DOI:
10.1039/B9PY00282K
(Paper)
Polym. Chem., 2010,
1, 97-101
Received
7th October 2009
, Accepted 16th November 2009
First published on
22nd December 2009
Abstract
It is shown that atom transfer radical polymerization (ATRP) with activators regenerated by electron transfer (ARGET) is a powerful approach for grafting of polymer brushes from surfaces of high-surface-area nanoporous hosts, allowing one to achieve controlled polymer loadings and film thicknesses while maintaining the pore accessibility and moderate or high surface area. The approach was illustrated on SBA-15 silicas with cylindrical pores of diameter 22 and 14 nm as the nanoporous supports, and poly(methyl methacrylate) (PMMA) and polystyrene (PS) as the grafted polymers. The polymer loadings up to 36 wt.% and the grafted layer thicknesses of up to at least 2 nm were achieved in the polymerization process carried out in small vials without using a vacuum line. The specific surface areas of the obtained silica-polymer composites were 60–190 m2 g−1. The polymer chains exhibited quite low polydispersity (1.18–1.32) for polymer loadings up to 29 wt.%, while a higher polydispersity was observed for higher loadings, in which case the pores of the support were filled to a significant extent with the polymer graft. Due to its simplicity, ARGET ATRP emerges as a powerful and yet surprisingly straightforward way to synthesize well-defined polymer brushes in nanopores, opening a convenient avenue to a wide range of porous high-surface-area silica-polymer composites.
Introduction
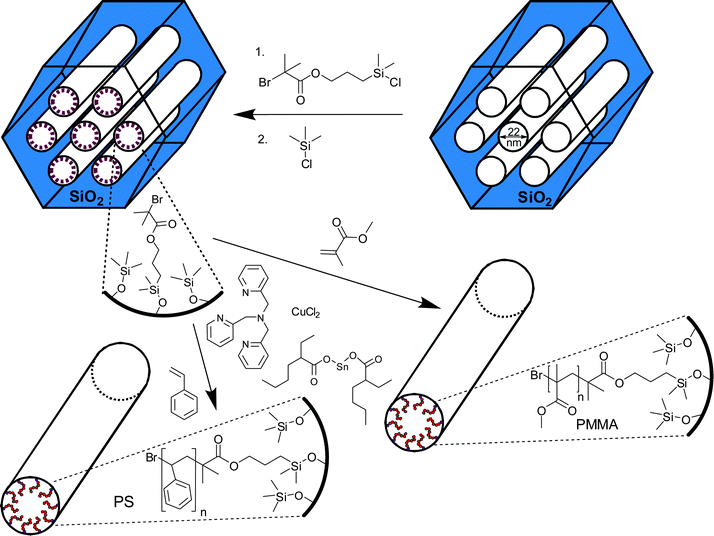
The functionalization of surfaces of inorganic solids with well-defined organic moieties has recently attracted much attention and led to the development of a remarkable variety of materials with desired mechanical properties, chemical functionality, surface properties and so forth.1–30 A major challenge is to develop powerful, general, and yet straightforward and environmentally friendly pathways to high-surface-area materials with designed surface properties. Porous silica-polymer composites30–38 constitute a highly promising group of such materials, because the structure of the high-surface-area silica imparts dimensional stability and can be readily adjusted in terms of the pore shape and diameter,6,8,9 while the polymer component imparts many desirable properties, including the chemical stability, adsorption or catalytic properties, and stimuli-responsive properties.1–5 Recently, well-defined porous silica-polymer composites with polymer moieties covalently bonded to silica framework were obtained through the dendrimer synthesis,31,32 the surface-initiated atom transfer radical polymerization (ATRP),33–39 and the attachment of polymers via the “click” chemistry.40 The surface-initiated controlled polymerization is highly attractive for the synthesis of high-surface-area nanoporous silica-polymer composites, because the polymer composition33–38 and molecular weight33 can be controlled. In addition, there are prospects for the tailoring of polymer architecture, and stimuli-responsive properties, as was extensively documented for polymer-grafted nanoparticles and flat surfaces2–5 and explored for nanoporous membranes.41–44 On the other hand, there are indications that the surface-initiated polymerization in nanopores may be more difficult to control than polymerizations initiated from flat surfaces and surfaces of nanoparticles.45,46 Moreover, there is a need to elaborate more convenient and environmentally friendly pathways to the considered composite materials.
Recently developed ATRP with activators regenerated by electron transfer (ARGET)47–51 provides a major improvement in both of the aforementioned areas, because it can be performed using a closed vial instead of using air-tight glassware and a vacuum line, and it involves low concentrations of copper catalyst (∼10–100 ppm vs. 1000–10000 ppm used in normal ATRP). In ARGET ATRP, a reducing agent (typically, tin(II) 2-ethylhexanoate, Sn(EH)2, or vitamin C) is used to convert Cu(II) to Cu(I), the latter being a component of the ATRP catalyst complex (activator), and to remove the oxygen (radical trap) through a cycle involving oxidation and reduction of copper species. ARGET ATRP has been successfully used to grow polymer brushes on flat surfaces49 and surfaces of nanoparticles.51 However, its applicability to graft thin polymer films on surfaces of high-surface-area nanoporous materials has not been demonstrated. This is a very challenging case, because dimensions of the nanopores are comparable to the length of extended chains even for low-molecular-weight polymers. Therefore, an uncontrolled growth of even a small fraction of chains to form high polymers may lead to the blocking of the nanopores. Moreover, a high-surface-area support may have a tendency to adsorb the ATRP catalyst,52 which may make an effort to decrease the concentration of the catalyst inherently difficult. Herein, it is demonstrated that ARGET ATRP (see Scheme 1) is fully applicable to grafting from the surfaces of nanoporous hosts, allowing one to achieve a controlled polymer loading and film thickness. Polymer chains exhibit quite low polydispersity, unless the polymerization process is prolonged until the nanopores are nearly completely filled, which resembles the performance of normal ATRP.33 The present work demonstrates new opportunities in the synthesis of well-defined high-surface-area silica-polymer composites through simple and benign chemistry.
Experimental section
Materials
SBA-15 silica with cylindrical pores of diameter ∼14 nm and ∼22 nm (as estimated using a geometrical relation employed in53) were synthesized as described elsewhere.53 For SBA-15 with ∼14 nm mesopores (d100 = 13.7 nm), 1.0 g TIPB per 2.4 g Pluronic P123 was used, the initial synthesis temperature was 17 °C and the hydrothermal treatment was performed for 2 days at 100 °C. For the SBA-15 with ∼22 nm mesopores (d100 = 22.4 nm), 2.0 g TIPB per 2.4 g Pluronic P123 was used, the initial synthesis temperature was 12.75 °C and the hydrothermal treatment was performed for 1 day at 130 °C. Methyl methacrylate (MMA, 99%) and styrene (St, 99%) were purchased from Aldrich and purified by passing through a column filled with basic alumina. The ligand tris[(2-pyridyl)methyl]amine (TPMA) was purchased from ATRP Solutions, Inc. and used as received. The initiator, 3-(chlorodimethylsilyl)propyl 2-bromoisobutyrate, was synthesized and grafted on the surface of silica as described elsewhere.54 All other chemicals including solvents, copper salt and reducing agent tin(II) 2-ethylhexanoate (Sn(EH)2) were used as received.
Synthetic procedures
A typical ARGET ATRP of MMA from surface of mesoporous silica was as follows: initiator-modified silica (30 mg, with 0.012 mmol initiator), MMA monomer (0.13 mL, 1.2 mmol), a pre-mixed solution of CuCl2 (0.033 mg, 0.00024 mmol) and TPMA ligand (0.69 mg, 0.0024 mmol) in anisole (0.17 mL) and anisole (3.10 ml) were added into a ∼5 mL glass vial. Then the vial was sealed with a rubber septum and the solution was stirred for 20 min before a solution of Sn(EH)2 (51 mg, 0.126 mmol) in anisole (0.3 mL) was injected. The molar ratio of reactants was as follows: initiator![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu(II):TPMA:MMA = 1:0.02:0.2:100. The amount of reducing agent Sn(EH)2, which is primarily related to free air space in the glass vial (∼1.6 ml), was determined earlier by gradually increasing the amount of the reducing agent until the polymerization proceeded at an appreciable rate. The mixtures for polymerization of MMA and styrene were placed in an oil bath at 40 °C and 80 °C respectively. The reaction was terminated by opening the vial and exposing the catalyst to air. The polymer/silica composites were isolated by filtration, washed with tetrahydrofuran (THF), acetone and methanol, and dried in a vacuum oven.
:Cu(II):TPMA:MMA = 1:0.02:0.2:100. The amount of reducing agent Sn(EH)2, which is primarily related to free air space in the glass vial (∼1.6 ml), was determined earlier by gradually increasing the amount of the reducing agent until the polymerization proceeded at an appreciable rate. The mixtures for polymerization of MMA and styrene were placed in an oil bath at 40 °C and 80 °C respectively. The reaction was terminated by opening the vial and exposing the catalyst to air. The polymer/silica composites were isolated by filtration, washed with tetrahydrofuran (THF), acetone and methanol, and dried in a vacuum oven.
Measurements
Nitrogen adsorption measurements at −196 °C were carried out using a Micromeritics ASAP 2020 volumetric adsorption analyzer. Samples were degassed under vacuum at 200 °C (for silicas) or 80 °C (for modified silicas) before adsorption measurements. The specific surface area was determined using the BET method.55 The total pore volume was calculated from the amount adsorbed at a relative pressure of 0.99.55 The pore size distribution was calculated using the BJH method for cylindrical mesopores.56 Thermogravimetric analysis (TGA) was performed under nitrogen with 5 °C min−1 ramping rate to 800 °C on Hi-Res 2950 thermogravimetric analyzer from TA Instruments. Molecular weight distributions were determined by gel permeation chromatography (GPC) on Alliance GPCV 2000 model with refractive index detector and polystyrene standards. The cleavage of polymers from silica support was performed using 48% HF water solution mixed with THF in the volume ratio of one to ten (similarly to conditions described in33). Small-angle X-ray scattering (SAXS) measurements were performed using a Bruker Nanostar small-angle/wide-angle X-ray scattering (SAXS/WAXS) instrument with a rotating anode X-ray source and Vantec-2000 two-dimensional detector.
Results and discussion
Ordered mesoporous silica support (SBA-15) with very large cylindrical pores53 (22 nm, as calculated using eqn 1 in ref. 53) was selected to allow us to follow the grafting process up to the polymer layer thicknesses (in dry state) of several nanometres. The surface of the silica was modified with 3-(2-bromoisobutyryloxy)propyldimethylsilyl groups (BiB, which serves as an ATRP initiator) and trimethylsilyl groups (see Scheme 1).33,54 The surface coverage of the initiator was about 0.98 groups per nm2, as estimated from TGA, which is consistent with the value reported earlier for regular SBA-15 (pore diameter ∼10 nm).54 Thus obtained initiator-functionalized SBA-15-BiB was placed in a glass vial to which monomer (methyl methacrylate, MMA), copper(II) chloride, tris[(2-pyridyl)methyl]amine (TPMA) ligand, and anisole (solvent) were added. Subsequently, the vial was sealed with a septum and a reducing agent Sn(EH)2 was added to convert Cu(II) to Cu(I). The molar ratio of reactants was: initiator:Cu(II):TPMA:MMA = 1:0.02:0.2:100. It should be noted that the initiator:Cu ratio in normal ATRP is commonly about 1:1, so the present composition corresponds to 50-fold lowering of Cu concentration. The mixture was placed in an oil bath at 40 °C. After a selected period of time, the reaction was terminated by opening the vial and exposing the catalyst to air to convert it to a deactivator form (that is, Cu(II) complex). The silica/PMMA composite was isolated by filtration, washed, and dried. The composites were white (see ESI Fig. S1†), being free from coloration due to residual Cu.
The products after 6, 11, 24 and 48 h of polymerization were analyzed by thermogravimetry (Fig. 1) and found to contain 13, 29, 36 and 48 wt.% of PMMA, respectively. Nitrogen adsorption isotherms (Fig. 2) showed that the mesopores were accessible for the polymer contents up to 36 wt.% (with BET specific surface area 60–140 m2 g−1, and total pore volume 0.23–0.66 cm3 g−1; see ESI Table S1†), while the sample with 48 wt.% of PMMA (content of organic moieties equal to 53 wt.%) had inaccessible mesopores. In the latter case, the inaccessibility of the pores is not surprising, because the composite contained the amount of organic moieties close to that needed to completely fill the mesopores of the silica support. Pore size distributions (PSDs) (Fig. 3) showed that the pore diameter systematically decreased as the loading of the polymer increased. The thickness of the polymer layer corresponds to the pore radius decrease and can be estimated on the basis of pore radius changes as 0.7, 1.7 and 3.3 nm for 13, 29 and 36 wt% PMMA loading (see Fig. 3). While the method used herein to calculate the diameter of cylindrical pores is known to overestimate it by 10–25% in the considered size range,53 thus leading to an overestimation of the polymer film thickness, it is clear that the thickness of the polymer layer increased with time and reached at least 2 nm. The latter is comparable with the highest thicknesses achieved using normal ATRP without blocking of the nanopores in the considered diameter range.33,35,36 Interestingly enough, the introduction of the loadings of the polymer up to 36 wt.%, the latter being higher than that achieved earlier using normal ATRP without pore blocking,33,35,36 did not lead to any prominent broadening of hysteresis loops on the nitrogen adsorption isotherms (Fig. 2), indicating the polymer film did not form any major constrictions in the mesopores.33 The grafted PMMA was detached from the surface of the silica support and analyzed by gel permeation chromatography (GPC) (see Fig. 4). The molecular weight was 4700, 7300, 8700 and 20800 g mol−1 for composites with 13, 29, 36 and 48 wt.% loading of PMMA, respectively. The polydispersity index (PDI) was 1.18, 1.28, 1.47 and 2.09, respectively. Clearly, the molecular weight of PMMA increased with the polymerization time thus providing strong evidence for the controlled growth of the polymer. PDI, which was initially quite low, became moderate or even quite high at high PMMA loadings. On the basis of GPC, TGA and gas adsorption data, the grafting density was calculated to be 0.06, 0.12, 0.14 and 0.09 chains per nm2, whereas the initiation efficiency was 6, 12, 14 and 9% for 4700, 7300, 8700 and 20800 g mol−1 PMMA, respectively. The apparent decrease in the grafting density and initiation efficiency for the highest-molecular-weight case may be related to the coupling of chains at later stages of the grafting process where the pores are nearly completely filled with the polymer and the deactivator cannot readily diffuse to convert the propagating chains to the dormant species.57 This contention is consistent with the more than two-fold increase in the molecular weight and the significant broadening of the molecular weight distribution at this stage. The grafting densities and initiation efficiencies were 2–3 times lower than those observed earlier for PAN grafted in nanopores via normal ATRP,33 but one should keep in mind that the repeating unit of PMMA occupies the volume about two times larger than that occupied by PAN repeating unit. As expected, the polymerization did not induce any structural changes in the mesoporous host (see SAXS data in ESI Fig. S2†).
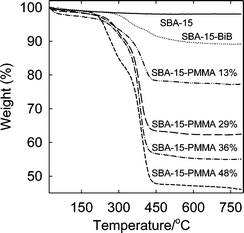 |
| | Fig. 1 Weight change patterns for SBA-15 silica before and after attachment of initiation sites and polymerization of methyl methacrylate (loading of PMMA in the composites is indicated in wt.%). | |
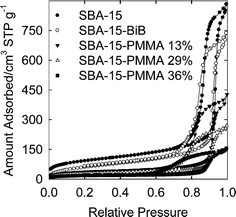 |
| | Fig. 2 Nitrogen adsorption isotherms for SBA-15 silica before and after attachment of initiation sites and polymerization of methyl methacrylate (loading of PMMA in the composites is indicated in wt.%). | |
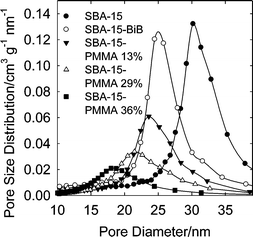 |
| | Fig. 3 Pore size distributions for SBA-15 silica before and after attachment of initiation sites and polymerization of methyl methacrylate (loading of PMMA in the composites is indicated in wt.%). | |
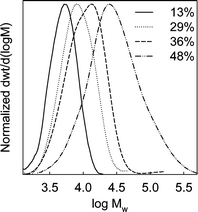 |
| | Fig. 4 Gel permeation chromatography (GPC) molecular weight distributions for poly(methyl methacrylate) cleaved from the surface of SBA-15/PMMA composites (loading of PMMA in the composites is indicated in wt.%). | |
Similar results were also obtained for PMMA grafting in 14 nm mesopores (see ESI Fig. S3–S7†). The loadings of 11, 22 and 34 wt.% were achieved after polymerization times of 4.5, 7.5 and 17 h. The mesopores were accessible for loadings up to 22 wt.%, and silica-PMMA composites with BET specific surface areas 80–190 m2 g−1, and total pore volumes 0.17–0.49 cm3 g−1 were obtained. The molecular weight of PMMA was 2700, 4300 and 6000 g mol−1, and PDI was 1.27, 1.32 and 1.50, respectively (see ESI Fig. S4 for molecular weight distributions†). The grafting density was calculated to be 0.06, 0.08, and 0.11 chains per nm2, whereas the initiation efficiency was 6, 8, and 11%, respectively. These data are similar to those for the larger-pore support discussed above.
ARGET ATRP is known to be applicable for a wide range of monomers polymerizable using ATRP.47–51 To verify the scope of this method in the context of functionalization of nanoporous materials, the polymerization of styrene was successfully performed. In this case, the polymerization temperature was 80 °C. After 24 and 30 h of polymerization, PS loadings of 14 and 19 wt.% were achieved with retention of accessible porosity (see Fig. 5 and 6, as well as ESI Fig. S8 and S9;† BET surface area 100–120 m2 g−1, and total pore volume 0.48–0.60 cm3 g−1). Molecular weights of 1150 and 2380 g mol−1 and PDIs of ∼1.3 were observed for the PS graft. The grafting density was calculated to be ∼0.24 chains per nm2, whereas the initiation efficiency was ∼25%, which are similar to those achieved for PAN grafted in nanopores using normal ATRP.33
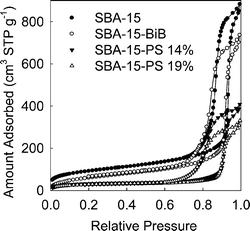 |
| | Fig. 5 Nitrogen adsorption isotherms for SBA-15 silica before and after attachment of initiation sites and polymerization of styrene (loading of PS in the composite is indicated in wt.%). | |
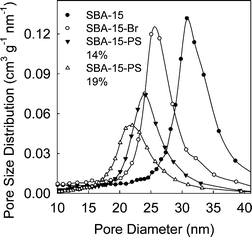 |
| | Fig. 6 Pore size distributions for SBA-15 silica before and after attachment of initiation sites and polymerization of styrene (loading of PS in the composite is indicated in wt.%). | |
Conclusions
One can conclude that ARGET ATRP is highly suitable for the synthesis of high-surface-area silica-polymer composites. Adjustable polymer loadings, including very high ones, and appreciable polymer film thicknesses can be achieved. Due to its simplicity, ARGET ATRP emerges as a powerful, yet surprisingly straightforward way to synthesize well-defined polymer brushes in nanopores, opening a convenient avenue to a wide range of high-surface-area silica-polymer composites.
Acknowledgements
M. K. acknowledges partial support from the Center for Engineered Polymeric Materials funded by NYSTAR. NSF is acknowledged for funding the SAXS/WAXS system through award CHE-0723028. Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research (Award PRF #49093-DNI5).
Notes and references
- K. Moller and T. Bein, Chem. Mater., 1998, 10, 2950 CrossRef CAS.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677 CrossRef CAS.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14 RSC.
- J. Ruhe, M. Ballauff, M. Biesalski, P. Dziezok, F. Grohn, D. Johannsmann, N. Houbenov, N. Hugenberg, R. Konradi, S. Minko, M. Motornov, R. R. Netz, M. Schmidt, C. Seidel, M. Stamm, T. Stephan, D. Usov and H. N. Zhang, Adv. Polym. Sci., 2004, 165, 79.
- Y. Tsujii, K. Ohno, S. Yamamoto, A. Goto and T. Fukuda, Adv. Polym. Sci., 2006, 197, 1 CAS.
- A. Stein, B. J. Melde and R. C. Schroden, Adv. Mater., 2000, 12, 1403 CrossRef CAS.
- D. Schmidt, D. Shah and E. P. Giannelis, Curr. Opin. Solid State Mater. Sci., 2002, 6, 205 CrossRef CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Froeba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef.
- B. G. Trewyn, I. I. Slowing, S. Giri, H.-T. Chen and V. S. Y. Lin, Acc. Chem. Res., 2007, 40, 846 CrossRef CAS.
- M. Chen, W. H. Briscoe, S. P. Armes and J. Klein, Science, 2009, 323, 1698 CrossRef CAS.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. G. Börner, A. Laschewsky, C. Duschl and J.-F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 5666 CrossRef CAS.
- I. Tan, Z. Zarafshani, J.-F. Lutz and M.-M. Titirici, ACS Appl. Mater. Interfaces, 2009, 1, 1869 Search PubMed.
- J. Huang, H. Murata, R. R. Koepsel, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2007, 8, 1396 CrossRef CAS.
- M. Husseman, E. E. Malmstrom, M. McNamara, M. Mate, D. Mecerreyes, D. G. Benoit, J. L. Hedrick, P. Mansky, E. Huang, T. P. Russell and C. J. Hawker, Macromolecules, 1999, 32, 1424 CrossRef.
- N. L. Jeon, I. S. Choi, G. M. Whitesides, N. Y. Kim, P. E. Laibinis, Y. Harada, K. R. Finnie, G. S. Girolami and R. G. Nuzzo, Appl. Phys. Lett., 1999, 75, 4201 CrossRef CAS.
- M. Husemann, M. Morrison, D. Benoit, K. J. Frommer, C. M. Mate, W. D. Hinsberg, J. L. Hedrick and C. J. Hawker, J. Am. Chem. Soc., 2000, 122, 1844 CrossRef CAS.
- R. R. Shah, D. Merreceyes, M. Husemann, I. Rees, N. L. Abbott, C. J. Hawker and J. L. Hedrick, Macromolecules, 2000, 33, 597 CrossRef CAS.
- B. Zhao, W. J. Brittain, W. S. Zhou and S. Z. D. Cheng, J. Am. Chem. Soc., 2000, 122, 2407 CrossRef CAS.
- D. Li, X. Sheng and B. Zhao, J. Am. Chem. Soc., 2005, 127, 6248 CrossRef CAS.
- C. D. H. Alarcon, T. Farhan, V. L. Osborne, W. T. S. Huck and C. Alexander, J. Mater. Chem., 2005, 15, 2089 RSC.
- S. Moya, O. Azzaroni, T. Farhan, V. L. Osborne and W. T. S. Huck, Angew. Chem., Int. Ed., 2005, 44, 4578 CrossRef CAS.
- Y. Mei, T. Wu, C. Xu, K. J. Langenbach, J. T. Elliott, B. D. Vogt, K. L. Beers, E. J. Amis and N. R. Washburn, Langmuir, 2005, 21, 12309 CrossRef CAS.
- X. Wei and S. M. Husson, Ind. Eng. Chem. Res., 2007, 46, 2117 CrossRef CAS.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Langmuir, 2007, 23, 9409 CrossRef CAS.
- M. M. Rahman, M. Czaun, M. Takafuji and H. Ihara, Chem.–Eur. J., 2008, 14, 1312 CrossRef CAS.
- P.-W. Chung, R. Kumar, M. Pruski and V. S.-Y. Lin, Adv. Funct. Mater., 2008, 18, 1390 CrossRef CAS.
- S.-i. Yamamoto and K. Matyjaszewski, Polym. J., 2008, 40, 496 CrossRef CAS.
- F. J. Xu, S. P. Zhong, L. Y. L. Yung, E. T. Kang and K. G. Neoh, Biomacromolecules, 2004, 5, 2392 CrossRef CAS.
- Y.-Z. You, K. K. Kalebaila, S. L. Brock and D. Oupicky, Chem. Mater., 2008, 20, 3354 CrossRef CAS.
- X. Huang and M. J. Wirth, Anal. Chem., 1997, 69, 4577 CrossRef CAS.
- E. J. Acosta, C. S. Carr, E. E. Simanek and D. F. Shantz, Adv. Mater., 2004, 16, 985 CrossRef CAS.
- J. P. K. Reynhardt, Y. Yang, A. Sayari and H. Alper, Chem. Mater., 2004, 16, 4095 CrossRef CAS.
- M. Kruk, B. Dufour, E. B. Celer, T. Kowalewski, M. Jaroniec and K. Matyjaszewski, Macromolecules, 2008, 41, 8584 CrossRef CAS.
- J. Moreno and D. C. Sherrington, Chem. Mater., 2008, 20, 4468 CrossRef CAS.
- L. Cao and M. Kruk, Polym. Prepr., 2008, 49(2), 294 CAS.
- L. Cao, B. Dufour, K. Matyjaszewski and M. Kruk, Polym. Prepr., 2009, 50, 348.
- C. Li, J. Yang, P. Wang, J. Liu and Q. Yang, Microporous Mesoporous Mater., 2009, 123, 228 CrossRef CAS.
- Z. Zhou, S. Zhu and D. Zhang, J. Mater. Chem., 2007, 17, 2428 RSC.
- M. Save, G. Granvorka, J. Bernard, B. Charleux, C. Boissiere, D. Grosso and C. Sanchez, Macromol. Rapid Commun., 2006, 27, 393 CrossRef CAS.
- L. Huang, S. Dolai, K. Raja and M. Kruk, Langmuir DOI:10.1021/la9026943.
- Q. Fu, G. V. R. Rao, S. B. Basame, D. J. Keller, K. Artyushkova, J. E. Fulghum and G. P. Lopez, J. Am. Chem. Soc., 2004, 126, 8904 CrossRef CAS.
- O. Schepelina and I. Zharov, Langmuir, 2007, 23, 12704 CrossRef CAS.
- A. Calvo, B. Yameen, F. J. Williams, O. Azzaroni and G. J. A. A. Soler-Illia, Chem. Commun., 2009, 2553 RSC.
- B. Yameen, M. Ali, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, Nano Lett., 2009, 9, 2788 CrossRef CAS.
- C. B. Gorman, R. J. Petrie and J. Genzer, Macromolecules, 2008, 41, 4856 CrossRef CAS.
- P. Pasetto, H. Blas, F. Audouin, C. Boissiere, C. Sanchez, M. Save and B. Charleux, Macromolecules, 2009, 42, 5983 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482 CrossRef CAS.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39 CrossRef CAS.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528 CrossRef CAS.
- K. Min, H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789 CrossRef CAS.
- L. Bombalski, H. Dong, J. Listak, K. Matyjaszewski and M. R. Bockstaller, Adv. Mater., 2007, 19, 4486 CrossRef CAS.
- J. V. Nguyen and C. W. Jones, Macromolecules, 2004, 37, 1190 CrossRef CAS.
- L. Cao, T. Man and M. Kruk, Chem. Mater., 2009, 21, 1144 CrossRef CAS.
- M. Kruk, B. Dufour, E. B. Celer, T. Kowalewski, M. Jaroniec and K. Matyjaszewski, J. Phys. Chem. B, 2005, 109, 9216 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- M. Kruk, M. Jaroniec and A. Sayari, Langmuir, 1997, 13, 6267 CrossRef CAS.
- M. Kruk, B. Dufour, E. B. Celer, T. Kowalewski, M. Jaroniec and K. Matyjaszewski, Polym. Mater. Sci. Eng. Preprints, 2007, 97, 274 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of characterization of the samples. See DOI: 10.1039/b9py00282k |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.