Specific interaction of DNA-functionalized polymer colloids
Rungsima
Chollakup
*a,
Wirasak
Smitthipong
a and
Arkadiusz
Chworos
*b
aKasetsart Agricultural and Agro-Industrial Product Improvement Institute (KAPI), Kasetsart University, 50 Phaholyotin Rd, Chatuchak, Bangkok, 10900, Thailand. E-mail: aaprmc@ku.ac.th; Fax: (+662) 9405634; Tel: (+662) 9428604
bDepartment of Physics, University of California, Santa Barbara, CA 93106, USA. E-mail: achworos@physics.ucsb.edu; Tel: (+805) 8933672
First published on 5th February 2010
Abstract
As a chemically based assembly system, DNA is a key player for use in the reversible and specific interactions of an advanced material. In this approach, we work with a one-component system where a number of DNA single strands are grafted on the surface of micro-sized particles. The interactions between the DNA-functionalized particles are controlled by the addition of DNA linker in solution, which has complementary sequences with the DNA strands grafted on the particles. The hydrogen bonding based on Watson-Crick pairing is the driving force to form double stranded DNA. Several techniques have been used to characterize this system; for example, optical microscopy, AFM, DLS and flow cytometry. The two DNA strands reversibly separate (denature) above a characteristic melting temperature of DNA's base pair sequences. The melting temperature of DNA in the study is in good agreement with that of calculation based on the thermodynamic point of view.
Introduction
The bottom-up construction of nano-architectures is one of the central aims in the development of new biomaterials. The recent synthesis of particle building blocks functionalized with specifically designed oligonucleotides has provided new possibilities for the assembly of networked materials.1–9 The varied applications of polymer microspheres benefit from precise control over surface chemistry. Grafted molecules or functional groups determine the rheology10 and stability of the colloids used in pastes and coatings, influence the self-organization of the dense suspensions used in materials synthesis, and provide specific biomolecular binding sites for biological assays and cell adhesion.11–13 Because of their chemical stability and high specificity, nucleic acids are key components in bottom-up materials development. DNA is a powerful and versatile tool for the directed self-assembly of structures with specific nanoscale features.14,15 Recently, researchers have shown that it is possible to create complex self-assembled nanoscopic structures by using the sequence specific binding properties of DNA.16,17 In addition, a number of groups have used the specificity of DNA hybridization for self-assembly of binary mixtures of colloids or nanoparticles.3,18–24 Nucleic acids can also be used as a bulk material for a new type of biomaterial based on DNA/RNA-surfactant solid films.4,5,25General assembly methods for DNA are based on the use of covalent linker molecules which possess alkanedithiol functionalities at opposing ends with chemical affinities towards gold particles.26,27 The thiol groups present at the end of the linker molecule are covalently attached to the gold colloidal particles and this facilitates aggregation upon binding to the complementary sequence. To prevent non-specific interaction, this method requires gold/silver surface coating using self-assembled monolayers of organic thiols. Although it is considered to be a covalent bond, the thiol–metal interaction is much weaker and DNA molecules are often replaced during the coating process. This method is also difficult to control and the assemblies are often unstable. If the material’s properties are to be fully exploited, new robust methods of assembly are needed.
In this study a grafting method was used to covalently attach single stranded DNA molecules to the polymer particles. Here we present results for DNA-functionalized polystyrene colloidal particle assembly through rational and reversible design. In this way, the molecular recognition properties of the oligonucleotides may be used to trigger the colloidal self-assembly process. The goal for the DNA/colloid hybrid materials reported herein is that the assemblies can be characterized easily by atomic force microscopy (AFM) and flow cytometry. In these studies, we investigate polymer particle formation of colloidal microstructures as a model system for programmable self-assembly.
Results and discussion
The first step to prepare these DNA-functionalized particles is to adsorb triblock copolymer surfactants (Pluronic F108) onto the surfaces of the polystyrene particles. While the original polystyrene particles aggregate rapidly in TE buffer solution, F108-adsorbed particles are stable. The stability of the particles is lost after centrifugation, indicating that the physical adsorption of F108 is reversible (data not shown here). Then, we swell the particles with a small amount of organic solvent, allowing the hydrophobic block (PPG) of the surfactant to penetrate the surface.28 The PPG hydrophobic blocks mix with the polystyrene core, becoming trapped in the glassy polystyrene matrix upon deswelling. After that, we deswell the particle by removing the solvent. The exposed hydrophilic blocks (PEG) of these surfactants can readily be labeled with other molecules, providing long spacers for displaying proteins, antibodies, etc.29 We find that many surfactants become permanently anchored to the surface after deswelling. In order to active the hydroxyl end groups of the PEG chains, F108-functionalized polystyrene particles are mixed with 4-nitrophenol chloroformate and triethylamine at room temperature (Fig. 1). Once the OH-end groups are activated, the DNA strands (Table 1) are added to the activated F108-functionalized polystyrene particles in TEAA buffer solution. Once the reaction is complete, the DNA strand is covalently linked to the F108 which has been anchored to the surface of polystyrene particles.
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
Prediction of a melting temperature (Tm) for a free DNA solution can be calculated from the thermodynamic data.30,31 The Tm is defined as the temperature at which half of the strands are in the double-helical state and half are in the “random-coil” state. For self-complementary oligonucleotide duplexes, the Tm is calculated from the predicted standard enthalpy ΔH° and standard entropy ΔS° and the total oligonucleotide strand concentration C, by using this equation:
| Tm = ΔH°/(ΔS° + R lnC/4) |
By using an optical microscope, control and reversible interactions of DNA-functionalized polystyrene particles are investigated. The control samples based on the DNA-grafted particles without DNA linker are prepared (Fig. 2a). The particles are well dispersed and do not form any aggregate structures in a wide temperature range, indicating that the negatively charged DNA-grafted polymers repel from each other. After single stranded complementary DNA linker molecules are added, functionalized polystyrene particles aggregate (Fig. 2b). The hybridization leads to the formation of a polymeric network. After the aggregate assemblies are heated to 60 °C, the aggregate particles dissociate to form a mono-dispersed solution (Fig. 2c). After the solution with dispersed particles is cooled down from 60 °C to 30 °C, they form aggregate structures again. This heating and cooling cycle is repeated several times, and results suggest that the process is homogeneous.
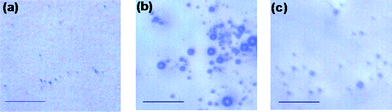 | ||
| Fig. 2 Optical microscope images of (a) DNA-grafted polystyrene particles, the scale bar is 50 μm. (b) After adding DNA linker and then incubating at 5 °C for 1 week all suspensions form aggregate structures. (c) Heating to 60 °C drives complete redispersion. | ||
The Tm of experimental DNA-functionalized particles is lower than that of calculated free DNA around 17 °C. This should be explained by the effect of triblock copolymer surfactants which can block or cause defects on double stranded DNA between DNA-functionalized particles and DNA linker. So grafting copolymers on microbeads induces a drastic change in the melting behavior of DNA-functionalized particles.34
The AFM image (Fig. 3a) presents a control sample of DNA-functionalized polystyrene particles in the absence of a complementary DNA linker. Particles are uniformly distributed without an aggregated structure. In the presence of DNA linker, polymer particles self-assemble and the aggregates are found (Fig. 3b). The particles are assembled in the presence of a DNA linker that is complementary to the DNA strands on the particles. So, they might appear as much more materials compared to the control sample.
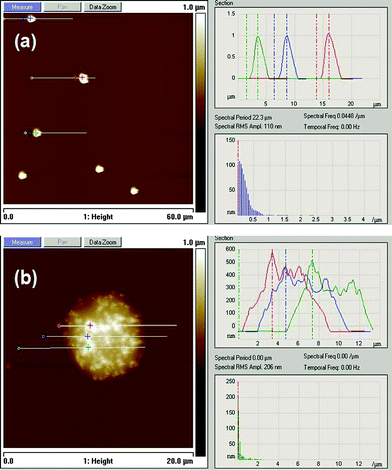 | ||
| Fig. 3 AFM height images of (a) DNA-functionalized polystyrene particles without linker and (b) self-assembly of particles in the presence of a complementary DNA linker. | ||
The size distribution of DNA-functionalized polystyrene particles can be determined using histogram analysis of dynamics light scattering (DLS) measurements. It is clear that the grafted particles have a uniform size distribution (Fig. 4a). The average diameter and polydispersity index of particles are determined to be 721 nm and 0.2 respectively by using the constrained regularisation (CONTIN) cumulant fit of DLS measurement.28 This suggests that the DNA-grafted particles are well-stabilized in suspension. It should be noted that the diameter of the polystyrene particle may be affected by the swelling/deswelling preparation method. When a complementary DNA sequence (linker) is added to the solution, the particles aggregate. The cumulative aggregates possess bigger diameter (Fig. 4b). Moreover, the polydispersity increases to be 2.5 that appears to be 10 times greater than that the one without DNA linker, indicating the formation of aggregated structures with a random size distribution. We are aware that some of bigger conglomerates might fall down to the bottom of the test tube during DLS measurements.
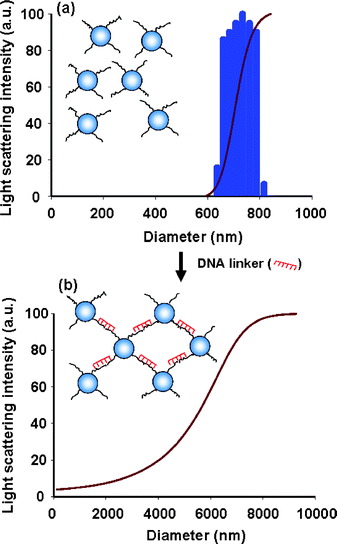 | ||
| Fig. 4 DLS histograms of (a) the uniform size distribution of DNA-functionalized polystyrene particle without linker and (b) the wide size distribution of aggregated particles in the presence of linker. | ||
One interesting question is how many DNA molecules are in fact grafted on each particle? We use flow cytometry measurements to determine the DNA packing density. For this purpose, the molecules of equivalent soluble fluorescence intensity are constructed and used for a calibration curve. Fig. 5a shows the calibration curve, formed by plotting the count (Y-axis) versus the peak channel (X-axis) for each of the four fluorescent intensity populations. Then, we compare the fluorescent intensity data from the DNA-labeled particles which can be converted to an approximate number of grafted DNA molecules per particle. We use fluorescent label FITC attached to the 3′-end of ssDNA molecules grafted on the surface of the particle (Table 1). The fluorescent intensities are translated into number of ssDNA molecules per particle using the calibration curve. The flow cytometry experiment indicates approximately 3000 DNA molecules per polystyrene particle. This result is consistent with the measurement for carboxylate-modified latex particles (4000 DNA molecules/particle) which used the same particle preparation method.3
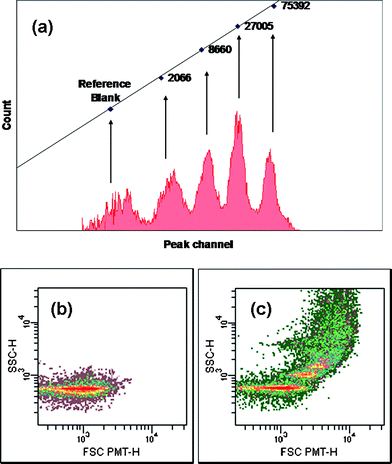 | ||
| Fig. 5 A flow cytometry histogram of (a) the calibration curve from standard FITC-particles (b) FITC-labeled DNA-functionalized polystyrene particles without linker and (c) FITC-labeled DNA-functionalized polystyrene particle in the presence of a DNA linker. | ||
The forward scatter (FSC) detector correlates with the particle volume and the side scatter (SSC) detector depends on the complexity of the particle (i.e. shape of the particle, the surface roughness).35,36 This flow cytometry technique also confirms that the DNA-functionalized particles do not bind to each other (Fig. 5b) unless through hybridization of complementary sequences of DNA linker (Fig. 5c). This result is in good agreement with the previous results obtained from optical microscopy, AFM and DLS.
Experimental
Materials
The DNA sequences are synthesized by Bioneer, Inc. (Table 1). Triblock copolymer surfactant [Pluronic F108: (PEG)129-(PPG)43-(PEG)129] is obtained from BASF. All other chemicals are obtained from Aldrich and used as received.Preparation of DNA-functionalized polystyrene particles
Polystyrene particles (1 μm) are grafted with the amine-functionalized DNA strand through PEG spacers using the polymer swelling/deswelling method according to the literature.3 Briefly, 100 μl of 10% w/w triblock copolymer, poly(ethylene glycol)-b-poly(propylene glycol)-b-poly(ethylene glycol) or F108, 100 μl of 10% w/w polystyrene particles and 10 μl of toluene are added in 1x TE buffer (10 mM Tris-HCl and 1 mM EDTA) at pH 8.0 to make up a final volume of 1 ml. Toluene is a good solvent for polystyrene and is slightly soluble in water. As it diffuses into the polymer particles, the chains dissolve and the interior becomes fluid. We remove the toluene by steam stripping. Then, the sample is washed five times by centrifugation and redispersion in 1x TE buffer, pH 8.0, to remove excess F108. After that, 100 μl of 10% w/w F108-functionalized particles (polystyrene-PEG spacer), 40 mg of 4-nitrophenol chloroformate, and 40 μl of triethylamine are added to methylene chloride to make up a final volume of 2 ml. The reaction, which activates the hydroxyl end groups of the PEG chains, proceeds at room temperature for 12 h, and the solvent is removed by evaporation. Once the hydroxyl end groups are activated, 20 μl of 2 μM 5′-amine-modified DNA are added to 20 μl of 10% w/w F108-functionalized particles in 50 mM TEAA buffer (triethylamine ammoniumphosphate) at pH 9.5 to make up a final volume of 50 μl. The mixtures are allowed to react for 4 h at room temperature with constant mixing.Characterization
Conclusions
We have shown that DNA-functionalized polystyrene particles can be conveniently prepared using a swelling/deswelling method with a PEG spacer and commercially available DNA molecules. We find that the specific interactions (hydrogen bonding) between the particles can be easily controlled by the addition of a complementary DNA linker. Aggregate particles assemble and disassemble according to the characteristic DNA melting temperature. In order to understand and control the hybridization of sequence guided particle self-assembly one can follow the dynamics of the assembly process using optical microscopy, AFM, DLS and flow cytometry. These results highlight nucleic acids as an excellent specific nanoscopic adhesive medium to guide the assembly of tailored materials. One can imagine designing the specific protein receptor which upon binding will trigger the aggregation of polystyrene coated metal particles and that can be used as a marker in medical diagnosis.Acknowledgements
We would like to gratefully acknowledge Professor Matthew Tirrell and Dr Krystyna R. Brzezinska from University of California (Santa Barbara) for scientific discussions.References
- N. C. Seeman, Nature, 2003, 421, 427 CrossRef.
- F. Huo, A. K. Lytton-Jean and C. A. Mirkin, Adv. Mater., 2006, 18, 2304 CrossRef CAS.
- A. J. Kim, P. L. Biancaniello and J. C. Crocker, Langmuir, 2006, 22, 1991 CrossRef CAS.
- W. Smitthipong, T. Neumann, A. Chworos, L. Jaeger and M. Tirrell, Macromol. Symp., 2008, 264, 13 CrossRef CAS.
- W. Smitthipong, A. Chworos, B. Lin, T. Neumann, S. Gajria, L. Jaeger and M. Tirrell, Mater. Res. Soc. Symp. Proc., 2008, 1094-DD06-05.
- A. Chworos, I. Severcan, A. Y. Koyfman, P. Weinkam, E. Oroudjev, H. G. Hansma and L. Jaeger, Science, 2004, 306, 2068 CrossRef CAS.
- K. Mougin, H. Haidara and G. Castelein, Colloids Surf., A, 2001, 193, 231–237 CrossRef CAS.
- H. Zhao and J. H. Waite, Biochemistry, 2005, 44, 15915–15923 CrossRef CAS.
- A. Vidyasagar, H. L. Smith, J. Majewski and R. G. Toomey, Soft Matter, 2009, 5, 4733 RSC.
- G. H. Fredrickson and P. Pincus, Langmuir, 1991, 7, 786 CrossRef CAS.
- M. Biesalski, A. Knabel, R. Tu and M. Tirrell, Biomaterials, 2006, 27, 1259 CrossRef CAS.
- K. Mougin, H. Haidara and J. Schultz, Langmuir, 2001, 17, 5952–5957 CrossRef CAS.
- J. H. Waite, Nat. Mater., 2008, 7, 8–9 CrossRef CAS.
- R. Bitton, J. Schmidt, M. Biesalski, R. Tu and M. Tirrell, Langmuir, 2005, 21, 11888 CrossRef CAS.
- D. S. Seferos, D. A. Giljohann, N. L. Rosi and C. A. Mirkin, ChemBioChem, 2007, 8, 1230 CrossRef CAS.
- N. C. Seeman, Mol. Biotechnol., 2007, 37, 246 Search PubMed.
- L. Jaeger and A. Chworos, Curr. Opin. Struct. Biol., 2006, 16, 531 CrossRef CAS.
- S. Y. Park, A. K. R. Lytton-Jean1, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553 CrossRef CAS.
- S. Y. Park, J. S. Lee, D. Georganopoulou, C. A. Mirkin and G. C. Schatz, J. Phys. Chem. B, 2006, 110, 12673 CrossRef CAS.
- P. H. Rogers, E. Michel, C. A. Bauer, S. Vanderet, D. Hansen, B. K. Roberts, A. Calvez, J. B. Crews, K. O. Lau, A. Wood, D. J. Pine and P. V. Schwartz, Langmuir, 2005, 21, 5562 CrossRef CAS.
- D. B. Lukatsky, B. M. Mulder and D. Frenkel, J. Phys.: Condens. Matter, 2006, 18, S567 CrossRef CAS.
- A. Elaissari, J. P. Chauvet, M. A. Halle, O. Decavallas, C. Pichot and P. Cros, J. Colloid Interface Sci., 1998, 202, 251 CrossRef CAS.
- F. Ganachaud, A. Elaissari, C. Pichot, A. Laayoun and P. Cros, Langmuir, 1997, 13, 701 CrossRef CAS.
- H. W. Walker and S. B. Grant, Langmuir, 1996, 12, 3151 CrossRef CAS.
- W. Smitthipong, T. Neumann, S. Gajria, Y. Li, A. Chworos, L. Jaeger and M. Tirrell, Biomacromolecules, 2009, 10, 221 CrossRef CAS.
- C. D. Bain and G. M. Whitesides, Angew. Chem., Int. Ed. Engl., 1989, 28, 506 CrossRef.
- L. H. Dubois and R. G. Nuzzo, Annu. Rev. Phys. Chem., 1992, 43, 437 CrossRef CAS.
- L. E. Dewalt, H. D. Ou-Yang and V. L. Dimonie, J. Appl. Polym. Sci., 1995, 58, 265 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459 CrossRef CAS.
- K. J. Breslauer, R. Franks, H. Blockers and L. A. Marky, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 3746 CAS.
- J. SantaLucia Jr., Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1460 CrossRef CAS.
- M.-P. Valignat, O. Theodoly, J. C. Crocker, W. B. Russel and P. M. Chaikin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4225 CrossRef CAS.
- A. K. R. Lytton-Jean and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 12754 CrossRef CAS.
- N. C. Harris and C.-H. Kiang, J. Phys. Chem. B, 2006, 110, 16393 CrossRef CAS.
- P. A. Hassan and S. K. Kulshreshtha, J. Colloid Interface Sci., 2006, 300, 744 CrossRef CAS.
- L. A. Sklar, Flow Cytometry for Biotechnology, Oxford University Press, 2005 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |