulfide bond targeting. Recently, Godwin, Lewis and co-workers reported the formation of PMPC polymers designed to possess a α-bis-sulfone terminal group for protein disulfide bond targeting.86 The ATRP synthesis of PMPC polymers was achieved from a α-bis-sulfide ATRP initiator (I11, Fig. 5) for specific conjugation to interferon-2α (IFN) disulfide bonds. A broad range of α-bis-sulfide PMPC molecular weights was obtained (Mn = 29.1–56.3 kDa) in a controlled manner (Mw/Mn < 1.23), followed by their reaction with oxone to afford ready-for-conjugation α-bis-sulfone PMPC polymers. The bioconjugation of the 2 disulfide bridges of IFN with α-bis-sulfone PMPC was undertaken at pH 8.2 in the presence of 1,4-dithiothreitol (DTT). SDS-PAGE revealed the formation of both mono- and di-PMPC-IFN bioconjugates which gave a marked resistance to antibody binding while keeping similar antiviral and antiproliferative activity compared to the native IFN. They also exhibited an increased pharmacokinetic profile when compared to their PEGylated counterparts.
3.1.1.4 Conjugation using click chemistry. Another coupling method that recently received a tremendous interest in bioconjugation is the copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction between an azide and an alkyne (CuAAC).103,104 This cycloaddition belongs to the class of chemical reactions, often referred as click chemistry, that share several very important features: (i) a very high efficiency in terms of both conversion and selectivity; (ii) mild experimental conditions; (iii) a simple workup and (iv) little or no by products.105–108
For example, Sumerlin and co-workers reported the bioconjugation reaction between azide α-functional PNIPAAm and alkyne functionalized BSA. NIPAAm was polymerized under AIBN initiation with 2-dodecylsulfanylthiocarbonylsulfanyl-2-methylpropionic acid 3-azidopropyl ester as a RAFT agent (RA7, Fig. 7).109 The corresponding well-defined PNIPAAm (Mn = 16.3 kDa, Mw/Mn = 1.06) was coupled to alkyne-containing BSA (obtained from coupling with propargyl maleimide) using CuSO4/sodium ascorbate as the catalyst in PBS. The formation of BSA-PNIPAAm was demonstrated by SDS-PAGE and SEC analysis. BSA was also reduced with TCEP prior to its derivatization with propargyl maleimide in order to increase the number of conjugation sites. Turbidimetry assays indicated that the conjugates retained their thermoresponsive behaviour.
In a recent study from Lecommandoux, Taton and co-workers, the coupling of poly(N-dimethylaminoethyl methacrylate) (PDMAEMA) with polypeptides synthesized from the polymerization of α-amino acid-N-carboxyanhydrides (NCAs) (see also section 3.4.3 Combination of NCA polymerization and CLRP) was achieved by a convergent CuAAC strategy (Fig. 10).110N-dimethylaminoethyl methacrylate (DMAEMA) was polymerized from azide or propargyl α-functional ATRP initiators (I12 and I13, Fig. 5) in the presence of CuBr/1,1,4,7,10,10 hexamethyltriethylenetetramine (HMTETA) catalytic system to afford the corresponding α-functional PDMAEMA (Mn = 8.8 or 10.4 kDa, Mw/Mn = 1.12 or 1.17). In parallel, poly(γ-benzyl-L-glutamate) (PBLG) was synthesized by NCA polymerization either from 3-azidopropylamine or from propargylamine to afford well-controlled azide or propargyl α-functional PBLG. The final step, consisting of a CuAAC reaction between azide α-functional PDMAEMA and propargyl α-functional PBLG (or vice versa), was catalyzed by CuBr/PMDETA and the PBLG-b-PDMAEMA bioconjugates were shown to maintain low PDIs (Mw/Mn < 1.18) with molecular weights in the 20.8–21.1 kDa range.
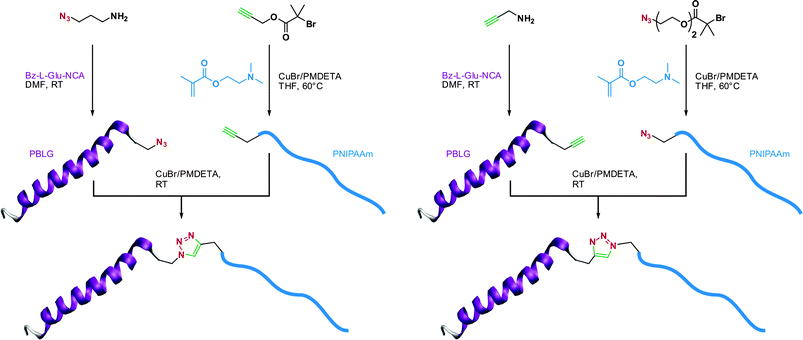 | ||
| Fig. 10 Convergent synthesis of bioconjugates by a combination of ATRP, NCA polymerization and click chemistry.110 | ||
3.1.2.1 Conjugation via an amine reactive group. Aldehyde terminated polymers (reductive amination). The synthesis of ω-aldehyde functionalized poly(N-vinylpyrrolidone) (PVP) by RAFT polymerization was recently developed by Klumperman and co-workers.111N-vinylpyrrolidone (NVP) was polymerized from a xanthate RAFT agent and the resulting ω-thiocarbonylthio end-group was hydrolyzed in water at 40 °C to yield ω-hydroxy PVP subsequently oxidized at high temperature to its aldehyde form with more than 90% yield. Coupling to lysozyme hydrochloride by the reductive amination pathway was then undertaken and successfully assessed by SDS-PAGE analysis.
3.1.2.2 Conjugation via a thiol reactive group. Maleimide terminated polymers. To the best of our knowledge, only one example112 was reported in this category and relied on the design of star polymer-protein conjugates, based on an extension of previous studies.113,114 From a tetrafunctionalized trithiocarbonate RAFT agent, a four-arm PNIPAAm (Mn = 5.47 and 51.6 kDa, Mw/Mn < 1.06) with approximately 95% retention of chain-end was subjected to radical cross-coupling with a furan-protected maleimido azo-initator114 and subsequently deprotected to display the maleimide moieties. Conjugation was achieved with T4L at pH 7.5 in the presence of EDTA and TCEP to afford multimeric T4L-PNIPAAm bioconjugates. A careful characterization indicated approximately 3 proteins per star polymer.
Pyridyl disulfide terminated polymers. RAFT polymerization offers the advantage of allowing the trithiocarbonate or the dithioester ω-chain end to be readily converted into a thiol moiety via aminolysis in the presence of an alkyl amine. The ω-thiol terminus can then react with a pyridyl disulfide group to afford the desired ω-functional polymer.
This pathway was illustrated by Davis and co-workers with several different polymers such as poly(methyl methacrylate) (PMMA), polystyrene (PS), poly(PEGA), poly(hydroxypropyl methacrylate) (PHPMA) and PNIPAAm using 4-cyanopentanoic acid dithiobenzoate (CDTB) or 3-(benzylsulfanylthiocarbonyl sulfanyl)-propionic acid (BSPA) as RAFT agents.115 In order to maintain a high chain-end fraction, low initiator over RAFT agent molar ratios and low monomer conversions were selected (60–75%, Mn = 1–12.5 kDa, Mw/Mn < 1.21). Depending on the polymer structure, 65–90% of trithiocarbonate or dithiobenzoate groups were still present after the polymerization. Subsequent aminolysis with hexylamine followed by reaction with 2,2′-dithiopyridine (DTP) yielded the corresponding PDS-terminated polymers. PDS ω-functional PNIPAAm and PHPMA were coupled to the NGR model peptide (GNGRGC), known to be a tumor-targeting peptide, in PBS at pH 8 with a yield of 85–92%.116
Vinyl sulfone terminated polymers. Maynard and co-workers recently developed a novel technique to target protein cysteine residues using vinyl sulfone-terminated polymers (Fig. 11).117 A poly(PEGA) prepared by RAFT (Mn = 6.7 kDa and Mw/Mn = 1.09) with a high fraction (99%) of dithiobenzoate end-groups was subjected to a reductive amination followed by the addition of divinylsulfone in the presence of TCEP (to avoid thiol oxidation). After only 30 min, 99 ± 6% of the poly(PEGA) contained a vinyl sulfone terminus. Bioconjugation experiments with BSA (previously reduced to display 3 thiol groups) were then performed with 20 eq. of vinyl sulfone ω-functional poly(PEGA). SDS-PAGE confirmed the formation of the desired BSA-poly(PEGA) bioconjugate that retained 92% of the initial activity of native BSA.
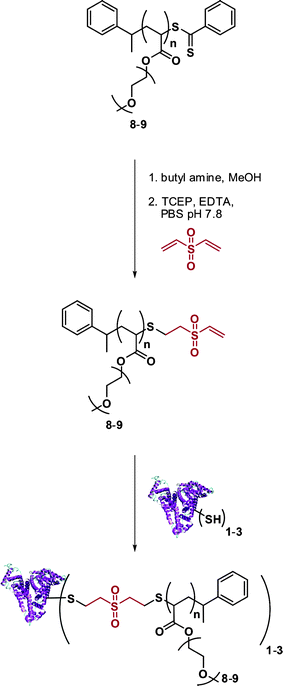 | ||
| Fig. 11 Synthesis of BSA-poly(PEGA) bioconjugates from vinyl sulfone-terminated polymers.117 | ||
3.1.2.3 Conjugation using click chemistry. The presence of a halide chain-end inherent to the ATRP process opened an avenue of opportunity for subsequent CuAAC reactions, especially from styrenic- and acrylate-based polymers.107 After polymerization, the bromine end-group can be indeed readily turned into the corresponding azide through a simple quantitative nucleophilic substitution in the presence of azide anions (N3−).
For example, Cornelissen and co-workers prepared BSA-PS giant amphiphiles118 by means of CuAAC. A PS block (Mn = 4.2 kDa, Mw/Mn = 1.15) prepared in anisole at 90 °C using CuBr/N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA) was reacted with azidotrimethylsilane and tetrabutylammonium fluoride. The bioconjugation was then undertaken with a small model peptide, Gly-Gly-Arg (GGR), tagged with 7-amino-methylcoumarin (AMC) and functionalized at the N-terminus with an alkyne function. The same ATRP catalyst was used for its coupling with the ω-azido PS to yield the corresponding PS-(GGR-AMC) biohybrid. The versatility of this approach was demonstrated with an alkyne-containing BSA for the construction of PS-BSA giant amphiphiles. Self-assembly of PS-b-(GGR-AMC) led to 150–2000 nm vesicles with a broad particle size distribution whereas PS-BSA bioconjugates yielded 30–70 nm micelles.
This synthetic methodology was expanded by Lutz and co-workers to the precise functionalization of RGD peptide (GGRGDG) with ω-azido poly(PEGA)119 and to the anchoring of TAT peptide120 (GGYGRKKRRQRRRG), a protein transduction domain (PTD) of the human immunodeficiency virus (HIV), on ω-azido PS. The RGD peptide was synthesized by solid-phase peptide synthesis (SPPS) followed by amidation of its N-terminus with pentynoic acid and cleavage from the support (see also section 3.4.1 Solid-phase peptide synthesis of peptide macroinitiators for CLRP). In parallel, a 6.85 kDa poly(PEGA) with low polydispersity index (Mw/Mn = 1.21) was treated with sodium azide. As a proof of concept, coupling reactions were demonstrated with low molecular weight alkyl azides using a CuBr/4,4′-di(5-nonyl)-2,2′-bipyridine (dNbpy) catalytic system. Finally, the alkyne functionalized RGD peptide was successfully attached onto ω-azido poly(PEGA) using CuBr/bpy. Similarly, a TAT peptide was functionalized with pentynoic acid prior to its reaction with ω-azido PS (Mn = 2.2 kDa, Mw/Mn = 1.21). The final CuAAC coupling between the protected oligopeptide and the polymer was performed using CuBr/bpy and afforded the PS-TAT bioconjugate in high yield.
In a similar approach previously used by Taton and Lecommandoux,110 He and co-workers reported the formation of ABC triblock polypeptide-polymer bioconjugates using the CuAAC methodology. In this study, azide ω-functional PEG-b-PS121 or PEG-b-PtBA122 diblock copolymers were synthesized by ATRP from a PEG-based ATRP macroinitiator using CuBr/PMDETA catalyst and subsequent azidation of the chain-end. In parallel, a series of α-propargyl PBLG (Mn = 2.7–26.6 kDa, Mw/Mn < 1.20) and α-propargyl PzLLys (Mn = 10.5–13.1 kDa, Mw/Mn < 1.27) was synthesized by ring-opening polymerization (ROP) (see also section 3.4.3. Combination of NCA polymerization and CLRP) of BLG-NCA (γ-benzyl-L-glutamate N-carboxyanhydride) and Z-L-Lys NCA (Nε-carbobenzoxy-L-lysine N-carboxyanhydride), respectively. Final CuAAC (CuBr/PMDETA) between azide and alkyne derivatives led to well-defined PBLG-b-PS-b-PEG and PzLLys-b-PAA-b-PEG triblock copolymer bioconjugates.
For instance, Davis and co-workers published the synthesis of heterotelechelic polymers for the bioconjugation of proteins using RAFT. An α-azide, ω-PDS heterotelechelic RAFT agent (RA8, Fig. 7) was synthesized and used to mediate the polymerization of five different monomers (MMA, HPMA, NIPAAm, PEGA and styrene) in the presence of AIBN as a source of radicals. The resulting difunctional polymers were obtained with good control over the molecular weight and the molecular weight distribution (Mn = 3.2–16.2 kDa, Mw/Mn = 1.08–1.7, conversion = 46–90%).123 Those α,ω-heterotelechelic polymers were then engaged in two consecutive conjugation steps with: (i) biotin amidopropyne clicked to the azido terminus of the polymers through CuAAC reaction using CuSO4/sodium ascorbate catalytic system (90% functionalization with HABA assay) and (ii) glutathione (γ-ECG), a model tripeptide containing a free cysteine or BSA via the PDS group. The coupling yield between ω-PDS PNIPAAm and γ-ECG was found to be 95 ± 5% whereas only ∼10% yield was obtained with BSA, assigned to the steric hindrance between the two macromolecules involved in the reaction.
Another strategy is based on the synthesis of bis-functionalized polymers for bioconjugation to cysteine-containing biomolecules using a post-polymerization dimerization procedure.124 In this work, a α-dimethyl fulvelene-protected maleimido ATRP initiator (I14, Fig. 5) initiated the polymerization of styrene at 80 °C in the presence of CuBr/CuBr2/PMDETA catalyst. The α-functional PS (Mn = 2.31 kDa and Mw/Mn = 1.15) was engaged in atom transfer radical dimerization reaction triggered by CuBr/PMDETA in the presence of 4 eq. of nano-Cu(0) at 70 °C. Bis fulvelene-protected maleimido dimeric PS (Mn = 4.2 kDa, Mw/Mn = 1.32) was then deprotected and successfully coupled at both sides with N-acetyl-L-cysteine methyl ester as a thiol-containing model compound.
A simple route towards heterotelechelic polymers for bioconjugation of two different proteins was also reported using the RAFT technique (Fig. 12). Biotin α-functional PNIPAAm (Mn = 10.9 kDa, Mw/Mn = 1.08) was obtained from a biotinylated trithiocarbonate RAFT agent (RA9, Fig. 7) and a protected maleimide was installed at the chain-end by radical cross coupling at 70 °C between the trithiocarbonate moiety (93% chain-end) and a protected maleimide azo-initiator.113 However, to avoid a significant loss of biotin end-group upon retro Diels–Alder deprotection at high temperature (120 °C), the RAFT agent must contain only amide groups (RA10, Fig. 7). The formation of BSA-PNIPAAm-SAv (see also section 4.1 Biotin/(Strep)avidin binding) heterodimer biohybrid was achieved in two steps: (i) first by reaction with BSA in PBS and subsequent purification to remove unreacted polymer and (ii) by using a fluorescently labeled streptavidin (SAv). The formation of the corresponding heterodimer was demonstrated by SDS-PAGE after visualization under UV light or after Coomassie Blue staining. Following the same pathway but with a symmetrical bistrithiocarbonate RAFT agent allowed homodimeric polymer-protein bioconjugate to be obtained.114 This was applied to the conjugation to T4L. By SDS-PAGE analysis, two new bands were observed at ∼48 kDa (21%) and ∼26 kDa (79%) corresponding respectively to dimeric and monomeric T4L-polymer conjugates.
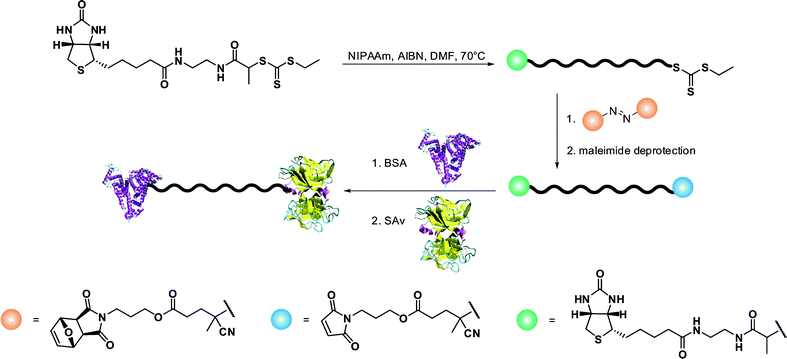 | ||
| Fig. 12 Synthetic approach to the formation of protein-heterodimer conjugates via the RAFT technique.113 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was successful as confirmed by SDS-PAGE in non-reducing conditions. The use of these midchain-functional polymers could be of great importance considering their “umbrella-like” shape allows a better protection against proteolytic degradation compared to the use of linear polymers.
:1) was successful as confirmed by SDS-PAGE in non-reducing conditions. The use of these midchain-functional polymers could be of great importance considering their “umbrella-like” shape allows a better protection against proteolytic degradation compared to the use of linear polymers.
The first example reported the synthesis of well-defined poly(N-methacryloyloxysuccinimide) (PNMS) by ATRP.126 The polymerization of N-methacryloyloxysuccinimide (NMS) from a classical ATRP initiator was carried out using CuBr/bpy catalyst during less than 15 min (80–96% conversion, Mn = 12.3–40.7 kDa, Mw/Mn = 1.13–1.20). The coupling reaction between PNMS and two model peptides (i.e. Gly-OMe and Gly-Gly-β-naphtylamide hydrobromide) was undertaken and revealed a good correlation with the stoichiometry of the model peptides added (the unreacted activated esters were further coupled with 1-amino-2-propanol). With Gly-Gly-β-naphtylamide hydrobromide as the model peptide, the conjugation resulted in a copolymer composed of HPMA biocompatible units89 and Gly-Gly peptidic pendant side chains. However, it was shown that sterically-hindered NHS side chains are prone to aminolytic ring-opening of the succinimide moiety and intramolecular attack by amides on neighbouring activated esters, leading to a non-negligible fraction of glutarimide residues.127
The same methodology was also developed for the copolymerization of HPMA and N-methacryloyloxysuccinimide by the RAFT process128 and its further multifunctionalization with HTSTYWWLDGAPK peptide, which is known to inhibit the assembly of anthrax toxin.129 Well-defined P(HPMA-co-NMS) random copolymers (Mn = 4.3–53.6 kDa, Mw/Mn < 1.22) obtained with a constant NMS ratio of 20 mol% were subjected to end-group removal (to favour side chains bioconjugation) and coupled to HTSTYWWLDGAPK peptide via its lysine residue. Subsequent addition of 1-amino-2-propanol to the reaction mixture ensured that all NHS esters have reacted. It was shown that the resulting bioconjugate exhibited much higher anthrax inhibition efficiency than the monovalent counterpart.
Two other functionalizable side-chain polymers for drug delivery purposes were successfully developed by Maynard’s group (Fig. 13).130,131p-Nitrophenyl methacrylate (pNPMA) and diethoxypropyl methacrylate (DEPMA) monomers were polymerized by RAFT from cumyl dithiobenzoate under AIBN initiation. The resulting poly(p-nitrophenyl methacrylate) (PpNPMA) polymer (Mn = 9.6 kDa, Mw/Mn = 1.15) was subjected to coupling with Gly-OMe as a model compound and yielded a high degree of functionalization (86%). Similarly, the acetal side chains of poly(diethoxypropyl methacrylate) (PDEPMA) (Mn = 7.7–14.4 kDa, Mw/Mn = 1.29–1.26) were subsequently turned into the corresponding aldehyde by acidic catalytic hydrolysis. The newly formed poly(3-formyl ethyl methacrylate) (PFEMA) polymer then allowed conjugation with aminooxy-RGD peptide to be readily performed.
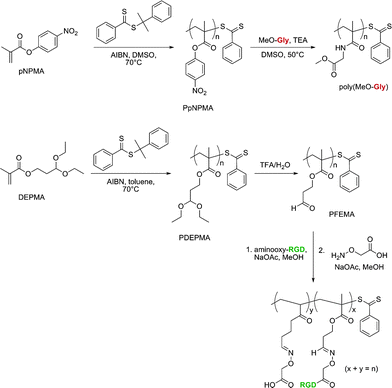 | ||
| Fig. 13 Development of polymers with activated ester and protected aldehyde side chains for bio-functionalization.130,131 | ||
3.2 Polymerization from protein/peptide macroinitiators: the “grafting from” method
The opposite pathway, which consists of growing a polymer chain by CLRP from a protein/peptide macroinitiator, the so-called “grafting from” method, has recently received significant interest due to its two main potential advantages: (i) a higher bioconjugation efficiency is anticipated due to a lower steric hindrance and (ii) the purification of the final materials is easier as only small molecules have to be removed such as unreacted monomer (and possibly ATRP catalyst or remaining radical initiator for RAFT), in contrast to preformed polymer for the “grafting to” approach.Matyjaszewski,132 Haddleton,133 Maynard84 and Velonia,134 investigated the “grafting from” strategy in combination with the ATRP process. From the reaction between amino groups of lysine residues and 2-bromoisobutyryl bromide, Matyjaszewski and co-workers were able to control the attachment of ATRP initiating moieties on α-chymotrypsin by varying the [ATRP initiator]0/[α-chymotrypsin]0 molar ratio.132 A ratio of 12:1 gave a single initiating site whereas increasing this ratio from 43:1 up to 85:1 resulted in the conjugation of respectively 3–7 and 7–10 initiator moieties per protein (catalytic activity was above 90% each time). These α-chymotrypsin ATRP macroinitiators initiated the polymerization of PEGMA in PBS at pH 6.0 with a CuBr/bpy catalyst. The almost monodisperse α-chymotrypsin-poly(PEGMA) bioconjugates were obtained with a good control whereas the typical “grafting to” method using either monomethoxy poly(ethylene glycol)-succinimidyl propionate (MePEG-SPA) or NHS-terminated poly(PEGMA) afforded a mixture of bioconjugates with higher PDI values. The authors also observed that the bioconjugates retained from 50 to 86% of the bioactivity of the native α-chymotrypsin.
Maynard and co-workers investigated the in situ formation of protein-polymer bioconjugates using either BSA or T4L as protein macroinitiators.84 BSA was first reduced with TCEP in order to maximize the number of free thiols. The resulting free cysteine residues were then coupled with a thiol-reactive PDS α-functional ATRP initiator (I10, Fig. 5) to afford a mixture of BSA macroinitiators with either 1 or 3 initiation sites. Then, NIPAAm was polymerized in water with CuBr/bpy in the presence or absence of 2-bromoisobutyrate-functionalized resin as a sacrificial initiator. The presence of the disulfide linkage allowed for further cleavage of PNIPAAm from BSA which was then analyzed by SEC (Mw/Mn = 1.34). The polymerization from T4L was also assessed and the bioconjugate activity was maintained after the polymerization step.
Haddleton and co-workers turned BSA and lysozyme into efficient protein macroinitiators with maleimido or NHS functionalized ATRP initiators, respectively (I1 and unprotected I9, Fig. 5).133 The corresponding BSA macroinitiator initiated the polymerization of PEGMA or DMAEMA using CuBr/N-(ethyl)-2-pyridylmethanimine as the catalytic system. In order to better monitor the bioconjugation reaction, small amounts of either hostasol methacrylate (HMA) or rhodamine methacrylate (RMA) fluorescent co-monomers were added. The resulting fluorescent bioconjugates were then successfully characterized by SDS-PAGE, SEC-HPLC and fluorescence spectroscopy. Conferring fluorescent properties to bioconjugates is a general approach that facilitates their detection during biomedical assays.
More recently, Velonia demonstrated the formation of amphiphilic BSA-PS bioconjugates by in situ ATRP and their further hierarchical self-assembly in aqueous solution to form bio-nanocontainers and nanoreactors.134 Following the work previously reported by Haddleton and co-workers,133 a BSA ATRP macroinitiator was prepared and engaged in emulsion polymerization (10% DMSO, PBS pH 7.4) of styrene triggered by addition of the CuBr/N-(propyl)-2-pyridylmethanimine catalyst. Well-defined BSA-PS bioconjugates (Mw/Mn < 1.2) were obtained from high monomer-to-macroinitiator ratios (1500:1 up to 3000:1). Enzymes entrapment experiments have also been performed with fluorescently tagged papain or Horse Radish Peroxidase (HRP). Further on, the catalytic activity of HRP was investigated based on the oxidation of 3,3′,5,5′- tetramethylbenzidine (TMB, substrate of HRP) in the presence of hydrogen peroxide (H2O2). The catalytic reaction took place in the nanoassemblies: whereas TMB was able to permeate into the superstructures (being catalytically transformed and then released in solution), catalytically active proteins (HRP, papain) were retained in the superstructures. This study highlighted a potential application in the area of biotechnology.
RAFT polymerization was also employed by Davis135,136 and Sumerlin137 for the in situ formation of polymer-protein bioconjugates. Davis and co-workers functionalized BSA with a PDS-based RAFT agent (RA12, Fig. 7) and used the resulting BSA macro-RAFT agent to mediate the aqueous polymerization of PEGA upon γ-irradiation to initiate the polymerization.135 The formation of BSA-poly(PEGA) bioconjugate was confirmed by SEC and by non-reducing PAGE. Aliquots from the polymerization medium were withdrawn at regular intervals of time and, after cleavage of the bioconjugates with TCEP aqueous solution, the poly(PEGA) was recovered and analyzed by SEC to show the linear evolution of poly(PEGA) molecular weight with conversion and the low polydispersity indexes obtained with the RAFT technique. NIPAAm and hydroxyethyl acrylate (HEA) were also polymerized from the BSA macro-RAFT agent in PBS at pH 6.0 (to avoid possible hydrolysis of RAFT agent and/or denaturation of the protein) using VA044 as a free radical initator (Fig. 14).136 The linear evolution of the logarithmic conversion was observed with time and SEC analysis revealed the formation of biomacromolecules with hydrodynamic diameters larger than that of native BSA.
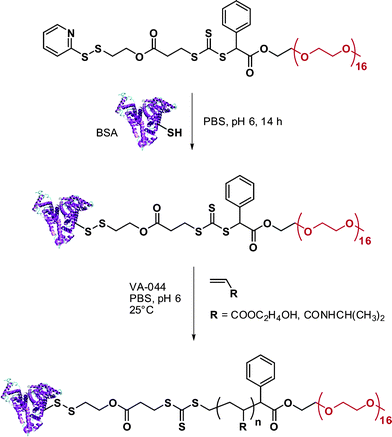 | ||
| Fig. 14 Synthesis of BSA-poly(HEA) and BSA-poly(NIPAAm) conjugates by RAFT polymerization using a BSA-macroRAFT agent.136 | ||
Sumerlin and co-workers also took advantage of the “grafting from” approach to achieve the synthesis of thermally responsive BSA-PNIPAAm bioconjugates with tunable bioactivity via the RAFT technique.137 After selective coupling of the BSA free cysteine to a maleimido RAFT agent (RA13, Fig. 7), NIPAAm was polymerized in PBS at pH 6 to give the corresponding BSA-PNIPAAm bioconjugates. The logarithmic monomer conversion, followed by both 1H NMR and gravimetry analysis, was linear and suggested a constant concentration of propagating radicals in the polymerization medium up to high monomer conversion. Aqueous SEC and SDS-PAGE permitted the formation of the bioconjugates to be assessed. TCEP protein degradation allowed to characterize the PNIPAAm polymer attached to the protein and revealed an efficient control up to 94% monomer conversion (Mn = 234 kDa and Mw/Mn = 1.38). Circular dichroism (CD) spectroscopy indicated that BSA retained its secondary structure even after coupling with the maleimide functionalized RAFT agent or after polymerization of NIPAAm. Interestingly, the responsive behaviour of the immobilized polymer facilitated the isolation of the bioconjugate and also allowed environmental modulation of its bioactivity.
Deriving from the work reported on the CLRP of nucleoside-containing monomers and/or initiators,138–141 Venkataraman and Wooley developed the synthesis of an amino-acid-based ATRP initiator for bioconjugates synthesis. O-protected L-valine was reacted with 2-bromopropionyl bromide to afford the resulting amino-acid-based ATRP initiator that initiated the sequential polymerization of tert-butyl acrylate (tBA) and styrene in the presence of CuBr/PMDETA catalyst.142 All the criteria of CLRP were obtained, leading to the formation of well-defined O-protected L-valine-PtBA-b-PS block copolymer (Mn = 22.5 kDa, Mw/Mn = 1.22).
An original strategy to prepare peptide-polymer conjugates with precise sites of attachment was reported by Maynard and co-workers (Fig. 15).143 The concept was to design an artificial amino acid containing an ATRP initiator moiety as a side chain, followed by its incorporation into a peptide sequence and the initiation of the polymerization from the resulting biomaterial. This is exemplified by: (i) Fmoc-protected tyrosine modified with 1-chloroethyl-phenyl group suitable for ATRP of styrene and (ii) Fmoc-Ser(OTrt)-OH modified with the 2-bromoisobutyrate group for ATRP of methacrylates. Kinetic studies under optimal conditions indicated a controlled polymerization with low PDI (≤ 1.25).
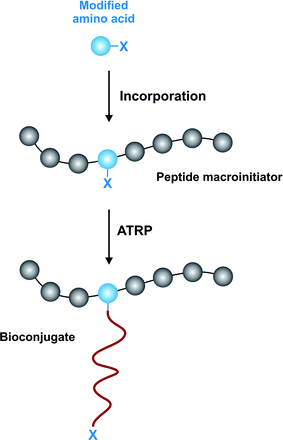 | ||
| Fig. 15 Incorporation of amino acid initiator for precise biohybrid synthesis.143 | ||
3.3 Polymerization of peptide-based monomers: the “grafting through” method
Another very interesting and promising alternative to the anchoring of proteins/peptides on the pendant side chains of polymers relies on the polymerization of peptide-based monomers.144,145 It offers the advantage over the “grafting to” method that the functionalization is quantitative and does not require additional post-polymerization steps.Van Hest brought a significant breakthrough in this domain,146 as his group synthesized several peptide-based (glutamic acid,147 VPGVG,147,148 AGAG,149,150 Gramicidin S151) methacrylates either by solution- or solid-phase peptide synthesis. These monomers were polymerized by ATRP in the presence of CuCl/bpy in DMSO (essential for efficient solubilization) to form hybrid polymers or hybrid diblock copolymers (Fig. 16). Glutamic acid-based methacrylate was synthesized upon DCC coupling of O-tBu, N-Boc protected glutamic acid with HEMA. The glutamic acid-based methacrylate (Glu-EMA) was then polymerized either from ethyl-2-bromo isobutyrate (EBIB) or di-α,ω-bromoisobutyrate-PEG macroinitiator to afford the corresponding homopolymer (Mn = 8.7 kDa, Mw/Mn = 1.11) and diblock copolymer (Mn = 10.9 kDa, Mw/Mn = 1.22).147 Similarly, the VPGVG thermoresponsive peptide sequence (upon LCST, a random coil to a type II β-turn transition is observed), which is predominantly present in tropoelastin, was transformed into the corresponding VPGVG-based methacrylate (VPGVG-MA) and polymerized with the same initiators and the same catalytic system. It yielded either a P(VPGVG-MA) homopolymer (Mn = 53.0 kDa, Mw/Mn = 1.25) or a P(VPGVG-MA)-b-PEG-b-P(VPGVG-MA) ABA triblock copolymer (Mn = 60.0 kDa, Mw/Mn = 1.24).147 More interestingly, those materials retained the thermally-responsive feature associated to the VPGVG peptidic sequence, as observed by CD spectroscopy and turbidity measurements. The authors observed that increasing the concentration of this biohybrid triblock copolymer or increasing the peptidic block length resulted in a decrease of the LCST whereas the increase of the pH solution induced lower LCST.148
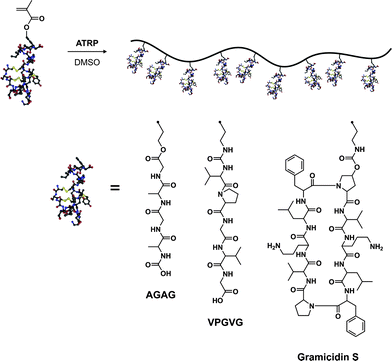 | ||
| Fig. 16 Polymerization of peptide-based monomers by ATRP.147–151 | ||
The polymerization of VPGVG-MA was also successfully undertaken by Cameron and co-workers using the RAFT polymerization with 4-cyanopentanoic acid dithiobenzoate and V-501 initiator at 70 °C.152 Surprisingly, the authors found that transition temperatures of those materials were much lower than those reported for the P(VPGVG-MA)-b-PEG-b-P(VPGVG-MA) triblock copolymer.148 This was assigned to the presence of the PEG central block that might affect the ability of the elastin-based polymers (EBPs) to phase separate and/or to the higher molecular weight obtained in the work of Cameron. The influence of the pH, the EBP concentration and the EBP molecular weight over the LCST of the polymer solutions was then investigated. A decrease of the LCST was observed by lowering the pH. Similarly, as expected from a previous report on elastin linear polymers (ELPs),153 a linear decrease of the LCST was observed when the concentration of the polymer was increased. Finally, a linear decrease of the LCST as a function of the increasing molecular weight of the EBPs was demonstrated.154,155
N-Boc protected AGAG-based methacrylate (Fig. 16), derived from the AGAG peptidic sequence (part of the silk protein) also known to induce β-sheets formation, and MMA were sequentially polymerized by ATRP in DMSO from 1,4-(2′-bromo-2′-methylpropionato)benzene (difunctional ATRP initiator) to afford a poly(methyl methacrylate)-b-poly(N-Boc-AGAG-MA)-b-poly(methyl methacrylate) triblock copolymer with good control (Mw/Mn = 1.19).149,150 FTIR investigations confirmed the capability of these AGAG-based block copolymers to form β-sheet structures. Van Hest also reported the possibility of polymerizing bulky cyclic peptide-based methacrylates such as Gramicidin S (cyclic decapeptide), an analogue of an antibiotic that had the propensity to form β-sheets (Fig. 16). Polymerization was performed under ATRP conditions from EBIB using CuCl/PMDETA as the catalyst, yielding the poly(Gramicidin S methacrylate) with a narrow MWD (Mw/Mn = 1.09) after 65% monomer conversion.151 As shown by FTIR analysis, the propensity to form intramolecular β-sheets was maintained.
3.4 In situ synthesis of polymer-peptide bioconjugates
In this section, the synthesis of both the polymer and the peptide is undertaken, which allows great flexibility over the overall design of the resulting bioconjugate. This can be achieved either by solid phase peptide synthesis or by the polymerization of NCA. Whereas, the first approach is generally limited to medium-sized peptides, the latter allows longer peptide sequences to be obtained in good yield and large quantities but without controlling the primary amino acid sequence.156,157SPPS in combination with ATRP were also employed by Van Hest and co-workers, for oligopeptide-polymer bioconjugates synthesis (Fig. 17).161 The protected Ser-Gly-Ala-Gly-Ala-Glu-Gly-Ala-Gly-Ala-Ser-Gly peptide was grown from Wang resin followed by deprotection and derivatization of the two Ser residues with 2-bromoisobutyryl bromide. After cleavage from the support, polymerization of MMA with CuCl/PMDETA complex was undertaken. A linear first-order kinetic plot and a molecular weight of 1.12 kDa were obtained. The two PMMA blocks were then recovered upon basic conditions revealing a PDI of 1.17. Self-assembly behaviour study of the PMMA-Ser-Gly-Ala-Gly-Ala-Glu-Gly-Ala-Gly-Ala-Ser-Gly-PMMA triblock-like copolymer bioconjugate indicated a propensity to form hollow polymersomes and large compound micelles in aqueous solution. Unfortunately, the initial structural conformation of the oligopeptide (β-hairpin) was not maintained upon polymerization of MMA. In contrast, it led to a random-coil structure, assigned to the steric hindrance of the PMMA blocks or to an aggregation process that was too fast.
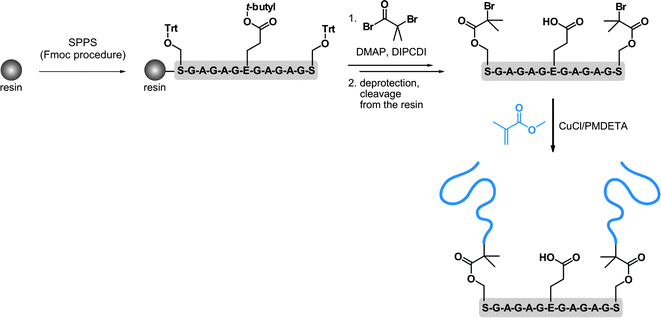 | ||
| Fig. 17 Synthesis of oligopeptide-polymer bioconjugates by a combination of solid-phase peptide synthesis (SPPS) and ATRP.161 | ||
Similarly, the RGD peptide (GRGDSP) was grown using Fmoc standard chemistry, deprotected and subsequently amidated with 2-bromo-2-methylpropionic acid. The released peptide-based ATRP initiator triggered the polymerization of dimethyl acrylamide (DMAA) in DMSO with the CuCl/tris[2-(dimethylamino)ethyl]amine (Me6TREN) catalyst. Even though side reactions led to non-linear first-order kinetic plots, GRGDSP-poly(dimethyl acrylamide) (GRGDSP-PDMAA) bioconjugates were obtained (Mn = 15–30 kDa) with rather low polydispersity indexes (1.4–1.6).162 The bioconjugates were then tethered on a silane surface through a photochemical immobilization process involving a benzophenone moiety already present at the surface of the glass slide. The authors were able to tune the apparent peptide film concentration by blending the GRGDSP-PDMAA with PDMAA homopolymer. The immobilized GRGDSP-PDMAA bioconjugate, containing a cell repelling polymer block and a cell-promoting adhesion polypeptide (RGD sequence), eventually showed a promoted cell adhesion feature of human skin fibroblasts (even at 0.02 wt% of peptide in the film). This strategy also allowed the synthesis of micropatterned peptide-polymer films that can be devoted to live-cell biochip applications.
Biesalski and co-workers took advantage of the ability of a cyclic octapeptide with an even number of alternating D- and L-amino-acids that self-assemble into well-defined nanotubes, to carry on the polymerization by ATRP of NIPAAm from these nanoarchitectures.163,164 The cyclooctapeptide was synthesized by standard Fmoc protocols on a Wang-Tentagel resin, difunctionalized on its 2 lysine residues by amidation with 2-bromo-N-butyl-2-methylpropanamide and cleaved from the resin. Following the self-assembly of these cyclic peptides by intermolecular hydrogen bonding,165–169 peptide nanotubes were formed and displayed at their periphery ATRP initiation sites suitable for subsequent surface-initiated ATRP of NIPAAm in aqueous dispersion.163 By tuning the polymerization time, the authors were able to tune the length of the polymer chains grown at the surface of the peptides nanotubes. More importantly, the length of the peptide nanotubes remained the same until a polymerization time of ∼5 h. After this time limit, small and uniform particles appeared and the concentration of the peptide-polymer hybrid nanotubes (PPNTs) dramatically decreased, which was assigned to a break-up of the PPNTs into smaller and well-defined nanoobjects.164 Complementary studies have been recently reported using a cyclic peptide modified at 3 distinct positions with ATRP initiation sites.170 AFM and FTIR investigations demonstrated that the PnBA-cyclic peptide adopted core-shell rod-like nanoarchitectures with an internal β-sheet structure surrounded by a soft PnBA coating.
Alkyne-terminated PNIPAAm (Mn = 2 kDa, Mw/Mn < 1.2) obtained from RAFT was successfully coupled via CuAAC to MUC1 VNTR peptide bearing N-terminal azide group, obtained from SPPS. By self-assembly of the conjugate in water, nearly uniform micelles of 60 ± 3 nm in diameter were obtained, presenting multiple copies of MUC1 VNTR peptide on its surface.171 Further functionalization of the thiol end-group was also undertaken with pyrene maleimide.
To demonstrate the versatility of this methodology, Wooley also reported the synthesis of bioconjugates from an antimicrobial peptide tritrpticin (VRRFPWWWPFLRR) using both NMP or ATRP.176 Tritrpticin was synthesized by Fmoc SPPS and coupled with fluorine-labeled TIPNO-based (N3, Fig. 2) alkoxyamine or with 2-bromoisobutyryl bromide for conducting NMP or ATRP, respectively. Sequential polymerization of tBA and styrene from the immobilized tritrpticin-derivatized macroinitiator yielded well-defined tritrpticin-PAA-b-PS block copolymers. It was observed that tritrpticin-PAA-b-PS was able to self-assemble into 51 nm micelles of narrow particle size distribution together with a small proportion of larger-scale aggregates. Micelles decorated with tritrpticin showed an enhancement of the antimicrobial activity against Staphylococcus aureus (S. aureus) and Echerichia coli (E. coli) when compared to the free peptide.
Washburn and co-workers also developed the solid-phase peptide synthesis of PHEMA-GRGDS bioconjugates by ATRP.177 The GRGDS peptide functionalized with an ATRP initiator triggered the polymerization of HEMA with CuCl/bpy at 50 °C. The acidic cleavage of the bioconjugate from the resin afforded rather well-defined GRGDS-PHEMA bioconjugates (Mw/Mn = 1.47). It was shown that the bioconjugates promoted the bioadhesion and spreading of mouse NIH-3T3 fibroblast cells.
By using sequential ATRP and NCA polymerization, Chaikof and co-workers prepared ABA poly(L-alanine)-b-poly(2-acryloyloxyethyl-lactoside)-b-poly(L-alanine) (PLA-b-PAEL-b-PLA) triblock bioconjugates.184,185 Dibromoxylene was used as a difunctional ATRP initiator for the polymerization of 2-acryloyloxyethylocta-acetyl-lactoside (AEL, G7, see Fig. 23) with CuBr/bpy. By tuning the [monomer]0/[initiator]0 molar ratio, a range of glycopolymers (see also section 4.3 Glycopolymers and sugar-protein interaction) with Mn from 9.3 to 38.2 kDa were prepared in a controlled fashion (Mw/Mn = 1.19–1.35). The bromine end-group was then converted into an amine to initiate the ring-opening polymerization (ROP) of Ala-NCA, leading to the formation of PLA-b-PAEL-b-PLA triblock hybrid copolymers after deprotection of the O-protecting acetyl groups. The same approach was used for the preparation, after removal of the benzyl group and deacetylation of the lactose units, of poly(L-glutamate)-b-poly(2-acryloyloxyethyl-lactoside)-b-poly(L-glutamate) triblock hybrid copolymers185 by NCA polymerization of β-benzyl-L-glutamate (BLG). When these amphiphilic triblock hybrid copolymers were aggregated in aqueous solution, FT-IR spectroscopy demonstrated that the α-helix/β-sheet ratio increased with an increase of the polypeptide block length.
A similar approach was adopted by Brzezinska and Deming for the synthesis by sequential ATRP and NCA polymerization of diblock poly(γ-benzyl-L-glutamate)-b-poly(methyl methacrylate) (PBLG-b-PMMA) hybrid copolymers.186 After ATRP of MMA, the bromine end-group of the resulting PMMA block (Mn = 3.8 kDa, Mw/Mn = 1.2) was converted into an amine moiety via a three-step reaction187 and subsequently transformed into the required macroinitiator to initiate the ROP of γ-benzyl-L-glutamate NCA. Well-defined diblock PBLG-b-PMMA copolymers were eventually obtained (Mw/Mn ≤ 1.2).
Along similar lines, this strategy was used by Taton and co-workers for the synthesis of AB2 miktoarm polystyrene-b-(poly(glutamic acid))2 (PS-b-(PGA)2) star copolymers.188 In this work, styrene was polymerized by ATRP before a specific chain-end modification step was achieved to introduce a gemini amino group to further initiate NCA polymerization. After ring-opening polymerization of γ-benzyl-L-glutamate (BLG), a PS-b-(PBLG)2 was obtained with low polydispersity (Mw/Mn ≤ 1.3) and with Mn in the 6.0–25.1 kDa range, followed by hydrolysis of the γ-benzyl to yield the corresponding PS-b-(PGA)2 miktoarm copolymer. Upon dispersion in aqueous solution, these PS-b-(PGA)2 copolymer self-assembled into well-defined micelles that exhibited pH-responsive properties. A random coiled structure at pH > 12 with RH = 16 nm was observed whereas at pH < 5, the PGA blocks took a compact α-helix conformation, leading to a slight decrease of the average particle size (RH = 11 nm). When a trifunctional ATRP initiator was used, well-defined PS-b-(PBLG)3 star copolymers were obtained (Mn = 10.5–28.1 kDa, Mw/Mn = 1.2–1.4).189 They exhibited a higher conformational stability than their linear counterparts, as observed by DSC and IR spectroscopy.
The utilization of a heterobifunctional initiator for both ATRP and ROP of NCA represents an alternative strategy to the chain-end modification method previously described. In this view, such a molecular initiator was developed by Menzel and co-workers for the sequential NCA polymerization of BLG and ATRP of MMA through a divergent chain growth. It comprised a nickel amido-amidate terminus for NCA polymerization and a classical ATRP initiating site (I15, Fig. 5).190 Even though poor initiation efficiencies were observed, a good control of the BLG polymerization was observed (Mw/Mn = 1.2–1.4) and the resulting PBLG block displayed the expected α-helical structure. ATRP of MMA catalyzed by CuBr/HMTETA or CuBr/bpy in DMF at 80–90 °C was then undertaken and led to Mn in the 41.0 to 110.0 kDa range with low polydispersities (Mw/Mn = 1.20–1.39).
In two more recent studies from the same group, NCA polymerization in combination with either NMP191,192 or ATRP191 was investigated to construct bioconjugates from difunctional initiators (Fig. 18). ROP of BLG-NCA was initiated from an amino TIPNO-based (N3, Fig. 2) alkoxyamine, followed by the NMP polymerization of styrene via a one-pot process to afford PBLG-b-PS bioconjugates with low PDI (Mw/Mn ∼ 1.1).192 In the second study, double-headed initiators were prepared: (i) I15 (Fig. 5) was used for the sequential ROP of BLG-NCA and ATRP of MMA while (ii) the second one, based on the TIPNO (N3, Fig. 2) alkoxyamine, was used for the sequential ROP of BLG-NCA and NMP polymerization of styrene.191 Well-defined PBLG-b-PMMA were obtained with rather-low PDI whereas the obtaining of PBLG-b-PS required a fine tuning of the NMP reaction conditions in order to ensure a good control.
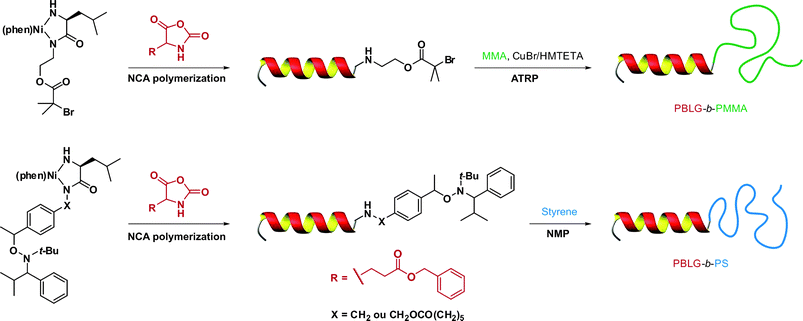 | ||
| Fig. 18 Synthesis of bioconjugates by NCA polymerization and subsequent ATRP or NMP.191,192 | ||
3.5 Bioconjugation to surfaces
CLRP methods also represent a convenient approach for the synthesis of well-defined polymer-grafted surfaces193–196 and have been naturally applied to design novel grafted polymer-protein/peptide bioconjugates.Kang and co-workers reported the synthesis of Si(111)-grafted poly(glycidyl methacrylate) (PGMA) brushes for subsequent glucose oxidase (GOD) immobilization by ATRP (Fig. 19).198 This procedure was first based on a covalent attachment of 4-vinylbenzyl chloride (VBC) on a Si(111) surface via radical-induced hydrosilylation. Subsequently, glycidyl methacrylate (GMA) was polymerized in a DMF–water mixture using a CuCl/CuCl2/bpy catalyst to obtain the Si-g-PGMA hybrid surface. In the final step, GOD was anchored on the Si-g-PGMA polymer by epoxy ring-opening of the PGMA with amine moieties of lysine residues, with a concentration estimated to be 0.17–0.23 mg cm−2. The authors observed that this concentration increased with the thickness of the grafted PGMA layer until a plateau where the accessibility of GOD to the surface was restricted by steric hindrance due to the polymer brushes. Activity enzyme tests revealed that the immobilized GOD displayed a good stability and a relative activity of ∼60% when compared to an equivalent amount of free native GOD, which is higher than with previously developed immobilization methods.199–202
 | ||
| Fig. 19 Design of Si(111)-grafted poly(glycidyl methacrylate) brushes by ATRP for glucose oxidase (GOD) immobilization.198 | ||
Not only inorganic but also biological surfaces can be of interest for bioconjugation investigations. For example, Haddleton and co-workers used hair as a biological surface due to keratin fibres that can be easily targeted with amine reactive functional polymers.203 Well-defined α-NHS fluorescently tagged poly(PEGMA) copolymers were prepared in toluene using CuBr/N-(ethyl)-2-pyridylmethanimine as the catalyst (Mn = 11.8–45.6 kDa, Mw/Mn = 1.08–1.48) from I2 (Fig. 5), followed by coupling with hair at ambient temperature and careful rinsing. CLSM showed intense fluorescence on the outer layer of the hair, in agreement with a covalent polymer coating. Besides, DSC experiments demonstrated a substantial increase of the denaturation temperature of the α-helical material of the keratin fibres upon bioconjugation.
Wooley and co-workers developed a method in which the TAT peptide was coupled to SCK nanoparticles.204,205 These nanoparticles were obtained by self-assembly of poly(acrylic acid)-b-poly(methyl acrylate) (PAA-b-PMA) prepared by sequential ATRP polymerization of tBA and MMA using CuBr/PMDETA and successive removal of tert-butyl groups. To ensure stability, a cross-linking reaction between carboxylic groups of the PAA shell and 2,2′-(ethylenedioxy) bis(ethylamine) via carbodiimide activation was performed. The TAT peptide extended by 4 additional glycine residues at the N-terminus was synthesized by SPPS and coupled within the nanoparticle shell with a carbodiimide-assisted coupling reaction. This study clearly demonstrated that the presence of the TAT peptide increased the intracellular uptake of the SCK nanoparticles. Besides this, in vivo and in vitro evaluations of these TAT-functionalized SCK nanoparticles were also undertaken and showed a preliminary assessment of their biocompatibility.205
The versatility of the method was demonstrated by the coupling of lysine-terminated A- or T-rich complementary peptide nucleic acid (PNA) sequences. In this way, PNA-decorated micelles that display different amounts of PNA moieties available at their surface (governed by the initial stoichiometry) were prepared. Due to the complementary bases tethered at their surface, A-rich and T-rich PNA-decorated micelles were able, by simple base pairing, to self-assemble into higher-ordered structures, as observed by AFM.206 It's worth mentioning that this strategy was also extended to the formation of nanoparticles functionalized with antigen for antibody-binding properties,207 saccharides for protein recognition,208 and folic acid for cancer-cell targeting,209 thus making it a powerful and truly versatile methodology.210
ATRP in combination with another transition-metal-catalyzed polymerization method, namely chain walking polymerization (CWP), has also been presented for the synthesis of polymer-protein colloidal bioconjugates.211 CWP is a powerful polymerization technique that offers great control over the macromolecular structure.212–215 A dendritic macroinitiator core was first synthesized by copolymerization of ethylene and a comonomer bearing an ATRP initiator moiety by CWP catalyzed by a chain walking palladium-α-diimine.212,216 ATRP of PEGMA was performed on the resulting dendritic macroinitiator at ambient temperature under CuBr/CuBr2/dNbpy catalysis and afforded core-shell nanoparticles. By changing the polymerization time, different copolymers were obtained with a broad range of molecular weights (Mn = 610–9180 kDa, Mw/Mn = 1.2–1.5) and with a radius of gyration, Rg, in the 19–64 nm range. The dendritic nanoparticles were then functionalized (49% yield) with N-acryloyloxysuccinimide by capping each methacrylate chain-end, for further conjugation to biomolecules through amidation reaction. The reactivity and bioavailability of the NHS-activated esters were assessed with fluorescein and ovalbumin (OB). The coupling reaction yield was found to be approximately 50% with fluorescein amine while the bioconjugation with OB afforded biohybrid superstructures with approximately 40 proteins per nanoparticle.
Van Hest reported the construction of clickable polymersomes from polystyrene-b-poly(acrylic acid) (PS-b-PAA) that were suitable for bioconjugation.217 To this end, the PS-b-PAA block copolymer was prepared by sequential ATRP of styrene and tBA (CuBr/PMDETA). The bromine terminal group was replaced by an azide upon treatment with azido trimethylsilane and tetrabutyl ammonium fluoride, followed by acidic hydrolysis of the tert-butyl groups. The resulting PS-b-PAA-N3 was able to self-assemble in aqueous solution into well-defined vesicles. The bioavailability of the azido groups was then successfully investigated by performing click reaction with a variety of alkyne ligands based on dansyl dye, biotin or enhanced green fluorescent protein (EGFP), using CuSO4/sodium ascorbate/tris-(benzyltriazolylmethyl) amine (TBTA) catalyst. Upon clicking, the nanoassemblies were shown to keep their initial morphology. However, due to the dense packing of the polymer chains in the vesicles, the optimum degree of functionalization was ∼25% with the dansyl alkyne.
Earlier work on clickable polymersomes from co-self-assembly of PS-b-PEG-N3 and PS-PIAT copolymers (where PIAT stands for L-isocyanoalanine(2-thiophen-3-yl-ethyl)amide) via ATRP was also reported.218,219 It was further extended to the immobilization of an active enzyme (Candida Antarctica lipase B, CalB) via its coupling with alkyne moieties displayed at the extremity of the PEG chains in the presence of CuSO4, sodium ascorbate and bathophenanthroline ligand. After purification of the CalB-decorated polymersomes, the structure of the hybrid vesicles remained unchanged and the enzymatic activity was maintained.220
Micelles of α,α′-PDS functional homotelechelic poly(PEGA)-b-PS-b-poly(PEGA) triblock copolymers (Mn ≈ 35.0 kDa, Mw/Mn < 1.25) obtained from RA14 (Fig. 7) under RAFT control, with 85% retention of the PDS end-groups, were also subjected to bioconjugation experiments.221 The PDS group accessibility was investigated using a thiol tethered rhodamine B or γ-ECG as a model tripeptide. In the latter case, 74% coupling was observed.
Adsorption of functional polymers onto silica particles was also investigated as a way of preparing coated colloidal objects suitable for bioconjugation. As presented by Zelikin and co-workers,222 a thiol-terminated PVP polymer from RAFT was adsorbed onto SiO2 particles by simple mixing leading to PVP-coated silica nanoparticles (Fig. 20). Thiol accessibility was demonstrated upon incubation with a thiol-reactive fluorophore, namely Alexa Fluor 488 (AF488) maleimide. These nanoparticles were stable in different aqueous solutions and organic solvents even though desorption of the polymer was observed for high concentration of DMF, DMSO or in the presence of polyethyleneimine (PEI) that induce electrostatic PEI-silica interactions. The authors finally coupled a fluorescent polypeptide (SIINFEKL), previously reacted with succinimidyl 3-(2-pyridyldithio)propionate (SPDP) to insert DTP moieties. Flow cytometry confirmed the formation of the covalent bond between the fluorescent peptide and the PVP-coated particles.
 | ||
| Fig. 20 End-functionalized poly(N-vinyl pyrrolidone) for bioconjugation and surface ligand immobilization onto coated silica nanoparticles.222 | ||
A recent study from Davis, Bulmus and co-workers proposed the formation of heterotelechelic bifunctional polymers for iron oxide nanoparticles (IONs) stabilization and biofunctionalization.223 In this work, a small library of α-dimethylphosphonate, ω-trithiocarbonate functionalized PS, PNIPAAm and poly(PEGA) polymers were prepared via RAFT with good control (Mn = 3.2–62 kDa, Mw/Mn < 1.24). After transformation of the α-dimethylphosphonate and aminolysis of the ω-trithiocarbonate with DTP, the resulting α-phosphonic acid, ω-PDS functionalized poly(PEGA)s were grafted onto IONs surface followed by bioconjugation experiments successfully performed with glutathione or NGR peptide via disulfide exchange to afford ION bioconjugates with 70 ± 10% yield for both peptide. Due to the PEG coating, IONs were also found to be resistant to protein adsorption.
4 Synthesis of polymer/peptide-protein bioconjugates using a non covalent approach
4.1 Biotin/(Strept)avidin binding
Biotin (5-(2-oxo-hexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoic acid), also known as vitamin H or B7, is a cofactor in the metabolism of fatty acids and leucine. It also plays a role in gluconeogenesis and helps to transfer carbone dioxide in several carboxylase enzymes. Only the D-(+)-biotin (among 8 isomers) is biologically active. It is also known that biotin has a strong affinity for avidin (Av, 64 kDa glycoprotein), neutravidin (NAv, 60 kDa non-glycosylated form of Av) and streptavidin (SAv, 53 kDa non-glycosylated protein). The avidin-biotin and streptavidin-biotin complexes represent the strongest, non-covalent, biological interaction known to date with respectively Ka = 1015 and 1013 M−1.224–227 This feature makes these complexes extremely stable even under harsh conditions with kinetics of dissociation exceptionally slow compared to the time scale of most of the experimental procedures. This explains why they have been widely used to functionalize proteins and more recently involved in the formation of protein-polymer bioconjugates in combination with CLRP techniques.Müller, Stayton, Hoffman and co-workers took advantage of the RAFT process to prepare a series of PNIPAAm polymers (Mn = 2.9–25.9 kDa) with narrow MWD (Mw/Mn = 1.08–1.16).230 NIPAAm was polymerized by RAFT from 1-pyrrolecarbodithioate using AIBN initiator. After cleavage of the dithiocarbamate end-group in basic conditions, the thiol terminated PNIPAAm was coupled to biotinamido-4-[4′-(maleimidomethyl)cyclohexanecarboxamido]butane (BMCC) to yield biotin functionalized PNIPAAm polymers that were further bound to SAv. However, steric hindrance only allowed the presence of two PNIPAAm chains per SAv protein.231 The PNIPAm-SAv bioconjugates displayed a LCST above which they aggregated into uniform and stable nanoparticles with diameters in the 250 to 900 nm range, whereas at 20 °C (below the LCST), they were fully water-soluble (Fig. 21).
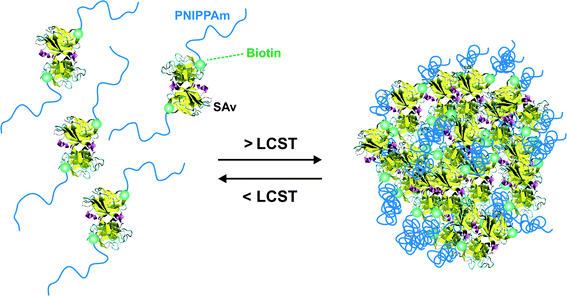 | ||
| Fig. 21 LCST-driven formation of poly(N-isopropyl acrylamide)-streptavidin (PNIPAAm-SAv) nanoparticles.230 | ||
Interestingly, the colloidal characteristics of these thermoresponsive nanoparticles could be tuned by playing with different factors such as the heating rate of the bioconjugate solution, the bioconjugate concentration and the molecular weight of the PNIPAAm block. In another complementary study, it was also observed that a SAv-PNIPAAm-b-PAA bioconjugate exhibited pH-dependent properties.232 Indeed, this bioconjugate was shown to form large aggregates at pH 4.0 both below and above the LCST (Dh = 540 and 700 nm respectively), whereas much smaller aggregates were observed below the LCST at pH 5.5 (Dh = 27 nm). At neutral pH, the SAv-PNIPAAm-b-PAA aggregation was prevented above the LCST due to the shielding effect of the PAA block. The aggregation phenomenon can thus be uncorrelated from the thermoresponsive behaviour of the PNIPAAm block due to the PAA segment.
4.1.4.1 Binding to planar surfaces. Choi and co-workers reported the immobilization of SAv onto biotinylated polymer brushes synthesized by a combination of ATRP and click chemistry.234 A disulfide-containing ATRP initiator was reacted with gold substrate for 12 h at room temperature to give the surface-immobilized ATRP initiator followed by surface-initiated polymerization of PEGMA using CuBr/bpy. After 1 h, the bromine terminus of the resulting poly(PEGMA) was substituted with an azide upon NaN3 treatment, before the click coupling of different alkyne-containing ligands (1-hexyne, 5-hexyn-1-ol, 4-pentynoic acid, propargyl benzoate, alkyne-containing biotin) using a CuSO4/sodium ascorbate catalyst. It showed that the poly(PEGMA)-coated gold surface avoided interaction with a number of model proteins such as BSA, fibrinogen, lysozyme, and RNase A. The biotin-functionalized polymer brushes showed a thickness decrease (−20 Å) upon exposure to SAv, which was ascribed to the specific interaction between SAv and biotin that induced a more condensed polymer layer. Finally, upon passing a SAv solution flow on the biotin-functionalized poly(PEGMA)-coated gold surface, a characteristic specific ligand-receptor interaction was found, demonstrating that the biotin was effectively attached at the poly(PEGMA) extremity.
4.1.4.2 Binding to nanoparticles. Wooley and co-workers successfully transformed their SCK nanoparticles into biotinylated colloidal scaffold from PAA-b-PMA block copolymers synthesized by ATRP from a biotinylated initiator (I17, Fig. 5).233 Stable and uniform biotinylated nanoparticles were obtained by mixing different ratios of biotinylated amphiphilic block copolymers (0–40%) and their non-biotinylated counterparts. By fluorescence correlation spectroscopy (FCS), they noticed that 10 to 25% of the theoretical amount of biotin was available at the surface of the nanoparticles.
Recently, Zhao, Liu and co-workers applied the CuAAC methodology to generate Av-polymer bioconjugates by using ATRP.235 In the first step, sequential ATRP of HEMA and MMA was initiated by a functional methoxy-PEG5000 ATRP macroinitiator, to give a PEG-b-PHEMA-b-PMMA (Mn = 13 kDa, Mw/Mn = 1.21). The PHEMA block was then modified by a mesylation followed by an azidation to obtain the PEG-b-PAzEMA-b-PMMA (PAzEMA stands for poly(azidoethyl methacrylate)) triblock copolymer suitable for further click reaction (Fig. 22). The functionalization of the PEG-b-PAzEMA-b-PMMA micelles (Dh = 41.4 nm) with alkynylated biotin was performed in a mixture tert-BuOH/water (1:4) in the presence of CuSO4/sodium ascorbate catalyst. Av/HABA assays confirmed the successful click coupling on the copolymer micelles and it was calculated that only ∼10% of biotin units were accessible to Av, probably due to steric hindrance or to the possibility that Av acted as a micelles cross-linking agent and thus induced the formation of micelles aggregates that would sterically hide remaining biotin units. This was demonstrated by incubating biotin-functionalized nanoparticles with Av (biotin/Av ratio of 2:1) where TEM highlighted the presence of both micelle aggregates and isolated micelles.
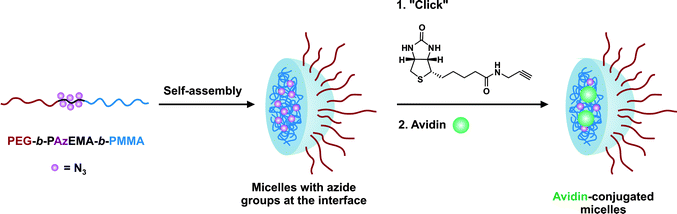 | ||
| Fig. 22 Bioconjugation of biotin to the interfaces of polymeric micelles by in situ click chemistry and further binding with avidin.235 | ||
Davis and co-workers used a α-biotinylated PEG macro-RAFT agent (RA15, Fig. 7) to mediate the polymerization of pyridyldisulfide ethylmethacrylate (PDSEMA) under AIBN initiation (Mn = 15.0–29.0 kDa, Mw/Mn = 1.24–1.39), resulting in a block copolymer able to self-assembled in methanol.236 An additional step of cross linking by disulfide formation in the presence of TCEP was undertaken,237 affording well-defined spherical 54 ± 4 nm micelles. SAv/HABA assays revealed that 75% of biotin were accessible at the surface of the core cross-linked micelles. TEM and DLS revealed the formation of higher-order structures with large diameters up to 1.8 ± 0.4 μm. However, the size of these particles could be lowered by using SAv with some of its 4 binding sites already pre-occupied with biotin.
4.2 Apoenzyme/cofactor reconstitution
Even though biotin/(S)Av couples certainly exhibit nearly ideal features in terms of binding and host guest recognition process, the apoenzyme/cofactor reconstitution strategy represents an interesting alternative to the construction of non-covalent polymer-protein bioconjugates. This has been elegantly illustrated by Nolte and co-workers who took advantage of the strong interactions involved in the reconstitution between an apoenzyme and its cofactor to create HRP-PEG-b-PS bioconjugates.238 The diblock precursor was synthesized in a controlled fashion by ATRP of styrene from a PEG based ATRP macroinitiator at 90 °C using CuBr/PMDETA as a catalyst. After nucleophilic substitution of the bromine terminus of the PS block with an azide, the resulting PEG-b-PS–N3 copolymer was coupled with an alkyne derivatized heme by CuAAC using the same catalyst. The reconstitution of apo-myoglobin (Mb) and apo-HRP was carried out at pH 7.5 and afforded Mb-PS-b-PEG and HRP-PS-b-PEG bioconjugates, respectively. After self-assembly, TEM and SEM analysis of the amphiphilic biomacromolecules showed a broad range of morphologies: micelles, micellar rods, octopi, figure eight, toroids and micellar aggregates.4.3 Glycopolymers and sugar-protein interaction
Carbohydrates are crucial for many biological processes, such as inflammation, cell-to-cell communication, fertilization and signal transmission.239–242 Indeed, sugars exhibit a great coding capacity as they are able to store biological information in oligosaccharide structures, glycoproteins as well as in glycolipids.243 Lectins, that are carbohydrate-binding proteins, can selectively recognise and decode the glycocode in oligosaccharides. Whereas individual protein-saccharide interactions are typically weak, the multivalent interactions employed in biological systems is characterized by high affinity (the so-called “cluster” glycoside effect)243,244 and high specificity. Due to their biomimetic properties, there is an increasing interest in synthetic glycopolymers, that are able to interact with lectins as multivalent ligands in a similar manner to natural glycoproteins.Synthetic glycopolymers can be obtained following two different strategies: (i) the direct polymerization of the corresponding glycomonomers (protected or not) or (ii) the synthesis of polymer scaffolds bearing pendant reactive sites subsequently functionalized by sugar moieties.245,246 The development of CLRP techniques has rendered it possible to produce tailor-made glycopolymers,247–270 well-reviewed in the recent literature.245,246 However, only examples investigating glycopolymers biofunctionality and/or reporting further bioconjugation experiments will be discussed in the following paragraphs (see Fig. 23 for the structure of the corresponding glycomonomers).
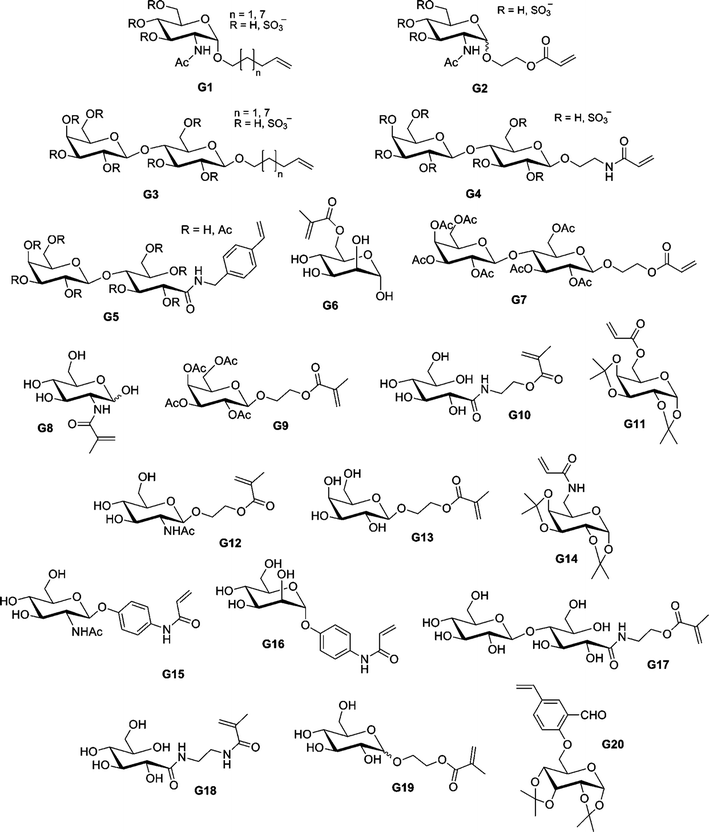 | ||
| Fig. 23 The structures of glycomonomers polymerized by CLRP techniques for bioconjugation purposes. | ||
In a similar synthetic way, nitroxide-mediated polymerization of styrene carrying acetylated lactose (G5 with R = Ac, Fig. 23) was undertaken using BST-TEMPO as the alkoxyamine in DMF at 130 °C.275 While NMP of the unprotected glycomonomer (G5 with R = H, Fig. 23) led to high PDI, the polymerization of the acetylated counterpart proceeded in a controlled fashion and yielded well-defined glycopolymers exhibiting narrow MWDs (Mw/Mn = 1.19–1.69) in the 9.7–21.7 kDa range. The resulting materials showed a strong CD spectra suggesting the stereospecific polymerization whereas lectin recognition exhibited a clear dependence with the number-average degree of polymerization: the higher the DPn of these multivalent glycoclusters, the stronger the affinity with lectin.
An original method, using chemo-enzymatic synthesis in the presence of Candida antarctica lipase (CAL, Novozyme 435), was used to generate 6-O-methacryloyl mannose (MaM, G6, Fig. 23) glycomonomer, followed by its subsequent aqueous RAFT polymerization yielding well-defined, linear poly(6-O-methacryloyl mannose) (PMaM) glycopolymers without the need for protecting group chemistry.276 RAFT polymerization kinetics using various initial monomer to chain transfer agent concentration ratios was investigated together with the protein binding ability of the generated glycopolymer using Concanavalin A (Con A).
As opposed to the direct polymerization of glycomonomers for tailor-made glycopolymers, an alternative route is to use a combination of CLRP and click chemistry, as recently reported by Haddleton and co-workers.277 Well-defined alkyne side-chain polymer scaffolds were obtained by polymerizing trimethylsilyl methacrylate by ATRP using CuBr/N-(ethyl)-2-pyridylmethanimine as the catalyst in toluene at 70 °C (Mn = 8.2–17.6 kDa, Mw/Mn = 1.09–1.17), allowed for further CuAAC with azidosugar derivatives (S1–3, Fig. 24). The versatility of the method was also demonstrated by using PEGMA or MMA as comonomers to produce well-defined random copolymers. CuAAC reactions were also performed via a C-6 or an α or β anomeric azide (S4 and S5, Fig. 24) to construct a library of mannose- and galactose-containing multidentate ligands. This was achieved following a coclicking reaction of appropriate mixture of mannose- and galactose-based azides leading to multivalent displays. The reactivity of these glycopolymers in the presence of model lectins able to selectively bind mannose (Con A) and galactose (Ricinus communis agglutinin, RCA I) moieties was assessed. The CuAAC was also used to attach α-mannoside, β-galactoside and β-lactoside derivatives (S6–9, Fig. 24) via an azide functionality bound directly to the sugar anomeric carbon to yield N-glycosyl 1,2,3-triazole functional polymers.278 These studies permitted not only the comparison of structurally identical ligands that only differ for the nature of the sugar epitopes, but also the preparation of glycopolymers that only differ from each other in the length and the nature of the linker connecting the carbohydrate units to the macromolecular backbone. Interestingly, it has been shown by the same group that CLRP and CuAAC could occur simultaneously, thus representing a new synthetic tool for the design of functional materials.279 This novel copper-catalyzed one-pot simultaneous CLRP-CuAAC process was then applied to the synthesis of glycopolymers from S8 (Fig. 24), thus yielding well-defined biomaterials in a simplified manner.
 | ||
| Fig. 24 Structure of sugar azides used for the synthesis of glycopolymers by a combination of CLRP and click chemistry. | ||
In the same spirit, the concept initially developed by Müller and co-workers126 was expended by Liu and co-workers for the synthesis of glycoconjugates.280 The bulk ATRP polymerization of N-acryloxy-succinimide (NAS) was catalyzed by CuBr/bpy. The NHS-activated side chains of the obtained poly(N-acryloxy-succinimide) (PNAS) were sequentially reacted with galactosamine and ethanolamine to obtained random coglycopolymers composed of variable ratios of pendant galactose and N-(2-hydroxypropyl) acrylamide (HPA) units.
By sequential ATRP and NCA polymerization, Chaikof and co-workers reported the synthesis of a poly(L-glutamate)-b-poly(2-acryloyloxyethyl lactoside)-b-poly(L-glutamate) and poly(L-alanine)-b-poly(2-acryloyloxyethyl lactoside)-b-poly(L-alanine) triblock copolymers from G7 (Fig. 23) and they noticed specific interactions with RCA I lectins.184,185
Interestingly, Stenzel and co-workers synthesized amphiphilic hybrid block copolymers based on 2-methacrylamido glucopyranose (MAG, G8, Fig. 23) and 5′-O-methacryloyl uridine (MAU) under RAFT control at low polymerization temperature (T = 60–70 °C) using (4-cyanopentanoic acid)-4-dithiobenzoate (CPADB) as a RAFT agent.282 The aim was to develop a new colloidal carrier for short antisense oligonucleotides (ASONs), which uses base pairing rather than electrostatic interactions to bind the gene. Very recently, NMP was successfully employed from 2-(2′,3′,4′,6′-tetra-O-acetyl-β-D-galactosyloxy)ethyl methacrylate (AcGalEMA) glycomonomer (G9, Fig. 23) to synthesize poly(2-(β-D-galactosyloxy)ethyl methacrylate-co-styrene)-b-polystyrene (P(GalEMA-co-S)-b-PS) amphiphilic block copolymers (Mn = 18.3–79.9 kDa, Mw/Mn = 1.26–1.50) under SG1 control (N2, Fig. 2), using either SG1-terminated P(AcGalEMA-co-S) or polystyrene macroinitiators, followed by deacetylation of AcGalEMA moieties.292 The self-assembling ability of PS-b-P(GalEMA-co-S) amphiphilic glycopolymers was then exploited to obtain micellar structures and honeycomb structured porous films of intact biofunctionality. The same group also reported the preparation of neoglycopolymers via the thiol-ene coupling reaction between ene-functional poly[di(ethylene glycol) methyl ether methacrylate-b-2-hydroxyethyl methacrylate)] diblock copolymer obtained from RAFT and glucothiose under photochemical conditions with 2,2-dimethoxy-2-phenylacetophenone (DMPA) as the photoinitiator.287 The glycopolymers were shown to undergo temperature-induced micellisation and to efficiently bind with Con A.
Combination of ROP of either ε-caprolactone (ε-CL)289 or BLG-NCA288 with ATRP has been undertaken by Dong and co-workers for the design of various biomimetic star-shaped glycopolymers (Fig. 25). A small library of four-arm poly(ε-caprolactone) and poly(BLG-NCA) were synthesized and readily transformed into ATRP macroinitiators for subsequent polymerization of unprotected D-gluconamidoethyl methacrylate (GAMA, G10, Fig. 23) glycomonomer. The length of each block was varied by playing with the initial stoichiometry of the reagents and in all case, a nice control of the polymerization process was reached exhibiting relatively narrow MWDs. Recognition properties of these star-shaped glycopolymers with Con A was also assessed.
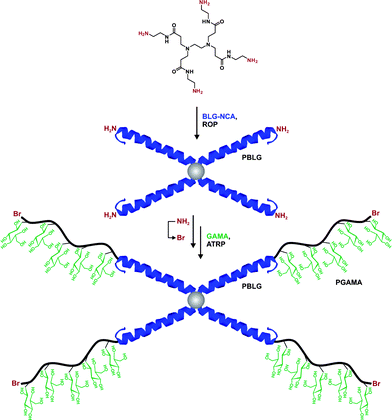 | ||
| Fig. 25 Synthesis of star-shaped poly(PBLG-b-PGAMA)4 biohybrids via a combination of ROP of BLG-NCA and ATRP of GAMA glycomonomer.288 | ||
Using a similar strategy, the same group used a difunctional poly(ε-caprolactone) ATRP macroinitiator for the preparation of a polypseudorotaxane/glycopolymer triblock copolymer, poly(D-gluconamidoethyl methacrylate)-polypseudorotaxane-poly(D-gluconamidoethyl methacrylate) (PGAMA-b-PPR-b-PGAMA), where inclusion complexation was performed with α-cyclodextrine (α-CD). After ATRP with CuBr/PMDETA as the catalyst in DMSO at 35 °C for 48 h, the resulting biomaterials exhibited controlled molecular weights and low PDIs (Mn = 33–39 kDa, Mw/Mn = 1.26–1.56) together with a specific biomolecular recognition with Con A in contrast as with BSA.291 Ring opening polymerization and subsequent RAFT polymerization were also employed for the synthesis of a poly(D-L-lactide)-b-poly(6-O-acryloyl-α-D-galactopyranose) block copolymer (Mn ≈ 60 kDa, Mw/Mn ≈ 1.2) from glycomonomer G11 (Fig. 23).286 Subsequent cross-linking and degradation of the core resulted in hollow sugar balls that could be useful for drug delivery purposes.
After promising results obtained for the preparation of polymers derived from serine- and tyrosine-based ATRP macroinitiator,143 Maynard and co-workers successfully undertaken the ATRP polymerization of glycomonomer G12 (Fig. 23) from the serine derivatized initiator (Mn = 15.4 kDa, Mw/Mn = 1.19). To demonstrate the versatility of their approach, the serine derived ATRP initiator was deprotected and coupled using SPPS to a model peptide, VMSVVQTK, which is well known to be O-GlcNAc modified in the human cellular factor (HCF) protein, an abundant chromatin-associated factor involved in cell proliferation and transcriptional regulation. Then, the C-terminus lysine of the polypeptide was replaced with a lysine derivative modified at the ε-position with a (7-methoxycoumarin-4-yl) acetyl (Mca) group for bioconjugation following purposes. ATRP of G12 from the polypeptide macroinitiator afforded a glycopolymer-peptide conjugate of 12.2 kDa after 93% conversion with a low PDI (Mw/Mn = 1.14). This work opens the door to the formation of tailor-made, well-defined polymer- or glycopolymer-peptide conjugates in which the conjugation site is accurately chosen.
Several authors took advantage of the fact that polymer end-groups prepared by RAFT can be readily converted to thiol moieties and anchored to gold substrates via the formation of Au–S covalent bonds.294–296 In their study, Cameron and co-workers prepared a poly(2-(β-D-galactosyloxy)ethyl methacrylate) (poly(GalEMA)) from glycomonomer G13 with CPADB as a RAFT agent and ACPA as the radical initiator, leading to a well defined glycopolymer (Mn = 24.1 kDa, Mw/Mn = 1.09).295 Formation of gold glyconanoparticles was observed upon addition of NaBH4 to a solution of glycopolymer in HAuCl2 (Fig. 26), the biological activity of which was demonstrated by agglomeration of peanut agglutinin (PNA)-coated agarose beads.
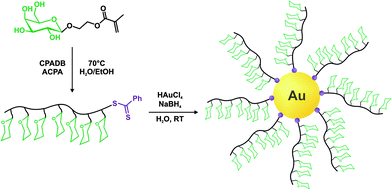 | ||
| Fig. 26 Preparation of glycopolymer-stabilized gold nanoparticles.295 | ||
Similar approaches were then reported for the design of either biotinylated poly(N-acryloylmorpholine-co-6-O-acrylamido-6-deoxy-1,2:3,4-di-O-isopropylidene-α-D-galactopyranose) (poly(NAM-co-GalAm)) glycopolymers from G14 (Fig. 23),296 or polyacrylamide-based macromolecular backbones with pendant sugar moieties, either N-acetyl-β-glucosamine (G15, Fig. 23) or α-mannoside (G16, Fig. 23),294 subsequently anchored at the surface of gold nanoparticles. In the latter example, molecular recognition was studied with E. coli, which induced aggregation of the nanoparticles at the cell periphery. Alternatively, by using a disulfide-carrying ATRP initiator (I18, Fig. 5), Kitano and co-workers successfully reported the coupling of galactose-containing glycopolymer brushes to colloidal gold monolayer deposited on a cover glass.297 The polymerization of lactobionamidoethyl methacrylate (LAMA, G17, Fig. 23) proceeded via a divergent chain-growth with CuBr/bpy as the catalyst. It yielded a series of glycopolymers (Mn = 5.5–16.0 kDa, Mw/Mn = 1.53–2.19) followed by their accumulation via Au–S bond on the gold substrate. The association and dissociation processes of galactose residues on the colloidal gold with a RCA I lectin were observed. The opposite strategy, which consists of making glycopolymer chains to grow from a gold surface, has also been investigated by Vamvakaki and co-workers.298 Surface-initiated ATRP (CuBr/bpy) of GAMA (G10, Fig. 23) and LAMA (G17, Fig. 23) resulted in a homogeneous increase in the dry film thickness with reaction time. It was also shown that the glycopolymer brushes exhibited strong binding interactions with specific lectins via the “glycocluster” effect.
Covalent immobilization of a range of carbohydrates derivatives onto polymer resin beads was recently described by Haddleton and co-workers using ATRP and CuAAC (Fig. 27).293 Two approaches were described: (i) the direct click coupling of mannose azides onto alkyne-derivatized Wang resin (i.e. immobilized monosaccharide) or (ii) the synthesis of glycopolymer chains displaying multiple copies of mannose epitopes grafted onto a Wang resin by surface-initiated ATRP (i.e. immobilized glycopolymer). These novel glyco-hybrid materials were able to efficiently recognize mannose-binding model lectins such as Con A.
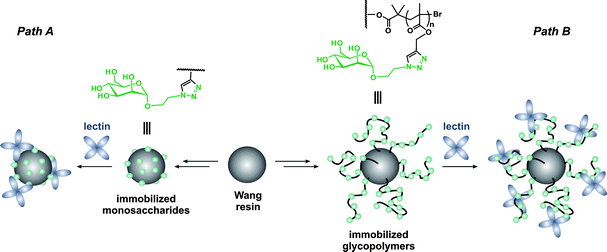 | ||
| Fig. 27 General approaches for the design of hybrid sugar supports: direct click coupling of mannose azides onto alkyne-derivatized Wang resin (Path A) or synthesis of glycopolymer chains displaying multiple copies of mannose epitopes grafted onto a Wang resin by surface-initiated ATRP (Path B).293 | ||
4.3.4.1 Functionalization with biotin. Chaikof and co-workers used two 4-aminobenzyl-biotinamide initiators (Fig. 28) to trigger the cyanoxyl-mediated copolymerization of 2-acrylaminoethyl lactoside (G4, Fig. 23) and acrylamide, giving well-defined glycopolymers with narrow MWDs.299,300 The binding efficiency between streptavidin and the biotinylated glycopolymer was assessed by SDS-PAGE. The production of glycopolymer-coated surfaces was presented from the reaction of a glycopolymer solution with streptavidin-derivatized poly(ethylene terephthalate) (PET) membranes, subsequently incubated with FITC-labeled galactose binding lectin.300
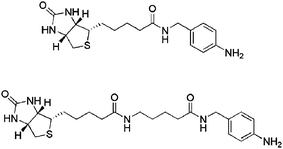 | ||
| Fig. 28 Structure of 4-aminobenzyl-biotinamide initiators. | ||
Similarly, ATRP initiators bearing a biotin moiety (I17 and I19, Fig. 5) have been successfully used at room temperature to prepare tailor-made biotin-terminated glycopolymers from LAMA (G17, Fig. 23)301 or from a methacrylate with a pendant N-acetylglucosamine (GlcNAc) unit (G12, Fig. 23)306 using CuBr/bpy or CuBr/Me6TREN, respectively. Affinity with streptavidin was then demonstrated by fluorescence displacement assays301 or surface plasmon resonance.306 In the latter case, an association constant, Ka, of ∼1015 M−1 was obtained, in good agreement with values reported in the literature.
With the RAFT technique, different strategies were employed to produce biotinylated glycopolymers: (i) the use of a biotin-functionalized RAFT agent;296,307 (ii) the copolymerization between a glycomonomer and a biotinylated monomer308 or (iii) via the thiol-ene click chemistry involving a biotin modified maleimide.302 By reaction of succinimido-2-[[2-phenyl-1-thioxo]thio]-propanoate with (+)-biotinyl-3,6-dioxaoctanediamine, Charreyre and co-workers were able to prepare a biotinylated RAFT309 agent (RA16, Fig. 7) further used for the copolymerization of NAM and GalAm (G14, Fig. 23) to yield biotin-terminated gradient glycopolymers in the 2–40 kDa range.296,307 Those materials were photochemically reduced in situ for the preparation of gold nanoparticles, at the surface of which the biotin was still accessible for bioconjugation to streptavidin.296 Terpolymerization between 2-gluconamidoethyl methacrylamide (GAEMA, G18, Fig. 23), biotinyl-2-aminoethyl methacrylamide hydrochloride (BAEMA) and 2-aminoethyl methacrylamide hydrochloride (AEMA) was undertaken in water at 70 °C in the presence of ACVA as a water-soluble radical initiator and S,S′-bis(R,R′-dimethyl-R′′-acetic acid) trithiocarbonate as a RAFT agent.308 The resulting glycopolymer (Mn = 12.9 kDa, Mw/Mn = 1.19) exhibiting pendant sugar, biotin and amine groups was further used for the surface functionalization of quantum dots (QDs), via standard EDC/NHS coupling chemistry, which then demonstrated excellent water-solubility and colloidal stability as well as lower cytotoxicity than uncoated QDs. Moreover, new activated esters based on pentafluorophenyl acrylate (FP-A) have been originally investigated as precursors to build well-defined biotinylated glycopolymers under RAFT control.302 Poly(pentafluorophenyl acrylate) in the 2.8–16.0 kDa range with PDI < 1.20 were modified in a nearly quantitative fashion by concomitant reaction with amino functional sugar (i.e.D-glucosamine or D-galactosamine) and in situ aminolysis of the RAFT end-group followed by its coupling with a biotin modified maleimide via thiol-ene click chemistry. Binding assays with specific lectin and Con A demonstrated the biofunctionality of these glycopolymers.
4.3.4.2 Functionalization leading to a covalent linkage. Functionalized ATRP initiators have been judiciously employed by several groups for the synthesis of glycopolymer bioconjugates. Finn and co-workers used an azide-containing initiator (I20, Fig. 5) for the polymerization of methacryloxyethyl glucoside (G19, Fig. 23) with CuBr/bpy at 25 °C to yield azido-terminated glycopolymer (Mn = 13 kDa, Mw/Mn = 1.3). In parallel, cowpea mosaic virus (CPMV) was derivatized with an azide bearing N-hydroxysuccinimide for a multi-site attachment. To form the virus-glycopolymer bioconjugate, the azido-terminated glycopolymer and the modified CPMV were then sequentially clicked onto a fluorescent dialkyne using a specific catalytic system for each moiety (CuSO4/sodium ascorbate and Cu-triflate/sulfonated bathophenanthroline ligand, respectively).305 The number of covalently bound polymer chains per particle was very close to the 150 CPMV available azide functionalities. A larger hydrodynamic radius and molecular weight than the native CPMV were also measured. Haddleton and co-workers took advantage of ATRP and CuAAC to construct well-defined neoglycopolymer-protein biohybrid materials as glycoprotein mimics using a protected-maleimide ATRP initiator (I9, Fig. 5). A small library of well-defined maleimide-terminated neoglycopolymers having multiple copies of D-mannose epitopes (Mn = 8–30 kDa, Mw/Mn = 1.20–1.28) or featuring different relative amounts of D-mannose and D-galactose binding epitopes following a co-clicking approach have been further coupled to BSA as a single thiol-containing model protein (Fig. 29).303 Surface plasmon resonance binding studies carried out using recombinant rat mannose-binding lectin (MBL) showed clear and dose-dependent MBL binding to glycopolymer-conjugated BSA. In addition, enzyme-linked immunosorbent assay (ELISA) revealed that the neoglycopolymer-protein materials described in this work possess significantly enhanced capacity to activate complement via the lectin pathway when compared with native unmodified BSA.
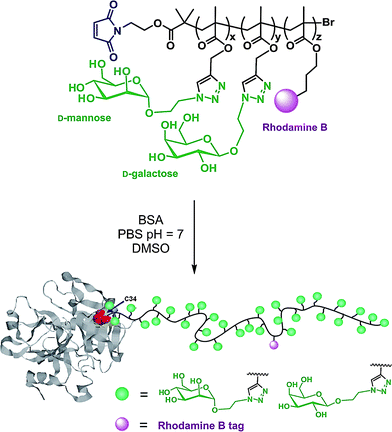 | ||
| Fig. 29 Synthesis of glycoprotein mimics by a combination of ATRP and click chemistry.303 | ||
Very recently, Maynard and co-workers used a pyridyl disulfide ATRP initiator (I10, Fig. 5)310 for the polymerization of N-acetyl-D-glucosamine (G12, Fig. 23) with CuBr/CuBr2/bpy as a catalyst in methanol–water mixture at 30 °C to design the corresponding end-functionalized glycopolymer (Mn = 10.2 kDa, Mw/Mn = 1.12), further employed to prepare a siRNA-conjugate.304 This may offer great potential regarding siRNA gene therapy (see also section 5 Synthesis of polymer-oligonucleotide bioconjugates).
The RAFT technique was employed under AIBN initiation in THF at 60 °C with a styrene-based glycomonomer, 1,2:3,4-di-O-isopropylidene-6-O-(2′-formyl-4′-vinylphenyl)-D-galactopyranose (IVDG, G20, Fig. 23), bearing an aldehyde function for further coupling with BSA as a model protein via the formation of Schiff base linkage.311 The polymerization exhibited a linear evolution of the logarithmic conversion with time as well as a linear increase of Mn with monomer conversion and narrow molecular weight distribution (Mn = 29 kDa, Mw/Mn ≈ 1.1). Nearly quantitative removal of protective isopropylidene groups yielded amphiphilic glycopolymers that self-assembled in water into well-defined micelles. Protein-bioconjugated nanoparticles were then successfully prepared by the immobilization of BSA onto aldehyde-functionalized micelles.
Bertozzi and co-workers recently developed an elegant approach for designing end-functionalized mucin-like glycopolymers, suitable for integration with current microarray technology platforms, involving RAFT polymerization from RA17 (Fig. 7), oxime linkages and click chemistry (Fig. 30).312 The strategy was to prepare a well-defined polymer intermediate which contained: (i) pendant ketones for the attachment of α- and β-aminooxy-GalNAc; (ii) a terminal pentafluorophenyl (PFP) ester that offers a reactive site for further reaction with propargyl amine to insert alkyne moieties and (iii) a trithiocarbonate group subsequently cleaved to release a free sulfhydryl group for conjugation to a fluorescent probe. The structurally uniform alkyne-terminated mucin mimetic glycopolymers were printed on azide-functionalized chips by microcontact printing in the presence of a copper catalyst by CuAAC. The surface-attached glycopolymers were shown to bind lectins in a ligand-specific manner.
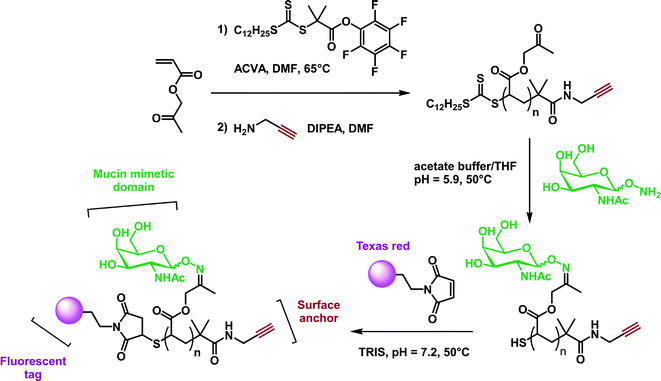 | ||
| Fig. 30 Synthesis of dual-end-functionalized mucin mimics using a polymer scaffold prepared by RAFT.312 | ||
5 Synthesis of polymer-oligonucleotide bioconjugates
Boasting an impressive range of well-defined polymer-protein/peptide bioconjugates from CLRP methods, investigations have been very recently extended to oligonucleotide bioconjugates.Oligonucleotides (ON) are short natural RNA (ribonucleic acid) or DNA (desoxiribonucleic acid) sequences, generally obtained by chemical synthesis, that contain genetic information. They have been extensively used for many years in gene therapies for the treatment of severe diseases because they can regulate the expression of a targeted protein or to replace a mutant allele. As they are not able to pass through the cell membrane (especially due to the strong negative charge of the phosphate backbone), researchers have thus elaborated “Trojan” systems consisting of viral, retroviral and adenoviral vectors to release the genetic material within the cell after the membrane crossing but also non viral vectors mainly consisting in polymers and especially polycationic polymers. Regarding this, the last couple of years have witnessed interesting studies devoted to the preparation of ON-polymer conjugates for therapeutic purposes using CLRP methodologies.
5.1 Direct bioconjugation to preformed polymers
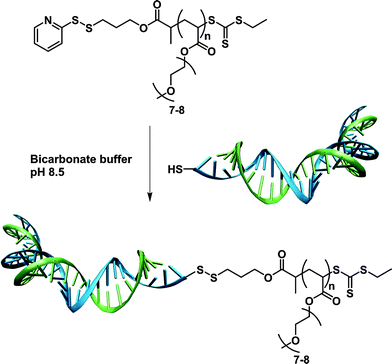 | ||
| Fig. 31 Synthesis of reversible siRNA–polymer conjugates by the RAFT technique.313 | ||
Very recently, aptamers with a disulfide protected thiol modification on the 3′ end, have been conjugated to maleimide activated branched PEGs of various molecular weights, obtained using I9 (Fig. 5) as an ATRP initiator. This work demonstrated an alternative approach to PEGylation of aptamers, and that the effect of PEG on the affinity for the target varied according to the structure and conformation of the synthetic polymer.314
This strategy was also exploited with a library of ω-PDS PNIPAAm polymers (Mn ranging from 7.0 to 22.2 kDa) conjugated to a 5′-thiol-modified oligonucleotide in PBS at pH 8.0 (ratio PNIPAAm/ON: 50/1). The achievement of the bioconjugation was demonstrated by agarose gel electrophoresis and the reaction yields were above 70% irrespective of the polymer molecular weight. A release study of the ON upon addition of DTT was undertaken and confirmed the attachment of the ON via a reversible disulfide bond.116
The thiol-ene reaction was recently demonstrated to be a powerful coupling method to covalently attach ON to polymers.316 In this study, HPMA and NIPAAm were polymerized by RAFT from dithioester and trithiocarbonate RAFT agents to afford PHPMA and PNIPAAm polymers in a controlled fashion. Then, after aminolysis reaction in the presence of 1,4-butanediol dimethacrylate, ene ω-functional PHPMA (Mn = 4.4 kDa, Mw/Mn = 1.04) and PNIPAAm (Mn = 13.5 kDa, Mw/Mn = 1.10) polymers were obtained and coupled to a thiol modified ON by thiol-ene click chemistry. The formation of the PNIPAAm-ON and PHPMA-ON bioconjugates was confirmed by agarose gel electrophoresis.
5.2 Bioconjugation to surfaces
The design of DNA-polymer bioconjugates on a planar solid support using surface-initiated RAFT polymerization has been reported by He.317A trithiocarbonate RAFT agent (RA19, Fig. 7) was attached to the distal point of a surface-immobilized oligonucleotide and initiation of the polymerization led to controlled growth of polymer chains. Growth kinetics of PEGMA or HEMA atop DNA molecules was investigated by monitoring the change of polymer film thickness as a function of reaction time. Comparing to polymer growth atop small molecules, the experimental results suggest that DNA molecules significantly accelerated polymer growth, which was speculated as a result of the presence of highly charged DNA backbones and purine/pyrimidine moieties surrounding the reaction sites.The immobilization of single strand deoxyribonucleic acid (ssDNA) on PVP coated SiO2 particles was also proposed by Zelekin and co-workers as an interesting method to prepare DNA biosensing systems (Fig. 20). Developing the same strategy as already reported earlier for the immobilization of SIINFEKL peptide at the surface of SiO2 particles, the authors coupled 5′-thiol terminated ssDNA using the disulfide exchange coupling on activated thiol PVP previously adsorbed at the surface of SiO2 particles.222
6 Conclusions
In this review, the huge potential of CLRP to prepare well-defined and original bioconjugates has been exposed. Since their discoveries, CLRP techniques (especially RAFT and ATRP) have brought about a clear breakthrough in this field. They allow the design of polymers with a high degree of control regarding the macromolecular architecture and the accurate insertion of functional groups in the polymer chains for further (covalent or non covalent) bioconjugation. Moreover, these polymerization techniques are easy to implement and can be performed with a broad range of monomers and functional initiators as well as in organic and aqueous solvents. All these important features make CLRP techniques valuable tools in the actual bioconjugation landscape. All kind of bioconjugates (polymer-protein/peptide, polymer-ON, glycoprotein mimics etc) described herein could have important benefits in diverse areas such as drug delivery purposes, biomaterials, bio- and nanotechnologies, gene therapy and much more in the near future.The interest of the medical field for bioconjugates can be observed by the numerous protein-polymer based therapeutics, including blockbuster PEG-protein drugs such as Neulasta®, Pegasys®, Pegintron®, and Mircera® that are already on the market for the treatment of Hepatitis C, cancers and others. As CLRP offers the possibility to accurately tune the macromolecular properties of the bioconjugates, it is likely that a forthcoming close interdisciplinary collaboration between macromolecular synthesis and the medical/pharmaceutical research area will result in the discovery of high value-added bioconjugates for therapeutic purposes and for other bio-related applications.
However, the development of this new class of biomaterials might be hampered by the limitations and drawbacks that CLRP techniques could potentially present. For instance with RAFT, complete removal of the RAFT end-group has to be efficiently performed prior to any biological application. With ATRP, the catalyst could be an issue as many proteins exhibit copper or iron binding pockets which may affect their biological integrities. Nevertheless, the majority of the examples detailed in this review showed a good conservation of the biological activity for polymer-protein conjugates or the ability for glycopolymers to efficiently bind lectins. It thus demonstrated the reliability and the efficiency of this approach regarding the exciting and rapidly expanding field of polymer science in therapeutics.
7 List of Abbreviations
| αCT | α-chymotrypsin |
| AA | acrylic acid |
| ACN | acetonitrile |
| ACVA | 4,4′-azobis (4-cyanovaleric acid) |
| AEL | 2-acryloylethyloctaacetyllactoside |
| AEMA | 2-aminoethyl methacrylamide hydrochloride |
| AF488 | Alexa Fluor 488 |
| AFM | atomic force microscopy |
| AGT | O 6-alkylguanine-DNA alkyltransferase |
| AIBN | 2,2′-azobisisobutyronitrile |
| Ala/A | alanine |
| AM | acrylamide |
| AMC | 7-amino-4-methylcoumarin |
| AN | acrylonitrile |
| Arg/R | arginine |
| ASON | antisense oligonucleotide |
| Asp/D | aspartic acid |
| ATRP | atom transfer radical polymerization |
| Av | avidin |
| AzEMA | azidoethyl methacrylate |
| BAEMA | biotinyl-2-aminoethyl methacrylamide hydrochloride |
| BDC | 4-(N,N-diethyl)dithiocarbamoylmethylbenzoic acid |
| BG | O 6-benzylguanine |
| Bpy | bipyridine |
| BLG-NCA | γ-benzyl-L-glutamate-N-carboxyanhydride |
| BMCC | 1-biotinamido-4-[4′-maleimidomethyl)cyclohexanecarboxamido]butane |
| Boc | tert-butyl carbonate |
| BSA | bovine serum albumin |
| BSPA | 3-(benzylsulfanylthiocarbonyl sulfanyl)-propionic acid |
| CalB | Candida Antarctica lipase B |
| CD | circular dichroism |
| CDTB | 4-cyanopentanoic acid dithiobenzoate |
| CHO | Chinese hamster ovary |
| CL | ε-caprolactone |
| CLRP | controlled living radical polymerization |
| CLSM | confocal laser scaning microscopy |
| Con A | Concanavalin A |
| CPADB | (4-cyanopentanoic acid)-4-dithiobenzoate |
| CP-ini | peptide nanotube macroinitiator |
| CPMV | cowpea mosaic virus |
| CTA | chain transfer agent |
| CuAAC | copper-catalyzed azide-alkyne cycloaddition |
| Cys/C | cysteine |
| CWP | chain walking polymerization |
| DCC | N-N′-dicyclohexyl carbodiimide |
| DEPMA | diethoxypropyl methacrylate |
| D h | hydrodynamic diameter |
| DHFR | dihydrofolate reductase |
| DIPEA | N,N-diisopropylethylamine |
| DLS | dynamic light scattering |
| DMAA | dimethyl acrylamide |
| DMAEMA | dimethylaminoethyl methacrylate |
| DMF | dimethyl formamide |
| DMSO | dimethyl sulfoxyde |
| dNbpy | 4,4′-di(5-nonyl)-2,2-bipyridine |
| DNA | desoxiribonucleic acid |
| DP n | number-average degree of polymerization |
| DSC | differential scanning calorimetry |
| DTP | 2,2′-dithiopyridine |
| DTT | 1,4-dithiothreitol |
| EBIB | ethyl-2-bromo isobutyrate |
| EBP | elastin-based polymer |
| E. coli | Echerichia coli |
| EDC | N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide |
| EDTA | ethylenediaminetetraacetic acid |
| ELP | elastin linear polymer |
| EPO | erythropoietin |
| f | initiation efficiency |
| f M0 | initial molar fraction of monomer M in a comonomer mixture |
| FCS | fluorescence correlation spectroscopy |
| FGF-2 | fibroblast growth factor-2 |
| FITC | fluoresceine isothiocyanate |
| Fmoc | fluorenylmethyloxycarbonyl |
| FPLC | fast protein liquid chromatography |
| FT-IR | fourier-transform infrared spectroscopy |
| GA | glutamic acid |
| GAEMA | 2-gluconamidoethyl methacrylamide |
| GalAm | 6-O-acrylamido-6-deoxy-1,2:3,4-di-O-isopropylidene-α-D-galactopyranose |
| GAMA | D-gluconamidoethyl methacrylate |
| GlcNAc | N-acetyl-D-glucosamine |
| Gln/Q | glutamine |
| Glu/E | glutamic acid |
| Glu-EMA | glutamic acid ethyl methacrylate |
| Gly/G | glycine |
| G-CSF | granulocyte colony stimulating factor |
| GMA | glycidyl methacrylate |
| GOD | glucose oxidase |
| GSH/γ-ECG | glutathione |
| HEMA | 2-hydroxyethyl methacrylate |
| His/H | histidine |
| HABA | 2-(4′-hydroxyazobenzene) benzoic acid |
| HCF | human cellular factor |
| HEA | hydroxyethyl acrylate |
| HIV | human immunodeficiency virus |
| HMA | hostasol methacrylate |
| HMTETA | 1,1,4,7,10,10-hexamethyltriethylene tetramine |
| HPA | N-(2-hydroxypropyl) acrylamide |
| HPLC | high performance liquid chromatography |
| HPMA | N-(2-hydroxypropyl)methacrylamide |
| HRP | horseradish peroxidase |
| IFN | interferon-2α |
| ION | iron oxide nanoparticle |
| IVDG | 1,2:3,4-di-O-isopropylidene-6-O-(2′-formyl-4′-vinylphenyl)-D-galactopyranose |
| LAMA | lactobionamidoethyl methacrylate |
| LCST | low critical solution temperature |
| Leu/L | leucine |
| CLRP | controlled/living radical polymerization |
| Lys/K | lysine |
| M | monomer |
| Mb | myoglobin |
| MBL | mannose-binding lectin |
| Mca | (7-methoxycoumarin-4-yl) acetyl |
| MA | methyl acrylate |
| Me6TREN | tris[2-(dimethylamino)ethyl]amine |
| MeOH | methanol |
| MMA | methyl methacrylate |
| M n | number-average molar mass |
| MPC | 2-methacryloyloxyethyl phosphorylcholine |
| M w | weight-average molar mass |
| MWD | molecular weight distribution |
| NAM | N-acryloylmorpholine |
| NAS | N-acryloyloxysuccinimide |
| NAv | neutravidin |
| nBA | n-butyl acrylate |
| NCA | α-amino acid-N-carboxyanhydride |
| NHS | N-hydroxysuccinimide |
| NIPAAm | N-isopropyl acrylamide |
| NMP | nitroxide-mediated polymeriazation |
| NMR | nuclear magnetic resonance |
| NMS | N-methacryloyloxysuccinimide |
| NPC | p-nitrophenyl chloroformate |
| NVP | N-vinyl pyrrolidone |
| OB | ovalbumin |
| ON | oligonucleotide |
| PAA | poly(acrylic acid) |
| PAEL | poly(2-acryloyloxyethyl-lactoside) |
| PBLG | poly(γ-benzyl-L-glutamate) |
| PBS | phosphate buffer solution |
| PCL | poly(ε-caprolactone) |
| PDEPMA | poly(diethoxypropyl methacrylate) |
| PDI | polydispersity index (Mw/Mn) |
| PDMAA | poly(dimethyl acrylamide) |
| PDMAEMA | poly(N-dimethylaminoethyl methacrylate) |
| PDS | pyridyl disulfide |
| PDSEMA | pyridyldisulfide ethylmethacrylate |
| PEG | poly(ethylene) glycol |
| PEGA | poly(ethylene glycol) methyl ether acrylate |
| PEGMA | poly(ethylene glycol) methyl ether methacrylate |
| PEI | polyethyleneimine |
| PET | poly(ethylene terephthalate) |
| PFEMA | poly(3-formyl ethyl methacrylate) |
| PFP | pentafluorophenyl |
| PGMA | poly(glycerol methacrylate) |
| Phe/P | phenylalanine |
| PHEMA | poly(2-hydroxyethyl methacrylate) |
| PHPMA | poly(N-(2-hydroxypropyl) methacrylamide) |
| PIAT | poly(L-isocyanoalanine(2-thiophen-3-yl-ethyl)amide) |
| PLA | poly(L-alanine) |
| PMA | propargyl methacrylate |
| PMDETA | N,N,N′,N′,N′′-pentamethyldiethylenetriamine |
| PMMA | poly(methyl methacrylate) |
| PMPC | poly(2-methacryloyloxyethyl phosphorylcholine) |
| PNA | peptide nucleic acid |
| PNA | peanut agglutinin |
| PNAS | poly(N-acryloxy-succinimide) |
| PnBA | poly(n-butyl methacrylate) |
| PNIPAAm | poly(N-isopropyl acrylamide) |
| PNMS | poly(N-methacryloyloxysuccinimide) |
| pNPMA | poly(p-nitrophenyl methacrylate) |
| PpNPMA | poly(p-nitrophenyl methacrylate) |
| PPNT | peptide-polymer hybrid nanotube |
| Pro/P | proline |
| PS | polystyrene |
| PtBA | poly(tert-butyl acrylate) |
| PTD | protein transduction domain |
| PVP | poly(N-vinyl pyrrolidone) |
| PzLLys | poly(Z-L-lysine) |
| QD | quantum dot |
| RAFT | reversible addition-fragmentation transfer |
| RCA I | ricinus communis agglutinin |
| RI | refractive index |
| ROP | ring-opening polymerization |
| ROMP | ring-opening metathesis polymerization |
| S | styrene |
| S. aureus | Staphylococcus aureus |
| SAv | streptavidin |
| SCK | shell-cross-linked |
| sCT | salmon calcitonin |
| SDS-PAGE | sodium dodecylsulfate polyacrylamide gel electrophoresis |
| SEC | size exclusion chromatography |
| SEC-HPLC | size exclusion high performance liquid chromatography |
| SEM | scanning electron microscopy |
| Ser/S | serine |
| SG1 | N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)] nitroxide |
| SPA | succinimidyl propionate |
| SPDP | succinimidyl 3-(2-pyridyldithio)propionate |
| SPR | surface plasmon resonance |
| R g | radius of gyration |
| R h | hydrodynamic radius |
| RMA | rhodamine B methacrylate |
| RNA | ribonucleic acid |
| siRNA | small interfering ribonucleic acid |
| SPA | succinimidyl propionate |
| SPPS | solid-phase peptide synthesis |
| ssDNA | single strand deoxyribonucleic acid |
| t | reaction time |
| T4L | V131C T4 lysozyme |
| tBA | tert-butyl acrylate |
| TBAF | tetrabutyl ammonium fluoride |
| TCEP | tris(2-carboxyethyl)phosphine |
| TEA | triethylamine |
| TEM | transmission electron microscopy |
| TEMPO | 2,2,6,6-tetramethylpiperidinyl-1-oxy |
| THF | tetrahydrofurane |
| Thr/T | threonine |
| TIPNO | N-tert-butyl-N-[1-phenyl-2-(methylpropyl)] nitroxide |
| TMS | trimethylsilyl |
| Trp/W | tryptophan |
| Tyr/Y | tyrosine |
| UV | ultra violet |
| Val/V | valine |
| VBC | 4-vinylbenzyl chloride |
| VO | 2-vinyl-4-4-dimethyl-5-oxazolone |
| zLLys-NCA | N ε -carbobenzoxy-L-lysine-N-carboxyanhydride |
Acknowledgements
We thank the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 212043 for funding (BLD). The French ministry of research and CNRS are also warmly acknowledged for financial support.References
- R. Mülhaupt, Angew. Chem., Int. Ed., 2004, 43, 1054 CrossRef.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347 CrossRef CAS.
- A. Favier and M. T. Charreyre, Macromol. Rapid Commun., 2006, 27, 653 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS.
- S. Yamago, Chem. Rev., 2009, 109, 5051 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270 CrossRef CAS.
- D. M. Haddleton and A. J. Limer, Prog. React. Kinet. Mech., 2004, 29, 187 CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402 CrossRef CAS.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45 RSC.
- J.-F. Lutz and H. G. Boerner, Prog. Polym. Sci., 2008, 33, 1 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, in Macromolecular Engineering, 2007, vol. 4, p. 2645 Search PubMed.
- H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1 CrossRef CAS.
- H.-A. Klok, Macromolecules, 2009, 42, 7990 CrossRef CAS.
- G. Hermanson, Bioconjugate Techniques, Academic Press, 1996 Search PubMed.
- F. M. Veronese, Biomaterials, 2001, 22, 405 CrossRef CAS.
- J. M. Antos and M. B. Francis, Curr. Opin. Chem. Biol., 2006, 10, 253 CrossRef CAS.
- M. Szwarc, Nature, 1956, 178, 1168 CrossRef CAS.
- K. Matyjaszewski and T. P. Davis, ed., Handbook of Radical Polymerization, John Wiley & Sons, Inc., Hoboken, 2002 Search PubMed.
- T. Otsu and M. Yoshida, Makromol. Chem. Rapid Commun., 1982, 3, 127 CrossRef CAS.
- T. Otsu, M. Yoshida and T. Tazaki, Makromol. Chem. Rapid Commun., 1982, 3, 133 CrossRef CAS.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121 CrossRef CAS.
- T. Otsu and A. Matsumoto, Adv. Polym. Sci., 1998, 136, 75 CAS.
- J. D. Druliner, Macromolecules, 1991, 24, 6079 CrossRef CAS.
- G. David, C. Boyer, J. Tonnar, B. Ameduri, P. Lacroix-Desmazes and B. Boutevin, Chem. Rev., 2006, 106, 3936 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156 CrossRef CAS.
- A. Debuigne, J.-R. Caille, C. Detrembleur and R. Jérôme, Angew. Chem., Int. Ed., 2005, 44, 3439 CrossRef CAS.
- A. Debuigne, J.-R. Caille and R. Jérôme, Angew. Chem., Int. Ed., 2005, 44, 1101 CrossRef CAS.
- J. D. Druliner M. Fryd 1991 p. 7 .
- D. Grande, R. Guerrero and Y. Gnanou, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 519 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987 CrossRef CAS.
- A. Studer, K. Harms, C. Knoop, C. Müller and T. Schulte, Macromolecules, 2004, 37, 27 CrossRef CAS.
- D. Benoit, S. Grimaldi, S. Robin, J.-P. Finet, P. Tordo and Y. Gnanou, J. Am. Chem. Soc., 2000, 122, 5929 CrossRef CAS.
- S. Grimaldi, J.-P. Finet, F. Le Moigne, A. Zeghdaoui, P. Tordo, D. Benoit, M. Fontanille and Y. Gnanou, Macromolecules, 2000, 33, 1141 CrossRef CAS.
- P. Lacroix-Desmazes, J.-F. Lutz and B. Boutevin, Macromol. Chem. Phys., 2000, 201, 662 CrossRef CAS.
- P. Lacroix-Desmazes, J.-F. Lutz, F. Chauvin, R. Severac and B. Boutevin, Macromolecules, 2001, 34, 8866 CrossRef CAS.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904 CrossRef CAS.
- D. Benoit, S. Grimaldi, J. P. Finet, P. Tordo, M. Fontanille and Y. Gnanou, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1997, 38, 729 CAS.
- D. Benoit, E. Harth, P. Fox, R. M. Waymouth and C. J. Hawker, Macromolecules, 2000, 33, 363 CrossRef CAS.
- D. Benoit, C. J. Hawker, E. E. Huang, Z. Lin and T. P. Russell, Macromolecules, 2000, 33, 1505 CrossRef CAS.
- E. Harth, B. Van Horn and C. J. Hawker, Chem. Commun., 2001, 823 RSC.
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260 CrossRef CAS.
- T. Diaz, A. Fischer, A. Jonquieres, A. Brembilla and P. Lochon, Macromolecules, 2003, 36, 2235 CrossRef CAS.
- K. Schierholz, M. Givehchi, P. Fabre, F. Nallet, E. Papon, O. Guerret and Y. Gnanou, Macromolecules, 2003, 36, 5995 CrossRef CAS.
- Y. Guillaneuf, D. Gigmes, S. R. A. Marque, P. Astolfi, L. Greci, P. Tordo and D. Bertin, Macromolecules, 2007, 40, 3108 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274 CrossRef CAS.
- J. Nicolas, L. Mueller, C. Dire, K. Matyjaszewski and B. Charleux, Macromolecules, 2009, 42, 4470 CrossRef CAS.
- J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7572 CrossRef CAS.
- J. Gromada and K. Matyjaszewski, Macromolecules, 2001, 34, 7664 CrossRef CAS.
- K. Min, H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2005, 127, 3825 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482 CrossRef CAS.
- S. V. Arehart and K. Matyjaszewski, Macromolecules, 1999, 32, 2221 CrossRef CAS.
- J. K. Oh, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 3161 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- D. H. Solomon, E. Rizzardo and P. Cacioli, Csiro, 1986 Search PubMed.
- P. Corpart, D. Charmot, T. Biadatti, S. Z. Zard and D. Michelet, Rhodia Chimie, 1998, p. 62 Search PubMed.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914 CrossRef CAS.
- F. Lecolley, L. Tao, G. Mantovani, I. Durkin, S. Lautru and D. M. Haddleton, Chem. Commun., 2004, 2026 RSC.
- L. McDowall, G. J. Chen and M. H. Stenzel, Macromol. Rapid Commun., 2008, 29, 1666 CrossRef CAS.
- D. Samanta, S. Mcrae, B. Cooper, Y. X. Hu and T. Emrick, Biomacromolecules, 2008, 9, 2891 CrossRef CAS.
- D. Miyamoto, J. Watanabe and K. Ishihara, Biomaterials, 2004, 25, 71 CrossRef CAS.
- D. Miyamoto, J. Watanabe and K. Ishihara, J. Appl. Polym. Sci., 2004, 91, 827 CrossRef CAS.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220 CrossRef CAS.
- S. M. Ryan, X. X. Wang, G. Mantovani, C. T. Sayers, D. M. Haddleton and D. J. Brayden, J. Controlled Release, 2009, 135, 51 CrossRef CAS.
- C. T. Sayers, G. Mantovani, S. M. Ryan, R. K. Randev, O. Keiper, O. I. Leszczyszyn, C. Blindauer, D. J. Brayden and D. M. Haddleton, Soft Matter, 2009, 5, 3038 RSC.
- L. Tao, J. Q. Liu, J. T. Xu and T. P. Davis, Org. Biomol. Chem., 2009, 7, 3481 RSC.
- K. L. Heredia, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2007, 40, 4772 CrossRef CAS.
- V. Vazquez-Dorbatt, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2009, 42, 7650 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966 CrossRef CAS.
- B. Le Droumaguet, G. Mantovani, D. M. Haddleton and K. Velonia, J. Mater. Chem., 2007, 17, 1916 RSC.
- E. Bays, L. Tao, C. W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 1777 CrossRef CAS.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955 CrossRef CAS.
- C. Boyer, J. Liu, L. Wong, M. Tippett, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207 CrossRef.
- A. Lewis, Y. Tang, S. Brocchini, J.-w. Choi and A. Godwin, Bioconjugate Chem., 2008, 19, 2144 CrossRef CAS.
- F. M. Veronese and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 453 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459 CrossRef CAS.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS.
- A. Abuchowski, T. Van Es, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578 CAS.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. van Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582 CAS.
- F. M. Veronese, C. Monfardini, P. Caliceti, O. Schiavon, M. D. Scrawen and D. Beer, J. Controlled Release, 1996, 40, 199 CrossRef CAS.
- F. M. Veronese, P. Caliceti and O. Schiavon, J. Bioact. Compat. Polym., 1997, 12, 196 CAS.
- K. Ishihara, T. Ueda and N. Nakabayashi, Polym. J., 1990, 22, 355 CrossRef CAS.
- K. Ishihara, T. Tsuji, Y. Sakai and N. Nakabayashi, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 859 CrossRef CAS.
- K. Ishihara, Trends Polymer Sci., 1997, 5, 401 Search PubMed.
- M. D. Bentley, M. J. Roberts and J. M. Harris, J. Pharm. Sci., 1998, 87, 1446 CrossRef CAS.
- L. Tao, J. Liu, J. Hu and T. P. Davis, Chem. Commun., 2009, 6560 RSC.
- M. G. J. ten Cate, N. Severin and H. G. Börner, Macromolecules, 2006, 39, 7831 CrossRef CAS.
- J. Hentschel and H. G. Börner, J. Am. Chem. Soc., 2006, 128, 14142 CrossRef CAS.
- J. Janatova, J. K. Fuller and M. J. Hunter, J. Biol. Chem., 1968, 243, 3612 CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172 CrossRef CAS.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653 CrossRef CAS.
- G. Pound, J. M. McKenzie, R. F. M. Lange and B. Klumperman, Chem. Commun., 2008, 3193 RSC.
- L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Macromolecules, 2009, 42, 8028 CrossRef CAS.
- K. L. Heredia, G. N. Grover, L. Tao and H. D. Maynard, Macromolecules, 2009, 42, 2360 CrossRef CAS.
- L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Chem. Commun., 2009, 2148 RSC.
- C. Boyer, J. Q. Liu, V. Bulmus and T. P. Davis, Aust. J. Chem., 2009, 62, 830 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493 CrossRef.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657 CrossRef CAS.
- A. J. Dirks, S. S. Van Berkel, N. S. Hatzakis, J. A. Opsteen, F. L. Van Delft, J. J. L. M. Cornelissen, A. E. Rowan, J. C. M. Van Hest, F. P. J. T. Rutjes and R. J. M. Nolte, Chem. Commun., 2005, 4172 RSC.
- J.-F. Lutz, H. G. Börner and K. Weichenhan, Macromolecules, 2006, 39, 6376 CrossRef CAS.
- J. F. Lutz, H. G. Börner and K. Weichenhan, Aust. J. Chem., 2007, 60, 410 CrossRef CAS.
- K. Wang, L. Liang, S. Lin and X. He, Eur. Polym. J., 2008, 44, 3370 CrossRef CAS.
- X. He, L. Zhong, K. Wang, S. Lin and S. Luo, React. Funct. Polym., 2009, 69, 666 CrossRef CAS.
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641 CrossRef CAS.
- Z. P. Tolstyka, J. T. Kopping and H. A. Maynard, Macromolecules, 2008, 41, 599 CrossRef CAS.
- L. Tao, J. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847 CrossRef CAS.
- A. Godwin, M. Hartenstein, A. H. E. Muller and S. Brocchini, Angew. Chem., Int. Ed., 2001, 40, 594 CrossRef CAS.
- S. R. A. Devenish, J. B. Hill, J. W. Blunt, J. C. Morrisb and M. H. G. Munroa, Tetrahedron Lett., 2006, 47, 2875 CrossRef CAS.
- M. J. Yanjarappa, K. V. Gujraty, A. Joshi, A. Saraph and R. S. Kane, Biomacromolecules, 2006, 7, 1665 CrossRef CAS.
- M. Mourez, R. S. Kane, J. Mogridge, S. Metallo, P. Deschatelets, B. R. Sellman, G. M. Whitesides and R. J. Collier, Nat. Biotechnol., 2001, 19, 958 CrossRef CAS.
- J. Y. Hwang, R. C. Li and H. D. Maynard, J. Controlled Release, 2007, 122, 279 CrossRef CAS.
- R. C. Li, J. Hwang and H. D. Maynard, Chem. Commun., 2007, 3631 RSC.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380 CrossRef CAS.
- J. Nicolas, V. San Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006, 4697 RSC.
- B. Le Droumaguet and K. Velonia, Angew. Chem., Int. Ed., 2008, 47, 6263 CrossRef CAS.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288 CrossRef CAS.
- A. Marsh, A. Khan, D. M. Haddleton and M. J. Hannon, Macromolecules, 1999, 32, 8725 CrossRef CAS.
- H. J. Spijker, A. J. Dirks and J. C. M. van Hest, Polymer, 2005, 46, 8528 CrossRef CAS.
- H. J. Spijker, A. J. Dirks and J. C. M. Van Hest, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242 CrossRef CAS.
- J.-F. Lutz, A. F. Thünemann and K. Rurack, Macromolecules, 2005, 38, 8124 CrossRef CAS.
- S. Venkataraman and K. L. Wooley, Macromolecules, 2006, 39, 9661 CrossRef CAS.
- R. M. Broyer, G. M. Quaker and H. D. Maynard, J. Am. Chem. Soc., 2008, 130, 1041 CrossRef CAS.
- D. W. P. M. Löwik, L. Ayres, J. M. Smeenk and J. C. M. Van Hest, Adv. Polym. Sci., 2006, 202, 19.
- F. Sanda and T. Endo, Macromol. Chem. Phys., 1999, 200, 2651 CrossRef CAS.
- J. C. M. van Hest and D. A. Tirrell, Chem. Commun., 2001, 1897 RSC.
- L. Ayres, M. R. J. Vos, P. J. H. M. Adams, I. O. Shklyarevskiy and J. C. M. Van Hest, Macromolecules, 2003, 36, 5967 CrossRef CAS.
- L. Ayres, K. Koch, P. H. H. M. Adams and J. C. M. Van Hest, Macromolecules, 2005, 38, 1699 CrossRef CAS.
- L. Ayres, P. H. H. M. Adams, D. W. P. M. Löwik and J. C. M. Van Hest, Biomacromolecules, 2005, 6, 825 CrossRef CAS.
- J. M. Smeenk, L. Ayres, H. G. Stunnenberg and J. C. M. Van Hest, Macromol. Symp., 2005, 225, 1 CrossRef CAS.
- L. Ayres, G. M. Grotenbreg, G. A. van der Marel, H. S. Overkleeft, M. Overhand and J. C. M. Van Hest, Macromol. Rapid Commun., 2005, 26, 1336 CrossRef CAS.
- F. Fernandez-Trillo, A. Dureault, J. P. M. Bayley, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Macromolecules, 2007, 40, 6094 CrossRef CAS.
- D. W. Urry, Methods Enzymol., 1982, 82, 673 Search PubMed.
- D. E. Meyer and A. Chilkoti, Biomacromolecules, 2004, 5, 846 CrossRef CAS.
- A. Chilkoti, M. R. Dreher, D. E. Meyer and D. Raucher, Adv. Drug Delivery Rev., 2002, 54, 613 CrossRef CAS.
- T. J. Deming, Adv. Drug Delivery Rev., 2002, 54, 1145 CrossRef CAS.
- T. J. Deming, Adv. Polym. Sci., 2006, 202, 1 CAS.
- H. Rettig, E. Krause and H. G. Börner, Macromol. Rapid Commun., 2004, 25, 1251 CrossRef CAS.
- M. G. J. ten Cate, H. Rettig, K. Bernhardt and H. G. Börner, Macromolecules, 2005, 38, 10643 CrossRef CAS.
- J. Hentschel, K. Bleek, O. Ernst, J.-F. Lutz and H. G. Boerner, Macromolecules, 2008, 41, 1073 CrossRef CAS.
- L. Ayres, P. Hans, J. Adams, D. W. P. M. Löwik and J. C. M. van Hest, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6355 CrossRef CAS.
- S. Loschonsky, K. Shroff, A. Worz, O. Prucker, J. Ruhe and M. Biesalski, Biomacromolecules, 2008, 9, 543 CrossRef CAS.
- J. Couet, J. D. Jeyaprakash, S. Samuel, A. Kopyshev, S. Santer and M. Biesalski, Angew. Chem., Int. Ed., 2005, 44, 3297 CrossRef CAS.
- J. Couet and M. Biesalski, Macromolecules, 2006, 39, 7258 CrossRef CAS.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324 CrossRef CAS.
- M. R. Ghadiri, K. Kobayashi, J. R. Granja, R. K. Chadha and D. E. McRee, Angew. Chem., Int. Ed. Engl., 1995, 34, 93 CrossRef CAS.
- R. Ghadiri, Adv. Mater., 1995, 7, 675 CrossRef.
- J. D. Hartgerink, J. R. Granja, R. A. Milligan and M. R. Ghadiri, J. Am. Chem. Soc., 1996, 118, 43 CrossRef CAS.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988 CrossRef CAS.
- S. Loschonsky, J. Couet and M. Biesalski, Macromol. Rapid Commun., 2008, 29, 309 CrossRef CAS.
- H. Kakwere, C. K. Y. Chun, K. A. Jolliffe, R. J. Payne and S. Perrier, Chem. Commun., 2010 10.1039/b924112d.
- J. Liu, Q. Zhang, E. E. Remsen and K. L. Wooley, Biomacromolecules, 2001, 2, 362 CrossRef CAS.
- M. L. Becker, J. Liu and K. L. Wooley, Chem. Commun., 2003, 180 RSC.
- Q. Zhang, E. E. Remsen and K. L. Wooley, J. Am. Chem. Soc., 2000, 122, 3642 CrossRef CAS.
- H. Huang, E. E. Remsen, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1999, 121, 3805 CrossRef.
- M. L. Becker, J. Liu and K. L. Wooley, Biomacromolecules, 2005, 6, 220 CrossRef CAS.
- Y. Mei, K. L. Beers, H. C. M. Byrd, D. L. VanderHart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472 CrossRef CAS.
- T. J. Deming, Nature, 1997, 390, 386 CrossRef CAS.
- T. J. Deming, J. Am. Chem. Soc., 1998, 120, 4240 CrossRef CAS.
- T. J. Deming, Macromolecules, 1999, 32, 4500 CrossRef CAS.
- K. R. Brzezinska and T. J. Deming, Macromolecules, 2001, 34, 4348 CrossRef CAS.
- I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944 RSC.
- W. Vayaboury, O. Giani, H. Cottet, A. Deratani and F. Schue, Macromol. Rapid Commun., 2004, 25, 1221 CrossRef CAS.
- C.-M. Dong, X.-L. Sun, K. M. Faucher, R. P. Apkarian and E. L. Chaikof, Biomacromolecules, 2004, 5, 224 CrossRef CAS.
- C.-M. Dong, K. M. Faucher and E. L. Chaikof, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5754 CrossRef CAS.
- K. R. Brzezinska and T. J. Deming, Macromol. Biosci., 2004, 4, 566 CrossRef CAS.
- V. Coessens, Y. Nakagawa and K. Matyjaszewski, Polym. Bull., 1998, 40, 135 CrossRef CAS.
- J. Babin, C. Leroy, S. Lecommandoux, R. Borsali, Y. Gnanou and D. Taton, Chem. Commun., 2005, 1993 RSC.
- S. Abraham, C.-S. Ha and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2774 CrossRef CAS.
- S. Steig, F. Cornelius, P. Witte, B. B. P. Staal, C. E. Koning, A. Heise and H. Menzel, Chem. Commun., 2005, 5420 RSC.
- S. Steig, F. Cornelius, A. Heise, R. J. L. Knoop, G. J. M. Habraken, C. E. Koning and H. Menzel, Macromol. Symp., 2007, 248, 199 CrossRef CAS.
- R. J. I. Knoop, G. J. M. Habraken, N. Gogibus, S. Steig, H. Menzel, C. E. Koning and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3068 CrossRef CAS.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677 CrossRef CAS.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14 RSC.
- Y. Tsujii, K. Ohno, S. Yamamoto, A. Goto and T. Fukuda, Adv. Polym. Sci., 2006, 197, 1 CAS.
- R. R. Bhat, M. R. Tomlinson, T. Wu and J. Genzer, Adv. Polym. Sci., 2006, 198, 51 CAS.
- S. Tugulu, A. Arnold, I. Sielaff, K. Johnsson and H.-A. Klok, Biomacromolecules, 2005, 6, 1602 CrossRef CAS.
- F. J. Xu, Q. J. Cai, Y. L. Li, E. T. Kang and K. G. Neoh, Biomacromolecules, 2005, 6, 1012 CrossRef CAS.
- T. Godjevargova, A. Dimov and D. Ivanova, J. Appl. Polym. Sci., 1998, 68, 323 CrossRef CAS.
- S. Wang, M. Yoshimoto, K. Fukunaga and K. Nakao, Biotechnol. Bioeng., 2003, 83, 444 CrossRef CAS.
- L. Cen, K. G. Neoh and E. T. Kang, Biosens. Bioelectron., 2003, 18, 363 CrossRef CAS.
- X. Liu, K. G. Neoh, L. Cen and E. T. Kang, Biosens. Bioelectron., 2004, 19, 823 CrossRef CAS.
- J. Nicolas, E. Khoshdel and D. M. Haddleton, Chem. Commun., 2007, 1722 RSC.
- M. L. Becker, E. E. Remsen, D. Pan and K. L. Wooley, Bioconjugate Chem., 2004, 15, 699 CrossRef CAS.
- M. L. Becker, L. O. Bailey and K. L. Wooley, Bioconjugate Chem., 2004, 15, 710 CrossRef CAS.
- J. L. Turner, M. L. Becker, X. Li, J.-S. A. Taylor and K. L. Wooley, Soft Matter, 2005, 1, 69 RSC.
- M. J. Joralemon, N. L. Smith, D. Holowka, B. Baird and K. L. Wooley, Bioconjugate Chem., 2005, 16, 1246 CrossRef CAS.
- M. J. Joralemon, K. S. Murthy, E. E. Remsen, M. L. Becker and K. L. Wooley, Biomacromolecules, 2004, 5, 903 CrossRef CAS.
- D. Pan, J. L. Turner and K. L. Wooley, Chem. Commun., 2003, 2400 RSC.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068 RSC.
- G. Chen, D. Huynh, P. L. Felgner and Z. Guan, J. Am. Chem. Soc., 2006, 128, 4298 CrossRef CAS.
- G. Chen and Z. Guan, J. Am. Chem. Soc., 2004, 126, 2662 CrossRef CAS.
- G. Chen, X. S. Ma and Z. Guan, J. Am. Chem. Soc., 2003, 125, 6697 CrossRef CAS.
- Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059 CrossRef CAS.
- Z. Guan, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3680 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414 CrossRef CAS.
- J. A. Opsteen, R. P. Brinkhuis, R. L. Teeuwen, D. W. Lowik and J. C. van Hest, Chem. Commun., 2007, 3136 RSC.
- D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. M. Christianen, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772 CrossRef CAS.
- D. M. Vriezema, A. Kros, R. de Gelder, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Macromolecules, 2004, 37, 4736 CrossRef CAS.
- S. F. M. van Dongen, M. Nallani, S. Schoffelen, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Macromol. Rapid Commun., 2008, 29, 321 CrossRef CAS.
- J. Q. Liu, H. Y. Liu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 899 CrossRef CAS.
- A. N. Zelikin, G. K. Such, A. Postma and F. Caruso, Biomacromolecules, 2007, 8, 2950 CrossRef CAS.
- C. Boyer, V. Bulmus, P. Priyanto, W. Y. Teoh, R. Amal and T. P. Davis, J. Mater. Chem., 2009, 19, 111 RSC.
- M. Wilchek and E. A. Bayer, Anal. Biochem., 1988, 171, 1 CrossRef.
- P. C. Weber, D. H. Ohlendorf, J. J. Wendoloski and F. R. Salemme, Science, 1989, 243, 85 CrossRef CAS.
- M. Wilchek and E. A. Bayer, in Methods Enzymol., ed. J. N. Abelson and M. I. Simon, San Diego, CA, 1990 Search PubMed.
- M. D. Savage, G. Mattson, S. Desai, G. W. Nielander, S. Morgensen and E. J. Conklin, Avidin-Biotin Chemistry: A Handbook, Pierce Chemical Co., 1992 Search PubMed.
- D. Bontempo, R. C. Li, T. Ly, C. E. Brubaker and H. D. Maynard, Chem. Commun., 2005, 4702 RSC.
- H. D. Maynard, K. L. Heredia, R. C. Li, D. P. Parra and V. Vazquez-Dorbatt, J. Mater. Chem., 2007, 17, 4015 RSC.
- S. Kulkarni, C. Schilli, A. H. E. Müller, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2004, 15, 747 CrossRef CAS.
- Z. Ding, R. B. Fong, C. J. Long, P. S. Stayton and A. S. Hoffman, Nature, 2001, 411, 59 CrossRef CAS.
- S. Kulkarni, C. Schilli, B. Grin, A. H. E. Müller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736 CrossRef CAS.
- K. Qi, Q. Ma, E. E. Remsen, C. G. Clark, Jr. and K. L. Wooley, J. Am. Chem. Soc., 2004, 126, 6599 CrossRef CAS.
- B. S. Lee, J. K. Lee, W. J. Kim, Y. H. Jung, S. J. Sim, J. Lee and I. S. Choi, Biomacromolecules, 2007, 8, 744 CrossRef CAS.
- X. Wang, L. Liu, Y. Luo and H. Zhao, Langmuir, 2009, 25, 744 CrossRef CAS.
- Z. Jia, J. Liu, C. Boyer, T. P. Davis and V. Bulmus, Biomacromolecules, 2009, 10, 3253 CrossRef CAS.
- Z. Jia, L. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106 CrossRef CAS.
- I. C. Reynhout, J. J. Cornelissen and R. J. Nolte, J. Am. Chem. Soc., 2007, 129, 2327 CrossRef CAS.
- A. Dove, Nat. Biotechnol., 2001, 19, 913 CrossRef CAS.
- R. A. Dwek, Chem. Rev., 1996, 96, 683 CrossRef CAS.
- L. L. Kiessling and C. W. Cairo, Nat. Biotechnol., 2002, 20, 234 CrossRef.
- D. A. Tirrell, Nature, 2004, 430, 837 CrossRef CAS.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593 RSC.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321 CrossRef CAS.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431 CrossRef CAS.
- S. G. Spain, M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2059 CrossRef CAS.
- L. Albertin, M. H. Stenzel, C. Barner-Kowollik, L. J. R. Foster and T. P. Davis, Macromolecules, 2005, 38, 9075 CrossRef CAS.
- J. Bernard, A. Favier, L. Zhang, A. Nilasaroya, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2005, 38, 5475 CrossRef CAS.
- J. Bernard, X. Hao, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Biomacromolecules, 2006, 7, 232 CrossRef CAS.
- Y. Chen and G. Wulff, Macromol. Chem. Phys., 2001, 202, 3273 CrossRef CAS.
- Y. Chen and G. Wulff, Macromol. Chem. Phys., 2001, 202, 3426 CrossRef CAS.
- Y. M. Chen and G. Wulff, Macromol. Rapid Commun., 2002, 23, 59 CrossRef CAS.
- H. Götz, E. Harth, S. M. Schiller, C. W. Frank, W. Knoll and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3379 CrossRef CAS.
- V. Ladmiral, L. Monaghan, G. Mantovani and D. M. Haddleton, Polymer, 2005, 46, 8536 CrossRef CAS.
- Z.-C. Li, Y.-Z. Liang, G.-Q. Chen and F.-M. Li, Macromol. Rapid Commun., 2000, 21, 375 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin and C. L. McCormick, Polymer, 2003, 44, 6761 CrossRef CAS.
- S. Muthukrishnan, G. Jutz, X. Andre, H. Mori and A. H. E. Müller, Macromolecules, 2005, 38, 9 CrossRef CAS.
- S. Muthukrishnan, M. Zhang, M. Burkhardt, M. Drechsler, H. Mori and A. H. E. Müller, Macromolecules, 2005, 38, 7926 CrossRef CAS.
- R. Narain and S. P. Armes, Chem. Commun., 2002, 2776 RSC.
- K. Ohno, Y. Tsujii and T. Fukuda, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2473 CrossRef CAS.
- K. Ohno, Y. Tsujii, T. Miyamoto, T. Fukuda, M. Goto, K. Kobayashi and T. Akaike, Macromolecules, 1998, 31, 1064 CrossRef CAS.
- M. Okada, Prog. Polym. Sci., 2001, 26, 67 CrossRef CAS.
- M. H. Stenzel, L. Zhang and W. T. S. Huck, Macromol. Rapid Commun., 2006, 27, 1121 CrossRef CAS.
- Q. Yang, J. Tian, M.-X. Hu and Z.-K. Xu, Langmuir, 2007, 23, 6684 CrossRef CAS.
- A. Housni, H. Cai and R. Narain, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2007, 48, 888 CAS.
- A. Housni, H. Cai, S. Liu, S. H. Pun and R. Narain, Langmuir, 2007, 23, 5056 CrossRef CAS.
- T.-Y. Guo, P. Liu, J.-W. Zhu, M.-D. Song and B.-H. Zhang, Biomacromolecules, 2006, 7, 1196 CrossRef CAS.
- C. Gao, S. Muthukrishnan, W. Li, J. Yuan, Y. Xu and A. H. E. Mueller, Macromolecules, 2007, 40, 1803 CrossRef CAS.
- Z. Özyürek, H. Komber, S. Gramm, D. Schmaljohann, A. H. E. Müller and B. Voit, Macromol. Chem. Phys., 2007, 208, 1035 CrossRef.
- M. Al-Bagoury, K. Buchholz and E.-J. Yaacoub, Polym. Adv. Technol., 2007, 18, 313 CrossRef CAS.
- S. Baskaran, D. Grande, X.-L. Sun, A. Yayon and E. L. Chaikof, Bioconjugate Chem., 2002, 13, 1309 CrossRef CAS.
- D. Grande, S. Baskaran, C. Baskaran, Y. Gnanou and E. L. Chaikof, Macromolecules, 2000, 33, 1123 CrossRef CAS.
- D. Grande, S. Baskaran and E. L. Chaikof, Macromolecules, 2001, 34, 1640 CrossRef CAS.
- X.-L. Sun, D. Grande, S. Baskaran, S. R. Hanson and E. L. Chaikof, Biomacromolecules, 2002, 3, 1065 CrossRef CAS.
- Y. Miura, D. Koketsu and K. Kobayashi, Polym. Adv. Technol., 2007, 18, 647 CrossRef CAS.
- A. M. Granville, D. Quemener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromol. Symp., 2007, 255, 81 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823 CrossRef CAS.
- J. Geng, J. Lindqvist, G. Mantovani, G. Chen, C. T. Sayers, G. J. Clarkson and D. M. Haddleton, QSAR Comb. Sci., 2007, 26, 1220 Search PubMed.
- J. Geng, J. Lindqvist, G. Mantovani and D. M. Haddleton, Angew. Chem., Int. Ed., 2008, 47, 4180 CrossRef CAS.
- Z. Hu, Y. Liu, C. Hong and C. Pan, J. Appl. Polym. Sci., 2005, 98, 189 CrossRef CAS.
- N. R. Cameron, S. G. Spain, J. A. Kingham, S. Weck, L. Albertin, C. A. Barker, G. Battaglia, T. Smart and A. Blanazs, Faraday Discuss., 2008, 139, 359 RSC.
- S. Pearson, N. Allen and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1706 CrossRef CAS.
- V. Ramiah, H. Matahwa, W. Weber, J. B. McLeary and R. D. Sanderson, Macromol. Symp., 2007, 255, 70 CrossRef CAS.
- Z. Deng, M. Ahmed and R. Narain, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 614 CrossRef CAS.
- L. Zhang and M. H. Stenzel, Aust. J. Chem., 2009, 62, 813 CrossRef CAS.
- S. R. S. Ting, A. M. Gregory and M. H. Stenzel, Biomacromolecules, 2009, 10, 342 CrossRef CAS.
- G. Chen, S. Amajjahe and M. H. Stenzel, Chem. Commun., 2009, 1198 RSC.
- S. Qiu, H. Huang, X.-H. Dai, W. Zhou and C.-M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2009 CrossRef CAS.
- X.-H. Dai and C.-M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 817 CrossRef CAS.
- L. Zhang, J. Bernard, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromol. Rapid Commun., 2008, 29, 123 CrossRef CAS.
- X.-H. Dai, C.-M. Dong and D. Yan, J. Phys. Chem. B, 2008, 112, 3644 CrossRef CAS.
- S. R. S. Ting, E. H. Min, P. Escalé, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422 CrossRef CAS.
- G. Chen, L. Tao, G. Mantovani, J. Geng, D. Nyström and D. M. Haddleton, Macromolecules, 2007, 40, 7513 CrossRef CAS.
- M. Toyoshima and Y. Miura, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1412 CrossRef CAS.
- S. G. Spain, L. Albertin and N. L. Cameron, Chem. Commun., 2006, 4198 RSC.
- R. Narain, A. Housni, G. Gody, P. Boullanger, M.-T. Charreyre and T. Delair, Langmuir, 2007, 23, 12835 CrossRef CAS.
- K. Mizukami, H. Takakura, T. Matsunaga and H. Kitano, Colloids Surf., B, 2008, 66, 110 CrossRef CAS.
- A. Mateescu, J. Ye, R. Narainc and M. Vamvakaki, Soft Matter, 2009, 5, 1621 RSC.
- S. Hou, X.-L. Sun, C.-M. Dong and E. L. Chaikof, Bioconjugate Chem., 2004, 15, 954 CrossRef CAS.
- X.-L. Sun, K. M. Faucher, M. Houston, D. Grande and E. L. Chaikof, J. Am. Chem. Soc., 2002, 124, 7258 CrossRef CAS.
- R. Narain, React. Funct. Polym., 2006, 66, 1589 CrossRef CAS.
- C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029 RSC.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156 CrossRef CAS.
- V. Vazquez-Dorbatt, Z. P. Tolstyka, C.-W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 2207 CrossRef CAS.
- S. S. Gupta, K. S. Raja, E. Kaltgrad, E. Strable and M. G. Finn, Chem. Commun., 2005, 4315 RSC.
- V. Vazquez-Dorbatt and H. D. Maynard, Biomacromolecules, 2006, 7, 2297 CrossRef CAS.
- G. Gody, P. Boullanger, C. Ladaviere, M.-T. Charreyre and T. Delair, Macromol. Rapid Commun., 2008, 29, 511 CrossRef CAS.
- X. Jiang, M. Ahmed, Z. Deng and R. Narain, Bioconjugate Chem., 2009, 20, 994 CrossRef CAS.
- M. Bathfield, F. D'Agosto, R. Spitz, M. T. Charreyre and T. Delair, J. Am. Chem. Soc., 2006, 128, 2546 CrossRef CAS.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372 CrossRef CAS.
- N.-Y. Xiao, A.-L. Li, H. Liang and J. Lu, Macromolecules, 2008, 41, 2374 CrossRef CAS.
- K. Godula, D. Rabuka, K. T. Nam and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 4973 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C. W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008, 3245 RSC.
- C. Da Pieve, P. Williams, D. M. Haddleton, R. M. J. Palmer and S. Missailidis, Bioconjugate Chem., 2009, 21, 169.
- J. T. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4302 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773 CrossRef CAS.
- P. He and L. He, Biomacromolecules, 2009, 10, 1804 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |