Non-α-hydroxylated aldehydes with evolved transketolase enzymes†
Armando
Cázares
a,
James L.
Galman
a,
Lydia G.
Crago
ab,
Mark E. B.
Smith
a,
John
Strafford
b,
Leonardo
Ríos-Solís
b,
Gary J.
Lye
b,
Paul A.
Dalby
b and
Helen C.
Hailes
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, UK WC1H 0AJ. E-mail: h.c.hailes@ucl.ac.uk; Fax: +44 (0)20 7679 7463; Tel: +44 (0)20 7679 7463
bDepartment of Biochemical Engineering, University College London, Torrington Place, London, UK WC1E 7JE
First published on 5th February 2010
Abstract
Transketolase mutants previously identified for use with the non-phosphorylated aldehyde propanal have been explored with a series of linear and cyclic aliphatic aldehydes, and excellent stereoselectivities observed.
Introduction
The use of biocatalysis as a sustainable, atom efficient strategy in organic synthesis is of increasing importance, and is particularly attractive due to the high stereoselectivities that can be achieved.1 Transketolase (TK) (EC 2.2.1.1) is an essential thiamine diphosphate (ThDP) dependent enzyme, which provides a link between the glycolytic and pentose phosphate pathways.2In vivo it catalyses the reversible transfer of a ketol unit to D-ribose-5-phosphate or D-erythrose-4-phosphate.2 The TK reaction is made irreversible using the donor β-hydroxypyruvate (HPA 1),3 which has been used with acceptors 2 such as α-hydroxyaldehydes where it is stereospecific for the (2R)-hydroxyaldehyde to give (S)-α,α′-dihydroxyketones 3 (Scheme 1).4,5 The substrate tolerance of TK towards a range α-hydroxyaldehydes has led to interest in industrial applications.6E. coli TK, which has been over-expressed,7 shows increased specific activity towards 1 compared to yeast and spinach TKs.8 | ||
| Scheme 1 Formation of α,α′-dihydroxy ketones (3S)-3 using TK. | ||
α,α′-Dihydroxyketone functionalities (3) are present in a range of natural products and are also important compounds for further conversion into other synthons, including ketosugars and 2-amino-1,3-diols.4d,9,10 TK shows high specificity towards the donor substrates but is more tolerant towards the acceptor aldehyde: several non-α-hydroxylated aldehydes have been used but lower relative rates of reaction (5–35% compared to hydroxylated aldehydes) were noted.5
With a view to enhancing the use of TK in synthetic applications with a wider range of aldehydes, we used saturation mutagenesis that was targeted to the TK active site residues. Mutants with improved activity towards glycolaldehyde (Scheme 1, R = CH2OH), and enhanced specificity to propanal 2a (R = CH2CH3) such as D469T, were identified.11 In addition, when propanal was used with wild-type (WT) TK, the ee of the product 3a (R = CH2CH3) was only 58% (Table 1) and therefore chiral assays were developed to identify mutants with improved stereoselectivities.12,13 Notable variants leading to high stereoselectivities were D469E (90% ee, 3S-isomer) and H26Y (88% ee, 3R-isomer), which remarkably with a single point active site mutation reversed the stereoselectivity.12
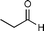
| Aldehyde | Product | WT-TK ee (yield) | D469E ee (yield) | D469T ee (yield) | D469K ee (yield) | D469L ee (yield) | H26Y ee (yield) | |
|---|---|---|---|---|---|---|---|---|
| 2a |
|
3a 12 | 58% (3S) (36%)12 | 90% (3S) (70%)12 | 64% (3S)12 (68%)17 | — | 12% (3S) (nd) | 88% (3R) (63%)12 |
| 2b |

|
3b | 75% (3S) (36%) | 98% (3S) (44%) | — | — | — | 92% (3R) (16%) |
| 2c |

|
3c | 84% (3S) (16%) | 97% (3S) (58%) | — | — | — | 84% (3R) (7%) |
| 2d |

|
3d | 85% (3S) (25%) | 97% (3S) (47%) | — | — | — | 84% (3R) (12%) |
| 2e |

|
3e | 74% (3S) (7%) | 86% (3S) (14%) | — | — | — | 78% (3R) (4%) |
| 2f |

|
3f | 66% (3S) (<3%) | 86% (3S) (18%) | — | — | — | 83% (3R) (21%) |
| 2g |

|
3g | 72% (1S) (<3%) | >99% (1S) (10%) | 99% (1S) (10%) | 99% (1S) (<3%) | 99% (1S) (<3%) | no reaction |
| 2h |
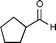
|
3h | 0% (<3%) | >99% (1S) (40%) | 99% (1S) (30%) | 25% (1S) (10%) | no reaction | 30% (1R) (<3%) |
| 2i |
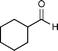
|
3i | 0% (<3%) | 97% (1S) (10%) | 99% (1S) (<3%) | 25% (1S) (<3%) | no reaction | no reaction |
The D469E mutant TK has also been reported to reduce the acceptance of glycolaldehyde and formaldehyde,14 and the D469 residue has been highlighted as a key residue involved in enantioselection with α-hydroxylated aldehydes: a yeast TK structure with the adjunct erythrose-4-phosphate, indicated it hydrogen bonds to the C-2 hydroxy group of 2-hydroxylated aldehydes in the active site.15 In view of the interesting substrate tolerances exhibited by the TK mutants, a more systematic study was carried out using linear and cyclic aliphatic aldehydes, with the aim of understanding substrate tolerance and limitations with selected mutants.
Results and discussion
Stereoselectivity of E. coli WT TK
Linear aliphatic aldehydes (C4–C8) and cyclopropane-, cyclopentane- and cyclohexanecarboxaldehyde were selected for use with WT-TK and TK mutants to determine the influence of chain length and ring size on reaction selectivities. Initially racemic α,α′-dihydroxyketones 3b–3i were prepared for chiral assay development. The commercially available aldehydes 2b–2i were converted into 3b–3i in yields of 2–35% using N-methylmorpholine and the previously described TK biomimetic reaction in water (Scheme 2).16 In general, lower yields were observed with the more lipophilic aldehydes, which may reflect poor substrate solubilities in water. | ||
| Scheme 2 Reagents and conditions: (i) N-methylmorpholine, pH 8, H2O; (ii) TK, ThDP, Mg2+, pH 7. | ||
Methods were established for the determination of ees in 3via monobenzoylation at the primary alcohol of 3g–3i and chiral HPLC. Compounds 3b–3f required dibenzoylation for satisfactory peak resolution by chiral HPLC. Then WT-TK and 1 were reacted with 2b–2f (C4–C8) to determine product stereoselectivities and yields (Table 1). As well as establishing ees via derivatisation and chiral HPLC, the selected ketodiols 3b and 3d were coupled to (S)-MTPACl to give the corresponding Mosher's esters: application of a recently reported NMR method indicated the major enantiomers formed were (3S)-3b and (3S)-3d.13
As in previous work using propanal, WT-TK leads to the formation of the (3S)-isomer as the major enantiomer, and the ees are given in Table 1: by analogy, the major isomers of 3c, 3e and 3f were assigned as (3S). Interestingly, the degree of stereoselectivity of the transformation varied with the length of the aliphatic chain of the aldehyde, reaching a maximum of 84% and 85% ee for pentanal and hexanal (Fig. 1), which are similar in size to the in vivo substrates of TK. In addition, increasing lipophilicity of the aldehyde acceptor resulted in lower conversion yields, probably due to decreasing aqueous solubilities.
 | ||
| Fig. 1 Variation in stereoselectivities for linear aliphatic aldehydes using WT-TK, D469E-TK and H26Y-TK. | ||
WT-TK was then used with the cyclic aldehydes 2g–2i. The conversion yields were low (<3%) and for the cyclopropane analogue 3g the ee was 72%. The major isomer formed was determined using the modified Mosher's ester method as again the (1S)-isomer.13 However, for the cyclopentane and cyclohexane analogues 3h and 3i, racemic products were formed. The substrate cyclopropane carboxaldehyde may be able to adopt a similar conformation to butanal in the active site leading to a similar level of stereoselectivity in the product; however, the larger-ringed cyclic analogues cannot.
Molecular modelling: WT TK
With a view to rationalising the stereoselectivities with WT-TK and the linear aliphatic aldehydes, which demonstrated a clear experimental relationship between chain length and ee in the product, molecular docking experiments were carried out using the X-ray crystal structure of WT-TK (PDB ID: 1QGD).18 Initially, in order to validate the method, direct docking of D-erythrose-4-phosphate, a natural substrate of TK, was carried out using AutoDock.19 The predominant conformations obtained were compared with an existing X-ray crystal structure of a noncovalent complex of E. coli TK with the aldose (PDB ID: 1NGS) and co-factor.20 Most docked conformations coincided with the conformation of the ligand in the crystal structure in terms of hydrogen bonding and consequently orientation. Despite the dynamic nature of proteins, the conformation generated in silico reproduced the experimental findings suggesting a molecular docking method would be a useful model to adopt.Direct docking of the linear aliphatic aldehydes (C3–C8) was performed into the TK active site containing a modelled ThDP-enamine intermediate and the results compared, firstly in terms of cluster populations, and then the hydrogen bonding and orientation. The conformations obtained populated one cluster preferentially in all cases, except for butanal, which occupied two energetically similar conformations. For all the C3–C8 aldehydes, docks were obtained in which the aldo oxygen atom is bound to two histidine residues (His26 and His261), bringing the substrates into close proximity with the coenzyme for nucleophilic attack.
The longer C5–C8 aldehydes docked with the aliphatic chain bound to a hydrophobic region around the lip of the entrance to the ThDP-containing cavity (Fig. 2, magenta model). This presented neither face of the aldehyde preferentially towards the ThDP enamine intermediate. Interestingly, the smallest aliphatic chain (C3) of 2a docked in an alternative orientation with the aliphatic chain occupying the narrow entrance to the cofactor containing cavity (Fig. 2, white model), marginally rendering the Re-face of the carbonyl exposed to the ThDP-enamine Cα atom.
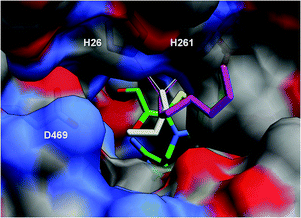 | ||
| Fig. 2 Aldehydes 2a (grey) and 2d (magenta) docked into the TK active site containing the modelled ThDP-enamine intermediate (green). A surface plot is shown with D469, H26 and H261 highlighted as sticks beneath the surface. | ||
This is reflected in the low ee (58%) in favour of the (3S)-isomer product for reaction with 2a. The slightly larger C4 aldehyde was found to dock in two different conformations, with the first similar to that for C3, and the second similar to the C5–C8 aldehydes. This transition of docking conformations at C4 in the series from C3 to C8 mirrors the observed increase in ee from 58% for C3, through 75% for C4 to 84% and 85% for C5 and C6 respectively. However, the increased ee resulting from the alternative conformations adopted by the C4–C8 aldehydes is not easily rationalised. One possibility is that as the aldo O-atom moves closer to H26 and H261 during the reaction, and the Van der Waals contacts between the aliphatic chains and the lipophilic region of the active site are weakened, allowing the chain to move further into the active site and more freedom to rotate the carbonyl with the Re-face of the carbonyl exposed to the ThDP-enamine Cα atom. The decrease in ee observed for C7 and C8 may be due to an increase in the Van der Waals interactions with the active-site wall and therefore, less flexibility in the re-orientation of the substrate.
Finally docking of the cyclic aldehydes 2g–2i was carried out; however, they docked preferentially in a lip at the entrance to the active site in non-productive conformations, which could explain the low yielding reactions observed. Only the cyclopropyl analogue 2g docked in a similar conformation to propanal 2a, which gave 3g in 72% ee. Modelling with the mutant TKs was not performed as we do not have the crystal structures of the mutants at present, and modelling both the protein mutation and the substrate binding simultaneously would be less informative.
Stereoselectivity of selected single-point TK mutants
In previous mutant screening studies using propanal as the acceptor, two active-site single-point mutant TKs exhibited marked improved and reversed stereoselectivity, Asp469Glu (D469E) and His26Tyr (H26Y). Variant D469E had led to the formation of (3S)-3a in 90% ee, while H26Y gave (3R)- 3a in 88% ee (Table 1).12 With the aim of understanding substrate tolerances, and enhancing and reversing stereoselectivities compared to WT-TK with the C4–C8 linear aldehydes, reactions were carried out using these high performing mutants: others screened with propanal were not explored due to the lower stereoselectivities achieved. D469E gave products in 14–58% isolated yield, with decreasing yield upon increasing chain length, but enhanced yields compared to WT-TK. Notably, high stereoselectivities (97–98% ee) were observed when using butanal, pentanal and hexanal, and the highest ees were when using aldehydes of a similar size to in vivo substrates for TK. As shown in Fig. 1, the greatest enhancement in ee between WT and D469E was for the C3 and C4 aldehydes. The use of H26Y gave products in generally lower yields (4–21%), with the highest (3R)-stereoselectivities observed in 3b using butanal (92% ee), decreasing to 78% ee for 3e. The reversal in ee was observed across the whole series of linear aldehydes used, although a smaller change in ee was observed in going from C3 to C8 (Fig. 1). The lower yields with some longer chain length analogues reflect a lower turnover of substrate and may be due to steric interactions in the active site.The selection of TK-mutants for use with cyclic aldehydes was less clear, and very low yields were observed using WT-TK. A recently developed tetrazolium red-based colorimetric assay was therefore used with the three substrates 2g–2i against the D469X library, which had led to the identification of several improved mutants when using propanal.12,21 From this, three mutants were selected, which were able to accept one or more of the cyclic aldehydes, D469T, D469K and D469L. These were used together with mutants D469E and H29Y, which gave high selectivities with the linear series. The results indicated that high stereoselectivities could be achieved with D469E: the cyclopropane and cyclopentane adducts, 3g and 3h, were formed in >99% ee, and cyclohexane analogue 3i formed in 97% ee (by HPLC) (Table 1). In all cases, the major isomer was the (1S)-product. When using D469T similar results were observed, with all products 3g-3i formed in 99% ee (the (1S)-isomer). Similar yields were observed with both mutants, with 3g and 3i formed in 3–10% yields, while the cyclopentane product was formed in a higher 30–40% yield. This possibly reflects the comparable ring size of 2h and the natural substrate, ribose-5-phosphate (furanose form). Mutants D469K and D469L gave the cyclopropane product in 99% ee in low yields, but when using 2h and 2i either low ees (D469K) or no reaction (D469L) were observed. The use of H26Y-TK gave no product with the cyclopropane and cyclohexane aldehydes, but when using cyclopentanecarboxaldehyde, 3h was formed in 30% ee, and the major isomer was the (1R)-product. Overall, these results suggest that when using cyclic substrates D469E and D469T may be suitable mutants to achieve bioconversions. The smallest cyclic substrate 2g appeared to have greater activity with a larger range of mutants (leading to high ees), presumably because it causes less steric problems. Most mutants gave (1S)-products, other than the low ee for the (1R)-isomer observed with H26Y-TK. The generally lower yields for the cyclic series may reflect poor substrate solubilities and/or product inhibition.
Initial relative rates were also determined for selected reactions using D469E, H26Y and D469T for the cyclic series (Table 2). Relative activities are given compared to propanal for each particular mutant: for comparison the wild-type specific activity towards glycolaldehyde was previously reported as 0.65 μmol mg−1min−1.11
| Aldehyde | Mutant | Initial rate/mM h−1 | TK/mg mL−1 | Spec. activity/μmol mg−1 min−1 | Initial rel. ratea |
|---|---|---|---|---|---|
| a Relative rates are given for substrates compared to propanal with the selected mutant. | |||||

|
D469E | 8.14 | 0.45 | 0.30 | 1 |

|
D469E | 2.14 | 0.45 | 0.08 | 0.3 |

|
D469E | 0.89 | 0.45 | 0.03 | 0.1 |

|
H26Y | 8.00 | 1.01 | 0.13 | 1 |

|
H26Y | 0.41 | 1.01 | 0.007 | 0.05 |

|
D469T | 12.5 | 0.30 | 0.69 | 1 |

|
D469T | 7.45 | 0.30 | 0.41 | 0.6 |

|
D469T | 4.47 | 0.30 | 0.25 | 0.4 |
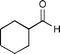
|
D469T | 3.46 | 0.30 | 0.19 | 0.3 |
Previous work highlighted that when using propanal, higher relative activities for D469E, H26Y and D469T compared to WT were observed. Here, relative rates decreased with increasing aldehyde size in all cases, although was most marked for H26Y. Interestingly, butanal had a higher initial rate with D469E than pentanal, although the isolated yield for the reaction was higher for the pentanal-derived product 3c. This may be due to product inhibition by 3b, limiting the overall reaction. For the cyclic aldehydes and D469T, increasing ring size also had less of an effect on initial rates, than increasing the linear chain length with D469E, although it decreased slightly as the ring size increased. This may result from lower conformational flexibilities of cyclic substrates, compared to aliphatic substrates, resulting in a smaller and more favourable entropy loss on binding. The small increase in conformational flexibility as the ring size increases may also lead to a less favourable loss of entropy upon binding.
Conclusions
Ideally, for use in synthesis, enzymes should have good substrate tolerance and demonstrate high stereoselectivities. In this work transketolase mutants able to accept lipophilic longer chain and cyclic aldehydes have been identified, which gives significant potential for further synthetic applications. With several substrate/mutant combinations very high stereoselectivities were observed, with moderate to low conversion yields. Future work is under way to investigate strategies to improve yields with lipophilic aldehyde acceptors.Experimental
General methods
Unless otherwise noted, solvents and reagents were reagent grade from commercial suppliers (Sigma-Aldrich) and used without further purification. Dry CH2Cl2 was obtained using anhydrous alumina columns.22 All moisture-sensitive reactions were performed under a nitrogen or argon atmosphere using oven-dried glassware. Reactions were monitored by TLC on Kieselgel 60 F254 plates with detection by UV, potassium permanganate and phosphomolybdic acid (PMA) [PMA hydrate (12 g) and ethanol (250 ml)] stains. Flash column chromatography was carried out using silica gel (particle size 40-63 μm). 1H NMR and 13C NMR spectra were recorded at the field indicated using Bruker AMX300 MHz, AMX400 Avance-500 MHz and Avance-600 MHz machines. Coupling constants are measured in Hertz (Hz) and unless otherwise specified, NMR spectra were recorded at 298 K. Mass spectra were recorded on a Thermo Finnegan MAT 900XP and Micro Mass Quattro LC electrospray mass spectrometers VG ZAB 2SE. Infrared spectra were recorded on a Shimadzu FTIR-8700 and Perkin Elmer Spectrum 100 FTIR spectrometer. Optical rotations were recorded on a Perkin Elmer model 343 polarimeter at 589 nm, quoted in deg cm2 g−1 and conc (c) in g/100 mL. Chiral HPLC analysis was performed on a Varian Prostar instrument equipped with a Chiracel AD chiral column (Daicel; Chiral Technologies Europe, France) 25 cm × 0.46 cm.Lithium hydroxypyruvate was synthesised as previously described.2 1,3-Dihydroxypentan-2-one 3a was prepared as previously described.12
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to a final concentration of 1 mg mL−1.
:1) to a final concentration of 1 mg mL−1.
:1) to give 3b as a white powder (47 mg, 12%). Mp 109–112 °C (EtOAc); νmax(neat)/cm−1 3413, 2960, 2935, 2874, 1719; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 20.1, CHHOH), 4.38 (1H, d, J 20.1, CHHOH), 4.31 (1H, dd, J 8.0 and 3.9, CHOH), 2.70 (br, OH), 1.75 (1H, m, 4-HH), 1.57 (1H, m, 4-HH), 1.46 (2H, m, CH2CH3), 0.95 (3H, t, J 7.3, CH3); 13C NMR (125 MHz; CDCl3) δ 211.9 (C-2), 74.8 (C-3), 65.6 (C-1), 36.3 (C-4), 18.1 (C-5), 13.8 (C-6); m/z (CI) 133 (MH+, 100%), 115 ([MH − H2O]+, 30), 85 (47); Found (HRCI) MH+ 133.08611. C6H12O3 requires 133.08647. Racemic 3b was dibenzoylated and HPLC analysis (97:3, 1 mL min−1) gave retention times of 20.4 min (R-isomer) and 24.5 min (S-isomer).
:1) to give 3c a white powder (152 mg, 35%). Mp 110–125 °C (EtOAc); νmax(neat)/cm−1 3430, 3263, 2956, 2929, 2872, 1720; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 19.4, CHHOH), 4.39 (1H, d, J 19.4, CHHOH), 4.31 (1H, dd, J 7.9 and 3.9, CHOH), 2.89 (br, OH), 1.80 (1H, m, 4-HH), 1.20–1.75 (5H, m, 4-HH, (CH2)2CH3), 0.91 (3H, t, J 6.6, CH3); 13C NMR (125 MHz; CDCl3) δ 211.7 (C-2), 75.0 (C-3), 65.6 (C-1), 34.0 (C-4), 26.8 (C-5), 22.5 (C-6), 13.9 (C-7); m/z (CI) 147 (MH+, 100%), 129 ([MH − H2O]+, 35), 85 (94); Found (HRCI) MH+ 147.10288. C7H14O3 requires 147.10212. Racemic 3c was dibenzoylated and HPLC analysis (97:3, 1 mL min−1) gave retention times of 16.6 min (R-isomer) and 20.0 min (S-isomer).
:1) to give 3d as a white powder (11 mg, 3%). Mp 106–110 °C (EtOAc); νmax(neat)/cm−1 3406, 2956, 2925, 2858, 1720; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 19.4, CHHOH), 4.38 (1H, d, J 19.4, CHHOH), 4.31 (1H, dd, J 7.9 and 3.9, CHOH), 2.89 (br, OH), 1.77 (1H, m, 4-HH), 1.58 (1H, m, 4-HH) 1.20–1.34 (6H, m, 5-H2, 6-H2, 7-H2), 0.89 (3H, t, J 6.9, CH3); 13C NMR (125 MHz; CDCl3) δ 211.7 (C-2), 75.0 (C-3), 65.6 (C-1), 34.3 (C-4), 31.6 (C-5), 24.4 (C-6), 22.5 (C-7), 14.0 (C-8); m/z (CI) 161 (MH+, 100%); Found (HRCI) MH+ 161.11823. C7H14O3 requires 161.11777. Racemic 3d was dibenzoylated and HPLC analysis (97:3, 1 mL min−1) gave retention times of 15.5 min (R-isomer) and 19.9 min (S-isomer).
:1) to give 3e as a white powder (22 mg, 4%). Mp 104–107 °C (EtOAc); νmax(neat)/cm−1 3406, 2955, 2925, 2857, 1718; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 19.4, CHHOH), 4.38 (1H, d, J 19.4, CHHOH), 4.30 (1H, dd, J 7.8, 3.9, CHOH), 2.96 (br s, OH), 1.78 (1H, m, 4-HH), 1.57 (1H, m, 4-HH), 1.27–1.50 (8H, m, 5-H2, 6-H2, 7-H2, 8-H2), 0.88 (3H, t, J 6.9, CH3); 13C NMR (125 MHz; CDCl3) δ 211.7 (C-2), 75.0 (C-3), 65.6 (C-1), 34.3 (C-4), 31.7 (C-5), 29.0 (C-6), 24.7 (C-7), 22.6 (C-8), 14.1 (C-9); m/z (CI) 175 (MH+, 100%), 139 (62), 113 (97), 97 (100); Found (HRCI) MH+ 175.13360. C9H19O3 requires 175.13342. Racemic 3e was dibenzoylated and HPLC analysis (97:3, 1.2 mL min−1) gave retention times of 11.6 min (R-isomer) and 15.4 min (S-isomer).
:1) to give 3f as a white powder (2%, 3 mg). Mp 100–103 °C (EtOAc); νmax(neat)/cm−1 3420, 2959, 2928, 2873, 1721; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 19.4, CHHOH), 4.38 (1H, d, J 19.4, CHHOH), 4.31 (1H, m, CHOH), 2.93 (br s, OH), 1.77 (1H, m, 4-HH), 1.20–1.61 (11H, m, 4-HH,5-H2, 6-H2, 7-H2, 8-H2, 9-H2), 0.88 (3H, t, J 7.0, CH3); 13C NMR (125 MHz; CDCl3) δ 211.7 (C-2), 75.0 (C-3), 65.6 (C-1), 34.3 (C-4), 31.8 (C-5), 29.3 (C-6), 29.1 (C-7), 24.7 (C-8), 22.7 (C-9), 14.1 (C-10); m/z (CI) 189 (MH+, 100%), 153 (93), 127 (97); Found (HRCI) MH+ 189.14971. C10H21O3 requires 189.14907. Racemic 3f was dibenzoylated and HPLC analysis (97:3, 1.2 mL min−1) gave retention times of 10.5 min (R-isomer) and 13.9 min (S-isomer).
:3, 1 mL min−1) gave retention times of 31.1 min (S-isomer) and 33.6 min (R-isomer).
:1) to give 3h as a white solid (0.119 g, 25%). Rf 0.21 (EtOAc–hexane, 1:1). Mp 110–112 °C (EtOAc–hexane, 1:1); νmax(KBr)/cm−1 3411, 2953, 2870, 1718; 1H NMR (300 MHz; CDCl3) δ 4.50 (1H, d, J 19.4, CHHOH), 4.36 (1H, d, J 19.4, CHHOH), 4.30 (1H, m, CHOH), 3.04 (2H, s, 2 × OH), 2.18 (1H, m), 1.31–1.75 (8H, m, 4 × CH2); 13C NMR (75 MHz; CDCl3) δ 211.5 (C-2), 76.9 (CHOH), 65.8 (CH2OH), 42.9, 29.0, 25.7 and 25.6; m/z (FTMS) found [M + NH4]+ 176.1281. C8H18O3N requires 176.1281. Racemic 3h was monobenzoylated and HPLC analysis (97:3, 1 mL min−1) gave retention times of 32.1 min (R-isomer) and 34.8 min (S-isomer).
:1) to give 3i as a white solid (0.088 g, 17%). Rf 0.21 (EtOAc–hexane, 1:1). Mp 115–118 °C (EtOAc–hexane, 1:1); νmax(KBr)/cm−1 3429, 2940, 1712; 1H NMR (300 MHz; CD3OD) δ 4.45 (1H, d, J 19.3, CHHOH), 4.34 (1H, d, J 19.3, CHHOH), 3.96 (1H, d, J 4.4, CHOH), 1.45–1.87 (6H, m), 1.16–1.30 (5H, m); 13C NMR (75 MHz; CD3OD) δ 214.3 (C-2), 80.7 (CHOH), 67.4 (CH2OH), 43.0, 30.3, 27.7, 27.3 (signals superimposed); m/z (CI) 173 (MH+, 100%), 155 (45), 137 (86), 95 (100); Found (HRCI) MH+ 173.11736. C9H17O3 requires 173.11722. Racemic 3i was monobenzoylated and HPLC analysis of the product (97:3, 1 mL min−1) gave retention times of 17.0 min (R-isomer) and 18.5 min (S-isomer).
:1) to a final concentration of 1 mg mL−1.
:1) to a final concentration of 1 mg mL−1.
:1) to give 3b as a white powder (47 mg, 12%). Mp 109–112 °C (EtOAc); νmax(neat)/cm−1 3413, 2960, 2935, 2874, 1719; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 20.1, CHHOH), 4.38 (1H, d, J 20.1, CHHOH), 4.31 (1H, dd, J 8.0 and 3.9, CHOH), 2.70 (br, OH), 1.75 (1H, m, 4-HH), 1.57 (1H, m, 4-HH), 1.46 (2H, m, CH2CH3), 0.95 (3H, t, J 7.3, CH3); 13C NMR (125 MHz; CDCl3) δ 211.9 (C-2), 74.8 (C-3), 65.6 (C-1), 36.3 (C-4), 18.1 (C-5), 13.8 (C-6); m/z (CI) 133 (MH+, 100%), 115 ([MH − H2O]+, 30), 85 (47); Found (HRCI) MH+ 133.08611. C6H12O3 requires 133.08647. Racemic 3b was dibenzoylated and HPLC analysis (97:3, 1 mL min−1) gave retention times of 20.4 min (R-isomer) and 24.5 min (S-isomer).
:1) to give 3c a white powder (152 mg, 35%). Mp 110–125 °C (EtOAc); νmax(neat)/cm−1 3430, 3263, 2956, 2929, 2872, 1720; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 19.4, CHHOH), 4.39 (1H, d, J 19.4, CHHOH), 4.31 (1H, dd, J 7.9 and 3.9, CHOH), 2.89 (br, OH), 1.80 (1H, m, 4-HH), 1.20–1.75 (5H, m, 4-HH, (CH2)2CH3), 0.91 (3H, t, J 6.6, CH3); 13C NMR (125 MHz; CDCl3) δ 211.7 (C-2), 75.0 (C-3), 65.6 (C-1), 34.0 (C-4), 26.8 (C-5), 22.5 (C-6), 13.9 (C-7); m/z (CI) 147 (MH+, 100%), 129 ([MH − H2O]+, 35), 85 (94); Found (HRCI) MH+ 147.10288. C7H14O3 requires 147.10212. Racemic 3c was dibenzoylated and HPLC analysis (97:3, 1 mL min−1) gave retention times of 16.6 min (R-isomer) and 20.0 min (S-isomer).
:1) to give 3d as a white powder (11 mg, 3%). Mp 106–110 °C (EtOAc); νmax(neat)/cm−1 3406, 2956, 2925, 2858, 1720; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 19.4, CHHOH), 4.38 (1H, d, J 19.4, CHHOH), 4.31 (1H, dd, J 7.9 and 3.9, CHOH), 2.89 (br, OH), 1.77 (1H, m, 4-HH), 1.58 (1H, m, 4-HH) 1.20–1.34 (6H, m, 5-H2, 6-H2, 7-H2), 0.89 (3H, t, J 6.9, CH3); 13C NMR (125 MHz; CDCl3) δ 211.7 (C-2), 75.0 (C-3), 65.6 (C-1), 34.3 (C-4), 31.6 (C-5), 24.4 (C-6), 22.5 (C-7), 14.0 (C-8); m/z (CI) 161 (MH+, 100%); Found (HRCI) MH+ 161.11823. C7H14O3 requires 161.11777. Racemic 3d was dibenzoylated and HPLC analysis (97:3, 1 mL min−1) gave retention times of 15.5 min (R-isomer) and 19.9 min (S-isomer).
:1) to give 3e as a white powder (22 mg, 4%). Mp 104–107 °C (EtOAc); νmax(neat)/cm−1 3406, 2955, 2925, 2857, 1718; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 19.4, CHHOH), 4.38 (1H, d, J 19.4, CHHOH), 4.30 (1H, dd, J 7.8, 3.9, CHOH), 2.96 (br s, OH), 1.78 (1H, m, 4-HH), 1.57 (1H, m, 4-HH), 1.27–1.50 (8H, m, 5-H2, 6-H2, 7-H2, 8-H2), 0.88 (3H, t, J 6.9, CH3); 13C NMR (125 MHz; CDCl3) δ 211.7 (C-2), 75.0 (C-3), 65.6 (C-1), 34.3 (C-4), 31.7 (C-5), 29.0 (C-6), 24.7 (C-7), 22.6 (C-8), 14.1 (C-9); m/z (CI) 175 (MH+, 100%), 139 (62), 113 (97), 97 (100); Found (HRCI) MH+ 175.13360. C9H19O3 requires 175.13342. Racemic 3e was dibenzoylated and HPLC analysis (97:3, 1.2 mL min−1) gave retention times of 11.6 min (R-isomer) and 15.4 min (S-isomer).
:1) to give 3f as a white powder (2%, 3 mg). Mp 100–103 °C (EtOAc); νmax(neat)/cm−1 3420, 2959, 2928, 2873, 1721; 1H NMR (500 MHz; CDCl3) δ 4.49 (1H, d, J 19.4, CHHOH), 4.38 (1H, d, J 19.4, CHHOH), 4.31 (1H, m, CHOH), 2.93 (br s, OH), 1.77 (1H, m, 4-HH), 1.20–1.61 (11H, m, 4-HH,5-H2, 6-H2, 7-H2, 8-H2, 9-H2), 0.88 (3H, t, J 7.0, CH3); 13C NMR (125 MHz; CDCl3) δ 211.7 (C-2), 75.0 (C-3), 65.6 (C-1), 34.3 (C-4), 31.8 (C-5), 29.3 (C-6), 29.1 (C-7), 24.7 (C-8), 22.7 (C-9), 14.1 (C-10); m/z (CI) 189 (MH+, 100%), 153 (93), 127 (97); Found (HRCI) MH+ 189.14971. C10H21O3 requires 189.14907. Racemic 3f was dibenzoylated and HPLC analysis (97:3, 1.2 mL min−1) gave retention times of 10.5 min (R-isomer) and 13.9 min (S-isomer).
:3, 1 mL min−1) gave retention times of 31.1 min (S-isomer) and 33.6 min (R-isomer).
:1) to give 3h as a white solid (0.119 g, 25%). Rf 0.21 (EtOAc–hexane, 1:1). Mp 110–112 °C (EtOAc–hexane, 1:1); νmax(KBr)/cm−1 3411, 2953, 2870, 1718; 1H NMR (300 MHz; CDCl3) δ 4.50 (1H, d, J 19.4, CHHOH), 4.36 (1H, d, J 19.4, CHHOH), 4.30 (1H, m, CHOH), 3.04 (2H, s, 2 × OH), 2.18 (1H, m), 1.31–1.75 (8H, m, 4 × CH2); 13C NMR (75 MHz; CDCl3) δ 211.5 (C-2), 76.9 (CHOH), 65.8 (CH2OH), 42.9, 29.0, 25.7 and 25.6; m/z (FTMS) found [M + NH4]+ 176.1281. C8H18O3N requires 176.1281. Racemic 3h was monobenzoylated and HPLC analysis (97:3, 1 mL min−1) gave retention times of 32.1 min (R-isomer) and 34.8 min (S-isomer).
:1) to give 3i as a white solid (0.088 g, 17%). Rf 0.21 (EtOAc–hexane, 1:1). Mp 115–118 °C (EtOAc–hexane, 1:1); νmax(KBr)/cm−1 3429, 2940, 1712; 1H NMR (300 MHz; CD3OD) δ 4.45 (1H, d, J 19.3, CHHOH), 4.34 (1H, d, J 19.3, CHHOH), 3.96 (1H, d, J 4.4, CHOH), 1.45–1.87 (6H, m), 1.16–1.30 (5H, m); 13C NMR (75 MHz; CD3OD) δ 214.3 (C-2), 80.7 (CHOH), 67.4 (CH2OH), 43.0, 30.3, 27.7, 27.3 (signals superimposed); m/z (CI) 173 (MH+, 100%), 155 (45), 137 (86), 95 (100); Found (HRCI) MH+ 173.11736. C9H17O3 requires 173.11722. Racemic 3i was monobenzoylated and HPLC analysis of the product (97:3, 1 mL min−1) gave retention times of 17.0 min (R-isomer) and 18.5 min (S-isomer).
Biocatalytic synthesis of 3b-3i
Determination of initial rates for selected reactions
Protein concentrations were determined using determined using a combined Bradford and SDS-PAGE densitometry method as previously described.11a
:3, 1.0 mL min−1). D469E-TK gave 3b (58 mg, 44%) in 98% ee (3S-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D−16.3 (c 1.4, CH3OH). H26Y-TK gave 3b (21 mg, 16%) in 92% ee (3R-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D +14.2 (c 1.6, CH3OH). The absolute stereochemistry of 3b generated using D469E-TK was determined using the Mosher’s derivatisation method.13
:1) to afford the Mosher's derivative as a colourless oil (4.6 mg, 43%). 1H NMR (600 MHz; CDCl3) δ 7.64 (2H, m, Ph), 7.45 (3H, m, Ph), 5.21 (d, J 18.0, CHHO (2R,3′R-trace)), 5.11 (1H, d, J 18.0, CHHO (2R,3′S)), 5.07 (1H, d, J 18.0, CHHO (2R,3′S)), 4,96 (1H, d, J 18.0, CHHO (2R,3′R-trace)), 4.33 (1H, m, CHOH), 3.66 (3H, s, OCH3), 2.90 (1H, d, J 5.4, OH), 1.79 (1H, m, CHH), 1.46–1.65 (3H, m, CHH and CH2), 0.97 (3H, t, J 7.0, CH2CH3); 13C NMR (150 MHz; CDCl3) δ 204.3 (C-2′), 166.2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester), 131.7, 129.9, 128.5, 127.5, 123.0 (q, JCF 290, CF3), 75.1 (CHOH), 67.0 (CH2OH), 55.8 (OCH3), 36.0, 29.7, 13.8 (CH2CH3); 19F NMR (282 MHz; CDCl3) δ -72.2; m/z (ES+) 371 (MNa+, 100%); Found (HRES) MNa+ 371.1096. C16H19O5F3Na requires 371.1106.
:3, 1.0 mL min−1). D469E-TK gave 3c (84 mg, 58%) in 97% ee (3S-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D−38.5 (c 1.4, CH3OH). H26Y-TK gave 3c (10 mg, 7%) in 84% ee (3R-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D +18.6 (c 1.4, CH3OH).
:3, 1.0 mL min−1). D469E-TK gave 3d (75 mg, 47%) in 97% ee (3S-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D−32.5 (c 1.4, CH3OH). H26Y-TK gave 3d (20 mg, 12%) in 84% ee (3R-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D +16.0 (c 1.4, CH3OH). The absolute stereochemistry of 3d generated using D469E-TK was determined using the Mosher’s derivatisation method.13
:1) to afford the Mosher's derivative as a colourless oil (3.2 mg, 28%). 1H NMR (600 MHz; CDCl3) δ 7.64 (2H, m, Ph), 7.45 (3H, m, Ph), 5.20 (d, J 16.8, CHHO (2R,3′R-trace)), 5.10 (1H, d, J 16.8, CHHO (2R,3′S)), 5.07 (1H, d, J 16.8, CHHO (2R,3′S)), 4,96 (1H, d, J 16.8, CHHO (2R,3′R-trace)), 4.32 (1H, dd, J 8.0 and 3.9, CHOH), 3.66 (3H, s, OCH3), 2.85 (1H, br s, OH), 1.05–1.80 (8H, m, 4 × CH2), 0.89 (3H, t, J 7.3, CH2CH3); 13C NMR (150 MHz; CDCl3) δ 204.3 (C-2′), 166.2 (CO ester), 131.7, 129.9, 128.5, 127.5, 75.3 (CHOH), 67.0 (CH2OH), 55.8 (OCH3), 33.9, 32.0, 29.5, 22.7, 14.0 (CH2CH3); 19F NMR (282 MHz; CDCl3) δ -72.2; m/z (ES+) 399 (MNa+, 100%); Found (HRES) MNa+ 399.1338. C18H23O5F3Na requires 399.1395.
:3, 1.2 mL min−1). D469E-TK gave 3e (25 mg, 14%) in 86% ee (3S-isomer) by HPLC (97:3, 1.2 mL min−1); [α]20D−22.7 (c 1.1, CH3OH). H26Y-TK gave 3e (6 mg, 4%) in 78% ee (3R-isomer) by HPLC (97:3, 1.2 mL min−1); [α]20D +36.7 (c 0.9, CH3OH).
:3, 1.2 mL min−1). D469E-TK gave 3f (34 mg, 18%) in 86% ee (3S-isomer) by HPLC (97:3, 1.2 mL min−1); [α]20D−13.3 (c 1.1, CH3OH). H26Y-TK gave 3f (39 mg, 21%) in 83% ee (3R-isomer) by HPLC (97:3, 1.2 mL min−1); [α]20D +9.1 (c 0.6, CH3OH).
:3, 1.0 mL min−1). D469E-TK gave 3g (13 mg, 10%) in >99% ee (1S-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D +70.0 (c 0.3, CHCl3). H26Y-TK gave no reaction. D469T-TK gave 3g (14 mg, 10%) in 99% ee (1S-isomer), D469K-TK gave 3g (3 mg, 2%) in 99% ee (1S-isomer), and D469L gave 3g (2 mg, 2%) in 99% ee (1S-isomer). The absolute stereochemistry of 3g generated using D469E-TK was determined using the Mosher’s derivatisation method.13
:1) to afford the Mosher's derivative as a colourless oil (0.013 g, 87%). Rf 0.45 (hexane–EtOAc; 1:1); [α]20D +45.0 (c 0.2, EtOH); νmax(KBr)/cm−1 3420, 2930, 2855, 1734; 1H NMR (300 MHz; CDCl3) δ 7.63 (2H, m, Ph), 7.43 (3H, m, Ph), 5.37 (1H, d, J 15.0, CHHO (2S,3′S)), 5.17 (d, J 18.0, CHHO (2S,3′R-trace)), 5.05 (d, J 18.0, CHHO (2S,3′R-trace)), 4.95 (1H, d, J 15.0, CHHO (2S,3′S)), 4.33 (1H, m, CHOH), 3.60 (3H, s, OCH3), 3.09 (1H, d, J 4.3 Hz), 1.03 (1H, m), 0.71 (2H, m), 0.55 (2H, m); 13C NMR (125 MHz; CDCl3) δ 203.0 (C-2′), 166.3 (CO ester), 131.8, 129.9, 128.5, 127.6, 78.1 (CHOH), 66.9 (CH2OH), 55.8 (OCH3), 14.7, 2.9 and 2.8; 19F NMR (282 MHz; CDCl3) δ -72.3; m/z (FTMS) found [M + NH4]+ 364.1364. C16H21F3O5N requires 364.1366.
:3, 1.0 mL min−1); [α]20D +33.0 (c 0.5, CHCl3). H26Y-TK gave 3h (3 mg, 2%) in 30% ee (1R-isomer), D469T-TK gave 3h (47 mg, 30%) in 99% ee (1S-isomer) and D469K-TK gave 3h (16 mg, 10%) in 25% ee (1S-isomer). The absolute stereochemistry of 3h generated using D469E-TK was determined using the Mosher’s derivatisation method.13
:1) to afford the Mosher's derivative as a colourless oil (0.018 g, 78%). Rf 0.50 (hexane–EtOAc; 4:1); [α]25D−60.0 (c 0.1, CHCl3); νmax(KBr)/cm−1 3429, 2930, 2855, 1733; 1H NMR (600 MHz; CDCl3) δ 7.62 (2H, m, Ph), 7.43 (3H, m, Ph), 5.20 (1H, d, J 16.9, CHHO (2S,3′S)), 4.92 (1H, d, J 16.9, CHHO (2S,3′S)), 4.31 (1H, d, J 3.6, CHOH), 3.65 (3H, s, OCH3), 2.91 (1H, m), 2.23 (1H, m), 1.27–1.85 (10H, m, 5 × CH2), no (2S,3′R) detected; 13C NMR (150 MHz; CDCl3) δ 204.0 (C-2′), 166.3 (CO ester), 131.9, 130.0, 128.6, 127.6, 123.2 (q, JCF 287, CF3), 84.7 (q, JCF 27, CCF3), 77.2 (CHOH), 67.4 (CH2OH), 55.9 (OCH3), 42.9, 29.1, 25.8 (signals superimposed); 19F NMR (282 MHz; CDCl3) δ -72.2; m/z (FTMS) found [M + NH4]+ 392.1678. C18H25F3O5N requires 392.1679.
:1) to afford the Mosher's derivative as a colourless oil (0.020 g, 87%). Rf 0.50 (hexane–EtOAc; 4:1); [α]25D +30.0 (c 0.1, CHCl3); νmax(KBr)/cm−1 3430, 2930, 1732; 1H NMR (600 MHz; CDCl3) δ 7.63 (2H, m, Ph), 7.43 (3H, m, Ph), 5.08 (1H, d, J 16.9, CHHO (2R,3′S)), 5.05 (1H, d, J 16.9, CHHO (2R,3′S)), 4.31 (1H, t, J 4.2, CHOH), 3.64 (3H, s, OCH3), 2.90 (1H, d, J 4.2, OH), 2.23 (1H, m), 1.34–1.78 (10H, m, 5 × CH2), no (2R,3′R) detected; 13C NMR (150 MHz; CDCl3) δ 204.0 (C-2′), 166.3 (CO ester), 131.9, 130.0, 128.6, 127.6, 123.2 (q, JCF 287, CF3), 84.7 (q, JCF 27, CCF3), 77.0 (CHOH), 67.4 (CH2OH), 55.9 (OCH3), 42.8, 29.1, 25.8 (signals superimposed); 19F NMR (282 MHz; CDCl3) δ−72.2.
:3, 1.0 mL min−1); [α]20D +33.0 (c 0.5, CHCl3). H26Y-TK gave no reaction. D469T-TK gave 3i (5 mg, 3%) in 99% ee (1S-isomer) and D469K-TK gave 3i (5 mg, 3%) in 25% ee (1S-isomer). The absolute stereochemistry with D469E-TK was determined using the Mosher’s derivatisation method.13
:1) to afford the Mosher's derivative as a colourless oil (0.018 g, 78%). Rf 0.45 (hexane–EtOAc; 4:1); [α]20D−26.0 (c 0.2, CHCl3); 1H NMR (300 MHz; CDCl3) δ 7.63 (2H, m, Ph), 7.43 (3H, m, Ph), 5.19 (1H, d, J 17.0, CHHO (2S,3′S)), 5.07 (d, J 17.0, CHHO (2S,3′R-trace)), 5.03 (d, J 17.0, CHHO (2S,3′R-trace)), 4.90 (1H, d, J 17.0, CHHO (2S,3′S)), 4.16 (1H, dd, J 5.2, 3.3, CHOH), 3.65 (3H, s, OCH3), 2.80 (1H, d, J 5.2, OH), 1.11–1.76 (11H, m); 13C NMR (125 MHz; CDCl3) δ 204.2 (C-2′), 166.2 (CO ester), 131.9, 129.9, 128.5, 127.5, 79.7 (CHOH), 67.8 (CH2OH), 55.8 (OCH3), 41.9, 29.6, 26.4, 25.9 (signal overlap), 25.5; 19F NMR (282 MHz; CDCl3) δ−72.2; m/z (FTMS) found [M + NH4]+ 406.1831. C19H27F3O5N requires 406.1836.
O ester), 131.7, 129.9, 128.5, 127.5, 123.0 (q, JCF 290, CF3), 75.1 (CHOH), 67.0 (CH2OH), 55.8 (OCH3), 36.0, 29.7, 13.8 (CH2CH3); 19F NMR (282 MHz; CDCl3) δ -72.2; m/z (ES+) 371 (MNa+, 100%); Found (HRES) MNa+ 371.1096. C16H19O5F3Na requires 371.1106.
:3, 1.0 mL min−1). D469E-TK gave 3c (84 mg, 58%) in 97% ee (3S-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D−38.5 (c 1.4, CH3OH). H26Y-TK gave 3c (10 mg, 7%) in 84% ee (3R-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D +18.6 (c 1.4, CH3OH).
:3, 1.0 mL min−1). D469E-TK gave 3d (75 mg, 47%) in 97% ee (3S-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D−32.5 (c 1.4, CH3OH). H26Y-TK gave 3d (20 mg, 12%) in 84% ee (3R-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D +16.0 (c 1.4, CH3OH). The absolute stereochemistry of 3d generated using D469E-TK was determined using the Mosher’s derivatisation method.13
:1) to afford the Mosher's derivative as a colourless oil (3.2 mg, 28%). 1H NMR (600 MHz; CDCl3) δ 7.64 (2H, m, Ph), 7.45 (3H, m, Ph), 5.20 (d, J 16.8, CHHO (2R,3′R-trace)), 5.10 (1H, d, J 16.8, CHHO (2R,3′S)), 5.07 (1H, d, J 16.8, CHHO (2R,3′S)), 4,96 (1H, d, J 16.8, CHHO (2R,3′R-trace)), 4.32 (1H, dd, J 8.0 and 3.9, CHOH), 3.66 (3H, s, OCH3), 2.85 (1H, br s, OH), 1.05–1.80 (8H, m, 4 × CH2), 0.89 (3H, t, J 7.3, CH2CH3); 13C NMR (150 MHz; CDCl3) δ 204.3 (C-2′), 166.2 (CO ester), 131.7, 129.9, 128.5, 127.5, 75.3 (CHOH), 67.0 (CH2OH), 55.8 (OCH3), 33.9, 32.0, 29.5, 22.7, 14.0 (CH2CH3); 19F NMR (282 MHz; CDCl3) δ -72.2; m/z (ES+) 399 (MNa+, 100%); Found (HRES) MNa+ 399.1338. C18H23O5F3Na requires 399.1395.
:3, 1.2 mL min−1). D469E-TK gave 3e (25 mg, 14%) in 86% ee (3S-isomer) by HPLC (97:3, 1.2 mL min−1); [α]20D−22.7 (c 1.1, CH3OH). H26Y-TK gave 3e (6 mg, 4%) in 78% ee (3R-isomer) by HPLC (97:3, 1.2 mL min−1); [α]20D +36.7 (c 0.9, CH3OH).
:3, 1.2 mL min−1). D469E-TK gave 3f (34 mg, 18%) in 86% ee (3S-isomer) by HPLC (97:3, 1.2 mL min−1); [α]20D−13.3 (c 1.1, CH3OH). H26Y-TK gave 3f (39 mg, 21%) in 83% ee (3R-isomer) by HPLC (97:3, 1.2 mL min−1); [α]20D +9.1 (c 0.6, CH3OH).
:3, 1.0 mL min−1). D469E-TK gave 3g (13 mg, 10%) in >99% ee (1S-isomer) by HPLC (97:3, 1.0 mL min−1); [α]20D +70.0 (c 0.3, CHCl3). H26Y-TK gave no reaction. D469T-TK gave 3g (14 mg, 10%) in 99% ee (1S-isomer), D469K-TK gave 3g (3 mg, 2%) in 99% ee (1S-isomer), and D469L gave 3g (2 mg, 2%) in 99% ee (1S-isomer). The absolute stereochemistry of 3g generated using D469E-TK was determined using the Mosher’s derivatisation method.13
:1) to afford the Mosher's derivative as a colourless oil (0.013 g, 87%). Rf 0.45 (hexane–EtOAc; 1:1); [α]20D +45.0 (c 0.2, EtOH); νmax(KBr)/cm−1 3420, 2930, 2855, 1734; 1H NMR (300 MHz; CDCl3) δ 7.63 (2H, m, Ph), 7.43 (3H, m, Ph), 5.37 (1H, d, J 15.0, CHHO (2S,3′S)), 5.17 (d, J 18.0, CHHO (2S,3′R-trace)), 5.05 (d, J 18.0, CHHO (2S,3′R-trace)), 4.95 (1H, d, J 15.0, CHHO (2S,3′S)), 4.33 (1H, m, CHOH), 3.60 (3H, s, OCH3), 3.09 (1H, d, J 4.3 Hz), 1.03 (1H, m), 0.71 (2H, m), 0.55 (2H, m); 13C NMR (125 MHz; CDCl3) δ 203.0 (C-2′), 166.3 (CO ester), 131.8, 129.9, 128.5, 127.6, 78.1 (CHOH), 66.9 (CH2OH), 55.8 (OCH3), 14.7, 2.9 and 2.8; 19F NMR (282 MHz; CDCl3) δ -72.3; m/z (FTMS) found [M + NH4]+ 364.1364. C16H21F3O5N requires 364.1366.
:3, 1.0 mL min−1); [α]20D +33.0 (c 0.5, CHCl3). H26Y-TK gave 3h (3 mg, 2%) in 30% ee (1R-isomer), D469T-TK gave 3h (47 mg, 30%) in 99% ee (1S-isomer) and D469K-TK gave 3h (16 mg, 10%) in 25% ee (1S-isomer). The absolute stereochemistry of 3h generated using D469E-TK was determined using the Mosher’s derivatisation method.13
:1) to afford the Mosher's derivative as a colourless oil (0.018 g, 78%). Rf 0.50 (hexane–EtOAc; 4:1); [α]25D−60.0 (c 0.1, CHCl3); νmax(KBr)/cm−1 3429, 2930, 2855, 1733; 1H NMR (600 MHz; CDCl3) δ 7.62 (2H, m, Ph), 7.43 (3H, m, Ph), 5.20 (1H, d, J 16.9, CHHO (2S,3′S)), 4.92 (1H, d, J 16.9, CHHO (2S,3′S)), 4.31 (1H, d, J 3.6, CHOH), 3.65 (3H, s, OCH3), 2.91 (1H, m), 2.23 (1H, m), 1.27–1.85 (10H, m, 5 × CH2), no (2S,3′R) detected; 13C NMR (150 MHz; CDCl3) δ 204.0 (C-2′), 166.3 (CO ester), 131.9, 130.0, 128.6, 127.6, 123.2 (q, JCF 287, CF3), 84.7 (q, JCF 27, CCF3), 77.2 (CHOH), 67.4 (CH2OH), 55.9 (OCH3), 42.9, 29.1, 25.8 (signals superimposed); 19F NMR (282 MHz; CDCl3) δ -72.2; m/z (FTMS) found [M + NH4]+ 392.1678. C18H25F3O5N requires 392.1679.
:1) to afford the Mosher's derivative as a colourless oil (0.020 g, 87%). Rf 0.50 (hexane–EtOAc; 4:1); [α]25D +30.0 (c 0.1, CHCl3); νmax(KBr)/cm−1 3430, 2930, 1732; 1H NMR (600 MHz; CDCl3) δ 7.63 (2H, m, Ph), 7.43 (3H, m, Ph), 5.08 (1H, d, J 16.9, CHHO (2R,3′S)), 5.05 (1H, d, J 16.9, CHHO (2R,3′S)), 4.31 (1H, t, J 4.2, CHOH), 3.64 (3H, s, OCH3), 2.90 (1H, d, J 4.2, OH), 2.23 (1H, m), 1.34–1.78 (10H, m, 5 × CH2), no (2R,3′R) detected; 13C NMR (150 MHz; CDCl3) δ 204.0 (C-2′), 166.3 (CO ester), 131.9, 130.0, 128.6, 127.6, 123.2 (q, JCF 287, CF3), 84.7 (q, JCF 27, CCF3), 77.0 (CHOH), 67.4 (CH2OH), 55.9 (OCH3), 42.8, 29.1, 25.8 (signals superimposed); 19F NMR (282 MHz; CDCl3) δ−72.2.
:3, 1.0 mL min−1); [α]20D +33.0 (c 0.5, CHCl3). H26Y-TK gave no reaction. D469T-TK gave 3i (5 mg, 3%) in 99% ee (1S-isomer) and D469K-TK gave 3i (5 mg, 3%) in 25% ee (1S-isomer). The absolute stereochemistry with D469E-TK was determined using the Mosher’s derivatisation method.13
:1) to afford the Mosher's derivative as a colourless oil (0.018 g, 78%). Rf 0.45 (hexane–EtOAc; 4:1); [α]20D−26.0 (c 0.2, CHCl3); 1H NMR (300 MHz; CDCl3) δ 7.63 (2H, m, Ph), 7.43 (3H, m, Ph), 5.19 (1H, d, J 17.0, CHHO (2S,3′S)), 5.07 (d, J 17.0, CHHO (2S,3′R-trace)), 5.03 (d, J 17.0, CHHO (2S,3′R-trace)), 4.90 (1H, d, J 17.0, CHHO (2S,3′S)), 4.16 (1H, dd, J 5.2, 3.3, CHOH), 3.65 (3H, s, OCH3), 2.80 (1H, d, J 5.2, OH), 1.11–1.76 (11H, m); 13C NMR (125 MHz; CDCl3) δ 204.2 (C-2′), 166.2 (CO ester), 131.9, 129.9, 128.5, 127.5, 79.7 (CHOH), 67.8 (CH2OH), 55.8 (OCH3), 41.9, 29.6, 26.4, 25.9 (signal overlap), 25.5; 19F NMR (282 MHz; CDCl3) δ−72.2; m/z (FTMS) found [M + NH4]+ 406.1831. C19H27F3O5N requires 406.1836.
Acknowledgements
CONACYT is thanked for a PhD studentship to A.C. and L.R.S. The UK Engineering and Physical Sciences Research Council (EPSRC) are thanked for DTA studentships to J.L.G. and L.G.C, and for support of the multidisciplinary Bioconversion Integrated with Chemistry and Engineering (BiCE) programme (GR/S62505/01). Financial support from the 12 industrial partners supporting the BiCE programme is also acknowledged. J. S. was supported on a Biotechnology and Biological Sciences Research Council (BBSRC) studentship (BBS/S/A/2004/10920). We thank the EPSRC National Mass Spectrometry Service Centre, Swansea University, for the provision of some high resolution MS data.Notes and references
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258 CrossRef CAS.
- E. Racker, in The Enzymes, ed. P. D. Boyer, H. Lardy and K. Myrzback, Academic Press, New York, 1961, Vol. 5 pp. 397 Search PubMed.
- P. Srere, J. R. Cooper, M. Tabachnick and E. Racker, Arch. Biochem. Biophys., 1958, 74, 295 CAS.
- (a) J. Bolte, C. Demuynck and H. Samaki, Tetrahedron Lett., 1987, 28, 5525 CrossRef CAS; (b) F. Effenberger, V. Null and T. Ziegler, Tetrahedron Lett., 1992, 33, 5157 CrossRef CAS; (c) L. Hecquet, J. Bolte and C. Demuynck, Tetrahedron, 1994, 50, 8677 CrossRef CAS; (d) Y. Kobori, D. C. Myles and G. M. Whitesides, J. Org. Chem., 1992, 57, 5899 CrossRef CAS; (e) R. K. Mitra, J. M. Woodley and M. D. Lilly, Enzyme Microb. Technol., 1998, 22, 64 CrossRef CAS.
- (a) C. Demuynck, J. Bolte, L. Hecquet and V. Dalmas, Tetrahedron Lett., 1991, 32, 5085 CrossRef; (b) G. R. Hobbs, M. D. Lilly, N. J. Turner, J. M. Ward, A. J. Willets and J. M. Woodley, J. Chem. Soc., Perkin Trans. 1, 1993, 165 RSC; (c) K. G. Morris, M. E. B. Smith, N. J. Turner, M. D. Lilly, R. K. Mitra and J. M. Woodley, Tetrahedron: Asymmetry, 1996, 7, 2185 CrossRef CAS.
- (a) N. J. Turner, Curr. Opin. Biotechnol., 2000, 11, 527 CrossRef CAS; (b) J. Bongs, D. Hahn, U. Schörken, G. A. Sprenger and C. Wandrey, Biotechnol. Lett., 1997, 19, 213 CrossRef CAS; (c) J. Shaeri, R. Wohlgemuth and J. M. Woodley, Org. Process Res. Dev., 2006, 10, 605 Search PubMed; (d) J. Shaeri, I. Wright, E. B. Rathbone, R. Wohlgemuth and J. M. Woodley, Biotechnol. Bioeng., 2008, 101, 761 CrossRef CAS.
- M. D. Lilly, R. Chauhan, C. French, M. Gyamerah, G. R. Hobbs, A. Humphrey, M. Isupov, J. A. Littlechild, R. K. Mitra, K. G. Morris, M. Rupprecht, N. J. Turner, J. M. Ward, A. J. Willetts and J. M. Woodley, Recombinant Dna Biotechnology Iii: the Integration of Biological and Engineering Sciences, 1996, 782, 513 Search PubMed.
- G. A. Sprenger and M. Pohl, J. Mol. Catal. B: Enzym., 1999, 6, 145 CrossRef CAS.
- D. C. Myles, P. J. Andrulis, III and G. M. Whitesides, Tetrahedron Lett., 1991, 32, 4835 CrossRef CAS.
- (a) C. U. Ingram, M. Bommer, M. E. B. Smith, P. A. Dalby, J. M. Ward, H. C. Hailes and G. J. Lye, Biotechnol. Bioeng., 2007, 96, 559 CrossRef CAS; (b) U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628 CrossRef CAS; (c) K. Smithies, M. E. B. Smith, U. Kaulmann, J. L. Galman, Ward and H. C. Hailes, Tetrahedron: Asymmetry, 2009, 20, 570 CrossRef CAS.
- (a) E. G. Hibbert, T. Senussi, S. J. Costelloe, W. Lei, M. E. B. Smith, J. M. Ward, H. C. Hailes and P. A. Dalby, J. Biotechnol., 2007, 131, 425 CrossRef CAS; (b) E. G. Hibbert, T. Senussi, M. E. B. Smith, S. J. Costelloe, J. M. Ward, H. C. Hailes and P. A. Dalby, J. Biotechnol., 2008, 134, 240 CrossRef CAS.
- M. E. B. Smith, E. G. Hibbert, A. B. Jones, P. A. Dalby and H. C. Hailes, Adv. Synth. Catal., 2008, 350, 2631 CrossRef CAS.
- J. Galman and H. C. Hailes, Tetrahedron: Asymmetry, 2009, 20, 1828 CrossRef CAS.
- U. Schörken, H. Sahm and G. A. Sprenger, in Biochemistry and Physiology of Thiamine Diphosphate Enzymes, ed. H. Bisswanger and A. Schellenberger, Intemann, Prien, 1996, ch. 6, pp 543–554 Search PubMed.
- U. Nilsson, L. Meshalkina, Y. Lindqvist and G. Schneider, J. Biol. Chem., 1997, 272, 1864 CrossRef CAS.
- M. E. B. Smith, K. Smithies, T. Senussi, P. A. Dalby and H. C. Hailes, Eur. J. Org. Chem., 2006, 1121 CrossRef CAS.
- M. E. B. Smith, B. H. Chen, E. G. Hibbert, U. Kaulmann, K. Smithies, J. L. Galman, F. Baganz, P. A. Dalby, H. C. Hailes, G. J. Lye, J. M. Ward, J. M. Woodley and M. Micheletti, Org. Process Res. Dev., 2010, 14, 99 Search PubMed.
- J. Littlechild, N. J. Turner, G. R. Hobbs and M. D. Lilly, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1995, 51, 1074 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639 CrossRef CAS.
- P. Asztalos, C. Parthier, R. Golbik, M. Kleinschmidt, G. Hübner, M. S. Weiss, R. Friedemann, G. Wille and K. Tittmann, Biochemistry, 2007, 46, 12037 CrossRef CAS.
- M. E. B. Smith, K. Smithies, U. Kaulmann, J. M. Ward and H. C. Hailes, Bioorg. Med. Chem., 2006, 14, 7062 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- E. Fiedler, S. Thorell, T. Sandalova, R. Golbik, S. Konig and G. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 591 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Colorimetric assay plates for the cyclic aldehydes. See DOI: 10.1039/b924144b |
| This journal is © The Royal Society of Chemistry 2010 |