Synthesis and biological applications of collagen-model triple-helical peptides
Gregg B.
Fields
*
University of Texas Health Science Center, Department of Biochemistry, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA. E-mail: fieldsg@uthscsa.edu
First published on 20th January 2010
Abstract
Triple-helical peptides (THPs) have been utilized as collagen models since the 1960s. The original focus for THP-based research was to unravel the structural determinants of collagen. In the last two decades, virtually all aspects of collagen structural biochemistry have been explored with THP models. More specifically, secondary amino acid analogs have been incorporated into THPs to more fully understand the forces that stabilize triple-helical structure. Heterotrimeric THPs have been utilized to better appreciate the contributions of chain sequence diversity on collagen function. The role of collagen as a cell signaling protein has been dissected using THPs that represent ligands for specific receptors. The mechanisms of collagenolysis have been investigated using THP substrates and inhibitors. Finally, THPs have been developed for biomaterial applications. These aspects of THP-based research are overviewed herein.
 Gregg B. Fields | Dr Gregg B. Fields received his B.S. and Ph.D. degrees in chemistry from the University of Florida and Florida State University, respectively. He was a Postdoctoral Scholar with Ken A. Dill at the University of California San Francisco. Dr Fields joined the faculty at the University of Minnesota in 1991 as an assistant professor, was promoted to associate professor in 1995 and then achieved the rank of full professor of chemistry & biochemistry at Florida Atlantic University in 1997. In 2008, Dr Fields became a Robert A. Welch Foundation Distinguished University Chair in Chemistry in the Department of Biochemistry at The University of Texas Health Science Center at San Antonio. Dr Fields' research interests are in the use of chemical approaches to better understand how protein three-dimensional structures influence cellular and enzymatic behaviors. |
1 Introduction
The collagen family is a diverse group of proteins made up of at least 28 members.1-5 Collagens are composed of three α chains of primarily repeating Gly-Xxx-Yyy triplets, which induce each α chain to adopt a left-handed polyPro II helix. Three left handed chains then intertwine to form a right-handed superhelix. The collagen triple-helix (Fig. 1) is important for the integrity and workings of multiple connective tissues, including skin, bone, cartilage, tendon and dentin. Most collagens assist in anchoring cells to the extracellular matrix and some function in cellular regulation.![“Ball and stick” computer generated models of (top) a continuous collagen triple-helix (peptide T3-785, 3[(Pro-Hyp-Gly)3-Ile-Thr-Gly-Ala-Arg-Gly-Leu-Ala-Gly-(Pro-Hyp-Gly)4]325) and (bottom) an unwound (heat denatured) version of the same sequence.](/image/article/2010/OB/b920670a/b920670a-f1.gif) | ||
| Fig. 1 “Ball and stick” computer generated models of (top) a continuous collagen triple-helix (peptide T3-785, 3[(Pro-Hyp-Gly)3-Ile-Thr-Gly-Ala-Arg-Gly-Leu-Ala-Gly-(Pro-Hyp-Gly)4]325) and (bottom) an unwound (heat denatured) version of the same sequence. | ||
Collagens have been classified according to their α chains.4 Homotrimeric collagens (i.e., types II and III) have three α chains of identical sequence. Heterotrimeric collagens have either two α chains of identical sequence (designated α1) and one α chain of differing sequence (designated α2), such as type I, or three α chains of differing sequence (designated α1, α2, and α3), such as type VI.6 Collagens are further classified into subfamilies, based on their quaternary structure. These subfamilies include fibrillar, fibril associated with interrupted triple-helices (FACIT), short chain, basement membrane, multiplexins, and membrane associated with interrupted triple-helices (MACIT).6 The most common collagens (types I, II, III, V, and XI) have fibrillar structures.
The triple-helical motif is also found in a variety of non-collagenous proteins, such as macrophage scavenger receptors types I and II and bacteria-binding receptor MARCO, complement component C1q, pulmonary surfactant apoproteins A and D, acetylcholinesterase, bovine conglutinin, collectin-43, ficolins, aggretin, ectodysplasm, and mannose binding protein.1,7,8
To fully investigate the structural and biological roles of collagen and collagen-like proteins, triple-helical peptides (THPs) or “mini-collagens” incorporating collagen-model sequences and three-dimensional structure have been constructed. The synthesis and application of triple-helical, collagen-model peptides was comprehensively reviewed in 1995-1996.7,9 Subsequent reviews have focused on collagen biochemistry captured via THP constructs.5,10-13 Thus, this perspective will primarily emphasize synthetic research conducted in the last decade and recent biological studies. Many of the details pertaining to THP construction and application are included herein. The topics covered include methodologies for assembling THPs (associated triple-helical peptides, templated triple-helical peptides, and triple-helical peptides of higher order structure), structural studies of THPs, biological studies of THPs (adhesion receptor binding, protease substrates and inhibitors, and lipoprotein-, glycosaminoglycan-, nucleic acid-, and other protein-collagen interactions), and miscellaneous applications of THPs.
2 Associated triple-helical peptides
It has long been noted that peptides containing repeating sequences of Gly-Pro-Pro or Gly-Pro-4R-Hyp [where 4R-Hyp is (2S,4R)-4-hydroxyproline] were capable of self-association in aqueous solution to form stable collagen-like triple-helical structures.9 The assembly of such sequences is now almost entirely based on stepwise solid-phase methodology using tert-butyloxycarbonyl (Boc) or 9-fluorenylmethoxycarbonyl (Fmoc) chemistries.14-20 To obtain THPs of reasonable stability, the self-association approach typically “sandwiches” a collagen-model sequence between repeats of Gly-Pro-Hyp (Fig. 2). Self-associated triple-helical peptides have often been used to study the structural aspects of collagen, where the peptide contains either a repeating tripeptide sequence or a “host-guest” sequence such as (Pro-Hyp-Gly)n-Xxx-Yyy-Gly-(Pro-Hyp-Gly)n.21-23![Modular structures of (top) sandwiched associated THP and (bottom) sandwiched associated triple-helical peptide-amphiphile. The associated THP features repeats of Gly-Pro-Hyp [(GPO)n] on both the N- and C-termini to induce or stabilize triple-helical structure, and a diverse collagen-like sequence [(GXY)n] in the middle for structural and/or biological studies. The peptide-amphiphile additionally possesses a pseudo-lipid attached to the N-terminus to further enhance triple-helical stability via hydrophobic interactions.](/image/article/2010/OB/b920670a/b920670a-f2.gif) | ||
| Fig. 2 Modular structures of (top) sandwiched associated THP and (bottom) sandwiched associated triple-helical peptide-amphiphile. The associated THP features repeats of Gly-Pro-Hyp [(GPO)n] on both the N- and C-termini to induce or stabilize triple-helical structure, and a diverse collagen-like sequence [(GXY)n] in the middle for structural and/or biological studies. The peptide-amphiphile additionally possesses a pseudo-lipid attached to the N-terminus to further enhance triple-helical stability via hydrophobic interactions. | ||
Although the majority of self-associated THPs have been homotrimers, a few examples of self-associated heterotrimers have been reported. In most cases, the heterotrimer was not isolated in pure form, but rather studied structurally as a mixture. Highly stable heterotrimers have been assembled from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 of (Asp-Hyp-Gly)10, (Pro-Lys-Gly)10, and (Pro-Hyp-Gly)10 (Tm = 65.0 °C) or (Asp-Hyp-Gly)10, (Pro-Arg-Gly)10, and (Pro-Hyp-Gly)10 (Tm = 54.0 °C).24,25 The stability of the former heterotrimer was comparable to (Pro-Hyp-Gly)10 homotrimer (Tm = 67.5 °C).25 NMR structural analysis revealed that the Asp and Lys side-chains formed a network of ionic hydrogen bonds, most likely accounting for the heterotrimer stability.26 A stable heterotrimer (Tm = 28 °C) was also obtained from a 1:2 mixture of (Pro-Pro-Gly)7 and (4S-Flp-4R-Flp-Gly)7 [where 4S-Flp is (2S,4S)-4-fluoroproline and 4R-Flp is (2S,4R)-4-fluoroproline].27 A mixture of (Pro-Hyp-Gly)10 and (Pro-Pro-Gly)10 (either 1:2 or 2:1) formed stable heterotrimers, with Tm dependent upon the scanning speed.28,29 A heterotrimer composed of a 2:1 mixture of (Gly-Pro-Hyp)3-Gly-Met-Hyp-Gly-Val-Gly-Glu-Lys-Gly-Glu-Hyp-Gly-Lys-Hyp-(Gly-Pro-Hyp)2-Gly-Tyr and Pro-Hyp-Gly-Asp-Hyp-(Gly-Pro-Hyp)2-Gly-Ile-Ser-Leu-Lys-Gly-Glu-Glu-Gly-Pro-Hyp-Gly-Pro-Ala-Gly-Pro-Hyp-Gly-Tyr-Hyp-Gly was found to have a Tm value of 14.5 °C.30 Additional heterotrimers have been created via 1:2 mixtures of diverse sequences.24
:1:1 of (Asp-Hyp-Gly)10, (Pro-Lys-Gly)10, and (Pro-Hyp-Gly)10 (Tm = 65.0 °C) or (Asp-Hyp-Gly)10, (Pro-Arg-Gly)10, and (Pro-Hyp-Gly)10 (Tm = 54.0 °C).24,25 The stability of the former heterotrimer was comparable to (Pro-Hyp-Gly)10 homotrimer (Tm = 67.5 °C).25 NMR structural analysis revealed that the Asp and Lys side-chains formed a network of ionic hydrogen bonds, most likely accounting for the heterotrimer stability.26 A stable heterotrimer (Tm = 28 °C) was also obtained from a 1:2 mixture of (Pro-Pro-Gly)7 and (4S-Flp-4R-Flp-Gly)7 [where 4S-Flp is (2S,4S)-4-fluoroproline and 4R-Flp is (2S,4R)-4-fluoroproline].27 A mixture of (Pro-Hyp-Gly)10 and (Pro-Pro-Gly)10 (either 1:2 or 2:1) formed stable heterotrimers, with Tm dependent upon the scanning speed.28,29 A heterotrimer composed of a 2:1 mixture of (Gly-Pro-Hyp)3-Gly-Met-Hyp-Gly-Val-Gly-Glu-Lys-Gly-Glu-Hyp-Gly-Lys-Hyp-(Gly-Pro-Hyp)2-Gly-Tyr and Pro-Hyp-Gly-Asp-Hyp-(Gly-Pro-Hyp)2-Gly-Ile-Ser-Leu-Lys-Gly-Glu-Glu-Gly-Pro-Hyp-Gly-Pro-Ala-Gly-Pro-Hyp-Gly-Tyr-Hyp-Gly was found to have a Tm value of 14.5 °C.30 Additional heterotrimers have been created via 1:2 mixtures of diverse sequences.24
Gly-Pro-Hyp repeats may not enhance triple-helical sequences with sufficient thermal stability for biological studies. Self-associated peptides have been further stabilized by the addition of lipophilic molecules at the N-terminus of the peptide. These “peptide-amphiphiles” (PAs) have incorporated an amino acid sequence with the propensity to form a triple-helix as the polar head group and a dialkyl or monoalkyl hydrocarbon chain as the non-polar tail (Fig. 2).14,15,31,32 Desirable peptide head group Tm values can be achieved for in vivo use, as triple-helical PAs have been constructed with Tm ranging from 30 to 70 °C.14,15,18,33-41
The stability of associated THPs has also been modulated by pH or photolysis. To create a triple-helix that was pH dependent, Hyp was modified by addition of a carboxylate by O-alkylation.42 Interestingly, the incorporation of 1 or 3 Hyp(CO2) residues within a Pro-Hyp-Gly template did not result in a pH-sensitive triple-helix. However, acetyl-[Pro-Hyp(CO2)-Gly]7-OH formed a triple-helix with a Tm = 17 °C at pH 2.7 but had no triple-helical structure at pH 7.2.42 pH-dependent triple-helical stability was also observed for (Pro-4R-Amp-Gly)6 sequences, where 4R-Amp is (2S,4R)-4-aminoproline.43,44 Modification of the sequence (Gly-Pro-Hyp)3-Gly-Cys-Hyp-Gly-Pro-Hyp-Gly-Pro-Cys-(Gly-Pro-Hyp)5-Gly-Gly-NH2 with an azobenzene bridge between the Cys residues resulted in a THP whose stability decreased up irradiation at λ = 330 nm at 27 °C.45 Unfortunately, the poor solubility of this peptide prohibited quantitative analysis of its stability.45
In all of the above cases, fairly standard solid-phase synthetic conditions have been utilized for THP assembly. Notable modifications include extended coupling times for Hyp and Pro derivatives and extended Fmoc deprotection times and/or the use of diaza(1,3)bicyclo[5.4.0]undecane (DBU) for deprotection [2% DBU plus 2% piperidine (to scavenge dibenzofulvene)].46-48 Although the use of Hyp without side-chain protection has been described,16,47,49 the commercial availability of Boc-Hyp(Bzl) and Fmoc-Hyp(tBu) and subsequent lack of potential interchain hydrogen-bonding and side-chain esterification during synthesis has lead to the predominant use of these derivatives.17,20,49,50 The synthesis and incorporation of 4R-Hyp analogs and derivatives, such as 3-Hyp, 4S-Hyp [(2S,4S)-Hyp; alloHyp; hyp], 4-cis-Hyp [(2R,4R)-Hyp], 4R-Flp, 4S-Flp (flp), 3S-Flp, 4,4-difluoroproline (Dfp), 4-oxoproline, 4R-Clp [(2S,4R)-4-chloroproline], 4S-Clp (clp), and 4R-Amp has been described.27,37,43,51-55 These secondary amino acids have been utilized primarily for studies on triple-helix stability (see below), although in one case Flp was used to examine the effects of triple-helix stability on melanoma cell binding.37 Finally, preparation of side-chain protected or glycosylated Fmoc-Hyl derivatives has been described,56-61 and Hyl(Gal) has been incorporated into a THP.62
3 Templated triple-helical peptides
Numerous approaches have been described by which THPs are nucleated and/or stabilized by covalent attachment to a template. In its simplest form, covalent attachment of three strands stabilizes a triple-helix.11,12,63 To study the nucleation step for the folding of collagen, a protocol was developed in the 1980s for the liquid phase synthesis by which three peptide strands were covalently linked via a C-terminal branch.64,65 The C-terminal branch was expected to align and entropically stabilize the C-terminus of the THP and thus enhance triple-helical thermal stability, and to provide a model of the disulfide-linked C-terminus of type III collagen. Branching was achieved by selective deprotection of Lys Nα- and ε-amino groups. Our laboratory evaluated systematically several general solid-phase methods for synthesizing covalently branched THPs that incorporate native collagen sequences. Branching of three peptide strands from one initial chain required three different protecting group strategies (Fig. 3): Nα-amino protection (A), Lys Nε-amino side-chain protection (B), which must be stable to the Nα-amino group removal conditions, and Cα-carboxyl protection (linker), which must be stable to both the Nα- and Nε-amino protecting group removal conditions. Four different synthetic schemes were employed, with the only common protecting group strategy being Fmoc for A.46,47,49 The combinations were: (a) B the Boc group, C the 2,6-dichlorobenzyl (Dcb) group, and the 4-hydroxymethylphenylacetic acid (PAM) linker; (b) B the Boc group, C the allyl group, and allyl-based 4-trityloxy-Z-but-2-enyloxyacetic acid linker; (c) B the allyloxycarbonyl (Aloc), C the tBu group, and the 4-hydroxymethylphenoxy (HMP) linker; and (d) B the 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl (Dde) group, C the tBu group, and 2-methoxy-4-alkoxybenzyl alcohol (SASRIN) linker. Tyr was incorporated prior to branching to provide a convenient chromophore for eventual concentration determination. In the case of (a), branching was achieved by synthesizing Fmoc-[Lys(Boc)]2-Tyr(Dcb)-Gly-PAM resin and deprotecting the Nα- and Nε-amino groups with TFA. The peptide-resin was cleaved with TFMSA. In the case of (b), branching was achieved by synthesizing Fmoc-[Lys(Boc)]2-Tyr(Al)-Gly-allyl resin and deprotecting the Nα- and Nε-amino groups with TFA. The THP was side-chain deprotected with TFA while still resin-bound, and cleaved with (Ph3P)4Pd. In the case of (c), both Aloc groups of [Lys(Aloc)]2-Tyr(tBu)-Gly-HMP resin were removed with (Ph3P)4Pd. In the case of (d), both Dde groups of [Lys(Dde)]2-Tyr(tBu)-SASRIN resin were removed with hydrazine. For all THPs, Ahx was incorporated onto all three amino termini to provide a flexible spacer.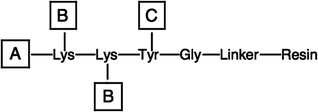 | ||
| Fig. 3 Template for C-terminal branching strategy. | ||
The four strategies indicated that C-terminal templates can be incorporated into THPs by a variety of approaches. Due to the mild conditions, we currently favor the method used to assemble the Fmoc-[Lys(Dde)]2-Tyr(tBu)-SASRIN branching template. We have subsequently improved yields by switching from the Dde group to the ivDde group, which has greater stability to the conditions of Fmoc chemistry.66 A total of 28 different branched THPs have been reported by our laboratory.34-36,46-48,50,62,67-70 The solid-phase branching strategy has also been used by other groups, based on Fmoc/Lys(Dde).71-74
Numerous other templated approaches have been described, including the introduction of a di-Lys or di-Glu template at the C- or N-terminal regions of the three peptide chains,75-77 a double disulfide “knot” at the C-terminal region of the three peptide strands,78,79 or a cis,cis-1,3,5-trimethylcyclohexane-1,3,5-tricarboxylic acid (Kemp triacid; KTA),80-83 cyclotriveratrylene,84 tris(2-aminoethyl)amine (TREN),85 macrocyclic,86 or Boc-β-Ala-tris(carboxyethoxymethyl)aminomethane [Boc-β-Ala-TRIS-(OH)3]87 template linked to the N-terminus of three peptide chains (Fig. 4).
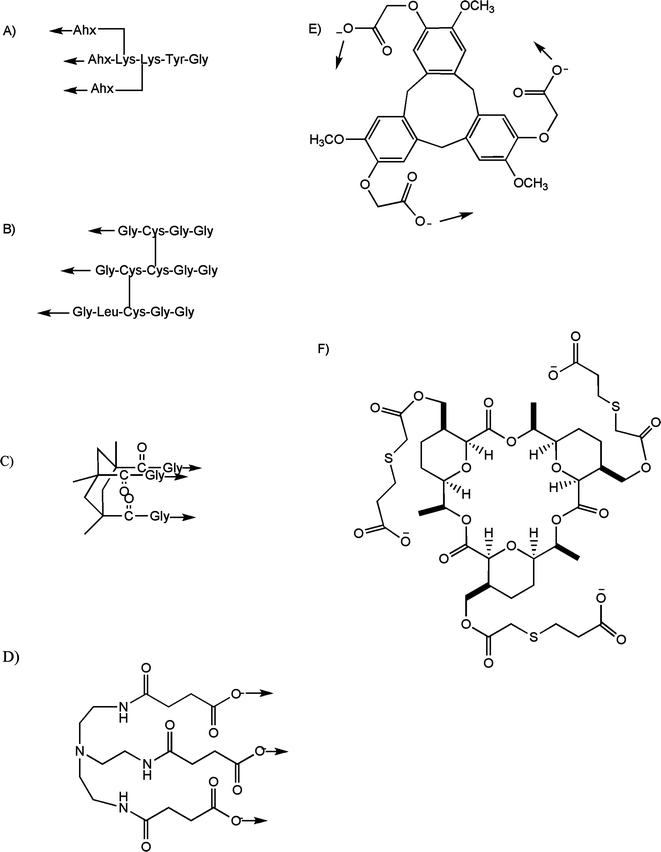 | ||
| Fig. 4 Templates used for the chemical synthesis of triple-helical peptides: (A) di-Lys branch after coupling to 6-aminohexanoic acid (Ahx); (B) disulfide bridge (cystine-knot); (C) cis,cis-1,3,5-trimethylcyclohexane-1,3,5-tricarboxylic acid (KTA) after coupling to Gly; (D) tris(2-aminoethyl)amine (TREN) after coupling to succinic acid; (E) cyclotriveratrylene (CTV) after coupling to α-bromoethanoic acid; and (F) macrocyclic. The arrows indicate the direction of collagen-like sequence incorporation. | ||
Tanaka et al.88 utilized chemoselective ligation to create a triple-helical di-Lys branched peptide in aqueous solution. Collagen-like sequences were synthesized with an N-terminal Cys residue, while the branch structure had three bromoacetic acid molecules coupled to the spacer.88 The individual chains were then ligated to the branch in NaHCO3/Gdn·HCl solution, forming thioether bonds.
Assembly of THPs using a Glu-Glu N-terminal branch required two peptide types, one containing a Glu α-thioester at its N-terminus and one without the Glu.75 Crosslinking was achieved by silver ion activation of the Glu thioester group, followed by nucleophilic attack of the N-terminal amino group from the other peptide. The process was repeated using the Glu α-thioester peptide to create a trimeric structure. Minimally protected peptides (restricted to amino group protection) allowed for high solubility and reactivity during crosslinking. The more simplified N-terminal di-Glu template Gly-Phe-Gly-Glu-Glu-Gly was assembled by solid-phase Fmoc methodology, isolated as Fmoc-Gly-Phe-Gly-Glu-Glu-Gly, and acylated to three peptide strands simultaneously on the solid-phase using 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU)/1-hydroxybenzotriazole (HOBt).77
The synthesis of an N- and C-terminal cross-linked THP has been described.76 Boc-Ser(tBu)-OSu was reacted with Fmoc-Lys-OH to obtain Fmoc-Lys[Boc-Ser(tBu)]-OH. Collagenous linear peptides of the sequence Cys-Gly-(Gly-Pro-Hyp)n-Lys(Ser)-NH2 (n = 3-10) (1) were then synthesized using Fmoc chemistry.76 The trivalent linker, tris-bromoacetylated Lys-Lys dimer 2, was prepared on MBHA resin. The tris-aminooxyacetylated branch-peptide 3 was synthesized by a similar method using Boc-aminooxyacetic acid instead of bromoacetic acid. Peptide 1 and the bromoacetyl peptide 2 were reacted to yield (Gly-Pro-Hyp)n[N] (4).76 Sodium periodate (3 equiv per Ser residue) was added to a solution of peptide 4 to convert the Ser residues into aldehydes, producing peptide 5. Peptide 5 and peptide 3 were dissolved in sodium acetate buffer, and after 3 h, the product (Gly-Pro-Hyp)n[NC] (6) was obtained.76
Extensive studies have been performed by Moroder and colleagues for creating C-terminally branched heterotrimeric THPs using disulfide bonding (cystine knots).3,17,78,89-91 In one representative example, the following peptide chains were constructed by solid-phase methodology: α1, which contained either 3 or 5 Gly-Pro-Hyp repeats on the N-terminus of Gly-Pro-Gln-Gly-Ile-Ala-Gly-Gln-Arg-Gly-Val-Val-Gly-Cys(Acm)-Gly-Gly-OH, where Acm is acetamidomethyl; α2, which contained 3 or 5 Gly-Pro-Hyp repeats on the N-terminus of Gly-Pro-Gln-Gly-Leu-Leu-Gly-Ala-Hyp-Gly-Ile-Leu-Gly-Cys(Acm)-Cys(StBu)-Gly-Gly-OH; and α1′, which contained either 3 or 5 Gly-Pro-Hyp repeats on the N-terminus of Gly-Pro-Gln-Gly-Ile-Ala-Gly-Gln-Arg-Gly-Val-Val-Gly-Leu-Cys(StBu)-Gly-Gly-OH. All three peptides were synthesized using Fmoc chemistry. Couplings were performed with HBTU/HOBt except for Cys residues, which were coupled as pentafluorophenyl esters to minimize racemization. The three chains were assembled into the heterotrimer by stepwise regioselective crosslinking. Peptide α1 was treated with 3-nitro-2-pyridine-sulfenyl chloride (Npys-Cl) to convert Cys(Acm) to Cys(Npys). Peptide α2 was treated with P(C4H9)3 to remove the StBu group. Peptides α1 and α2 were reacted at pH 4.5 to form a dimer. The dimer was treated with Npys-Cl to convert the α2 chain Cys(Acm) to Cys(Npys). The α1′ chain was treated with P(C4H9)3 to remove the StBu group. Peptide α1′ and the dimer were reacted at pH 4.5 to form a trimer. The heterotrimer containing 3 Gly-Pro-Hyp repeats was obtained in 72% yield over all crosslinking reactions.78,89 A similar strategy was utilized to construct heterotrimeric THPs possessing N-terminal cystine knots.92
A more simplified, oxidative approach has been utilized for the assembly of cystine knot homotrimeric THPs.93,94 Oxidation with aldrithiol has been utilized for the assembly of heterotrimeric THPs possessing cystine knots, with isolation of the desired heterotrimer achieved by size-exclusion chromatography in combination with RP-HPLC, MS, and NMR spectroscopic analyses.95
Two synthetic routes have been used to prepare KTA template-assembled collagen-based structures. The first method utilized solid-phase methodology exclusively. Boc-(Gly-Pro-Hyp)n-MBHA resin was prepared and the Boc group removed. KTA-(Gly-OH)3 was coupled using DIPCDI and HOBt for 3 d.96 Structures containing 3 or 6 Gly-Pro-Nleu repeats (where Nleu is N-isobutylglycine) or (Gly-Nleu-Pro)n were also synthesized using this approach.82,83 The KTA-(Gly-OH)3 coupling was allowed to proceed for 1-2 d.
The second method to connect the KTA template and peptide-peptoid chains involved peptide bond formation in solution. Peptide-peptoid chains were assembled by solid-phase methods and cleaved from the resin as N-terminal free amine and C-terminal amidated forms. The KTA-(Gly-OH)3 template and free-amine peptide-peptoid chains were then coupled in solution using EDC and HOBt.82
The N-terminal TREN template was prepared by coupling tris(2-aminoethyl)amine to monobenzylated succinate, and removing the benzyl ester groups by hydrogenation.85 TREN-(suc-OH)3 is then acylated to three peptide strands simultaneously on the solid-phase.85 The macrocyclic template was coupled to the N-termini of collagen-model peptides in solution using PyBOP and DIEA.86 In similar fashion, the cyclotriveratrylene template was coupled in solution using BOP.84 Boc-β-Ala-TRIS-(OH)3 was coupled to peptides in solution using 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT).87
The effect of templates is often to enhance the thermal stability of collagen-like sequences. For example, the THP acetyl-Gly-Gly-(Pro-Hyp-Gly)5-NH2 has a Tm value of 9.2 °C, while templated versions of the same sequence, using either (+)CTV or KTA, had Tm values of 58 and 62 °C, respectively.84 Similarly, (Gly-Pro-Hyp)6 has a Tm = 25.4 °C, (Pro-Hyp-Gly)6 with a C-terminal branch has a Tm = 39.4 °C, (Gly-Pro-Hyp)6 with an N-terminal branch has a Tm = 56.2 °C, and (Gly-Pro-Hyp)6 with both an N- and C-terminal branch has a Tm = 69.7 °C.73,76 An N-terminal macrocyclic template induced triple-helical structure into the sequence Gly-(Pro-Pro-Gly)7-NH2 with a Tm = 39.9 °C, whereas the non-templated sequence was not triple-helical.86 Triple-helices containing peptoid residues, such as N-isobutylglycine (Nleu), have been stabilized by templates. Acetyl-(Gly-Nleu-Pro)6-NH2 has a Tm = 26 °C, while KTA-[Gly-(Gly-Nleu-Pro)6-NH2]3, TREN-[suc-(Gly-Nleu-Pro)6-NH2]3, and Boc-β-Ala-TRIS-[(Gly-Nleu-Pro)6-OCH3]3 had Tm values of 36, 46, and 33 °C, respectively.83,85,87 The thermal stability of the α2β1 integrin binding sequence (Gly-Pro-Hyp)3-Gly-Phe-Hyp-Gly-Glu-Arg-(Gly-Pro-Hyp)3 was enhanced by an N-terminal Gly-Phe-Gly-Glu-Glu-Gly template, resulting in an increase in Tm from 25 to 44 °C.77 Acetyl-(Pro-Hyp-Gly)5-Pro-Cys(StBu)-Cys(StBu)-Gly-Gly-Gly-NH2 has a Tm = 20.3 °C, while the double disulfide linked [acetyl-(Pro-Hyp-Gly)5-Pro-Cys-Cys-Gly-Gly-Gly-NH2]3 has a Tm = 68.1 °C.93 Formation of the cystine knot in (Gly-Pro-Pro)3-Gly-Pro-Arg-Gly-Glu-Lys-Gly-Glu-Arg-Gly-Pro-Arg-(Gly-Pro-Pro)3-Gly-Pro-Cys-Cys-Gly increases Tm from 35 to 43 °C.94
“Templated” triple-helices have also been constructed using metals. The addition of dopamine on the N-terminus of (Gly-Nleu-Pro)6, followed by incubation with Fe3+, resulted in a non-triple-helical peptide becoming triple-helical with a Tm value of 28 °C.97 Addition of 2,3-dihydroxybenzoic acid on the C-terminus of Boc-β-Ala-TRIS-[(Gly-Nleu-Pro)6]3, followed by incubation with Fe3+, resulted in a stabilized triple-helical structure (22 °C increase in Tm).97 In similar fashion, addition of hydroxamic acid on the C-terminus of Boc-β-Ala-TRIS-[(Gly-Pro-Nleu)6]3, followed by incubation with Fe3+, resulted in a stabilized triple-helical structure (7 °C increase in Tm).98
4 Triple-helical peptides of higher order structure
Collagen triple-helices are well known to interact and form higher order structures, such as fibrils. THPs have been found to form higher order structures based the presence of secondary amino acids and an appropriate distribution of hydrophobic and elżectrostatic residues.99 Consequently, a number of approaches have been described by which triple-helical peptides are directed to form higher order structures. Longer THPs have been constructed using THP self-assembly driven by hydrophobic interactions at the N- and C-termini, for either pentafluoroPhe-(Gly-Pro-Hyp)10-Phe or Phe-(Gly-Pro-Hyp)3-Gly-Gln-Hyp-Gly-Leu-Hyp-Gly-Leu-Hyp-(Gly-Pro-Hyp)4-Gly-Tyr.100-102 The Phe-pentafluoroPhe stacking interaction resulted in THP aggregates of 260 nm in diameter which were fibril-like in nature.100 Micron-length fibers of ∼70 nm in diameter have been produced following thermal annealing of (Pro-Arg-Gly)4-(Pro-Hyp-Gly)4-(Glu-Hyp-Gly)4.103 In similar fashion, electrostatic interactions of the cystine knot THP (Gly-Pro-Pro)3-Gly-Pro-Arg-Gly-Glu-Lys-Gly-Glu-Arg-Gly-Pro-Arg-(Gly-Pro-Pro)3-Gly-Pro-Cys-Cys-Gly resulted in the formation of nanorods and microfibrils of 6 μm in length and 130 nm in width.94 Metal coordination has been utilized to form microflorettes from THPs.104 The (Pro-Hyp-Gly)9 was modified by the addition of His2 at the C-terminus and nitriloacetic acid (NTA) at the N-terminus. Introduction of Zn2+, Cu2+, Ni2+, or Co2+ resulted in the formation of higher order structures; in the case of Zn2+, microflorettes were observed with diameters >5 μm.104To study platelet aggregation, THPs have been crosslinked via N- and/or C-terminal Cys or Lys residues to obtain higher order structures.19,71,74,105-114 Cys crosslinking has been achieved via 3-(2-pyridyldithio)-propionic acid N-hydroxysuccinimide ester,19,74,106,108-111,113,114 while Lys crosslinking has utilized glutaraldehyde or disuccinimidyl glutarate.19,106,108
Polymerization of THPs via native chemical ligation has created nanofibers of 10-20 nm.115 In similar fashion, disulfide-linked trimeric peptides possessing self-complementary sequences have been designed and produced fibrils of >400 nm in length or supramolecules of up to 14 μm in diameter.116,117 Self-complementary sequences have also been utilized to produce collagen-like gels from THPs.118 The most straightforward construction of higher order structures was achieved by direct polycondensation of (Pro-Hyp-Gly)n with 1-ethyl-3-(3-dimethyl-aminopropyl)-carbodiimide and HOBt.119 Final molecular weights of over 10 kDa were obtained for polycondensation of (Pro-Hyp-Gly)5 and (Pro-Hyp-Gly)10 in DMSO for 48 h at 20 °C, and similar results were obtained in phosphate buffer (pH = 7.4) using 50 mg ml−1 Pro-Hyp-Gly.119 The poly(Pro-Hyp-Gly)10 aggregates formed nanofiber-like structures of 10 nm in diameter.119
Radial growth of THPs has been achieved by metal-triggered assembly. Hyp was replaced with a bipyridyl-modified Lys (Byp) to create (Pro-Hyp-Gly)4-Pro-Byp-Gly-(Pro-Hyp-Gly)4, and association of THPs promoted by addition of Fe(II).120 Fibers were obtained of 3-5 μm in length and ∼10 nm in width. Combining the internal incorporation of Byp with His2 at the C-terminus and nitriloacetic acid at the N-terminus [NTA-(Pro-Hyp-Gly)4-Pro-Byp-Gly-(Pro-Hyp-Gly)4-His-His-NH2] produced a THP capable of forming a metal-triggered three-dimensional collagen-like network.121
A host-guest approach was used to examine supramolecular assemblies formed by THPs.122 The host peptide was (Glu)5-(Gly-Xxx-Hyp-Gly-Pro-Hyp)6-(Glu)5, and the guest residues were Ala, Pro, Ser, and Val. The authors have found that no supramolecular order occurs when Xxx = Ser, a similar, banded spherulite order was seen for Xxx = Ala or Pro, and a nematic order was observed for Xxx = Val. In addition, the (Glu)5 ends were required for assembly formation. The supramolecular structures corresponded to those observed for type I collagen in vitro.
THP dendrimers have been constructed from N-(benzyloxycarbonyl)-tris(carboxyethoxymethyl)aminomethane (Z-TRIS[OH]3). Z-TRIS[OH]3 was coupled to the N-terminus of THPs in solution, the Z group removed, and the dendrimer formed by addition of trimesoyl chloride.123 THP dendrimers have also been assembled using a poly(amidoamine) dendrimer, with subsequent crosslinking by tissue transglutaminase.124
5 Structural stability studies
The use of THPs for X-ray crystallographic analysis of triple-helix conformation has been reviewed recently,125 and thus will not be covered here. Of particular note is the continued evaluation of the collagen triple-helix as being either of 7/2-helical symmetry, 10/3-helical symmetry, or a combination of both.126-128 As proposed based on structural analysis,129 interstrand hydrogen bonds formed between the GlyN-H and XxxC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O are critical for triple-helix stability, as replacement of a central amide bond of (Pro-Pro-Gly)10 with either an ester or (E)-alkene, or replacement of the central Pro-Pro with a Pro-trans-Pro isostere, substantially destabilizes the THP.130-132
O are critical for triple-helix stability, as replacement of a central amide bond of (Pro-Pro-Gly)10 with either an ester or (E)-alkene, or replacement of the central Pro-Pro with a Pro-trans-Pro isostere, substantially destabilizes the THP.130-132
One of the most intensive areas of THP research is the role of hydrogen bonding versus inductive effects in triple-helix stabilization.5 The non-native amino acid 4R-Flp has been shown to induce hyperstability in the triple-helix of (Pro-4R-Flp-Gly)10 compared to (Pro-4R-Hyp-Gly)10.133,134 Alternatively, 4S-Flp destabilizes the triple-helix of (Pro-4S-Flp-Gly)7 compared to (Pro-4R-Hyp-Gly)7.135 More specifically, substitution of all of the 4R-Hyp residues in (Pro-4R-Hyp-Gly)10 or (Pro-4R-Hyp-Gly)7 by 4R-Flp increased Tm by 22 and 9 °C, respectively.133-135 Conversely, an analogous substitution in (Pro-4R-Hyp-Gly)7 by 4S-Flp dramatically decreased Tm by greater than 26 °C.135 The relative effects of 4R-Hyp, 4R-Flp, and 4S-Flp coincide with their propensity for forming trans-peptide bonds compared to cis-peptide bonds.135,136 A similar correlation has been found using additional Pro and Hyp analogs.137,138 This “inductive effect,” based on the electronegativity and stereochemistry of the 4-substituent in the pyrrolidine ring system, is an important contributor to triple-helix stability.133,134,136,139Ktrans/cis, the pucker of the pyrrolidine ring (exoversusendo), and the phi and psi torsion angles are interdependent parameters.137,140 Consideration of these parameters provides a more complete understanding of the forces that contribute to the stability of the triple-helix.125,141
Thermodynamic analyses of Hyp- and Flp-containing THPs indicated that the enhanced stability of (Pro-Hyp-Gly)10 compared to (Pro-Pro-Gly)10 is enthalpically driven, while the enhanced stability of (Pro-4R-Flp-Gly)10 compared to (Pro-Pro-Gly)10 is entropically driven.142,143 Based on these results, it was proposed that stabilization of triple-helices by Flp and Hyp are mechanistically distinct, with the former driven by hydrophobic effects and the latter by hydrogen bonding. However, studies with Dfp [which has a similar hydrophobicity as 4R-Flp but a Ktrans/cis = 3.6, similar to Pro (4.6) but much lower than 4R-Flp (6.7)] demonstrated similar Tm values for THPs containing Dfp or Pro in the Yyy position, which were 6 °C lower than that observed for the 4R-Flp-containing THP.55 Thus, hydrophobicity is not the dominant driving force in the stability of Flp-containing THPs.
The effects of single site substitution of naturally occurring amino acids on triple-helix stability have been examined previously using a host-guest approach, where either Pro in the Xxx or Hyp in the Yyy position of a Gly-Pro-Hyp triplet is replaced by the “guest” residue.21-23 A comprehensive ranking of triple-helix propensity was established based on homotrimeric host-guest THPs.11,22,23 Of the 20 natural amino acids studied, none provided enhanced stability compared with Gly-Pro-Hyp. The most stable residues in the Yyy position were Hyp > Arg > Met, while for the Xxx position Pro > charged residues > Ala > Gln.22,23,125 An algorithm for the prediction of THP stability based on sequence was subsequently developed.144 Surprisingly, 4R-Flp in the Yyy position was slightly destabilizing compared with Hyp within a host-guest THP, in contrast to (Pro-4R-Flp-Gly)n and (Pro-Hyp-Gly)n THPs.145 It was suggested that different mechanisms are in place when Pro-4R-Flp-Gly is inserted within (Pro-Hyp-Gly)n compared with (Pro-4R-Flp-Gly)n.145
THP host-guest studies revealed that electrostatic effects can contribute favorably to the stability of THPs.125,144,146 Favorable electrostatic interactions were observed for Gly-Lys-Asp and Gly-Arg-Asp within the acetyl-(Gly-Pro-Hyp)3-Gly-Xxx-Yyy-(Gly-Pro-Hyp)4-Gly-Gly-NH2 host.147 Conversely, guests Gly-Arg-Lys, Gly-Lys-Arg, and Gly-Glu-Asp exhibited charge repulsion.147 The sequence Gly-Pro-Lys-Gly-[Asp/Glu]-Hyp was found to be as stabilizing as (Gly-Pro-Hyp)2, presumably due to interchain ion pairs.148 Arg in the Yyy position can offer high stability, possibly based on its ability to form a hydrogen bond with CO in a neighboring strand.125,149 Heterotrimeric host-guest peptide studies demonstrated that an increased number of Arg residues decreased thermal stability for an acetyl-(Gly-Pro-Hyp)3-Gly-Pro-Yyy-(Gly-Pro-Hyp)4-Gly-Gly-NH2 sequence.92 For example, if Yyy = Hyp in all three chains, Tm = 47.2 °C, whereas replacement of Hyp by Arg in one chain, two chains, or three chains resulted in Tm = 44.5, 40.8, and 37.5 °C, respectively. The role of electrostatic effects for stabilizing the collagen-like domain of Streptococcus pyogenes cell surface protein Scl2 was examined using the THP acetyl-(Gly-Pro-Hyp)3-Gly-Lys-Asp-Gly-Lys-Asp-Gly-Gln-Asn-Gly-Lys-Asp-Gly-Pro-Leu-(Gly-Pro-Hyp)4-Gly-Tyr-NH2.150 The Gly-Lys-Asp-Gly-Lys-Asp motif was found to contribute greater stability to the triple-helix than what was predicted based on host-guest studies (see above), and stability was pH dependent, indicating that electrostatic stabilization was an important mechanism for the Scl2 triple-helix.150
The effects of D-amino acids on triple-helical stability was examined using the guest triplets Gly-Asp-Hyp and Gly-Asp-Ala with either D- or L-Asp.151 The D-Asp was significantly destabilizing for the triple-helix, but could be accommodated.151 Triple-helical peptide-amphiphile substrates were also able to accommodate fluorogenic D-amino acids.152
The non-native amino acid trans-4-amino-L-proline is more stabilizing than Hyp for Phe-(Pro-Yyy-Gly)6 triple-helices.43 Interestingly, β-D-galactose glycosylation of Thr in the Yyy position of (Gly-Pro-Yyy)10 greatly enhances triple-helical stability compared to Thr.153-155 In turn, replacement of the central Gly residue of (Pro-Hyp-Gly)7 with either D-Ala or D-Ser resulted in no triple-helix formation in an aqueous environment.156
A host-guest approach was used in which (Pro-Hyp-Gly)7 was the control peptide, to examine the role of tertiary amides for the stability of triple-helical structure.157 The middle Pro-Hyp-Gly triplet was substituted by N-methylalanine (meAla) in the Xxx or Yyy position, Ala in the Xxx or Yyy positions, or Pro in the Yyy position. Overall, meAla was found to be more destabilizing in either the Xxx or Yyy position than Ala, leading the authors to conclude that the presence of a tertiary amide is not sufficient for conformational stabilization of the triple-helix. These conclusions were at odds with prior results where N-isobutylglycine (Nleu) did not dramatically destabilize triple-helical structure, and in fact enhanced it compared with Pro. The authors proposed that Nleu may have conformational preferences that are similar to Pro and Hyp, while N-methylglycine (Sar) and N-methylalanine (meAla) do not. In a related study, acetyl-(Gly-Nleu-Pro)3-Gly-Xxx-Pro-(Gly-Nleu-Pro)3-NH2 was used as a host, with the guest being a variety of alkyl and aralkyl peptoid residues.158 Interestingly, virtually all peptoid residues could be incorporated and stable triple-helices maintained.
Mizuno et al. have further examined the stability of triple-helices incorporating 4R-Hyp in the Xxx position.159 For many years, it was believed that peptides containing 4R-Hyp in the Xxx position of Gly-Xxx-Yyy repeats did not form collagen-like triple-helices. This group previously demonstrated that acetyl-(Gly-4R-Hyp-Thr)10-NH2 forms a stable triple-helix.154 The Yyy position was varied to Ser, Val, Ala, and alloThr to evaluate the specific interactions that stabilize Gly-4R-Hyp-Yyy triple-helices. Only the Thr and Val containing peptides formed stable triple-helices in water. Thus, the contributions to triple-helix stability appear to be both the hydroxyl and methyl groups of Thr, as well as their stereochemical configuration. Thermodynamic parameters further indicated that the hydroxyl group of Thr alone does not account for increased triple-helix stability. Molecular modeling showed that the Thr methyl group covers the interchain hydrogen bond between the carbonyl group of Hyp of the adjacent chain and the amino group of the next Gly residue in the same chain. The distance between the Thr hydroxyl group and the Hyp hydroxyl group is longer than found in typical hydrogen bonds.
Host-guest THPs incorporating either Gly-Trp-Hyp or Gly-Pro-Trp have been studied by a variety of fluorescence measurements.160 Side chains in the Xxx position were found to be more solvent accessible and considerably more mobile than side chains in the Yyy position.160
The host-guest approach has also been used to examine triple-helix nucleation.23,161 The fast folding THPs were those with Hyp in the Yyy position of (Gly-Pro-Hyp)3-Gly-Xxx-Yyy-(Gly-Pro-Hyp)4.161 Folding kinetics have also been found to be affected by chain register in heterotrimeric sequences.162
Multiple hereditary connective tissue diseases have been linked to collagen mutations.63 These mutations most often disrupt collagen folding, resulting in its defective structure and function.6,8,163-165 Mutations of type I collagen genes have been identified in both osteogenesis imperfecta (OI) and Ehlers-Danlos syndrome (EDS).1,6,8,163-167 OI dominant-negative mutations can occur in either gene that encode the α chains of type I collagen and are typically missense mutations that change the Gly codons in the triple-helical motifs. Gly substitutions result in different effects on helix stability, depending on their location and the newly substituted amino acid.
THPs have been designed to house various Gly substitutions, and NMR and CD spectroscopic studies of such peptides permit the folding upstream, downstream and at the mutation site to be examined.164,168 By using mutant THPs, steps in the normal nucleation and propagation of triple-helical structures have been elucidated.169-175 NMR, CD, and X-ray crystallographic studies lend insight into the genotype-phenotype relationship of different collagen mutations with severity of associated disease.129,176-178 For example, host-guest studies utilizing the THP acetyl-(Gly-Pro-Hyp)3-Zzz-Pro-Hyp-(Gly-Pro-Hyp)4-Gly-Gly-NH2 indicated that destabilization by individual residues compared to Zzz = Gly correlated to the severity of OI mutations, based on the α1(I) chain.178
(Pro-Hyp-Gly)4-Pro-Hyp-Ala-(Pro-Hyp-Gly)5, which contains a single interruption of the Gly-Pro-Hyp repeat (Ala for Gly mutation), has a Tm = 28 °C compared to Tm = 60 °C for (Pro-Hyp-Gly)10.169,171 A combination of 1H-NMR and X-ray crystallographic experiments has shown (Pro-Hyp-Gly)4-Pro-Hyp-Ala-(Pro-Hyp-Gly)5 to be only 70-75% triple-helical, with the “interrupted” region a distortion of the triple-helix.129,169,170 Direct interstrand hydrogen bonds were replaced by interstitial water bridges at the site of this interruption, and hence thermal destabilization of this peptide was due solely to entropic factors.129,169 Flanking sequences have substantial effects on the relative destabilization of Gly mutations.177,179 Additional studies have been performed with THPs that model Gly to Ala, Ser, or Cys mutations. Triple-helix nucleation was found to occur on either side of such mutations, but that different nucleation mechanisms may be invoked.180 The ability to nucleate the triple-helix on the N-terminal side of a mutation depends on sequence, with Hyp-rich sequences favoring nucleation.181
Analysis of host-guest heterotrimeric constructs have revealed that the greatest decrease in triple-helix stability occurs upon the first mutation of a Gly residue in a single chain, with subsequent mutations in the other two chains only slightly decreasing stability.182 NMR studies of a Gly to Ala or Ser substitution revealed that hydrogen bonding at the mutation site may involve only one chain, and greater conformational flexibility was observed C-terminal to the substitution.183
The effects of Gly mutations on mineralization were examined using self-assembled monolayers of THPs. Within the sequence Cys-Gly-Lys-Hyp-(Gly-Pro-Hyp)2-Gly-Glu-Hyp-(Gly-Pro-Hyp)2-Gly-Arg-Hyp-Gly-Pro-Hyp-Gly-Pro-Hyp-Gly-Asp-Hyp-(Gly-Pro-Hyp)5, replacement of Gly by Ala had little effect on mineral thickness, but replacement by Val or Asp resulted in thinner calcium phosphate minerals.184 The Ala substitution did produce a lower Ca/P ratio (similar to that observed in OI), while Val or Asp substitution produced higher Ca/P ratios.184
Other types of interruptions, such as Gly or Hyp deletions, may have different molecular mechanisms then Gly mutations for their destabilizing effects.171 THPs have been utilized to study natural breaks in the Gly-Xxx-Yyy repeat that occur in collagens, such as type IV. The most well studied is the Gly-Pro-Hyp-Gly-Ala-Ala-Val-Met-Gly-Pro-Hyp-Gly-Pro-Hyp sequence found in the α5(IV) chain (residues 386-399), where 4 residues are found between Gly residues in the break (G4G interruption). It was observed that the Gly-Pro-Hyp regions that flank the break conform to normal triple-helical structure, and hydrophobic interactions replace the Gly packing within the break a preserve a pseudo-triple-helix.185,186 Replacement of Gly-Ala-Ala at the site of the break by Gly-Pro-Hyp further decreases the stability of the triple-helix.185 This suggests that secondary amino acids are unfavorable within a break,185 a trend that was observed with other triple-helical break sequences of G1G interruptions.187 However, one distinction of G4G versus G1G interruptions is that the hydrophobic residue in the former packs near the helix central axis, while the hydrophobic residue in the latter is found on the helix surface.187 G1G interruptions create a local region of flexibility within the triple-helix, a process which has been suggested to be important for molecular recognition.188
The collagenous domain of mannose binding lectin possesses a Gly54Asp mutation at a frequency as high as 30%.189 Studies with THPs demonstrated that the mutation only slightly decreased the stability of the triple-helix (Tm difference of 2 °C), but the sequence N-terminal to the mutation was disordered compared to the parent THP.189 The mutation occurs at a junction between highly ordered and weakly ordered helical regions, and thus the mutation is less destabilizing than Gly mutations observed in OI.189
6 Adhesion receptor binding to collagen
A great variety of cell surface receptors have been recognized for their collagen binding activity. Cell binding is often facilitated by triple-helical conformation.190 Identification of collagen binding sites has been aided by THP libraries, including one that covers charge clustered sites in the α1(I) collagen chain48 and the types II and III collagen Toolkits.191 The best-characterized cell surface adhesion molecules are integrins, which are heterodimeric proteins composed of one α and one β subunit. The collagen binding integrins include α1β1, α2β1, α3β1, α10β1, and α11β1.192,193 In an effort to better understand the roles of individual integrins, numerous triple-helical binding sites for the collagen binding integrins have been identified (Table 1). The Gly-Phe-Hyp-Gly-Glu-Arg motif, in triple-helical conformation, has been shown to bind to the α2β1 and α11β1 integrins and recombinant α1 and α2 A-domains.38,77,109,194-198 The co-crystal structure of a THP incorporating Gly-Phe-Hyp-Gly-Glu-Arg and the α2 A-domain revealed that the Glu residue directly coordinated the α2 A-domain metal ion (Mg2+), the Arg residue formed a salt bridge with an Asp residue in the α2 A-domain, and Phe was involved in hydrophobic interactions with the receptor.195 Changes in receptor conformation upon ligand binding appear initially induced by changes in metal ion coordination.195,199 Binding also results in changes in the triple-helix main-chain conformation and bending of the triple-helix.200 The Gly-Phe-Hyp-Gly-Glu-Arg motif is found within type I collagen at α1(I)502-507 and type IV collagen at α1(IV)385-390. A THP model of α1(I)502-516 binds to platelets, a model of α1(I)496-507 binds to endothelial cells, and models of α1(IV)382-393 bind to HT-1080 and melanoma cells.38,109,201,202| Receptor | Binding site location | Sequence |
|---|---|---|
| For type IV collagen, sequence numbers are based on the human α1(IV) and α2(IV) genes.324 | ||
| α1β1, α2β1, α11β1 | α1(I)502-507 | GFOGER |
| α1β1, α2β1 | α1(III)64-69 | GROGER |
| α1β1, α2β1 | α1(IV)382-393 | GAOGFOGERGEK |
| α1β1, α2β1 | α1(I)127-135 | GLOGERGRO |
| α1β1 | α1(IV)437-448 | GPPGDQGPPGIP |
| α2(IV)454-465 | GAKGRAGFPGLP | |
| α2β1 | α1(I)433-438 | GADGEA |
| α2β1 | α1(III)115-120 | GLOGEN |
| α2β1 | α1(III)522-528 | GGPOGPR |
| α3β1 | α1(IV)531-543 | GEFYFDLRLKGDK |
| α2β1, α11β1 | Scl1 collagen-like region | GLPGER |
| GpVI, LAIR-1 | α1(III)517-543 | GAOGLRGGAGPOGPEGGKGAAGPOGPO |
| CD44 | α1(IV)1263-1277 [IV-H1] | GVKGDKGNPGWPGAP |
| DDR2 | α1(III)397-408 | GPRGQOGVMGFO |
Other collagen binding motifs have been identified for the α2β1 integrin. Gly-Leu-Hyp-Gly-Glu-Arg from type I collagen has been identified as a ligand for the α1β1 integrin, α1 and α2 A-domains, and α2 I-domain,38,196 while the Gly-Arg-Hyp-Gly-Glu-Arg motif from type III collagen binds the α1 and α2 I-domains (Table 1).203,204 Gly-Leu-Pro-Gly-Glu-Arg from the collagen-like region of Scl1 has been shown to bind the α2β1 and α11β1 integrins (Table 1).205 Gly-Gly-Pro-Hyp-Gly-Pro-Arg from α1(III)522-528 and Gly-Leu-Hyp-Gly-Glu-Asn from α1(III)115-120 binds the α2β1 integrin (Table 1).71,72,204 Based on the model of type I collagen fibers, the Gly-Phe-Hyp-Gly-Glu-Arg and Gly-Leu-Hyp-Gly-Glu-Arg sequences are accessible for integrin binding.206
The α1β1 integrin, either isolated from human placenta or recombinant, simultaneously binds Asp441 from two α1(IV) chains and Arg458 from the α2(IV) chain.207-209 This type IV collagen region is believed to be a higher affinity α1β1 integrin binding site than the Gly-Phe-Hyp-Gly-Glu-Arg motif found in type IV collagen,194 although the KD value for integrin binding to the heterotrimeric THP was 20 μM while binding to homotrimeric Gly-Phe-Hyp-Gly-Glu-Arg occurred with KD = 1.45 μM.209-211 Proline hydroxylation is required for α1β1 integrin binding.212
The platelet α2β1 integrin binds to the Asp-Gly-Glu-Ala motif in some THP constructs213 but not others.109 The melanoma cell α3β1 integrin binds to α1(IV)531-543.67,69,214,215 Binding of the α3β1 integrin to collagen-derived substrate is not dependent upon triple-helical conformation,67,214,215 unlike the α1β1 and α2β1 integrins.38
Another collagen-binding receptor is the proteoglycan CD44. The overexpression of CD44 is found on a variety of tumor cells,216 and elevated CD44 expression by 4 to 6-fold is associated with tumor growth and metastasis.217 CD44 in the chondroitin sulfate proteoglycan (CSPG) modified form is among the receptors uniquely overexpressed in metastatic melanoma.216 Interestingly, CD44 has recently been revealed as a cancer stem cell marker for at least 6 different tumor types.218-220 A theory is emerging that CD44 positive cells within a tumor display true stem cell properties such that one cell can give rise to an entire tumor.218 Additionally, CD44 interaction with hyaluronan induces ankyrin binding to MDR1 (P-glycoprotein), resulting in the efflux of chemotherapeutic agents and chemoresistance in tumor cells.221
The type IV collagen α1(IV)1263-1277 sequence (Table 1; designated [IV-H1]) promotes melanoma cell adhesion, spreading, and signaling.33,37,46,62,222-224 Affinity chromatography studies with single-stranded and triple-helical [IV-H1] peptides resulted in the isolation of melanoma cell CD44 receptors, in the chondroitin sulfate proteoglycan (CSPG) form.62,225,226 Loss of triple-helical structure dramatically reduces melanoma cell adhesion, spreading, and signaling modulated by this ligand.33,37,46 Melanoma cell responses to the triple-helical [IV-H1] sequence have been compared for the galactosylated versus non-galactosylated ligands.62 Galactosylation was found to strongly modulate adhesion and spreading, both of which were dramatically decreased due to the presence of a single sugar. This study was the first demonstration of the prophylactic effects of ligand glycosylation on tumor cell interaction with the basement membrane, while related reports have shown that (a) tumor cell surface sialic acid reduced binding to type IV collagen227 and (b) decreased laminin binding glycans results in increased prostate and breast carcinoma motility.228
There is a family of receptor tyrosine kinases (RTKs) that have extracellular domain motifs homologous to the dictyostelium discoideium protein discoidin-I.229 The RTKs with discoidin-I homology have been named discoidin domain receptors (DDRs).230 Two distinct DDRs have been characterized. DDR1 is expressed in tumor cells themselves, while DDR2 has been detected in the stromal cells surrounding the tumor.229 Numerous breast carcinoma cell lines, as well as other carcinomas, have been shown to highly express DDR1.229,231,232
DDR1 and DDR2 are activated by fibrillar collagens, i.e. Tyr phosphorylation of DDR is substantially increased.230,232,233 Triple-helical conformation is required for collagen to serve as a ligand for DDR1 and DDR2.230,233,234 The specificity of DDR interaction with collagen is also dependent upon the ligand carbohydrate content. Deglycosylation of collagen by treatment with sodium m-periodate resulted in significant reduction of collagen-induced stimulation of DDR.230 Thus, collagen requires both an intact triple-helical conformation and glycosylation for optimum induction of DDR.
The type III collagen Toolkit was utilized to identify the Gly-Pro-Arg-Gly-Gln-Hyp-Gly-Val-Met-Gly-Phe-Hyp binding site for DDR2 (Table 1).191,235 Autophosphorylation of DDR2 was induced by this THP.235 Interestingly, this site bound to DDR2 and induced activation even though it does not contain a glycosylated Hyl residue, and thus appears at conflict with prior studies (see above).
THPs containing Gly-Pro-Hyp repeats alone have been shown to bind platelet glycoprotein VI (GpVI), with two repeats of Gly-Pro-Hyp representing the smallest binding motif.74,110,111,113 Proline hydroxylation is required for GpVI binding.212 However, it has been noted that simple Gly-Pro-Hyp repeating sequences are not found in native collagens, and thus GpVI interaction with collagens is more complex than THP models initially revealed.206 The GpVI binding sequence Gly-Ala-Hyp-Gly-Leu-Arg-Gly-Gly-Ala-Gly-Pro-Hyp-Gly-Pro-Glu-Gly-Gly-Lys-Gly-Ala-Ala-Gly-Pro-Hyp-Gly-Pro-Hyp from type III collagen was recently identified using the Toolkit (Table 1).114
Leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1) binds types I and III collagen with high affinity (KD = 14.2 and 16.2 nM, respectively).236 THP models showed LAIR-1 binding to (Gly-Pro-Hyp)10 but not (Gly-Pro-Pro)10.236 Use of the types II and III collagen Toolkits resulted in several binding sites identified for LAIR-1 and LAIR-2 (Table 1).237 Although a common motif was not observed, the presence of 2-4 Gly-Pro-Hyp repeats within binding sites was found.237
Staphylococcus aureus possesses a microbial surface component recognizing adhesive matrix molecule (MSCRAMM), referred to as CNA, that binds to collagen. CNA binds to several THPs, and a crystal structure was obtained for the complex of CNA31-344 and (Gly-Pro-Hyp)4-Gly-Pro-Arg-Gly-Arg-Thr-(Gly-Pro-Hyp)4.238 The interactions between the CNA and the THP occurred in the Gly-Pro-Hyp regions, primarily due to hydrophobic interactions with Pro residues and hydrogen bonding with Hyp residues.238 In similar fashion, the Yersinia enterocolitica adhesion A (YadA), a collagen binding trimeric autotransporter, binds to (Pro-Hyp-Gly)10 with KD = 0.17-0.28 μM.239 Specific sequence specificity for YadA binding was not observed.
The binding affinities of receptors for THPs have been determined in several studies. The α1β1 integrin binds to α1(I)127-138 and α1(I)496-507 THPs with KD = 7.0 and 1.7 μM, respectively.38 The α2 I-domain binds to α1(I)127-138 and α1(I)496-507 THPs with KD = 0.40 and 1.1 μM, respectively.38 The Gly-Arg-Hyp-Gly-Glu-Arg motif is bound by the α1 and α2 I-domains with KD = 23 and 283 nM, respectively.203 As mentioned above, the α1β1 integrin binds to [α2(IV)454-465][α1(IV)437-448][α1(IV)437-448] THP with KD = 20 μM.209 THP models of α3β1 integrin and CD44/CSPG binding sites have [cell adhesion]50 values in the range of 0.5-5 μM.37,46,48,62,67 Two triple-helical peptides derived from type I collagen α1(I) chain sequences were found to inhibit α2 A-domain binding to type I collagen with IC50 values of ∼5-10 μM.196 The recombinant α2 A-domain was shown to bind to type I collagen with KD∼ 6-10 μM.196 The α1 A-domain has two classes of binding sites within type I collagen, with the higher affinity site exhibiting a KD = 0.11 μM.196 Thus, THPs have been shown to have cell binding affinities in the same range as native collagens.
The binding of cell surface receptors to extracellular matrix proteins induces intracellular signaling and ultimately alterations of cellular phenotypes. THPs have been utilized to elucidate collagen-induced cell signaling pathways.33,38,69,107,110,198,202,206,240-242 Induction of Tyr phosphorylation of focal adhesion kinase (p125FAK) was enhanced and the time of induction was shortened when the α3β1 integrin ligand was in triple-helical conformation.69 The clustered THP ligand induced more rapid paxillin Tyr phosphorylation than the single-stranded ligand. In addition, paxillin bound directly to pp125FAK. Overall, these studies showed that (a) a model of an isolated sequence from type IV collagen, α1(IV)531-543, can induce α3β1 integrin-mediated signal transduction in melanoma cells and (b) ligand conformation (secondary, tertiary, and/or quaternary structure) can directly influence several α3β1 integrin-mediated signal transduction events.69 Melanoma cell signal transduction via CD44/CSPG has also been studied.33 A combination of ligand (and hence cell surface receptor) clustering and triple-helical conformation was required for maximum induction of p125FAK phosphorylation by [IV-H1].
THPs were used to examine the expression of matrix metalloproteinases (MMPs) via outside-in melanoma signaling.201,202 Gene expression and protein production of matrix metalloproteinase-1 (MMP-1), MMP-2, MMP-3, MMP-13, and MT1-MMP were modulated with the α2β1-specific THP, whereas the CD44-specific THP yielded significant stimulation of MMP-8 and lower levels of modulation of MMP-1, MMP-2, MMP-13, and MT1-MMP. The profile seen in response to α2β1 integrin engagement is consistent with high invasion potential, in that the MMPs upregulated can participate in the dissolution of basement membrane (type IV) and type I collagen. These results were indicative of specific activation events that tumor cells undergo upon binding to select regions of basement membrane collagen. THP ligands were found to provide a general approach for monitoring the regulation of proteolysis in cellular systems.
The α2β1 integrin THP ligand derived from type I collagen residues α1(I)496-507 was examined for induction of human aortic endothelial cell (HAEC) activation.198 In addition, a “mini-extracellular matrix” (mini-ECM) composed of a mixture of the α1(I)496-507 ligand and a second, α-helical ligand incorporating the endothelial cell proliferating region of SPARC (secreted protein acidic and rich in cysteine) was studied for induction of HAEC activation. Following HAEC adhesion to α1(I)496-507, mRNA expression of E-selectin-1, vascular cellular adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and monocytic chemoattractant protein-1 (MCP-1) was stimulated, while that of endothelin-1 was inhibited. ELISA analysis demonstrated that E-selectin-1 and MCP-1 protein expression was also stimulated, whereas endothelin-1 protein expression diminished. Engagement of the α2β1 integrin initiated a HAEC response similar to that of TNFα-induced HAECs, but was not sufficient to induce an inflammatory response. Addition of the SPARC119-122 region had only a slight effect on HAEC activation. Other cell-ECM interactions appear to be required in order to elicit an inflammatory response in HAECs.
The α1(I)496-507 THP was also found to inhibit tube formation, as induced by human umbilical vein endothelial cell (HUVEC) binding to type I collagen.38 Tube formation, which is a model for angiogenesis, was found to proceed via the α2β1 integrin through the p38 MAPK pathway.
The crosslinked THPs Gly-Lys-Hyp-(Gly-Pro-Hyp)10-Gly-Lys-Hyp-Gly and Gly-Cys-Hyp-(Gly-Pro-Hyp)10-Gly-Cys-Hyp-Gly bind to platelet GpVI (see earlier discussion), resulting in activation of the αIIbβ3 integrin and platelet aggregation.71,106,240 Downstream events linked to GpVI binding include Ca2+ mobilization, phosphatidylinositol-3-kinase induced activation of phopholipase C (PLC), activation of protein kinase C, and release of arachidonic acid.71,106,110,240,242 The (Gly-Pro-Hyp)10 motif can induce Tyr phosphorylation of Syk and PLCγ2, but crosslinking or higher order THP structures are required for 5-hydroxytryptamine secretion and platelet activation.101,107,111 (Gly-Pro-Hyp)10 has been shown to induce platelet aggregation when linked to amino-functionalized latex nanoparticles, but acetyl-(Gly-Pro-Hyp)7-Gly does not.243
7 Triple-helical substrates
Alterations in activities of one family of proteases, the MMPs (Fig. 5), have been implicated in primary and metastatic tumor growth, angiogenesis, and pathological degradation of extracellular matrix components, such as collagen and laminin.244-246 The “collagenolytic” MMPs [MMPs that catalyze the hydrolysis of one or more of the interstitial collagens (types I-III) within their triple-helical domain] include the secreted proteases MMP-1, MMP-2, MMP-8, MMP-9, and MMP-13 and the membrane-bound proteases MT1-MMP and MT2-MMP.247-250 In the cases of MMP-1, MMP-8, MMP-13, MT1-MMP, and MT2-MMP, efficient collagenolytic activity for the isolated enzyme requires both the catalytic (CAT) and hemopexin-like (HPX) domains.251-256 The linker region between these domains also participates in collagenolysis, either by direct binding of substrate257 or by allowing for the proper orientation of the catalytic and hemopexin-like domains.258 The gelatinase members of the MMP family (MMP-2 and MMP-9) possess three fibronectin type II (FN II) inserts within their CAT domains, and these inserts possess similar type I collagen binding sites.259,260 While collagenolytic MMPs possess common domain organizations, there are subtle differences in their processing of triple-helical substrates.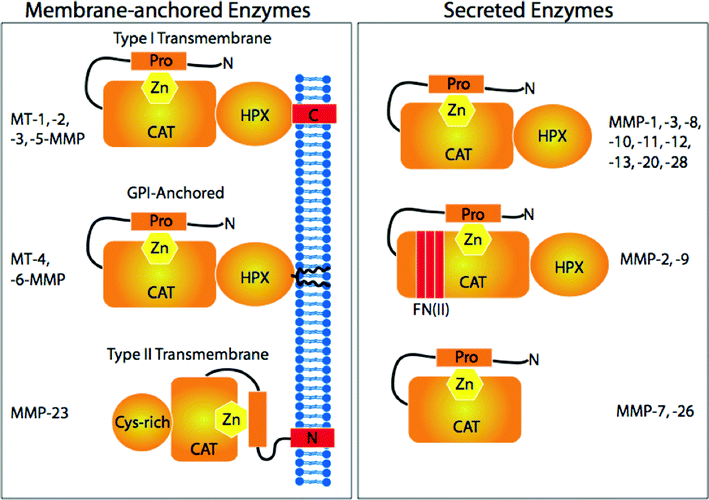 | ||
| Fig. 5 MMP family members and their structural domains. Reproduced from reference 285 by permission of the American Society for Biochemistry and Molecular Biology. | ||
Our laboratory has previously described a number of fluorescence resonance energy transfer (FRET) THP substrates (fTHPs) based on a consensus types I-III collagen 769-783 region that are either suitable for most collagenolytic MMPs or selective for different collagenolytic MMPs (Table 2).36,39-41152,201,202,256,261-263 In addition, we have developed a selective MMP-2/MMP-9/MMP-12 THP substrate, α1(V)436-447 fTHP [(Gly-Pro-Hyp)5-Gly-Pro-Lys(Mca)-Gly-Pro-Pro-Gly∼Val-Val-Gly-Glu-Lys(Dnp)-Gly-Glu-Gln-(Gly-Pro-Hyp)5-NH2].201 In all cases, FRET is achieved by incorporating (7-methoxycoumarin-4-yl)acetyl (Mca) as the fluorophore in the P5 position and 2,4-dinitrophenyl (Dnp) as the quencher in the P5′ position (Fig. 6). Due to the differing sequences of the fTHPs, their relative triple-helical thermal stabilities can vary (Table 2). In order to obtain fTHPs that are stable under near-physiological conditions, as well as to examine the effects of substrate thermal stability on MMP activity, we utilized our previously described peptide-amphiphile approach.14,15,31,32 In addition, the Lys branching protocol has been utilized for the construction of triple-helical MMP substrates in the Fields laboratory,34-36,264 while the Moroder laboratory has utilized the double disulfide knot to create triple-helical MMP substrates.3,78,79,89,90
| fTHP | P2-P1∼P1′-P2′-P3′-P4 Sequence | Peptide T m /°C | C6-Peptide Tm/°C | C10-Peptide Tm/°C |
|---|---|---|---|---|
| Substitutions relative to fTHP-4 are indicated in bold. ND = not determined, Mob = 4-methoxybenzyl. | ||||
| fTHP-4 | Gln-Gly∼Leu-Arg-Gly-Gln | 36.5 | ND | 43.0 |
| fTHP-9 | Gln-Gly∼Cys(Mob)-Arg-Gly-Gln | 47.2 | ND | 60.0 |
| fTHP-10 | Orn-Gly∼Leu-Arg-Gly-Gln | 56.6 | ND | 62.0 |
| fTHP-11 | Orn-Gly∼Cys(Mob)-Arg-Gly-Gln | 44.2 | ND | 47.0 |
| fTHP-12 | Leu-Gly∼Met-Arg-Gly-Gln | 45.0 | 51.0 | ND |
| fTHP-13 | Val-Asn∼Phe-Arg-Gly-Gln | 39.0 | 50.0 | ND |
| fTHP-14 | Val-Asn∼Phe-Arg-Gly-Pro | ND | 46.0 | ND |
![Space-filling (top) and “ball and stick” (bottom) computer generated models of fTHP-4, 3[(Gly-Pro-Hyp)5-Gly-Pro-Lys(Mca)-Gly-Pro-Gln-Gly∼Leu-Arg-Gly-Gln-Lys(Dnp)-Gly-Val-Arg-(Gly-Pro-Hyp)5-NH2]. For the space-filling model, the individual peptide strands are red, blue, and yellow, while the fluorophore (Mca) and quencher (Dnp) are green. Reproduced from reference 268 by permission of Wiley & Sons.](/image/article/2010/OB/b920670a/b920670a-f6.gif) | ||
| Fig. 6 Space-filling (top) and “ball and stick” (bottom) computer generated models of fTHP-4, 3[(Gly-Pro-Hyp)5-Gly-Pro-Lys(Mca)-Gly-Pro-Gln-Gly∼Leu-Arg-Gly-Gln-Lys(Dnp)-Gly-Val-Arg-(Gly-Pro-Hyp)5-NH2]. For the space-filling model, the individual peptide strands are red, blue, and yellow, while the fluorophore (Mca) and quencher (Dnp) are green. Reproduced from reference 268 by permission of Wiley & Sons. | ||
Through the use of the substrates described in Table 2, we can assign unique triple-helical peptidase behaviors to most of the collagenolytic MMPs.34-36,39-41201 MMP-1 has the weakest triple-helical peptidase activity, and has great difficulty in hydrolyzing more thermally stable substrates. MMP-1 does not tolerate long chain, hydrophobic residues interacting with its S1′ subsite nor positively charged residues interacting with the S2 subsite. MMP-2 possesses many of the characteristics described for MMP-1, but additionally will cleave Gly-Gln bonds. Unlike MMP-1, MMP-2 prefers the combination of Leu in the substrate P2 subsite and Met in the P1′ subsite compared to Gln and Leu in these respective subsites. MMP-8 is a robust triple-helical peptidase that favors long chain, hydrophobic residues interacting with its S1′ subsite. MMP-9 activity is similar to that of MMP-2, except that cleavage of Gly-Gln bonds is not observed. MMP-13 processes thermally stable sequences efficiently, with little effect of sequence. In contrast to MMP-1, MMP-8, and MT1-MMP, MMP-13 prefers a positively charged residue interacting with the S2 subsite, and may favor certain interrupted triple-helical sequences over uninterrupted ones. MT1-MMP is the most robust triple-helical peptidase, and is reasonably sensitive to substrate thermal stability. It disfavors positively charged residues interacting with the S2 subsite, and is also relatively ineffectual at processing interrupted triple-helical sequences that are cleaved by soluble MMPs. MT2-MMP behaves similarly to MT1-MMP, except that it is less active.
THPs have been utilized recently to identify residues in the HPX domain that participate in collagenolysis.263 MMP-1 HPX residues Ile290 and Arg291 were found to contribute to recognition of triple-helical structure, and facilitate both the binding and catalysis of the triple-helix.263 MMP-1 CAT and HPX domains collaborate in collagen catabolism by properly aligning the triple-helix and coupling conformational states to facilitate hydrolysis.263 The HPX domain is not critical for triple-helical peptidase activity of shorter substrates (such as fTHP-4).34,36,263 However, as the THP substrate becomes longer and more “collagen-like”, the HPX domain becomes more significant for facilitating proteolysis.79,89,263 The C-terminally extended THP used in reference 263 includes Arg-Gly-Glu-Arg residues in subsites P14′-P17′; molecular modeling of fibrillar collagen found this sequence to interact with the MMP-1 HPX domain.265
Mammalian collagenases cleave interstitial (types I-III) collagen into 1/4 and 3/4 length fragments. Peptide substrate studies have shown that collagen primary structure is not the basis for discriminatory MMP collagenolytic behavior.266 For example, MMP-3 cleaves a single-stranded peptide model of the collagen cleavage site at a rate comparable to MMP-1, but MMP-3 does not cleave this sequence in native collagen.266 Studies with THPs have allowed for the further refinement of a previously described model of the MMP cleavage sites in interstitial collagens.267,268 This model suggested that all of the information necessary for efficient hydrolysis of collagen is contained in a 24 residue stretch.267 Cleavage site regions are distinguished by a low content (<10%) of charged residues in addition to being “tightly” triple-helical (high secondary amino acid content) prior to the cleavage site and “loosely” triple-helical (low secondary amino acid content) following the cleavage site (Fig. 7).267,269,270 One of the implications of this model is that low secondary amino acid content creates a triple-helical region that is distinct, and more flexible, than a high secondary amino acid content region.
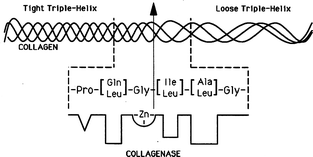 | ||
| Fig. 7 Model of the collagenase cleavage site in interstitial collagens. The four triplet region that precedes the scissile (Gly∼Ile/Leu) bond is rich in secondary amino acids (50% of the Xxx + Yyy residues, Pro always found in subsite P3), and has a low average side-chain molal volume (<45 ml). The four triplet region that follows the scissile bond is secondary amino acid deficient (a maximum of 2 secondary amino acid residues, not in neighboring triplets). The overall 25 amino acid residue region is hydrophobic, containing a maximum of 2 charged residues. Reproduced from reference 261 by permission of Wiley & Sons. | ||
Evidence to support the MMP cleavage site model includes the detection of triple-helical backbone mobilities by one- and two-dimensional NMR experiments using 15N-labeled THPs.32,173,271 NMR studies of a triple-helical peptide model of the type I collagen cleavage site have shown a reduced triple-helical content in the secondary amino acid poor region.270,272 Free energy landscape computational analysis indicated the secondary amino acid poor region of the cleavage site can form a structure complementary to the MMP active site.273 This complementary behavior was not observed for other regions possessing potential cleavage sites.273
The lack of charged residues, particularly in the “loosely” triple-helical region, constitutes another mechanism by which the stability of the triple-helix is compromised. Studies with both homotrimeric and heterotrimeric triple-helical peptides have demonstrated enhanced stability based on charged pairs (Asp and Lys).25,26
X-Ray crystallographic analysis has indicated that hydration patterns are different in the secondary amino acid poor region of the type III collagen cleavage site compared with secondary amino acid rich regions, in that more ordered water is found in the secondary amino acid poor region.274 X-Ray crystallographic analysis of a series of host guest peptides revealed that the 1st hydration shell in triple-helical peptides, found at 2.75 Å, is where water molecules directly link to peptide atoms by hydrogen bonds, whereas the 2nd hydration shell, found at 3.55 Å, is where water molecules interact with each other.275 The introduction of Hyp (as occurs in the “tightly” triple-helical region) causes an increase in the ratio between the number of water molecules in the 1st and 2nd hydration shells.275 Thus, more peptide-bound water occurs in the “tightly” triple-helical region, and comparatively less in the “loosely” triple-helical region. In addition, NMR hydration experiments revealed that the 1st hydration shell is kinetically labile (upper limits for H2O residence times in the nanosecond to sub-nanosecond range),276 and thus hydration-dehydration is probably not rate-limiting during collagenolysis.
Overall, there are numerous structural and hydration differences between secondary amino acid rich and poor regions within triple-helices. The proposed collagen cleavage site model would only be valid if collagenases had extended active or substrate binding sites. Studies using mutant collagens showed that MMP-1, MMP-8, and MMP-13 had active or substrate binding sites that extended at least S2 through S8′ and conformational restriction of the cleavage site in collagen (by introducing Pro residues in the P′ subsites) eliminated MMP activity.277-279 THP substrates that are extended by 6 residues in the N-terminal or 9 residues in the C-terminal direction compared with those given in Table 2 have significantly improved rates of hydrolysis, demonstrating MMP interactions with substrate subsites as far as P13 and P17′.263
8 THP transition-state analogs
The activity of MMPs can be regulated in numerous ways including gene expression, activation of the zymogen precursors, association with endogenous inhibitors such as the tissue inhibitors of metalloproteinases (TIMPs), or the physical removal of enzyme via proteolysis or cellular internalization. One regulatory approach has focused on inhibiting MMPs using THPs. Our laboratory prefers this approach as it affects only active protease populations and does not disturb the balance of native protease-inhibitor complexes (such as the natural distribution of MMP-TIMP complexes).Metallo(zinc)-proteases use the nucleophilic attack of a water molecule as one of the steps of amide bond hydrolysis.280 The tetrahedral intermediate that results from water addition to the amide carbonyl has been the focus of many protease inhibitor designs. Phosphinic peptides/phosphinates [Ψ{PO2H-CH2}] have been shown to behave as transition state analog inhibitors of MMPs.281 Phosphinate THP inhibitors (THPIs; Fig. 8) have several potential advantages over other inhibitor constructs. These analogs incorporate specificity elements for both the S and S′ subsites of the enzyme, while the triple-helical structure interacts with both the active site and secondary substrate binding sites (exosites).244,282,283
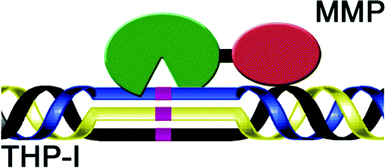 | ||
| Fig. 8 Model structure of a triple-helical transition state analog MMP inhibitor. The MMP CAT domain is green, the MMP HPX domain is red, and the non-hydrolyzable PO2H-CH2 bond is indicated in purple. | ||
In order to create the desired phosphinate transition state analogs, we prepared protected building blocks GlyΨ{PO2H-CH2}Val 1a and GlyΨ{PO2H-CH2}Leu 1b (Scheme 1).284-286 Briefly, this route consisted of a mild silylation of Fmoc-aminophosphinic acid (5)287 with TMS-Cl and DIEA to give the trivalent phosphinate, which reacted in a Michael-type reaction with an allyl acrylate288 to afford phosphinic acid 7 as the racemate (Scheme 1). This phosphinate dipeptide analog was protected on phosphorus as the adamantyl ester and the allyl ester was deprotected using CpRu(CH3CN)3PF6 catalysis to give the protected phosphinate dipeptide mimic 1a [(R,S)-2-isopropyl-3-((1-(N-(Fmoc)amino)-methyl)-adamantyloxyphosphinyl)propanoic acid] and its Leu analog 1b.
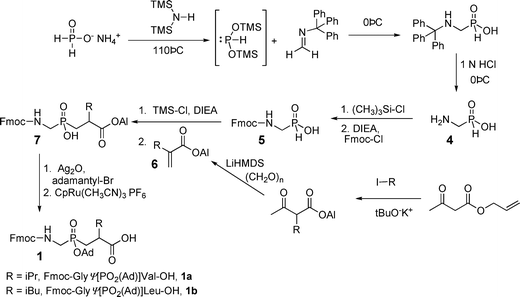 | ||
| Scheme 1 Synthesis of protected phosphinate amino acids and phosphinodipeptides. | ||
To assemble THPIs, dipeptide mimics 1a and 1b were incorporated into peptides by Fmoc solid-phase synthesis. Fmoc-phosphinodipeptide 1a was utilized to create C6-(Gly-Pro-Hyp)4-Gly-Pro-Pro-GlyΨ{PO2H-CH2}(R,S)Val-Val-Gly-Glu-Gln-Gly-Glu-Gln-Gly-Pro-Pro-(Gly-Pro-Hyp)4-NH2 [designated α1(V)GlyΨ{PO2H-CH2}Val THPI].284
The α1(V)GlyΨ{PO2H-CH2}Val THPI (which contains the S configuration in the P1′ position, equivalent to an L-amino acid) was initially tested against MMP-2 and MMP-9 (Table 3). Due to the low melting temperature of the potential inhibitor (Tm∼ 25 °C), Ki values were first determined at 10 °C. α1(V)GlyΨ{PO2H-CH2}Val THPI was found to be a very effective inhibitor of MMP-2 and MMP-9, with Ki values of 4 and 2 nM, respectively. When inhibition assays were repeated at 37 °C, the Ki value increased for MMP-2 but not for MMP-9 (Table 3). Thus, triple-helical structure modulated inhibition of MMP-2 but not MMP-9.
| Enzyme | Inhibitor | T/°C | K i (app) /nM |
|---|---|---|---|
| MMP-2 | α1(V)GlyΨ{PO2H-CH2}Val THP | 10 | 4.14 ± 0.47 |
| ″ | ″ | 37 | 19.23 ± 0.6 |
| ″ | MMP inhibitor III | 10 | 3.17 ± 0.23 |
| ″ | ″ | 37 | 0.83 ± 0.03 |
| ″ | α1(I-III)GlyΨ{PO2H-CH2}Leu THP | 10 | 0.18 ± 0.00 |
| ″ | ″ | 37 | 0.08 ± 0.01 |
| MMP-9 | α1(V)GlyΨ{PO2H-CH2}Val THP | 10 | 1.76 ± 0.05 |
| ″ | ″ | 37 | 1.29 ± 0.00 |
| ″ | α1(I-III)GlyΨ{PO2H-CH2}Leu THP | 10 | 0.02 ± 0.01 |
| ″ | ″ | 37 | 0.09 ± 0.00 |
| MMP-1 | α1(I-III)GlyΨ{PO2H-CH2}Leu THP | 10 | 7.83 ± 1.03 |
| ″ | ″ | 37 | 26.70 ± 5.2 |
| ″ | MMP inhibitor III | 10 | 2.48 ± 0.35 |
| ″ | ″ | 37 | 4.72 ± 0.38 |
To determine if an increase in Ki as a function of temperature was a general trend for inhibition of MMP-2, inhibition of MMP-2 by MMP inhibitor III (a hydroxamic acid-Leu-homoPhe dipeptide) was examined. At 10 °C, the Ki value for MMP-2 inhibition was 3 nM (Table 3). Increasing the temperature to 37 °C decreased the Ki to 0.8 nM (Table 3). Thus, for a small molecule inhibitor, an increase in temperature slightly increased the affinity towards MMP-2, most likely due to enhanced hydrophobic interactions. This further suggested that the decreased inhibition of MMP-2 by α1(V)GlyΨ{PO2H-CH2}Val THPI as a function of increasing temperature is due to unfolding of the inhibitor triple-helical structure.
MMP-1, MMP-3, MMP-8, MMP-13, and MT1-MMP were tested for inhibition by α1(V)GlyΨ{PO2H-CH2}Val THPI. No inhibition of MMP-1, MMP-3, or MT1-MMP was observed up to an α1(V)GlyΨ{PO2H-CH2}Val THPI concentration of 25 μM. MMP-8 and MMP-13 were inhibited weakly, with Ki values in the range of 50 and 10 μM, respectively. Thus, this study utilized a GlyΨ{PO2H-CH2}Val transition state analog to bind selectively at the S1-S1′ site of MMP-2 and MMP-9. Selective inhibition of these MMPs is desirable, as MMP-2 has been validated as an anticancer drug target, while MMP-9 inhibition may be useful in treating early-stage cancers.282
Our second inhibitor design utilized a THP substrate mimicking α1(II)769-783, which is hydrolyzed by MMP-1, MMP-2, MMP-8, MMP-9, MMP-13, and MT1-MMP.39,40 The P1-P1′ subsites of the triple-helical peptide, which incorporate Gly-Leu in the substrate, were substituted by a GlyΨ{PO2H-CH2}Leu transition state analog. Because the Tm value for α1(V)GlyΨ{PO2H-CH2}Val THPI was low (see above),284 the α1(I-III)GlyΨ{PO2H-CH2}Leu THPI incorporated (4R)-Flp to enhance triple-helicity.37,133,134 Thus, the sequence of this inhibitor was C6-Gly-Pro-Flp-(Gly-Pro-Hyp)4-Gly-Pro-Gln-GlyΨ{PO2H-CH2}(R,S)Leu-Ala-Gly-Gln-Arg-Gly-Ile-Arg-(Gly-Pro-Hyp)4-Gly-Pro-Flp-NH2, and it exhibited a Tm value of 30 °C.285 Studies revealed low nM Ki values for inhibition of MMP-1, MMP-2, and MMP-9 (Table 3). Our second transition state analog inhibitor appears to be effective against a broader range of collagenolytic MMPs than the first inhibitor. Interestingly, MMP-1 was sensitive to the triple-helical structure of the inhibitor (Ki increased ∼4 times when the inhibitor was thermally unwound), but neither MMP-2 nor MMP-9 was. This contrasts with the sensitivity of MMP-2 to the triple-helical structure of α1(V)GlyΨ{PO2H-CH2}Val THPI (Table 3), and indicates that there is a sequence-dependent sensitivity to triple-helical structure for some MMPs.
While the eight-step synthesis of the dipeptide analog (3 steps required for fragment 2) was a fairly succinct route, overall yields starting from fragment 2 were approximately 16% mainly due to the inefficiencies of the last two steps. It appeared that reaction conditions in both these steps are not particularly compatible with the Fmoc group leading to significant deprotection and side-product formation. We reasoned that benzyl ester protection of the C-terminus would potentially allow for its removal under the same hydrogenation conditions required for unveiling of the amine.289 To test our hypothesis, we prepared benzyl ester fragment 7a (Scheme 2). Catalytic hydrogenation of 7a with 5% Pd/C under hydrogen in the presence of Fmoc-OSu afforded the desired dipeptide mimic 1a in good yields (Scheme 2).289 This one-pot reaction involved the simultaneous removal of the Cbz and benzyl protecting groups and re-protection of N-terminus with Fmoc. Importantly, this method does not appear to be sensitive to the steric environment of the ester group. The Gly-Leu dipeptide mimic 7b was also prepared and underwent efficient bis-deprotection and Fmoc addition to give 1b (Scheme 2).289
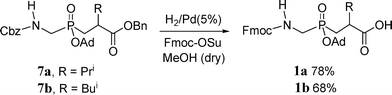 | ||
| Scheme 2 Tandem bis-deprotection and Fmoc attachment leading to protected phosphinodipeptides. | ||
A single-stranded peptide model of the α1(I)715-721 collagen sequence has been identified as a ligand for the MMP-2 FN II insert and inhibited MMP-2 gelatinolysis.290 Our laboratory assembled a triple-helical version of this ligand [α1(I)715-721 THP], and evaluated it for the ability to inhibit MMP-2 and MMP-9 triple-helical peptidase and gelatinase activities.285α1(I)715-721 THP inhibited type V collagen-model triple-helical peptidase activity but not interstitial collagen-model triple-helical peptidase activity. To our knowledge, this demonstrated the first use of an exosite binder to selectively inhibit one collagen-based MMP activity but not another.
9 THP models for studying lipoprotein-, glycosaminoglycan-, nucleic acid-, and other protein-collagen interactions
THPs have been used to examine macromolecular interactions in addition to those previously mentioned. Macrophage scavenger receptor (MSR) binds to a variety of polyanionic molecules, including proteins, phospholipids, polysaccharides, and nucleic acids. MSRs contain a transmembrane segment, a coiled-coil α-helical region, and a triple-helical domain. THP models of MSR-1 were used to study binding interactions of acetylated low-density lipoproteins (Ac-LDLs).88,291 Initially, Ac-LDL was found to bind to a THP model of receptor residues 323-340 [(Gly-Pro-Hyp)4-Gly-Lys-Thr-Gly-Lys-Pro-Gln-Leu-Asn-Gly-Gln-Lys-Gly-Gln-Lys-Gly-Glu-Lys-(Gly-Pro-Hyp)3] (Table 4).88 Binding did not occur to a single-stranded model of this sequence, nor to a THP in which Lys337 was changed to Ala. The binding affinity (KD) of Ac-LDL for the MSR THP and mouse peritoneal macrophages was 31 and 3.2 μg mL−1, respectively.291 Ac-LDL, but not LDL, bound to the MSR THP, and binding was inhibited by dextran sulfate, fucoidan, sulfatides, poly(G), and poly(I), but not poly(C). These binding behaviors were analogous to those exhibited by the native receptor.| Biomolecule | Binding site location | Sequence |
|---|---|---|
| For type IV collagen, sequence numbers are based on the human α1(IV) and α2(IV) genes.324 | ||
| Ac-LDL | MSR323-340 | GKTGKPGLNGQKGQKGEK |
| poly(I) | MSR333-341 | PKGQKGEKG |
| heparin | α1(I)82-93 | GLPGMKGHRGFS |
| heparin | α1(I)925-937 | GDRGIKGHRGFSG |
| heparin, heparan sulfate | α1(V)904-918 | GKOGPRGQRGPTGPR |
| heparin, heparan sulfate | AChE146-161 | GROGRKGROGVRGPR |
| heparin, heparan sulfate | AChE251-266 | GROGKRGKTGLKGDI |
| vWF, SPARC | α1(III)399-408 | RGQOGVMGFO |
MSR has been shown to specifically mediate the uptake of poly(I) and poly(G), but not poly(C), poly(U), or poly(A). A THP model of MSR-1 residues 333-341 [(Pro-Hyp-Gly)3-Pro-Lys-Gly-Gln-Lys-Gly-Glu-Lys-Gly-(Pro-Hyp-Gly)4] was used to study binding interactions of tetraplex nucleic acids.292 The MSR-1 THP preferably bound tetraplex poly(I) over double-stranded and single-stranded forms (Table 4). The MSR-1 THP interaction with poly(I) was also sequence specific.
THPs have been used to identify and characterize glycosaminoglycan binding sites in collagen and acetylcholinesterase (AChE). A series of THPs was screened to find the high affinity binding site for low MW heparin in type I collagen.70 The highest affinities were found for α1(I)82-93 THP (KD = 1.7 μM) and α1(I)925-937 THP (KD = 1.6 μM) (Table 4), which was a factor of 10 worse than heparin binding to type I collagen (KD = 165 nM). This result was not surprising, as type I collagen has multiple heparin recognition sites that may contribute to cooperatively bind heparin. A THP incorporating α1(V)904-918 bound heparin and heparan sulfate, with significant contributions from Lys905, Arg909, and triple-helical structure (Table 4).293
The AChE collagen-like tail has two heparin binding sites of differing affinities, one located in the N-terminal region of the tail (residues 149-152, Gly-Arg-Lys-Gly-Lys) and one in the C-terminal region (residues 254-257, Gly-Lys-Arg-Gly-Lys). THPs were constructed incorporating AChE residues 146-161 [Pro-Hyp-Gly-Arg-Hyp-Gly-Arg-Lys-Gly-Arg-Hyp-Gly-Val-Arg-Gly-Pro-Arg-Gly-(Pro-Hyp-Gly)4] and 251-266 [Pro-Hyp-Gly-Arg-Hyp-Gly-Lys-Arg-Gly-Lys-Thr-Gly-Leu-Lys-Gly-Asp-Ile-Gly-(Pro-Hyp-Gly)4] (Table 4).294 Heparin, heparan sulfate, and chondroitin sulfate bound to both AChE THPs, resulting in increased Tm values. However, the increase in Tm due to chondroitin sulfate binding was lower, as were the THP Δ[Θ]225 nm values, suggesting a different mode of interaction for chondroitin sulfate compared with heparin and heparan sulfate. Binding of heparin was dependent on triple-helical conformation.295 A subsequent study utilized THPs starting from the two heparin binding motifs.296 Triplets were systematically added within a (Gly-Xxx-Yyy)8 template and the effects on thermal stability and heparin binding quantified. Heparin binding was modulated by a combination of sequence and triple-helix stability. The higher heparin binding affinity of the C-terminal region (compared with the N-terminal region) was achieved when a minimum of 9 residues from that region were incorporated into the THP. Calorimetry indicated a lack of significant enthalpy change as a result of heparin binding, suggesting that binding was largely due to electrostatic interactions.
Heat shock protein 47 (HSP47) is a molecular chaperone that facilitates normal procollagen biosynthesis. HSP47 has been proposed to stabilize correctly folded triple-helical intermediates and/or prevent lateral association of procollagen in the ER. The determinants for binding of HSP47 were evaluated using THPs.297 HSP47 specificity was found to reside in triple-helical (Pro-Hyp-Gly)5-Xxx-Arg-Gly-(Pro-Hyp-Gly)4 sequences, and the role of Arg in binding was further confirmed by chemical modification of collagen. The binding affinities of HSP47 to type I collagen and Arg-containing THPs were comparable. Subsequent studies demonstrated that triple-helical structure was critical for HSP47 binding, and that only a single Arg residue was needed to promote a specific interaction between a heterotrimeric THP and HSP47.298 The best binding to HSP47 was observed when Thr or Pro was positioned 3 residues N-terminal to Arg and in the same chain.299 Localization of THP binding to HSP47 was achieved by crosslinking via photolysis of p-benzoyl-L-Phe (Bpa) residues incorporated within the THP.298
A high affinity binding site for von Willebrand factor (vWF) was found using the type III collagen THP Toolkit (Table 4).300 As the collagen-binding A3 domain of vWF was found to rely on different interactions with collagen than the α2β1 integrin,301,302 it was expected that the vWF binding motif would differ from the α2β1 integrin binding site found in type III collagen (see Table 1). Although this binding site was the same as identified for DDR2, binding of vWF is dependent upon the Arg, first Hyp, and Val residues, while the Met residue is important for DDR2 binding.191 The Gly-Val-Met-Gly-Phe-Hyp motif is also bound by SPARC/osteonectin/BM-40 (Table 4).303 X-Ray crystallographic structural analysis of the SPARC/THP complex revealed that, in contrast to integrin binding, SPARC does not distort the triple-helix, but rather rearranges to form a deep pocket to accommodate the Phe residue from the trailing THP chain.303
The THP Gly-Cys-Hyp-(Gly-Pro-Hyp)10-Gly-Cys-Hyp-Gly-NH2, which binds to platelet GpVI (see earlier discussion), is also bound by keratinocyte growth factor (KGF).304 The non-hydroxylated form of the THP is bound with less avidity by KGF, while a THP possessing the integrin binding Gly-Phe-Hyp-Gly-Glu-Arg motif is not bound by KGF.304
THPs possessing (Pro-Hyp-Gly)n and (Pro-Pro-Gly)n motifs have been shown to bind to the collagen-binding domain (CBD) of Clostridium histolyticum type I collagenase, while non-triple-helical peptides do not.305 Gly-(Pro-Hyp-Gly)8 was subsequently utilized to map the residues within the CBD that facilitate binding to triple-helices.306 The most significant contributions to binding activity were observed for residues Thr957, Tyr970, Leu992, Tyr994, and Tyr996.306 The importance of side-chain hydroxyl groups, specifically that of Tyr994, for interacting with THPs is reminiscent of a similar effect observed via mutation of Tyr189 in MMP-8.40
10 Miscellaneous applications of THPs
The use of THPs for biomaterial applications is encouraged by the serum protease stability of (Gly-Pro-Pro)10.307 A PA THP incorporating the α2β1 integrin binding site α1(I)496-507 supported human aortic endothelial cell adhesion and spreading at levels similar to type I collagen.18 Inclusion of a second peptide-amphiphile, which incorporated residues 119-122 from SPARC, provided a mixed PA surface that promoted comparable adhesion and spreading to type I collagen.18 As discussed earlier, the α1(I)496-507 THP induced minimal endothelial cell activation.18,198 The endothelial cell adhesion, spreading, and activation properties of α1(I)496-507 THP were favorable where promotion of wound healing is desired.Corneal epithelial cell and fibroblast attachment and growth has been studied for surface-bound peptoid-containing THPs.308 (Gly-Pro-Nleu)10-Gly-Pro-NH2, when immobilized to ethylene propylene copolymer film, supported cell adhesion and growth. No effect was seen for (Gly-Nleu-Pro)10 nor analogous non-triple-helical sequences, suggesting a specific interaction based on peptoid location and triple-helical conformation. Adhesion was inhibited by the linear Lys-Asp-Gly-Glu-Ala peptide, indicative of α2β1 integrin involvement (see Table 1). However, the affinity of adhesion was not evaluated, as concentration dependencies were not performed nor was adhesion compared to native collagen.
Immobilization of (Gly-Pro-Pro)5-Gly-Phe-Hyp-Gly-Glu-Arg-(Gly-Pro-Pro)5-Gly (CMP1) onto poly(3-hydroxybutyrate-co3-hydroxyvalerate) (PHBV) microspheres promoted Hep3B liver cell growth and spreading.309 In addition, cellular activities such as albumin secretion and P-450 activity were maintained for 14 d, suggesting application for liver tissue engineering.309 In similar fashion, (Gly-Pro-Hyp)7-Tyr was copolymerized with poly(ethylene oxide) diacrylate to create PEG hydrogels; chondrocyte encapsulation resulted in increased glycosaminoglycan and collagen production compared with unmodified PEG hydrogels.310
Cys-(Gly-Pro-Hyp)7 has been attached to gold nanoparticles and used to visualize type I collagen fibers via binding to the gap regions.311
The collagen-model peptide acetyl-(Pro-Pro-Gly)5 and ethylene-diamine-core polyamidoamine dendrimer were combined to create a collagen-mimetic delivery vehicle.312 Although only 5 repeats, the Pro-Pro-Gly sequence appeared to possess triple-helical character, presumably due to surface clustering.312 Delivery of the model drug rose bengal was temperature dependent, correlating with the triple-helical content of the THP.312
The addition of ferrocene (Fc) to the N-terminus of (Pro-Hyp-Gly)n-Cys was found to increase the peptide triple-helical stability.313 In turn, Fc-CO-(Pro-Hyp-Gly)n-Cys, when prepared as films on gold surfaces, exhibited a linear but weakly distance dependent electron transfer.314
The THP acetyl-(Pro-Hyp-Gly)5-Pro-Lys-Gly-(Pro-Hyp-Gly)5-Ala-NH2 was used as a model for analysis of glycation end products following exposure to glucose, ribose, and glyoxal.20 Carboxymethyl-lysine was observed following all treatments, while treatment of the THP with glyoxal resulted in the formation of glyoxal lysine dimer.20 The THP was a good model for collagen advanced glycation end products.
Discussion and conclusions
Due to the application of THPs, collagen-based research has made significant strides in the past decade. The use of THP models has greatly advanced knowledge of both collagen structure and biological activities. Analyses of THPs possessing non-native amino acids reopened discussion on the forces that modulate triple-helical structure and stability. In addition, the unique contributions toward triple-helical stability within heterotrimeric systems have been unveiled. Development of THP model systems has included the implementation of a variety of templates, which serve to further stabilize triple-helical structure. THP templates may ultimately allow for a reduction in the number of Gly-Pro-Hyp or Gly-Pro-Flp repeats required for appropriate stability and a more tractable THP synthesis overall. All of the above studies have provided the foundation for development of THP-based biomaterials.Identification of collagen binding sites has rapidly progressed with the use of the collagen Toolkits.191 Similarly, THP substrates have allowed for a better mechanistic understanding of collagenolytic proteases. Analysis of receptor and enzyme binding to triple-helices have indicated that such processes have been achieved by convergent evolutionary mechanisms.306,315
MMP inhibitor programs began in earnest in the 1980s, using the destruction of ECM components as a model for inhibitor design.316 Initial clinical trials with MMP inhibitors were disappointing, with one of the problematic features being a lack of inhibitor selectivity.244,282,283,317,318 Selectivity appears to be a crucial aspect for future MMP inhibitor design, as several MMPs have been validated as targets in certain cancers (MMP-2, MMP-9, MT1-MMP) while others have been found to be host-beneficial and thus anti-targets (MMP-3, MMP-8).282,319 The development of THPIs has provided truly selective and potent compounds for modulating MMP activities. These results are most promising, as recent studies indicate that inhibition of MMP collagenolytic activity does not led to musculoskeletal syndrome (MSS),320 a common side effect of prior, small molecule MMP inhibitors. THP substrates also allow for high-throughput screening and identification of exosite binding metalloproteinase inhibitors.321-323 The use of exosite binding may well represent an important new step for selective inhibitor development.
While many advances have been made, there are some concerns with THP based research. It has been noted that structural studies with THP models may have been biased due to high secondary amino acid content.125 Identification of binding sites and receptor-mediated signaling has not fully accounted for ligand glycosylation and the role of heterotrimeric structure. While higher order structures have been obtained from THPs, precise control of these macrostructures is not always readily achieved. Nonetheless, the promise of THP based research is great, and the aforementioned shortcomings represent further areas of exploration.
Acknowledgements
I gratefully acknowledge the National Institutes of Health (CA98799, EB000289, and MH078948), the Robert A. Welch Foundation, and the Texas Higher Education Science and Technology Acquisition and Retention (STAR) Award for support of my laboratory's research on metalloproteases. I am also most grateful to Drs. Janelle L. Lauer-Fields, Trista Robichaud, and Margaret Ndinguri for constructing the new figures in this manuscript.References
- J. Myllyharju and K. I. Kivirikko, Ann. Med., 2001, 33, 7–21 Search PubMed.
- T. Hashimoto, T. Wakabayashi, A. Watanabe, H. Kowa, R. Hosoda, A. Nakamura, I. Kanazawa, T. Arai, K. Takio, D. M. A. Mann and T. Iwatsubo, EMBO J., 2002, 21, 1524–1534 CrossRef CAS.
- C. Boulégue, H.-J. Musiol, M. G. Götz, C. Renner and L. Moroder, Antioxidants Redox Signaling, 2008, 10, 113–125 Search PubMed.
- M. K. Gordon and R. A. Hahn, Cell Tissue Res., 2010, 339, 247–257 CrossRef CAS.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS.
- W. G. Cole, Prog. Nucl. Acid Res. Mol. Biol., 1994, 47, 29–80 Search PubMed.
- B. Brodsky and N. K. Shah, FASEB J., 1995, 9, 1537–1546 CAS.
- B. R. Olsen and Y. Ninomiya, in Guidebook to the Extracellular Matrix, Anchor, and Adhesion Proteins, 2nd Edition, ed., T. Kreis and R. Vale, Oxford University Press, Oxford, U.K., 1999, pp. 380–383 Search PubMed.
- G. B. Fields and D. J. Prockop, Biopolymers (Pept. Sci.), 1996, 40, 345–357 CrossRef CAS.
- C. L. Jenkins and R. T. Raines, Nat. Prod. Rep., 2002, 19, 49–59 RSC.
- T. Koide, Connective Tissue Res., 2005, 46, 131–141 CrossRef CAS.
- T. Koide, Phil. Trans. R. Soc. B, 2007, 362, 1281–1291 CrossRef CAS.
- B. Brodsky, G. Thiagarajan, B. Madhan and K. Kar, Biopolymers, 2008, 89, 345–353 CrossRef CAS.
- Y.-C. Yu, P. Berndt, M. Tirrell and G. B. Fields, J. Am. Chem. Soc., 1996, 118, 12515–12520 CrossRef CAS.
- Y.-C. Yu, M. Tirrell and G. B. Fields, J. Am. Chem. Soc., 1998, 120, 9979–9987 CrossRef CAS.
- J. Ottl, H. J. Musiol and L. Moroder, J. Pept. Sci., 1999, 5, 103–110 CrossRef CAS.
- B. Saccá and L. Moroder, J. Pept. Sci., 2002, 8, 192–204 CrossRef CAS.
- N. B. Malkar, J. L. Lauer-Fields, D. Juska and G. B. Fields, Biomacromolecules, 2003, 4, 518–528 CrossRef CAS.
- C. G. Knight, C. M. Onley and R. W. Farndale, in Methods in Molecular Biology, Volume 273 (Platelets and Megakaryocytes, Volume 2: Perspectives and Techniques), ed., J. M. Gibbins and M. P. Mahaut-Smith, Humana Press, Totowa, NJ, 2004, pp. 349–363 Search PubMed.
- H. Yamada, T. Sasaki, S. Niwa, T. Oishi, M. Murata, T. Kawakami and S. Aimoto, Bioorg. Med. Chem. Lett., 2004, 14, 5677–5680 CrossRef CAS.
- N. K. Shah, J. A. M. Ramshaw, A. Kirkpatrick, C. Shah and B. Brodsky, Biochemistry, 1996, 35, 10262–10268 CrossRef CAS.
- A. V. Persikov, J. A. M. Ramshaw and B. Brodsky, Biopolymers (Pept. Sci.), 2000, 55, 436–450 CrossRef CAS.
- A. V. Persikov, J. A. M. Ramshaw, A. Kirkpatrick and B. Brodsky, Biochemistry, 2000, 39, 14960–14967 CrossRef CAS.
- V. Guaba and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 2683–2690 CrossRef CAS.
- V. Gauba and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 15034–15041 CrossRef CAS.
- J. A. Fallas, V. Gauba and J. D. Hartgerink, J. Biol. Chem., 2009, 284, 26851–26859 CrossRef CAS.
- J. A. Hodges and R. T. Raines, J. Am. Chem. Soc., 2005, 127, 15923–15932 CrossRef CAS.
- D. A. Slatter, C. A. Miles and A. J. Bailey, J. Mol. Biol., 2003, 329, 175–183 CrossRef CAS.
- R. Berisio, V. Granata, L. Vitagliano and A. Zagari, Biopolymers, 2004, 73, 682–688 CrossRef CAS.
- B. Madhan, J. Xiao, G. Thiagarajan, J. Baum and B. Brodsky, J. Am. Chem. Soc., 2008, 130, 13520–13521 CrossRef CAS.
- P. Berndt, G. B. Fields and M. Tirrell, J. Am. Chem. Soc., 1995, 117, 9515–9522 CrossRef CAS.
- Y.-C. Yu, V. Roontga, V. A. Daragan, K. H. Mayo, M. Tirrell and G. B. Fields, Biochemistry, 1999, 38, 1659–1668 CrossRef CAS.
- G. B. Fields, J. L. Lauer, Y. Dori, P. Forns, Y.-C. Yu and M. Tirrell, Biopolymers, 1998, 47, 143–151 CrossRef CAS.
- J. L. Lauer-Fields, K. A. Tuzinski, K. Shimokawa, H. Nagase and G. B. Fields, J. Biol. Chem., 2000, 275, 13282–13290 CrossRef CAS.
- J. L. Lauer-Fields, H. Nagase and G. B. Fields, J. Chromatogr. A., 2000, 890, 117–125 CrossRef CAS.
- J. L. Lauer-Fields, T. Broder, T. Sritharan, H. Nagase and G. B. Fields, Biochemistry, 2001, 40, 5795–5803 CrossRef CAS.
- N. B. Malkar, J. L. Lauer-Fields, J. A. Borgia and G. B. Fields, Biochemistry, 2002, 41, 6054–6064 CrossRef CAS.
- S. M. Sweeney, G. DiLullo, S. J. Slater, J. Martinez, R. V. Iozzo, J. L. Lauer-Fields, G. B. Fields and J. D. San Antonio, J. Biol. Chem., 2003, 278, 30516–30524 CrossRef CAS.
- D. Minond, J. L. Lauer-Fields, H. Nagase and G. B. Fields, Biochemistry, 2004, 43, 11474–11481 CrossRef CAS.
- D. Minond, J. L. Lauer-Fields, M. Cudic, C. M. Overall, D. Pei, K. Brew, R. Visse, H. Nagase and G. B. Fields, J. Biol. Chem., 2006, 281, 38302–38313 CrossRef CAS.
- D. Minond, J. L. Lauer-Fields, M. Cudic, C. M. Overall, D. Pei, K. Brew, M. L. Moss and G. B. Fields, Biochemistry, 2007, 46, 3724–3733 CrossRef CAS.
- S.-G. Lee, J. Y. Lee and J. Chmielewski, Angew. Chem., Int. Ed., 2008, 47, 8429–8432 CrossRef CAS.
- I. R. Babu and K. N. Ganesh, J. Am. Chem. Soc., 2001, 123, 2079–2080 CrossRef CAS.
- M. Umashankara, I. R. Babu and K. N. Ganesh, Chem. Commun., 2003, 2606–2607 RSC.
- U. Kusebauch, S. A. Cadamuro, H.-J. Musiol, L. Moroder and C. Renner, Chem.–Eur. J., 2007, 13, 2966–2973 CrossRef CAS.
- C. G. Fields, D. J. Mickelson, S. L. Drake, J. B. McCarthy and G. B. Fields, J. Biol. Chem., 1993, 268, 14153–14160 CAS.
- C. G. Fields, C. M. Lovdahl, A. J. Miles, V. L. Matthias-Hagen and G. B. Fields, Biopolymers, 1993, 33, 1695–1707 CrossRef CAS.
- B. Grab, A. J. Miles, L. T. Furcht and G. B. Fields, J. Biol. Chem., 1996, 271, 12234–12240 CrossRef CAS.
- G. B. Fields and C. G. Fields, in Innovation and Perspectives in Solid Phase Synthesis: Peptides, Polypeptides, and Oligonucleotides, ed., R. Epton, Intercept, Andover, UK, 1992, pp. 153–162 Search PubMed.
- C. G. Fields, B. Grab, J. L. Lauer and G. B. Fields, Anal. Biochem., 1995, 231, 57–64 CrossRef CAS.
- C. L. Jenkins, L. E. Bretscher, I. A. Guzei and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 6422–6427 CrossRef CAS.
- K. M. Thomas, D. Naduthambi, G. Tririya and N. J. Zondlo, Org. Lett., 2005, 7, 2397–2400 CrossRef CAS.
- N. Jiravanichanun, N. Nishino and K. Okuyama, Biopolymers, 2006, 81, 225–233 CrossRef CAS.
- M. D. Shoulders, I. A. Guzei and R. T. Raines, Biopolymers, 2007, 89, 443–454.
- M. D. Shoulders, K. J. Kamer and R. T. Raines, Bioorg. Med. Chem. Lett., 2009, 19, 3859–3862 CrossRef CAS.
- J. Broddefalk, J. Bäcklund, F. Almqvist, M. Johansson, R. Holmdahl and J. Kihlberg, J. Am. Chem. Soc., 1998, 120, 7676–7683 CrossRef CAS.
- J. Broddefalk, M. Forsgren, I. Sethson and J. Kihlberg, J. Org. Chem., 1999, 64, 8948–8953 CrossRef CAS.
- B. Holm, J. Broddefalk, S. Flodell, E. Wellner and J. Kihlberg, Tetrahedron, 2000, 56, 1579–1586 CrossRef CAS.
- N. B. Malkar, J. L. Lauer-Fields and G. B. Fields, Tetrahedron Lett., 2000, 41, 1137–1140 CrossRef CAS.
- J. Marin, A. Violette, J.-P. Briand, G. Guichard and Eur, J. Org. Chem., 2004, 3027–3039 CAS.
- M. Cudic, J. L. Lauer-Fields and G. B. Fields, J. Pept. Res., 2005, 65, 272–283 CrossRef CAS.
- J. L. Lauer-Fields, N. B. Malkar, G. Richet, K. Drauz and G. B. Fields, J. Biol. Chem., 2003, 278, 14321–14330 CrossRef CAS.
- D. Baronas-Lowell, J. L. Lauer-Fields and G. B. Fields, J. Liq. Chromatogr. Rel. Technol., 2003, 26, 2225–2254 CrossRef CAS.
- S. Thakur, D. Vadolas, H. P. Germann and E. Heidemann, Biopolymers, 1986, 25, 1081–1086 CrossRef CAS.
- H. P. Germann and E. Heidermann, Biopolymers, 1988, 27, 157–163 CrossRef CAS.
- S. R. Chhabra, B. Hothi, D. J. Evans, P. D. White, B. W. Bycroft and W. C. Chan, Tetrahedron Lett., 1998, 39, 1603–1606 CrossRef CAS.
- A. J. Miles, A. P. N. Skubitz, L. T. Furcht and G. B. Fields, J. Biol. Chem., 1994, 269, 30939–30945 CAS.
- C. G. Fields, B. Grab, J. L. Lauer, A. J. Miles, Y.-C. Yu and G. B. Fields, Lett. Pept. Sci., 1996, 3, 3–16 CrossRef CAS.
- J. L. Lauer, C. M. Gendron and G. B. Fields, Biochemistry, 1998, 37, 5279–5287 CrossRef CAS.
- S. M. Sweeney, C. A. Guy, G. B. Fields and J. D. San Antonio, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7275–7280 CrossRef CAS.
- M. J. Barnes, C. G. Knight and R. W. Farndale, Biopolymers (Pept. Sci.), 1996, 40, 383–397 CrossRef CAS.
- L. F. Morton, A. R. Peachey, C. G. Knight, R. W. Farndale and M. J. Barnes, J. Biol. Chem., 1997, 272, 11044–11048 CrossRef CAS.
- W. Henkel, T. Vogl, H. Echner, W. Voelter, C. Urbanke, D. Schleuder and J. Rauterberg, Biochemistry, 1999, 38, 13610–13622 CrossRef CAS.
- C. G. Knight, L. F. Morton, D. J. Onley, A. R. Peachey, T. Ichinohe, M. Okuma, R. W. Farndale and M. J. Barnes, Cardiovascular Res., 1999, 41, 450–457 Search PubMed.
- H. Hojo, Y. Akamatsu, K. Yamauchi, M. Kinoshita, S. Miki and Y. Nakamura, Tetrahedron, 1997, 53, 14263–14274 CrossRef CAS.
- Y. Tanaka, K. Suzuki and T. Tanaka, J. Pept. Res., 1998, 51, 413–419 CAS.
- S. T. Khew and Y. W. Tong, Biochemistry, 2008, 47, 585–596 CrossRef CAS.
- J. Ottl and L. Moroder, J. Am. Chem. Soc., 1999, 121, 653–661 CrossRef CAS.
- J. Ottl, D. Gabriel, G. Murphy, V. Knäuper, Y. Tominaga, H. Nagase, M. Kröger, H. Tschesche, W. Bode and L. Moroder, Chem. Biol., 2000, 7, 119–132 CrossRef CAS.
- M. Goodman, Y. Feng, G. Melacini and J. P. Taulane, J. Am. Chem. Soc., 1996, 118, 5156–5157 CrossRef CAS.
- M. Goodman, G. Melacini and Y. Feng, J. Am. Chem. Soc., 1996, 118, 10928–10929 CrossRef CAS.
- Y. Feng, G. Melacini, J. P. Taulane and M. Goodman, Biopolymers, 1996, 39, 859–872 CrossRef CAS.
- Y. Feng, G. Melacini and M. Goodman, Biochemistry, 1997, 36, 8716–8724 CrossRef CAS.
- E. T. Rump, D. T. S. Rijkers, H. W. Hilbers, P. G. de Groot and R. M. J. Liskamp, Chem.–Eur. J., 2002, 8, 4613–4621 CrossRef CAS.
- J. Kwak, A. De Capua, E. Locardi and M. Goodman, J. Am. Chem. Soc., 2002, 124, 14085–14091 CrossRef CAS.
- J. C. Horng, A. J. Hawk, Q. Zhao, E. S. Benedict, S. D. Burke and R. T. Raines, Org. Lett., 2006, 8, 4735–4738 CrossRef CAS.
- W. Cai, D. Wong, G. A. Kinberger, S. W. Kwok, J. P. Taulane and M. Goodman, Bioorg. Chem., 2007, 35, 327–337 CrossRef CAS.
- T. Tanaka, A. Nishikawa, Y. Tanaka, H. Nakamura, T. Kodama, T. Imanishi and T. Doi, Protein Eng., 1996, 9, 307–313 CrossRef CAS.
- J. Ottl, R. Battistuta, M. Pieper, H. Tschesche, W. Bode, K. Kühn and L. Moroder, FEBS Lett., 1996, 398, 31–36 CrossRef CAS.
- J. C. D. Müller, J. Ottl and L. Moroder, Biochemistry, 2000, 39, 5111–5116 CrossRef CAS.
- B. Saccá, S. Fiori and L. Moroder, Biochemistry, 2003, 42, 3429–3436 CrossRef CAS.
- T. Koide, Y. Nishikawa and Y. Takahara, Bioorg. Med. Chem. Lett., 2004, 14, 125–128 CrossRef CAS.
- D. Barth, O. Kyrieleis, S. Frank, C. Renner and L. Moroder, Chem.–Eur. J., 2003, 9, 3703–3714 CrossRef CAS.
- O. D. Krishna and K. L. Kiick, Biomacromolecules, 2009, 10, 2626–2631 CrossRef CAS.
- D. A. Slatter, L. A. Foleya, A. R. Peacheya, D. Nietlispacha and R. W. Farndale, J. Mol. Biol., 2006, 359, 289–298 CrossRef CAS.
- Y. Feng, G. Melacini, J. P. Taulane and M. Goodman, J. Am. Chem. Soc., 1996, 118, 10351–10358 CrossRef CAS.
- W. Cai, S. W. Kwok, J. P. Taulane and M. Goodman, J. Am. Chem. Soc., 2004, 126, 15030–15031 CrossRef CAS.
- G. A. Kinberger, J. P. Taulane and M. Goodman, Inorg. Chem., 2006, 45, 961–963 CrossRef CAS.
- K. Kar, Y.-H. Wang and B. Brodsky, Protein Sci., 2008, 17, 1086–1095 CrossRef CAS.
- M. A. Cejas, W. A. Kinney, C. Chen, G. C. Leo, B. A. Tounge, J. G. Vinter, P. P. Joshi and B. E. Maryanoff, J. Am. Chem. Soc., 2007, 129, 2202–2203 CrossRef CAS.
- M. A. Cejas, W. A. Kinney, C. Chen, J. G. Vinter, H. R. Almond Jr., K. M. Balss, C. A. Maryanoff, U. Schmidt, M. Breslav, A. Mahan, E. Lacy and B. E. Maryanoff, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8513–8518 CrossRef CAS.
- K. Kar, S. Ibrar, V. Nanda, T. M. Getz, S. P. Kunapuli and B. Brodsky, Biochemistry, 2009, 48, 7959–7968 CrossRef CAS.
- S. Rele, Y. Song, R. P. Apkarian, Z. Qu, V. P. Conticello and E. L. Chaikof, J. Am. Chem. Soc., 2007, 129, 14780–14787 CrossRef CAS.
- M. M. Pires and J. Chmielewski, J. Am. Chem. Soc., 2009, 131, 2706–2712 CrossRef CAS.
- L. F. Morton, I. Y. McCulloch and M. J. Barnes, Thrombosis Res., 1993, 72, 367–372 CrossRef CAS.
- L. F. Morton, P. G. Hargreaves, R. W. Farndale, R. D. Young and M. J. Barnes, Biochem. J., 1995, 306, 337–344 CAS.
- J. Asselin, J. M. Gibbins, M. Achison, Y. H. Lee, L. F. Morton, R. W. Farndale, M. J. Barnes and S. P. Watson, Blood, 1997, 89, 1235–1242 CAS.
- M. W. Verkleij, L. F. Morton, C. G. Knight, P. G. de Groot, M. J. Barnes and J. J. Sixma, Blood, 1998, 91, 3808–3816 CAS.
- C. G. Knight, L. F. Morton, D. J. Onley, A. R. Peachey, A. J. Messent, P. A. Smethurst, D. S. Tuckwell, R. W. Farndale and M. J. Barnes, J. Biol. Chem., 1998, 273, 33287–33294 CrossRef CAS.
- J. W. M. Heemskerk, P. Siljander, W. M. J. Vuist, G. Breikers, C. P. M. Reutelingsperger, M. J. Barnes, C. G. Knight, R. Lassila and R. W. Farndale, Thromb. Haemost., 1999, 81, 782–792 Search PubMed.
- J. Asselin, C. G. Knight, R. W. Farndale, M. J. Barnes and S. P. Watson, Biochem. J., 1999, 339, 413–418 CrossRef CAS.
- M. W. Verkleij, M. J. W. Ijsseldijk, G. J. Heijnen-Snyder, E. G. Huizinga, L. F. Morton, C. G. Knight, J. J. Sixma, P. G. de Groot and M. J. Barnes, Thromb. Haemost., 1999, 82, 1137–1144 Search PubMed.
- P. A. Smethurst, D. J. Onley, G. E. Jarvis, M. N. O'Connor, C. G. Knight, A. B. Herr, W. H. Ouwehand and R. W. Farndale, J. Biol. Chem., 2007, 282, 1296–1304 CAS.
- G. E. Jarvis, N. Raynal, J. P. Langford, D. J. Onley, A. Andrews, P. A. Smethurst and R. W. Farndale, Blood, 2008, 111, 4986–4996 CrossRef CAS.
- S. E. Paramonov, V. Gauba and J. D. Hartgerink, Macromolecules, 2005, 38, 7555–7561 CrossRef CAS.
- T. Koide, D. L. Homma, S. Asada and K. Kitagawa, Bioorg. Med. Chem. Lett., 2005, 15, 5230–5233 CrossRef CAS.
- F. W. Kotch and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3028–3033 CrossRef CAS.
- C. M. Yamazaki, S. Asada, K. Kitagawa and T. Koide, Biopolymers (Pept. Sci.), 2008, 90, 816–823 CrossRef CAS.
- T. Kishimoto, Y. Morihara, M. Osanai, S.-i. Ogata, M. Kamitakahara, C. Ohtsuki and M. Tanihara, Biopolymers, 2005, 79, 163–172 CrossRef CAS.
- D. E. Przybyla and J. Chmielewski, J. Am. Chem. Soc., 2008, 130, 12610–12611 CrossRef CAS.
- M. M. Pires, D. E. Przybyla and J. Chmielewski, Angew. Chem., Int. Ed., 2009, 48, 7813–7817 CrossRef CAS.
- R. L. Martin, L. Waldmann and D. L. Kaplan, Biopolymers, 2003, 70, 435–444 CrossRef CAS.
- G. A. Kinberger, W. Cai and M. Goodman, J. Am. Chem. Soc., 2002, 124, 15162–15163 CrossRef CAS.
- S. T. Khew, Q. J. Yang and Y. W. Tong, Biomaterials, 2008, 29, 3034–3045 CrossRef CAS.
- R. Berisio, A. De Simone, A. Ruggiero, R. Improta and L. Vitagliano, J. Pept. Sci., 2008, 15, 131–140.
- K. Okuyama, G. Wu, N. Jiravanichanun, C. Hongo and K. Noguchi, Biopolymers (Pept. Sci.), 2006, 84, 421–432 CrossRef CAS.
- K. Okuyama, X. Xu, M. Iguchi and K. Noguchi, Biopolymers (Pept. Sci.), 2006, 84, 181–191 CrossRef CAS.
- S. P. Boudko, J. Engel, K. Okuyama, K. Mizuno, H. P. Bächinger and M. A. Schumacher, J. Biol. Chem., 2008, 283, 32580–32589 CrossRef CAS.
- J. Bella, M. Eaton, B. Brodsky and H. M. Berman, Science, 1994, 266, 75–81 CAS.
- C. L. Jenkins, M. M. Vasbinder, S. J. Miller and R. T. Raines, Org. Lett., 2005, 7, 2619–2622 CrossRef CAS.
- N. Dai, X. J. Wang and F. A. Etzkorn, J. Am. Chem. Soc., 2008, 130, 5396–5397 CrossRef CAS.
- N. Dai and F. A. Etzkorn, J. Am. Chem. Soc., 2009, 131, 13728–13732 CrossRef CAS.
- S. K. Holmgren, K. M. Taylor, L. E. Bretscher and R. T. Raines, Nature, 1998, 392, 666–667 CrossRef CAS.
- S. K. Holmgren, L. E. Bretscher, K. M. Taylor and R. T. Raines, Chem. Biol., 1999, 6, 63–70 CrossRef CAS.
- L. E. Bretscher, C. L. Jenkins, K. M. Taylor, M. L. DeRider and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 777–778 CrossRef CAS.
- E. S. Eberhardt, N. Panasik, Jr. and R. T. Raines, J. Am. Chem. Soc., 1996, 118, 12261–12266 CrossRef CAS.
- M. L. DeRider, S. J. Wilkens, M. J. Waddell, L. E. Bretscher, F. Weinhold, R. T. Raines and J. L. Markley, J. Am. Chem. Soc., 2002, 2497–2505 CrossRef CAS.
- C. L. Jenkins, G. Lin, J. Duo, D. Rapolu, I. A. Guzei, R. T. Raines and G. R. Krow, J. Org. Chem., 2004, 69, 8565–8573 CrossRef CAS.
- N. Panasik Jr., E. S. Eberhardt, A. S. Edison, D. R. Powell and R. T. Raines, Int. J. Pept. Protein Res., 1994, 44, 262–269 CAS.
- L. Vitagliano, R. Berisio, A. Mastrangelo, L. Mazzarella and A. Zagari, Protein Sci., 2001, 10, 2627–2632 CAS.
- R. T. Raines, in Peptides for Youth - Proceedings of the Twentieth American Peptide Symposium, June 25-30, 2007, Montreal, Canada, ed., E. Escher, W. D. Lubell and S. Del Valle, Springer, New York, 2009, pp. xci–xcviii Search PubMed.
- M. Doi, Y. Nishi, S. Uchiyama, Y. Nishiuchi, T. Nakazawa, T. Ohkubo and Y. Kobayashi, J. Am. Chem. Soc., 2003, 125, 9922–9923 CrossRef CAS.
- Y. Nishi, S. Uchiyama, M. Doi, Y. Nishiuchi, T. Nakazawa, T. Ohkubo and Y. Kobayashi, Biochemistry, 2005, 44, 6034–6042 CrossRef CAS.
- A. V. Persikov, J. A. M. Ramshaw and B. Brodsky, J. Biol. Chem., 2005, 280, 19343–19349 CrossRef CAS.
- A. V. Persikov, J. A. M. Ramshaw, A. Kirkpatrick and B. Brodsky, J. Am. Chem. Soc., 2003, 125, 11500–11501 CrossRef CAS.
- M. G. Venugopal, J. A. M. Ramshaw, E. Braswell, D. Zhu and B. Brodsky, Biochemistry, 1994, 33, 7948–7956 CrossRef CAS.
- A. V. Persikov, J. A. M. Ramshaw, A. Kirkpatrick and B. Brodsky, J. Mol. Biol., 2002, 316, 385–394 CrossRef.
- A. V. Persikov, J. A. M. Ramshaw, A. Kirkpatrick and B. Brodsky, Biochemistry, 2005, 44, 1414–1422 CrossRef CAS.
- W. Yang, V. C. Chan, A. Kirkpatrick, J. A. M. Ramshaw and B. Brodsky, J. Biol. Chem., 1997, 272, 28837–28840 CrossRef CAS.
- A. Mohs, T. Silva, T. Yoshida, R. Amin, S. Lukomski, M. Inouye and B. Brodsky, J. Biol. Chem., 2007, 282, 29757–29765 CrossRef CAS.
- N. K. Shah, B. Brodsky, A. Kirkpatrick and J. A. M. Ramshaw, Biopolymers, 1999, 49, 297–302 CrossRef CAS.
- J. L. Lauer-Fields, P. Kele, G. Sui, H. Nagase, R. M. Leblanc and G. B. Fields, Anal. Biochem., 2003, 321, 105–115 CrossRef CAS.
- J. G. Bann, D. H. Peyton and H. P. Bächinger, FEBS Lett., 2000, 473, 237–240 CrossRef.
- J. G. Bann and H. P. Bächinger, J. Biol. Chem., 2000, 275, 24466–24469 CrossRef CAS.
- J. G. Bann, H. P. Bächinger and D. H. Peyton, Biochemistry, 2003, 42, 4042–4048 CrossRef CAS.
- J.-C. Horng, F. W. Kotch and R. T. Raines, Protein Sci., 2007, 16, 208–215 CAS.
- E. A. Kersteen and R. T. Raines, Biopolymers, 2001, 59, 24–28 CrossRef CAS.
- J. Kwak, E. A. Jefferson, M. Bhumralkar and M. Goodman, Bioorg. Med. Chem., 1999, 7, 153–160 CrossRef CAS.
- K. Mizuno, T. Hayashi and H. P. Bachinger, J. Biol. Chem., 2003, 278, 32373–32379 CrossRef CAS.
- K. Simon-Lukasik, A. V. Persikov, B. Brodsky, J. A. M. Ramshaw, W. R. Laws, J. B. A. Ross and R. D. Ludescher, Biophys. J., 2003, 84, 501–508 CrossRef CAS.
- M. S. Ackerman, M. Bhate, N. Shenoy, K. Beck, J. A. M. Ramshaw and B. Brodsky, J. Biol. Chem., 1999, 274, 7668–7673 CrossRef CAS.
- B. Saccá, C. Renner and L. Moroder, J. Mol. Biol., 2002, 324, 309–318 CrossRef CAS.
- D. J. Prockop and K. I. Kivirikko, Ann. Rev. Biochem., 1995, 64, 403–434 CrossRef CAS.
- J. Baum and B. Brodsky, Curr. Opin. Struct. Biol., 1999, 9, 122–128 CrossRef CAS.
- H. Kuivaniemi, G. Tromp and D. J. Prockop, Human Mutation, 1997, 9, 300–315 CrossRef CAS.
- G. A. Di Lullo, S. M. Sweeney, J. Körkkö, L. Ala-Kokko and J. D. San Antonio, J. Biol. Chem., 2002, 277, 4223–4231 CrossRef CAS.
- M. M. Cohen Jr., Am. J. Med. Gen., 2002, 112, 304–313 Search PubMed.
- B. Brodsky and J. Baum, Nature, 2008, 453, 998–999 CrossRef CAS.
- C. G. Long, M. H. Li, J. Baum and B. Brodsky, J. Mol. Biol., 1992, 225, 1–4 CrossRef CAS.
- B. Brodsky, N. H. Li, C. G. Long, J. Apigo and J. Baum, Biopolymers, 1992, 32, 447–451 CrossRef CAS.
- C. G. Long, E. Braswell, D. Zhu, J. Apigo, J. Baum and B. Brodsky, Biochemistry, 1993, 32, 11688–11695 CrossRef CAS.
- X. Liu, D. L. Siegel, P. Fan, B. Brodsky and J. Baum, Biochemistry, 1996, 35, 4306–4313 CrossRef CAS.
- X. Liu, S. Kim, Q.-H. Dai, B. Brodsky and J. Baum, Biochemistry, 1998, 37, 15528–15533 CrossRef CAS.
- A. V. Buevich, Q.-H. Dai, X. Liu, B. Brodsky and J. Baum, Biochemistry, 2000, 39, 4299–4308 CrossRef CAS.
- M. Bhate, X. Wang, J. Baum and B. Brodsky, Biochemistry, 2002, 41, 6539–6547 CrossRef CAS.
- J. Bella, B. Brodsky and H. M. Berman, Connect. Tissue Res., 1996, 35, 401–406 CrossRef CAS.
- W. Yang, M. L. Battineni and B. Brodsky, Biochemistry, 1997, 36, 6930–6935 CrossRef CAS.
- K. Beck, V. C. Chan, N. Shenoy, A. Kirkpatrick, J. A. M. Ramshaw and B. Brodsky, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4273–4278 CrossRef CAS.
- D. L. Bodian, B. Madhan, B. Brodsky and T. E. Klein, Biochemistry, 2008, 47, 5424–5432 CrossRef CAS.
- A. V. Buevich, T. Silva, B. Brodsky and J. Baum, J. Biol. Chem., 2004, 279, 46890–46895 CrossRef CAS.
- T. J. Hyde, M. Bryan, B. Brodsky and J. Baum, J. Biol. Chem., 2006, 281, 36937–36943 CrossRef CAS.
- V. Gauba and J. D. Hartgerink, J. Am. Chem. Soc., 2008, 130, 7509–7515 CrossRef CAS.
- Y. Li, B. Brodsky and J. Baum, J. Biol. Chem., 2009, 284, 20660–20667 CrossRef CAS.
- P. Xu, J. Huang, P. Cebe and D. L. Kaplan, Biomacromolecules, 2008, 9, 1551–1557 CrossRef CAS.
- A. Mohs, M. Popiel, Y. Li, J. Baum and B. Brodsky, J. Biol. Chem., 2006, 281, 17197–17202 CrossRef CAS.
- Y. Li, B. Brodsky and J. Baum, J. Biol. Chem., 2007, 282, 22699–22706 CrossRef CAS.
- G. Thiagarajan, Y. Li, A. Mohs, C. Strafaci, M. Popiel, J. Baum and B. Brodsky, J. Mol. Biol., 2008, 376, 736–748 CrossRef CAS.
- J. Bella, J.-J. Liu, R. Kramer, B. Brodsky and H. Berman, J. Mol. Biol., 2006, 362, 298–311 CrossRef CAS.
- A. Mohs, Y. Li, E. Doss-Pepe, J. Baum and B. Brodsky, Biochemistry, 2005, 44, 1793–1799 CrossRef CAS.
- G. B. Fields, Connect. Tissue Res., 1995, 31, 235–243 CrossRef CAS.
- R. W. Farndale, T. Lisman, D. Bihan, S. Hamaia, C. S. Smerling, N. Pugh, A. Konitsiotis, B. Leitinger, P. G. de Groot, G. E. Jarvis and N. Raynal, Biochem. Soc. Trans., 2008, 36, 241–250 CrossRef CAS.
- K. Kühn and J. Eble, Trends Cell Biol., 1994, 4, 256–261 CrossRef CAS.
- A. Van Der Flier and A. Sonnenberg, Cell Tissue Res., 2001, 305, 285–298 CrossRef CAS.
- C. G. Knight, L. F. Morton, A. R. Peachey, D. S. Tuckwell, R. W. Farndale and M. J. Barnes, J. Biol. Chem., 2000, 275, 35–40 CrossRef CAS.
- J. Emsley, C. G. Knight, R. W. Farndale, M. J. Barnes and R. C. Liddington, Cell, 2000, 101, 47–56 CrossRef CAS.
- Y. Xu, S. Gurusiddappa, R. L. Rich, R. T. Owens, D. R. Keene, R. Mayne, A. Höök and M. Höök, J. Biol. Chem., 2000, 275, 38981–38989 CrossRef CAS.
- W.-M. Zhang, J. Käpylä, J. S. Puranen, C. G. Knight, C.-F. Tiger, O. T. Pentikäinen, M. S. Johnson, R. W. Farndale, J. Heino and D. Gullberg, J. Biol. Chem., 2003, 278, 7270–7277 CrossRef CAS.
- D. Baronas-Lowell, J. L. Lauer-Fields and G. B. Fields, J. Biol. Chem., 2004, 279, 952–962 CAS.
- J. Bella and H. M. Berman, Structure, 2000, 8, R121–R126 CrossRef CAS.
- J. Emsley, C. G. Knight, R. W. Farndale and M. J. Barnes, J. Mol. Biol., 2004, 335, 1019–1028 CrossRef CAS.
- J. L. Lauer-Fields, T. Sritharan, M. S. Stack, H. Nagase and G. B. Fields, J. Biol. Chem., 2003, 278, 18140–18145 CrossRef CAS.
- D. Baronas-Lowell, J. L. Lauer-Fields, J. A. Borgia, G. F. Sferrazza, M. Al-Ghoul, D. Minond and G. B. Fields, J. Biol. Chem., 2004, 279, 43503–43513 CrossRef CAS.
- J. K. Kim, Y. Xu, X. Xu, D. R. Keene, S. Gurusiddappa, X. Liang, K. K. Wary and M. Höök, J. Biol. Chem., 2005, 280, 32512–32520 CrossRef CAS.
- N. Raynal, S. W. Hamaia, P. R.-M. Siljander, B. Maddox, A. R. Peachey, R. Fernandez, L. J. Foley, D. A. Slatter, G. E. Jarvis and R. W. Farndale, J. Biol. Chem., 2006, 281, 3821–3831 CrossRef CAS.
- C. C. Caswell, M. Barczyk, D. R. Keene, E. Lukomska, D. Gullberg and S. Lukomski, J. Biol. Chem., 2008, 283, 36168–36175 CrossRef CAS.
- A. B. Herr and R. W. Farndale, J. Biol. Chem., 2009, 284, 19781–19785 CrossRef CAS.
- J. Eble, R. Golbik, K. Mann and K. Kuhn, EMBO J., 1993, 12, 4795–4802 CAS.
- R. Golbik, J. A. Eble, A. Ries and K. Kühn, J. Mol. Biol., 2000, 297, 501–509 CrossRef CAS.
- B. Saccá, E.-K. Sinner, J. Kaiser, C. Lübken, J. A. Eble and L. Moroder, ChemBioChem, 2002, 9, 904–907 CrossRef.
- C. Renner, B. Saccá and L. Moroder, Biopolymers (Pept. Sci.), 2004, 76, 34–47 CrossRef CAS.
- L. Barth, E. K. Sinner, S. A. Cadamuro, C. Renner, D. Oesterhelt and L. Moroder, in Peptides For Youth: The Proceedings of the 20th American Peptide Symposium, ed., E. Escher, W. D. Lubell and S. Del Valle, Springer Science, New York, 2009, pp. 295–296 Search PubMed.
- S. Perret, J. A. Eble, P. R.-M. Siljander, C. Merle, R. W. Farndale, M. Theisen and F. Ruggiero, J. Biol. Chem., 2003, 278, 29873–29879 CrossRef CAS.
- S. A. Santoro, M. M. Zutter, J. E. Wu, W. D. Staatz, E. U. M. Saelman and P. J. Keely, Meth. Enzymol., 1994, 245, 147–183 CAS.
- A. J. Miles, J. R. Knutson, A. P. N. Skubitz, L. T. Furcht, J. B. McCarthy and G. B. Fields, J. Biol. Chem., 1995, 270, 29047–29050 CrossRef CAS.
- C. Li, J. B. McCarthy, L. T. Furcht and G. B. Fields, Biochemistry, 1997, 36, 15404–15410 CrossRef CAS.
- D. Naor, S. Nedvetzki, I. Golan, L. Melnik and Y. Faitelson, Crit. Rev. Clin. Lab. Sci., 2002, 39, 527–579 Search PubMed.
- M. Goebeler, D. Kaufmann, E. B. Brocker and C. E. Klein, J. Cell Sci., 1996, 109, 1957–1964 CAS.
- M. E. Prince, R. Sivanandan, A. Kaczorowski, G. T. Wolf, M. J. Kaplan, P. Dalerba, I. L. Weissman, M. F. Clarke and L. E. Ailles, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 973–978 CrossRef CAS.
- M. P. Ponnusamy and S. K. Batra, J. Ovarian Res., 2008, 1, 4 Search PubMed.
- L. Du, H. Wang, L. He, J. Zhang, B. Ni, X. Wang, H. Jin, N. Cahuzac, M. Mehrpour, Y. Lu and Q. Chen, Clin. Cancer Res., 2008, 14, 6751–6760 CrossRef CAS.
- L. Y. W. Bourguigon, K. Peyrollier, W. Xia and E. Gilad, J. Biol. Chem., 2008, 283, 17635–17651 CrossRef.
- M. K. Chelberg, J. B. McCarthy, A. P. N. Skubitz, L. T. Furcht and E. C. Tsilibary, J. Cell Biol., 1990, 111, 261–270 CrossRef CAS.
- K. H. Mayo, D. Parra-Diaz, J. B. McCarthy and M. Chelberg, Biochemistry, 1991, 30, 8251–8267 CrossRef CAS.
- Y.-C. Yu, T. Pakalns, Y. Dori, J. B. McCarthy, M. Tirrell and G. B. Fields, Meth. Enzymol., 1997, 289, 571–587 CAS.
- D. J. Mickelson, A. E. Faassen and J. B. McCarthy, J. Cell Biol., 1991, 115, 287a.
- J. R. Knutson, G. B. Fields, J. Iida, A. J. Miles and J. B. McCarthy, Proc. Am. Assoc. Cancer Res., 1995, 36, 68.
- J. Dennis, C. Waller, R. Timpl and V. Schirrmacher, Nature, 1982, 300, 274–276 CrossRef CAS.
- X. Bao, M. Kobayashi, S. Hatakeyama, K. Angata, D. Gullberg, J. Nakayama, M. N. Fukuda and M. Fukuda, Proc. Natl. Acad. Sci. U. S. A., 2009 Search PubMed in press.
- F. Alves, W. Vogel, K. Mossie, B. Millauer, H. Höfler and A. Ulinch, Oncogene, 1995, 10, 609–618 CAS.
- W. Vogel, G. D. Gish, F. Alves and T. Pawson, Mol. Cell, 1997, 1, 13–23 CrossRef CAS.
- J. L. Perez, S. Q. Jing and T. W. Wong, Oncogene, 1996, 12, 1469–1477 CAS.
- B. Leitinger, J. Biol. Chem., 2003, 278, 16761–16769 CrossRef CAS.
- A. Shrivastava, C. Radziejewski, E. Campbell, L. Kovac, M. McGlynn, T. E. Ryan, S. Davis, M. P. Goldfarb, D. J. Glass, G. Lemke and G. D. Yancopoulos, Mol. Cell, 1997, 1, 25–34 CrossRef CAS.
- G. Agarwal, L. Kovac, C. Radziejewski and S. J. Samuelsson, Biochemistry, 2002, 41, 11091–11098 CrossRef CAS.
- A. D. Konitsiotis, N. Raynal, D. bihan, E. Hohenester, R. W. Farndale and B. Leitinger, J. Biol. Chem., 2008, 283, 6861–6868 CrossRef CAS.
- R. J. Lebbink, T. de Ruiter, J. Adelmeijer, A. B. Brenkman, J. M. van Helvoort, M. Koch, R. W. Farndale, T. Lisman, A. Sonnenberg, P. J. Lenting and L. Meyaard, J. Exp. Med., 2006, 203, 1419–1425 CrossRef CAS.
- R. J. Lebbink, N. Raynal, T. de Ruiter, D. G. Bihan, R. W. Farndale and L. Meyaard, Matrix Biol., 2009, 28, 202–210 CrossRef CAS.
- Y. Zong, Y. Xu, X. Liang, D. R. Keene, A. Hook, S. Gurusiddappa, M. Hook and S. V. L. Narayana, EMBO J., 2005, 24, 4224–4236 CrossRef CAS.
- J. C. Leo, H. Elovaara, B. Brodsky, M. Skurnik and A. Goldman, Protein Eng. Des. Sel., 2008, 21, 475–484 CrossRef CAS.
- M. Achison, C. Joel, P. G. Hargreaves, S. O. Sage, M. J. Barnes and R. W. Farndale, Blood Coagulation Fibrinolysis, 1996, 7, 149–152 CrossRef CAS.
- J. C. Mountford, S. K. Melford, C. M. Bunce, J. Gibbins and S. P. Watson, Thromb. Haemost., 1999, 82, 1153–1159 Search PubMed.
- J.-M. Pasquet, R. Bobe, B. Gross, M.-P. Gratacap, M. G. Tomlinson, B. Payrastre and S. P. Watson, Biochem. J., 1999, 342, 171–177 CrossRef CAS.
- M. A. Cejas, C. Chen, W. A. Kinney and B. E. Maryanoff, Bioconj. Chem., 2007, 18, 1025–1027 CrossRef CAS.
- C. M. Overall and C. Lopez-Otin, Nat. Rev. Cancer, 2002, 2, 657–672 CrossRef CAS.
- M. Egeblad and Z. Werb, Nat. Rev. Cancer, 2002, 2, 161–174 CrossRef CAS.
- B. Fingleton, Curr. Pharm. Design, 2007, 13, 333–346 CrossRef CAS.
- L. J. McCawley and L. M. Matrisian, Curr. Opin. Cell Biol., 2001, 13, 534–540 CrossRef CAS.
- C. M. Overall, Mol. Biotech., 2002, 22, 51–86 Search PubMed.
- C. J. Morrison and C. M. Overall, J. Biol. Chem., 2006, 281, 26528–26539 CrossRef CAS.
- H. F. Bigg, A. D. Rowan, M. D. Barker and T. E. Cawston, FEBS J., 2007, 274, 1246–1255 CrossRef CAS.
- I. N. Clark and T. E. Cawston, Biochem. J., 1989, 263, 201–206 CAS.
- V. Knäuper, S. Cowell, B. Smith, C. Lopez-Otin, M. O'Shea, H. Morris, L. Zardi and G. Murphy, J. Biol. Chem., 1997, 272, 7608–7616 CrossRef CAS.
- V. Knäuper, A. Osthues, Y. A. DeClerk, K. E. Langley, J. Bläser and H. Tschesche, Biochem. J., 1993, 291, 847–854.
- G. Murphy, J. A. Allan, F. Willenbrock, M. I. Cockett, J. P. O'Connell and A. J. P. Docherty, J. Biol. Chem., 1992, 267, 9612–9618 CAS.
- E. Ohuchi, K. Imai, Y. Fujii, H. Sato, M. Seiki and Y. Okada, J. Biol. Chem., 1997, 272, 2446–2451 CrossRef CAS.
- D. R. Hurst, M. A. Schwartz, M. A. Ghaffari, Y. Jin, H. Tschesche, G. B. Fields and Q.-X. A. Sang, Biochem. J., 2004, 377, 775–779 CrossRef CAS.
- S. J. De Souza, H. M. Pereira, S. Jacchieri and R. R. Brentani, FASEB J., 1996, 10, 927–930 CAS.
- S. Iyer, R. Visse, H. Nagase and K. R. Acharya, J. Mol. Biol., 2006, 362, 78–88 CrossRef CAS.
- B. Steffensen, U. M. Wallon and C. M. Overall, J. Biol. Chem., 1995, 270, 11555–11566 CrossRef CAS.
- X. Xu, Z. Chen, Y. Wang, Y. Yamada and B. Steffensen, Biochem. J., 2005, 392, 127–134 CrossRef CAS.
- J. L. Lauer-Fields and G. B. Fields, Biol. Chem., 2002, 383, 1095–1105 CrossRef CAS.
- J. L. Lauer-Fields, H. Nagase and G. B. Fields, J. Biomolecular Techniques, 2004, 15, 305–316 Search PubMed.
- J. L. Lauer-Fields, M. J. Chalmers, S. A. Busby, D. Minond, P. R. Griffin and G. B. Fields, J. Biol. Chem., 2009, 284, 24017–24024 CrossRef CAS.
- K. A. Tuzinski, H. Nagase and G. B. Fields, in Peptides: Frontiers of Peptide Science, ed., J. P. Tam and P. T. P. Kaumaya, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1999, pp. 743–744 Search PubMed.
- S. Perumal, O. Antipova and J. P. R. O. Orgel, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2824–2829 CrossRef CAS.
- H. Nagase and G. B. Fields, Biopolymers, 1996, 40, 399–416 CrossRef CAS.
- G. B. Fields, J. Theor. Biol., 1991, 153, 585–602 CrossRef CAS.
- J. L. Lauer-Fields, D. Juska and G. B. Fields, Biopolymers (Pept. Sci.), 2002, 66, 19–32 CrossRef CAS.
- C. M. Stultz, J. Mol. Biol., 2002, 319, 997–1003 CrossRef CAS.
- S. Fiori, B. Saccá and L. Moroder, J. Mol. Biol., 2002, 319, 1235–1242 CrossRef CAS.
- P. Fan, M. H. Li, B. Brodsky and J. Baum, Biochemistry, 1993, 32, 13299–13309 CrossRef CAS.
- S. Fiori and L. Moroder, in Peptides: The Wave Of The Future (Proceedings of the Seventeenth American Peptide Symposium, June 9-14, 2001, San Diego, CA), ed., R. A. Houghton and M. Lebl, American Peptide Symposium, San Diego, 2001, pp. 355–356 Search PubMed.
- R. Salsas-Escat and C. M. Stultz, Proteins: Struct. Funct. Bioinform., 2010, 78, 325–335 Search PubMed.
- R. Z. Kramer and H. M. Berman, J. Biomol. Struct. Dynamics, 1998, 16, 367–380 CAS.
- K. Okuyama, C. Hongo, G. Wu, K. Mizuno, K. Noguchi, S. Ebisuzaki, Y. Tanaka, N. Nishino and H. P. Bächinger, Biopolymers, 2009, 91, 361–372 CrossRef CAS.
- G. Melacini, A. M. J. J. Bonvin, M. Goodman, R. Boelens and R. Kaptein, J. Mol. Biol., 2000, 300, 1041–1048 CrossRef CAS.
- H. Wu, M. H. Byrne, A. Stacey, M. B. Goldring, J. R. Birkhead, R. Jaenisch and S. M. Krane, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5888–5892 CrossRef CAS.
- K. A. Hasty, H. Wu, M. Byrne, M. B. Goldring, J. M. Seyer, R. Jaenisch, S. M. Krane and C. L. Mainardi, Matrix, 1993, 13, 181–186 Search PubMed.
- K. E. Williams and D. R. Olsen, Matrix Biol., 2009, 28, 373–379 CrossRef CAS.
- R. E. Babine and S. L. Bender, Chem. Rev., 1997, 97, 1359–1472 CrossRef.
- A. L. Gall, M. Ruff, R. Kannan, P. Cuniasse, A. Yiotakis, V. Dive, M. C. Rio, P. Basset and D. Moras, J. Mol. Biol., 2001, 307, 577–586 CrossRef CAS.
- C. M. Overall and O. Kleifeld, Nat. Rev. Cancer, 2006, 6, 227–239 CrossRef CAS.
- C. M. Overall and O. Kleifeld, Br. J. Cancer, 2006, 94, 941–946 CrossRef CAS.
- J. L. Lauer-Fields, K. Brew, J. K. Whitehead, S. Li, R. P. Hammer and G. B. Fields, J. Am. Chem. Soc., 2007, 129, 10408–10417 CrossRef CAS.
- J. L. Lauer-Fields, J. K. Whitehead, S. Li, R. P. Hammer, K. Brew and G. B. Fields, J. Biol. Chem., 2008, 283, 20087–20095 CrossRef CAS.
- J. L. Lauer-Fields and G. B. Fields, in Peptides: Breaking Away (Proceedings of the Twenty-First American Peptide Symposium), ed., M. Lebl, American Peptide Society, Albuquerque, NM, 2009, pp. 239–241 Search PubMed.
- S. Li, J. K. Whitehead and R. P. Hammer, J. Org. Chem., 2007, 72, 3116–3118 CrossRef CAS.
- K. M. Huntington, T. Yi, Y. Wei and D. Pei, Biochemistry, 2000, 39, 4543–4551 CrossRef CAS.
- M. Bhowmick, R. R. Sappidi, G. B. Fields and S. D. Lepore, Biopolymers (Pept. Sci.), 2010 Search PubMed submitted.
- X. Xu, Z. Chen, Y. Wang, L. Bonewald and B. Steffensen, Biochem. J., 2007, 406, 147–155 CrossRef CAS.
- K. Yamamoto, N. Nishimura, T. Doi, T. Imanishi, T. Kodama, K. Suzuki and T. Tanaka, FEBS Lett., 1997, 414, 182–186 CrossRef CAS.
- S. S. Mielewczyk, K. J. Breslauer, R. B. Anachi and B. Brodsky, Biochemistry, 1996, 35, 11396–11402 CrossRef CAS.
- S. Ricard-Blum, M. Beraud, N. Raynal, R. W. Farndale and F. Ruggiero, J. Biol. Chem., 2006, 281, 25195–25204 CrossRef CAS.
- E. Doss-Pepe, P. Deprez, N. C. Inestrosa and B. Brodsky, Biochemistry, 2000, 39, 14884–14892 CrossRef CAS.
- P. Deprez, E. Doss-Pepe, B. Brodsky and N. C. Inestrosa, Biochem. J., 2000, 350, 283–290 CrossRef CAS.
- E. Doss-Pepe, P. Deprez, T. Silva, N. C. Inestrosa, A. Kirkpatrick, J. A. M. Ramshaw and B. Brodsky, Biochim. Biophys. Acta, 2004, 1698, 187–195 CAS.
- T. Koide, Y. Takahara, S. Asada and K. Nagata, J. Biol. Chem., 2002, 277, 6178–6182 CrossRef CAS.
- T. Koide, S. Asada, Y. Takahara, Y. Nishikawa, K. Nagata and K. Kitagawa, J. Biol. Chem., 2006, 281, 3432–3438 CAS.
- T. Koide, Y. Nishikawa, S. Asada, C. M. Yamazaki, Y. Takahara, D. L. Homma, K. Otaka, N. Wakamiya, K. Nagata and K. Kitagawa, J. Biol. Chem., 2006, 281, 11177–11185 CrossRef CAS.
- T. Lisman, N. Raynal, D. Groeneveld, B. Maddox, A. R. Peachey, E. G. Huizinga, P. G. de Groot and R. W. Farndale, Blood, 2006, 108, 3753–3756 CrossRef CAS.
- R. A. Romijn, E. Westein, B. Bouma, M. E. Schiphorst, J. J. Sixma, P. J. Lenting and E. G. Huizinga, J. Biol. Chem., 2003, 278, 15035–15039 CrossRef CAS.
- N. Nishida, H. Sumikawa, M. Sakakura, N. Shimba, H. Takashi, H. Terasawa, E.-i. Suzuki and I. Shimada, Nat. Struct. Biol., 2003, 10, 53–58 CrossRef CAS.
- E. Hohenester, T. Sasaki, C. Giudici, R. W. Farndale and H. P. Bächinger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18273–18277 CrossRef CAS.
- M. Ruehl, R. Somasundaram, I. Schoenfelder, R. W. Farndale, C. G. Knight, M. Schmid, R. Ackermann, E. O. Riecken, M. Zeitz and D. Schuppan, J. Biol. Chem., 2002, 277, 26872–26878 CrossRef CAS.
- O. Matsushita, T. Koide, R. Kobayashi, K. Nagata and A. Okabe, J. Biol. Chem., 2001, 276, 8761–8770 CrossRef CAS.
- J. J. Wilson, O. Matsushita, A. Okabe and J. Sakon, EMBO J., 2003, 22, 1743–1752 CrossRef CAS.
- C.-Y. Fan, C.-C. Huang, W.-C. Chiu, C.-C. Lai, G.-G. Liou, H.-C. Li and M.-Y. Chou, FASEB J., 2008, 22, 3795–3804 CrossRef CAS.
- G. Johnson, M. Jenkins, K. M. McLean, H. J. Griesser, J. Kwak, M. Goodman and J. G. Steele, J. Biomed. Mater. Res., 2000, 51, 612–624 CrossRef CAS.
- S. T. Khew, X. H. Zhu and Y. W. Tong, Tissue Eng., 2007, 13, 2451–2463 CrossRef CAS.
- H. J. Lee, J.-S. Lee, T. Chansakul, C. Yu, J. H. Elisseeff and S. M. Yu, Biomaterials, 2006, 27, 5268–5276 CrossRef CAS.
- X. Mo, Y. An, C.-S. Yun and S. M. Yu, Angew. Chem., Int. Ed., 2006, 45, 2267–2270 CrossRef CAS.
- C. Kojima, S. Tsumura, A. Harada and K. Kono, J. Am. Chem. Soc., 2009, 131, 6052–6053 CrossRef CAS.
- S. K. Dey and H.-B. Kraatz, Bioconj. Chem., 2006, 17, 84–89 CrossRef CAS.
- S. K. Dey, Y.-T. Long, S. Chowdhury, T. C. Sutherland, H. S. Mandal and H.-B. Kraatz, Langmuir, 2007, 23, 6475–6477 CrossRef CAS.
- J. L. Lauer-Fields, T. Sritharan, M. Kashiwagi, H. Nagase and G. B. Fields, J. Biol. Chem., 2007, 282, 142–150 CAS.
- M. Whittaker, C. D. Floyd, P. Brown and A. J. H. Gearing, Chem. Rev., 1999, 99, 2735–2776 CrossRef CAS.
- A. Saghatelian, N. Jessani, A. Joseph, M. Humphrey and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10000–10005 CrossRef CAS.
- B. G. Rao, Curr. Pharm. Design, 2005, 11, 295–322 CrossRef CAS.
- R. Roy, J. Yang and M. A. Moses, J. Clin. Oncol., 2009, 27, 5287–5297 CrossRef CAS.
- V. M. Baragi, G. Becher, A. M. Bendele, R. Biesinger, H. Bluhm, J. Boer, H. Deng, R. Dodd, M. Essers, T. Feuerstein, B. M. Gallagher Jr., C. Gege, M. Hochgürtel, M. Hofmann, A. Jaworski, L. Jin, A. Kiely, B. Korniski, H. Kroth, D. Nix, B. Nolte, D. Piecha, T. Powers, F. Richter, M. Schneider, C. Steeneck, I. Sucholeiki, A. Taveras, A. Timmermann, J. Van Veldhuizen, J. Weik, X. Wu and B. Xia, Arthritis Rheum., 2009, 60, 2008–2018 CrossRef CAS.
- J. L. Lauer-Fields, T. P. Spicer, P. S. Chase, M. Cudic, G. D. Burstein, H. Nagase, P. Hodder and G. B. Fields, Anal. Biochem., 2008, 373, 43–51 CrossRef CAS.
- J. L. Lauer-Fields, D. Minond, P. S. Chase, P. E. Baillargeon, S. A. Saldanha, R. Stawikowska, P. Hodder and G. B. Fields, Bioorg. Med. Chem., 2009, 17, 990–1005 CrossRef CAS.
- M. Cudic, G. D. Burstein, G. B. Fields and J. Lauer-Fields, Chem. Biol. Drug Design, 2009, 74, 473–482 Search PubMed.
- S. L. Hostikka and K. Tryggvason, J. Biol. Chem., 1988, 263, 19488–19493 CAS.
- R. Z. Kramer, J. Bella, P. Mayville, B. Brodsky and H. M. Berman, Nature Struct. Biol., 1999, 6, 454–457 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |