Living spontaneous gradient copolymers of acrylic acid and styrene: one-pot synthesis of pH-responsive amphiphiles†
Simon
Harrisson
*,
Francesca
Ercole
and
Benjamin W.
Muir
CSIRO Molecular and Health Technologies, Bayview Ave, Clayton, VIC 3168, Australia. E-mail: simon.harrisson@csiro.au; Fax: +61 3 9545 2446; Tel: +61 3 9545 2499
First published on 22nd December 2009
Abstract
RAFT polymerization was used to prepare copolymers of acrylic acid (AA) and styrene (STY) with mole fractions of STY (FSTY) ranging from 0.1 to 0.3 and targeted degrees of polymerization between 50 and 150. The high reactivity of AA-terminal radicals towards STY in this system (rAA = 0.082) resulted in the spontaneous formation of composition gradients, resulting in polymers with block-like structures comprising a STY-rich segment, a relatively short transitional segment, and a segment of AA homopolymer. Atomic force microscopy analysis of thin films of the copolymer revealed phase separated structures which developed after exposure to water. Dynamic light scattering measurements showed pH-responsive amphiphilicity that resulted in dissolved polymer at neutral and basic pH and self-assembly in weakly acidic solutions.
Introduction
The ability to prepare block copolymers is one of the most useful attributes of living polymerization. By combining two or more distinct segments within a single polymer chain, it is possible to prepare an enormous variety of functional materials, including compatibilisers for polymer blends,1 thermoplastic elastomers,2 and the components of supramolecular assemblies such as polymersomes and polymeric micelles.3 Living radical polymerization techniques such as nitroxide-mediated polymerization (NMP),4 atom transfer radical polymerization (ATRP),5,6 and reversible addition–fragmentation chain transfer (RAFT) polymerization7 are relatively insensitive to contaminants such as water and oxygen, and are compatible with a wide range of functionalities. This has allowed the production of an enormous variety of functional block copolymers.There remain two major drawbacks to current block copolymer preparation techniques via living radical polymerization. The first is that two separate polymerization reactions are required to produce a diblock copolymer. These reactions, with their preparation and workup stages, are time-consuming, with the result that it typically takes several days to produce a single block copolymer. Second, the nature of living radical polymerization, with low but unavoidable levels of termination, requires that polymerizations be stopped before reaching full conversion in order to limit the formation of dead polymer chains. This leads to lower reproducibility in their synthesis, as it is difficult to stop a polymerization at a predetermined target conversion. As a result, the preparation of block copolymers by living radical polymerization can be time-consuming, expensive, and poorly reproducible.
Spontaneous gradient copolymers
Spontaneous gradient copolymers present the possibility of a functionally equivalent alternative to conventional block copolymers. In a copolymerization of two monomers, M1 and M2, with reactivity ratios, r1 and r2, the relative rate of consumption of each monomer (df1/df2) is given by the Mayo–Lewis equation:2| df1/df2 = (r1f12 + f1f2)/(r2f22 + f1f2) = F1/F2 | (1) |
In most cases, F1/F2 is different to f1/f2, resulting in composition drift given by the integrated copolymerization composition equation:2
 | (2) |
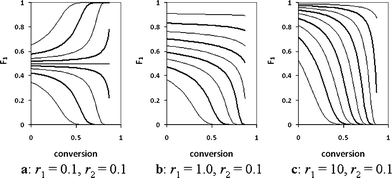
In a living polymerization, this causes the average composition of the polymer chains to vary as a function of chain length. Some copolymer composition curves are shown in Fig. 1, illustrating the variation of composition along an average polymer chain as a function of the initial mole fraction of M1 for values of r1 ranging from 0.1 to 10 and r2 equal to 0.1.
 | ||
| Fig. 1 Copolymer composition trajectories for various r1 and r2 = 0.1. F1 represents the instantaneous mole fraction of M1 in the copolymer. Initial fraction of M1 ranges from 0.1 to 0.9 in steps of 0.1. | ||
In each case shown in Fig. 1, it is possible to find a trajectory that comprises an initial region of relatively stable composition, a relatively short transitional region of rapidly changing composition, and a final section of homopolymer. As a general rule, such a trajectory occurs whenever one monomer shows a strong tendency to alternate (r < 0.2), and when the initial monomer ratio is chosen so as to maximize the difference between F1/F2 and f1/f2. The resulting polymers show block-like variation in composition as a function of chain length, and might be expected to show properties similar to those of block copolymers. They can be prepared by a one-pot synthesis, and their composition is entirely determined by the initial monomer ratio and total conversion. Thus, these polymers should display many of the useful properties of block copolymers, with greater reproducibility and at lower cost.
Gradient copolymers have been the subject of a significant body of research, the bulk of which has focused on copolymers with smoothly varying composition.8,9 Only a few examples of gradient polymers with block-like structures have been presented in the literature, the best known being the styrene (STY)–maleic anhydride system10–18 and related STY–maleimide systems (e.g. STY–N-phenyl maleimide19). In these cases, both monomers have reactivity ratios close to zero, resulting in polymers with composition profiles similar to those of Fig. 1a. Another system that has received some attention is the copolymerization of vinyl acetate with various acrylate monomers (e.g. methyl acrylate19 or tert-butyl acrylate20) in which the reactivity ratio of vinyl acetate is typically close to zero and the reactivity ratio of the acrylate is ≫1, leading to composition trajectories similar to those of Fig. 1c. Polymerizations of monomers with such widely varying reactivities can be difficult to control. The intervening region, represented by Fig. 1b, has received little or no attention, although block-like structures can be prepared by appropriate selection of initial monomer ratios. In Fig. 1b, polymerizations with initial f1 = 0.2–0.4 will yield polymers with block-like structures.
In this report, we demonstrate the preparation of copolymers showing block-like properties of microphase separation and self-assembly from the STY–acrylic acid (AA) monomer system. In bulk at 50 °C, the reactivity ratios of these monomers have been reported as rSTY = 0.38 and rAA = 0.13.21 Thus, AA shows a strong tendency to alternate with STY, while STY has only a weak tendency to alternate with AA. The expected copolymer composition trajectories for these reactivity ratios are shown in Fig. 2.
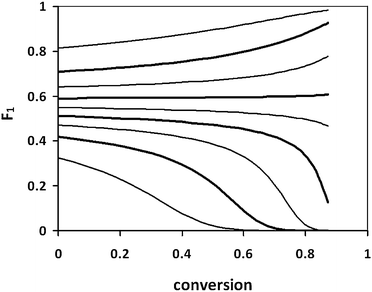 | ||
| Fig. 2 Instantaneous copolymer composition trajectories for bulk copolymerization of STY and AA at 50 °C. Reactivity ratios: rSTY = 0.38 and rAA = 0.13.21 Initial mole fraction of STY ranges from 0.1 to 0.9 in steps of 0.1. | ||
It is apparent that copolymers prepared from mixtures of AA and STY with initial fSTY approximately 0.2–0.3 will contain an initial segment of poly(STYx-ran-AA(1−x)), where x is close to 0.5, followed by a transitional segment, and a final segment of polyAA. The initial segment would be expected to be more hydrophobic than the final segment, but both segments should be soluble in sufficiently alkaline solution. These properties might lead to pH-responsive amphiphiles, with self-assembly in weakly acidic solutions.
The living copolymerization of STY and AA has previously been reported using NMP.22–24 The reactivity ratios of the STY–AA system are unusually sensitive to temperature and solvent (due to the formation of hydrogen-bonded AA dimers), so under the conditions used (120 °C, 1,4-dioxane solution, rSTY = 0.72 ± 0.04, rAA = 0.27 ± 0.07),23 relatively weak gradients were formed. The use of RAFT (Scheme 1) allowed us to conduct the polymerization at lower temperatures and in bulk, and enabled us to access block-like structures.
 | ||
| Scheme 1 RAFT copolymerization of styrene and acrylic acid. | ||
Experimental
Materials
STY and AA were obtained from Aldrich and flash distilled before use. S-Butyl-S′-2-propionic acid trithiocarbonate was synthesized according to a published procedure.25 2,2′-Azobis(isobutyronitrile) (AIBN) was obtained from TCI-EP and recrystallized from MeOH before use.Polymerizations
Mixtures of AA and STY were prepared containing 10, 20 or 30 mol% STY on a scale of 5 g (10 mol% and 30 mol% STY) or 10 g (20 mol% STY). To each mixture was added sufficient RAFT agent, S-butyl-S′-2-propionic acid trithiocarbonate, to give a molar ratio of monomer : RAFT of 50, 100 or 150, and 0.2 mg AIBN per gram solution (∼0.1 mol% relative to monomer). The solutions were each divided into three parts, placed in ampoules, freeze–thaw degassed (3×) and flame-sealed, and placed in an oil bath at 60 °C. Ampoules were removed from the oil bath after 2–24 h. A sample of the polymerization mixture was analyzed by 1H NMR to determine overall conversion and residual monomer ratio. The polymer was separated from the remaining mixture by precipitating from methanol. Characteristics of the polymerization mixtures and isolated polymers for each polymerization are shown in Table 1.| Solution | f STY,initial | [M]/[RAFT] | t/h | Conv. (%) | f STY | F STY | M n,theo/g mol−1 | M n (gpc) | PDI |
|---|---|---|---|---|---|---|---|---|---|
| a n.d.: insufficient polymer was recovered for methylation and further analysis. | |||||||||
| 1a | 0.1 | 50 | 2 | 4.57 | 0.08 | n.d.a | 218 | n.d.a | n.d.a |
| 6 | 24.1 | 0.04 | 0.32 | 1100 | 1090 | 1.15 | |||
| 24 | 76.3 | 0.00 | 0.13 | 3380 | 2830 | 1.19 | |||
| 1b | 0.1 | 150 | 2 | 17.6 | 0.06 | 0.36 | 2390 | 1790 | 1.22 |
| 6 | 37.5 | 0.01 | 0.21 | 5094 | 5410 | 1.24 | |||
| 24 | 86.1 | 0.00 | 0.12 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
9360 | 1.44 | |||
| 2 | 0.2 | 100 | 2 | 12.4 | 0.17 | 0.43 | 1170 | 1340 | 1.22 |
| 4 | 29.0 | 0.11 | 0.43 | 2710 | 2960 | 1.12 | |||
| 24 | 85.8 | 0.00 | 0.22 | 7760 | 5300 | 1.14 | |||
| 3a | 0.3 | 50 | 2 | 13.1 | 0.27 | 0.46 | 624 | 860 | 1.22 |
| 6 | 63.4 | 0.08 | 0.46 | 2980 | 2820 | 1.21 | |||
| 24 | 88.0 | 0.00 | 0.32 | 4050 | 4150 | 1.35 | |||
| 3b | 0.3 | 150 | 2 | 18.4 | 0.26 | 0.48 | 2610 | 3400 | 1.56 |
| 6 | 80.7 | 0.02 | 0.33 | 11200 |
9060 | 1.35 | |||
| 24 | 87.5 | 0.00 | 0.35 | 12100 |
13100 |
1.37 |
Methylation of polymers
Methylation was carried out according to the procedure of Hutson et al.26 0.1 g of polymer was dissolved or suspended in 1 g THF. 0.9 g of 20% tetramethylammonium hydroxide in methanol was added and then 0.3 g of methyl iodide. The samples were shaken at room temperature for at least 24 h. Methylated samples were analyzed by 1H NMR (to determine copolymer composition) and GPC (to determine molecular weight).GPC
Gel permeation chromatography (GPC) was performed on a Waters 515 HPLC pump and Waters 717 Plus Autosampler equipped with Waters 2414 refractive index detector and 3× Mixed-C (7.5 mm × 300 mm, 5 µm particle size, linear molecular weight range 200–2000000) and 1 Mixed E PLgel column (7.5 mm × 300 mm, 3 µm particle size, linear molecular weight range up to 30000) from Polymer Laboratories. Tetrahydrofuran (THF) with a flow rate of 1.0 mL min−1 was used as eluent at 30 °C. Molecular weights were calculated via calibration with narrow polydispersity polystyrene standards (Polymer Laboratories) ranging from 600 to 7.5 × 106 g mol−1. Number (Mn) and weight-average (Mw) molecular weights were calculated using Waters Millennium/Empower software. A third order polynomial was used to fit the log Mnvs. time calibration curve.
NMR
1H NMR (200 MHz) spectra were obtained with a Bruker Av200 spectrometer at 25 °C. Spectra were recorded for samples dissolved in CDCl3 and chemical shifts are reported as parts per million from external tetramethylsilane. Monomer fractions were obtained from the NMR spectra by taking the ratio of the areas of the resonances corresponding to the vinylic hydrogens of each monomer. Monomer conversions were determined by the ratio of the area of vinylic hydrogens to the area of aliphatic hydrogen resonances (from backbone protons). Copolymer compositions were determined from the relative ratios of aromatic (5H) and methoxy (3H) protons in the methylated copolymer, normalized for the number of protons.Differential scanning calorimetry
Thermal analysis of the STY–AA copolymers was performed using a differential scanning calorimeter (Mettler-Toledo DSC 821) using the procedure of Wong et al.27 Temperature and heat flow were calibrated using indium and zinc as reference samples. The copolymer samples were first heated at 40 °C min−1 to 200 °C and held at constant temperature for 20 min to erase thermal history and dry the samples of adsorbed water. Samples were then quenched at −40 °C min−1 to 0 °C before being reheated to 200 °C at 10 °C min−1. Glass transition temperature (Tg) measurements were obtained from the second heating scan.Dynamic light scattering
1 mg mL−1 solutions of polymers 1–3 were prepared by dissolving in water containing one equivalent of NaOH, then diluting with buffer solutions to prepare 0.1 mg mL−1 solutions of polymer in 50 mM buffer at pHs varying between 3.5 and 6.4 (citrate buffer) or 7.9 (phosphate buffer). The resulting solutions were passed through a 0.45 µm filter before analysis in a Zetasizer-Nano instrument (Malvern, UK). The analysis was performed at 25 °C and for each sample, the mean diameter of 6 determinations was calculated.Atomic force microscopy
An Asylum Research MFP-3D atomic force microscope (Santa Barbara, CA, USA) was used for all surface analysis in tapping mode with ultrasharp silicon nitride tips (NSC15 noncontact silicon cantilevers, MikroMasch, Spain). The tips used in this study had a typical force constant of 40 N m−1 and a resonant frequency of 320 kHz. Typical scan settings involved the use of an applied piezo deflection voltage of 0.8 V at a scan rate of 0.3 Hz. Films of polymer were prepared on ultra-flat single crystal, silicon wafers (〈100〉, 1 cm2 × 0.5 mm thick, M.M.R.C P/L). Silicon wafers were cleaned by immersion in a 1% RBS-35 surfactant (Pierce)–10% ethanol solution and ultrasonication for 1 h followed by extended rinsing in Milli-Q water and blow drying. To produce thin polymer films on silicon for AFM analysis, a 0.5 wt% solution of polymer was prepared in 90 : 10 dioxane–water. A drop of solution was placed on a silicon wafer and allowed to evaporate at room temperature. The film was then annealed in a vacuum oven at 130 °C for 24 h. Annealed polymer-coated silicon wafers were subsequently immersed in neutral (unbuffered) Milli-Q water overnight to enhance phase contrast. Samples were blow dried with ultra high purity nitrogen gas immediately before imaging.Determination of reactivity ratios
Point estimates and joint confidence intervals for reactivity ratios were determined using the visualization of the sum of squares method developed by van den Brink et al.28 The integrated copolymerization equation was fitted to conversion, monomer fraction and copolymer composition data, assuming non-negligible errors in all variables. As noted by van den Brink et al., this method is computationally inefficient but simple to implement, and was performed on Microsoft Excel 2003 software.Results and discussion
Synthesis of STY–AA gradient copolymers
RAFT polymerizations were carried out using mixtures of STY and AA containing 10, 20 or 30 mol% STY (fSTY = 0.1, 0.2, and 0.3), with target degrees of polymerization of 50, 100 or 150. The polymerizations were performed in bulk at 60 °C for 24 h, using the RAFT agent S-butyl-S′-2-propionic acid trithiocarbonate and AIBN as initiator. The properties of the resulting polymers are summarized in Table 2.| Polymer | f STY,initial | [M]/[RAFT] | Conversion (%) | M n (PDI) | M n,theo/g mol−1 | F STY | T g/°C | x | a | b | c |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Approximate structure: (STYx-ran-AA(1−x))a-block-(STY-grad-AA)b-block-AAc. | |||||||||||
| 1a | 0.1 | 50 | 76.3 | 2830 (1.19) | 3380 | 0.13 | 120 | 0.34 | 5 | 11 | 22 |
| 1b | 0.1 | 150 | 86.1 | 9360 (1.44) | 11400 |
0.12 | 122 | 0.34 | 15 | 39 | 75 |
| 2 | 0.2 | 100 | 85.6 | 5300 (1.14) | 7800 | 0.22 | 128 | 0.41 | 28 | 25 | 33 |
| 3a | 0.3 | 50 | 88.0 | 4150 (1.35) | 4050 | 0.32 | 132 | 0.45 | 24 | 10 | 10 |
| 3b | 0.3 | 150 | 87.5 | 13100 (1.37) |
12100 |
0.35 | 140 | 0.45 | 70 | 27 | 34 |
Additional polymerizations were carried out for shorter periods of time (2–6 h) in order to track the changes in monomer composition and evolution of the molecular weight distributions as a function of conversion. The polymerizations were moderately well-controlled, with polydispersities less than 1.6 in all cases (average polydispersity 1.27), and good agreement between measured and theoretical Mn, as shown in Fig. 3.
 | ||
| Fig. 3 M n and PDI vs. theoretical Mn for STY–AA polymerizations. | ||
Best results were obtained for the polymerizations with lower target degree of polymerization (DPn) and fSTY. Thus polymerizations of 1a and 2, with fSTY of 0.1 and 0.2, respectively, and target DPn of 50 and 100, were well-controlled throughout, with PDI consistently less than 1.2. Polymerization of 3b, with fSTY of 0.3 and target DPn of 150, was relatively poorly controlled, with PDI greater than 1.35, and polymerizations of 1b and 3a were intermediate, with PDI ≈ 1.2 in the early stages of polymerization, and some loss of control in the final stages causing an increase in polydispersity of the final polymer to 1.44 and 1.37, respectively. The loss of control at higher molecular weight may be a result of chain transfer, which has previously been noted in living polymerizations of AA.29
Measurement of the comonomer composition at various conversions showed that STY was consumed more rapidly than AA. The separated polymers were exhaustively methylated with methyl iodide, and 1H NMR analysis was used to determine the copolymer composition by measuring the relative proportions of methyl acrylate and STY units. Both the comonomer and the copolymer composition results were then fitted to the integrated copolymer composition equation using the visualization of the sum of squares space method developed by van den Brink et al.,28 resulting in point estimates for rAA and rSTY of 0.082 and 0.21, respectively. The point estimates and 95% joint confidence interval are shown in Fig. 4. It can be seen that the 95% confidence interval for rAA is quite narrow (0.06–0.11) while that of rSTY is broad (0–0.6). This reflects the very limited effect of rSTY on the copolymer composition when fSTY is small. As the point estimate of 0.21 for rSTY was reasonably close to previously reported values (obtained from the Polymer Handbook30), it was used for subsequent modeling (e.g. in Fig. 5 and 6). Values of rSTY ranging from 0 to 0.6 can be substituted without significantly affecting the results.
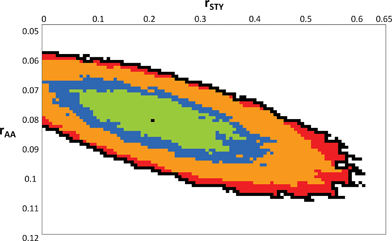 | ||
| Fig. 4 95% Joint confidence interval (JCI) for reactivity ratios of AA and STY with point estimate (rAA = 0.082, rSTY = 0.21). Internal contours indicate 50%, 70%, and 90% JCIs. | ||
 | ||
| Fig. 5 (a) Modeled and measured monomer compositions (solid line and open symbols), and cumulative copolymer compositions (dashed line and filled symbols) for fSTY,initial = 0.1 (△), 0.2 (◇), and 0.3 (□). Model parameters: rAA = 0.082 and rSTY = 0.21. (b) Modeled instantaneous copolymer compositions for fSTY,initial = 0.1, 0.2 and 0.3 as a function of conversion (solid lines) and best fit approximations as a copolymer comprising a copolymer block of constant composition, a gradient block, and a homopolymer block (dashed lines). | ||
 | ||
| Fig. 6 Simulated polymer sequences (10 each) for 1a, 3a, 1b, 3b and 2. The direction of polymerization is from left to right. AA–AA dyads are shown in blue, AA–STY and STY–AA dyads in green, and STY–STY dyads in red. The bottom set has the same average composition gradient as 2 but a statistical sequence distribution, in which the probability of finding a particular monomer is independent of the previous monomer. | ||
The comonomer and copolymer composition results are shown in Fig. 5a, with the modeled values for reactivity ratios rAA = 0.082 and rSTY = 0.21. These reactivity ratios were used to predict the average copolymer composition profile of each polymer chain, shown in Fig. 5b. These profiles can be approximated as an initial STY-rich segment of nearly constant composition, followed by a transitional gradient segment, and finally a segment of AA homopolymer, giving an approximate structure poly(STYx-ran-AA(1−x))a-block-(STY-grad-AA)b-block-AAc (Fig. 5b). Values of x, a, b, and c for each polymer are given in Table 2.
While Fig. 5b accurately describes the average properties of the polymer chains, each individual chain is an assembly of discrete monomer units of STY or AA. The smooth variations in composition shown in Fig. 5b are not meaningful on this scale. Fig. 6 provides a schematic representation of the copolymer composition gradient on the scale of an individual chain. Ten chains are presented for each polymer synthesized. Each chain consists of a number of units equal to the number-average degree of polymerization of the polymer, while each unit is colored according to whether the monomer represented is the first unit of an AA–AA (blue), AA–STY or STY–AA (green) or STY–STY (red) dyad. The probability that a given unit is assigned to AA or STY is determined by the identity of the previous unit and the monomer ratio at the conversion corresponding to that chain length:
 | (3) |
The simulated chains shown in Fig. 6 are characterized by long alternating AA–STY sequences with occasional AA–AA defects. The frequency of AA–AA defects gradually increases until an AA homopolymer is reached. STY–STY dyads are rare. All chains have clearly defined STY-rich and STY-poor segments, but the random variation in each chain makes it difficult to define a gradient segment in an individual chain. The strong tendency of AA-terminal radicals to react with STY monomer (rAA ≪ 1) results in a more ordered copolymer composition than would be expected from the composition gradient alone. This is illustrated by the final set of chains in Fig. 6, which has the same composition gradient as 2, but in which the probability of finding AA or STY at a particular location is independent of the preceding unit. In this case, the long alternating AA–STY sequences found in 2 are absent.
Consideration of the average polymer composition (Fig. 5) and the composition on the level of individual chains (Fig. 6) shows that the polymers display an initial block-like segment of approximately alternating STY–AA copolymer and a final segment of AA homopolymer, separated by a less well-defined transitional region. Polymers 2, 3a, and 3b more closely approximate a block copolymer structure, while polymers 1a and 1b display a relatively short initial block and long transitional segment.
Block-like properties of copolymers
We hypothesized that the block-like structure of the polymers would result in behavior characteristic of block copolymers such as microphase separation. DSC of the polymers showed only a single glass transition temperature, between 120 and 140 °C (Table 2) depending on the polymer composition and molecular weight (Tg = 110 °C + (66 × fSTY) + (0.00067 × Mn) ± 1 °C), and thin films of the copolymers cast onto silicon wafers for AFM analysis gave flat, featureless surfaces. Thus no evidence of microphase separation was observed in the bulk polymer, suggesting that the STY-rich segment is compatible with the AA homopolymer. The transitional segment may also compatibilize the two blocks.The cast polymer films were then incubated in Milli-Q water for 24 h, and reanalyzed by AFM. Fig. 7 shows tapping mode AFM phase contrast images of the incubated copolymer films. Phase contrast imaging via AFM monitors the phase lag of the cantilever oscillation, relative to the signal on the piezoelectric driver. This allows mapping of the phase features of a material and can be used to detect changes in the surface composition of a polymer. Distinct phase contrast is visible in the films of polymers 2, 3a, and 3b, while little phase contrast is evident in the films of 1a and 1b. The images clearly show regions of high and low phase contrast in the polymers containing 20% STY or greater. AFM height images (see ESI†) reveal that these polymers are also rougher, with RMS roughness ranging from 0.5 to 0.8 nm while the polymers 1a and 1b have RMS roughnesses of 0.2 and 0.3 nm, respectively. Polymers 2 and 3a display the greatest phase contrast with phase separated features of 10–50 nm in size. We hypothesize that exposure to water causes the less hydrophilic STY-rich segments to segregate from the AA homopolymer, leading to the observed microphase separation. This is observed most clearly in polymers 2, 3a and 3b, which contain a greater proportion of STY and have a more defined block-like structure than polymers 1a and 1b.
 | ||
| Fig. 7 AFM tapping mode phase contrast images of annealed and hydrated block copolymers (1a, 1b, 2, 3a and 3b) on silicon wafers (scan size: 1 µm, phase scale: 5°). | ||
pH-responsive amphiphilicity
Polyacrylic acid is a weak polyelectrolyte, which is neutral at pH less than about 3 and fully dissociated at pH greater than about 7.31 The presence of STY units in one segment of the chain should increase the hydrophobicity of that segment, causing it to become insoluble at low pH while the AA segment remains soluble. Thus the STY–AA gradient copolymers should show pH-responsive amphiphilicity.Dynamic light scattering (DLS) measurements (summarized in Table 3) of 1 mg mL−1 solutions of the polymers in 50 mM buffer solutions of pH ranging from 3 to 8 showed that at pH 6.4 and above, all polymers were present in solution as unimers or poorly defined aggregates. At pH 5.3, polymers 2, 3a and 3b formed well-defined micelles. Polymers 3a and 3b precipitated from solution at pH 4.5, while 2 remained in solution as micelles. At pH 3.5, polymer 1b formed a micellar solution, but all other polymers precipitated.
| pH | 1a | 1b | 2 | 3a | 3b |
|---|---|---|---|---|---|
| a Polymer precipitated. b A multimodal distribution was observed, comprising unimers (<10 nm) and loose aggregates (>100 nm). | |||||
| 3.5 | ppta | 25.0 (0.45) | ppta | ppta | ppta |
| 4.5 | mmb | 15.5 (0.55) | 25 (0.09) | ppta | ppta |
| 5.3 | mmb | mmb | 18.7 (0.23) | 34.2 (0.12) | 50.8 (0.13) |
| 6.4 | mmb | mmb | mmb | mmb | mmb |
| 7.9 | mmb | mmb | mmb | mmb | mmb |
The pH-responsive amphiphilicity is also demonstrated by the change in number-average hydrodynamic diameter Dh as a function of pH (Fig. 8). The number-average Dh has been chosen to emphasize the smallest diameter component of the solution in the case of multimodal distributions. For polymers 2, 3a and 3b, Dh increases dramatically below pH 6, while polymers 1a and 1b show an increase in Dh below pH 5. Intensity-weighted size distributions for all polymers are shown in the ESI†.
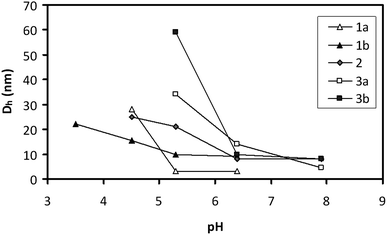 | ||
| Fig. 8 Number-average hydrodynamic diameter, Dh, of all polymers as a function of pH. | ||
The clearest evidence of amphiphilicity was observed for polymers 2, 3a, and 3b, which formed micelles with reasonably narrow size distributions (polydispersity ∼0.10) at pH 4.5 (2) and 5.3 (2, 3a, and 3b). While polymers 1a and 1b showed some evidence of aggregation at pH ≤ 4.5, the size distributions of these aggregates were broad (1b) or multimodal (1a). The broader distributions of these polymers may be related to their less-ordered structure (Fig. 6). The pH of the transitions from unimeric solutions to micelles and from micelles to precipitate depended on FSTY, with 3a and 3b (FSTY = 0.32 and 0.35, respectively) forming micelles at higher pH (5.3), 2 (FSTY = 0.22) at intermediate pH (4.5–5.3), and 1b (FSTY = 0.12) at low pH (3.5–4.5).
Conclusions
Mixtures of STY and AA containing 10–30% STY have been polymerized via RAFT polymerization. Preferential consumption of STY during the polymerization leads to the formation of copolymers with STY content that varies as a function of chain length. The polymers contain a STY-rich segment (containing 36–46% STY) of nearly constant composition, a transitional segment, and an AA homopolymer segment. The relative proportions of each segment are determined by the initial mole fraction of STY. The copolymers display properties characteristic of block copolymers, such as microphase separation (observed by AFM) and self-assembly in a selective solvent (measured by DLS). The presence of a substantial fraction of AA in the STY-rich segment of the polymers imparts pH-responsive properties to the copolymers, such that both segments are soluble in alkaline or neutral solutions, while micellar solutions are formed at lower pH. The pH at which assembly takes place is determined by the composition of the polymer. The copolymers are produced in a single step, without the chain extension step required by conventional block copolymerizations; self-assembly is accomplished by dissolving the monomer in alkaline solution, then adjusting pH, with no requirement for organic solvents or dialysis. Thus these copolymers offer the potential for significant reductions in time and cost of synthesis and assembly compared to conventional amphiphilic block copolymers.Acknowledgements
Support from CSIRO Molecular and Health Technologies and the Cooperative Research Center for Polymers (SH and FE) is gratefully acknowledged.Notes and references
- L. M. Robeson, Polymer Blends: A Comprehensive Review, Hanser-Verlag, Munich, 2007 Search PubMed.
- G. G. Odian, Principles of Polymerization, Wiley-Interscience, Hoboken, NJ, 4th edn, 2004 Search PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef CAS.
- K. Matyjaszewski, M. J. Ziegler, S. V. Arehart, D. Greszta and T. Pakula, J. Phys. Org. Chem., 2000, 13, 775 CrossRef.
- U. Beginn, Colloid Polym. Sci., 2008, 286, 1465 CrossRef CAS.
- S. Harrisson and K. L. Wooley, Chem. Commun., 2005, 3259 RSC.
- M.-Q. Zhu, L.-H. Wei, M. Li, L. Jiang, F.-S. Du, Z.-C. Li and F.-M. Li, Chem. Commun., 2001, 365 RSC.
- H. de Brouwer, M. A. J. Schellekens, B. Klumperman, M. J. Monteiro and A. L. German, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3596 CrossRef CAS.
- X. Hao, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis and E. Evans, Polymer, 2004, 45, 7401 CrossRef CAS.
- E. Chernikova, P. Terpugova, C. Bui and B. Charleux, Polymer, 2003, 44, 4101 CrossRef CAS.
- F. S. Du, M.-Q. Zhu, H. Q. Guo, Z.-C. Li, F.-M. Li, M. Kamachi and A. Kajiwara, Macromolecules, 2002, 35, 6739 CrossRef CAS.
- D. Benoit, C. J. Hawker, E. E. Huang, Z. Lin and T. P. Russell, Macromolecules, 2000, 33, 1505 CrossRef CAS.
- E. S. Park, M.-N. Kim, I.-M. Lee, H.-S. Lee and J.-S. Yoon, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2239 CrossRef CAS.
- M. C. Davies, J. V. Dawkins and D. J. Hourston, Polymer, 2005, 46, 1739 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- G. Moad, R. T. A. Mayadunne, E. Rizzardo, M. Skidmore and S. Thang, Macromol. Symp., 2003, 192, 1 CrossRef CAS.
- S. Wang and G. W. Poehlein, J. Appl. Polym. Sci., 1993, 49, 991 CrossRef CAS.
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260 CrossRef CAS.
- L. Couvreur, B. Charleux, O. Guerret and S. Magnet, Macromol. Chem. Phys., 2003, 204, 2055 CrossRef CAS.
- C. Lefay, B. Charleux, M. Save, C. Chassenieux, O. Guerret and S. Magnet, Polymer, 2006, 47, 1935 CrossRef CAS.
- B. T. T. Pham, D. Nguyen, C. J. Ferguson, B. S. Hawkett, A. K. Serelis and C. H. Such, Macromolecules, 2003, 36, 8907 CrossRef CAS.
- L. Hutson, J. Krstina, C. L. Moad, G. Moad, G. R. Morrow, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2004, 37, 4441 CrossRef CAS.
- C. L. H. Wong, J.-K. Kim and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2842 CrossRef CAS.
- M. van den Brink, A. M. van Herk and A. L. German, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3793 CrossRef CAS.
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, John Wiley and Sons, New York, NY, 4th edn, 1999, vol. 1 Search PubMed.
- E. Högfeldt, T. Miyajima, J. A. Marinsky and M. Muhammed, Acta Chem. Scand., 1989, 43, 496 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: GPC traces, AFM height images and intensity-weighted size distributions (DLS) for polymers 1–3. See DOI: 10.1039/b9py00301k |
| This journal is © The Royal Society of Chemistry 2010 |