DOI:
10.1039/B9NR00240E
(Paper)
Nanoscale, 2010,
2, 107-113
Ag nanoparticle sheet as a marker of lateral remote photocatalytic reactions†
Received
25th August 2009
, Accepted 23rd October 2009
First published on 17th November 2009
Introduction
The photocatalytic activity of TiO2 in the decomposition of organic and inorganic materials has been studied intensively in the past several decades.1–3 Some of these studies have resulted in industrial applications such as self-cleaning glasses and antibacterial tiles. Meanwhile the development of new materials with higher photocatalytic efficiency remains an important research subject.4–16 Recently, the doping of N, S, C and transition metal ions into TiO2 was found to narrow the large bandgap of TiO2 and improve photocatalytic activity under visible-light irradiation.4–7 The control of TiO2nanostructure has also been studied to increase the active surface area.8 Investigations into TiO2 nanotubes have shown high mobility of electrons along the long-axis of the tubular structure, therefore they are regarded as potential materials for various optoelectronic device applications.9–12 Recently, Majima et al.13 reported highly efficient charge separation in TiO2 nanotubes compared with TiO2 powder. Significant photocatalytic enhancements by the combination of TiO2 nanotubes and Ag or Au or semiconductor particles were also reported in very recent studies as hot topics in this field.14
At present, the photocatalytic activity of TiO2 is understood as a combination of two kinds of reactions; one is directoxidationvia photoexcited electrons and holes on TiO2, and another is indirect (remote) redox reactions viareactive oxygen species such as hydroxyl radicals (HO˙) and superoxide radical anions, (O2˙−), produced in the presence of humid air.2,15 However, it is not easy to distinguish between these individual contributions to the total photocatalytic activity, especially for nanosized new materials given the limited characterization tools available for the nanoscale. Tatsuma et al.16 have conducted measurements of remoteoxidation by use of photocatalyst flow cells, as well as TiO2 nanoparticulate membranes. Haick et al. and Kawahara et al.17 have characterized lateral remoteoxidation (the indirect oxidation progressing on substrates in the lateral direction) by use of surfaces modified with self-assembled monolayers (SAMs). As a further attempt, in this study we investigate photocatalytic activity of TiO2 nanotubes synthesized by anodization in a perchloric acid-based electrolyte, focusing on lateral remoteoxidation using a ‘Ag nanosheet’ as a marker. The Ag nanosheet is a two-dimensional crystalline film composed of myristate-capped silver nanoparticles (d = 5 nm) having an extremely sharp localized plasmon absorption band (as sharp as a dispersion in a good solvent) at λmax = 470 nm.18,19 The sheet is fabricated at the air–water interface as a 10 mm sized homogeneous film with nm thickness (Fig. 1). We deposit this silver nanosheet ontop of a TiO2 nanotube-modified substrate at 10–20% surface coverage, and then monitor the photodegradation process under UV irradiation. Here the lateral remote photocatalytic reaction produced by TiO2 nanotubes is characterized by the change in the plasmon absorption band (i.e., the red-shift of the plasmon absorption band due to the fusion of nanoparticles after decomposition of organic capping molecules), in combination with surface morphological studies. X-ray photoelectron spectroscopy (XPS) is also conducted to confirm the degradation of the myristate capping molecules. The lateral remote photocatalytic reaction of the TiO2 nanotubes is qualitatively compared with that of commercial TiO2 particles. For other recent applications based on the surface plasmon absorption of metallic nanoparticles, see ref. 20.
Experimental
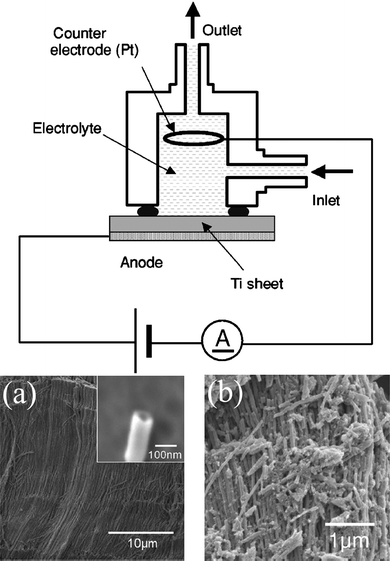
A titanium foil (purity: 99.5%, Japan Metal Service, 20 × 16 × 0.2 mm) was anodized in a solution cell made of Teflon with a platinum counter electrode (Fig. 2). A mixture of 60% perchloric acid and 99.5% ethanol at the volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 was used as an electrolyte solution in continuous flow, during the anodization process. An anodic potential of 40 V (vs.counter electrode) was applied for 5 min by a stabilized power supply. This procedure produces TiO2 nanotubes with a diameter of 70 nm and a length of more than 10 μm as shown in Fig. 2(a).12TiO2 nanotubes are still amorphous when these are synthesized by anodization, however, we can change them to anatase crystals by annealing at 400 °C for 1 h and at 500 °C for 30 min.12TiO2 anatase crystals are quite brittle as shown in Fig. 2(b).
:15 was used as an electrolyte solution in continuous flow, during the anodization process. An anodic potential of 40 V (vs.counter electrode) was applied for 5 min by a stabilized power supply. This procedure produces TiO2 nanotubes with a diameter of 70 nm and a length of more than 10 μm as shown in Fig. 2(a).12TiO2 nanotubes are still amorphous when these are synthesized by anodization, however, we can change them to anatase crystals by annealing at 400 °C for 1 h and at 500 °C for 30 min.12TiO2 anatase crystals are quite brittle as shown in Fig. 2(b).
The TiO2 nanotube-modified quartz or a naturally oxidized Si(100) substrates (2 mm × 2 mm) were fabricated by spontaneous adsorption of ultrasonicated amorphous TiO2 nanotubes in pure water (9.6 mg mL−1) for a defined time. The excess solution was then removed by the ‘spin-drying’ technique using a spin coater (200 rpm 5 s and 600 rpm 5 s) and the substrates were annealed under the conditions described above to obtain TiO2 anatase crystals. We used these TiO2 anatase crystals in all experiments.
The absorbances at λ = 250 nm of the TiO2 nanotube layers were 0.047 (1 min), 0.081 (3 min), 0.107 (6 min) and 0.173 (3 min + 3 min21). The substrates modified with commercial TiO2 particles (NDH-510C Nippon Soda Co., Ltd and ST-01 Ishihara Sangyo Kaisha Ltd) were fabricated by the same procedure as that of TiO2 nanotubes from 1.5 mg ml−1 aqueous solutions. The absorbances at λ = 250 nm of the TiO2 particle layers were 0.025 (1 min), 0.075 (3 min), 0.103 (6 min) and 0.228 (10 min).
The Ag nanosheet was formed at the air–water interface as a Langmuir film and transferred to the hydrophilic TiO2-modified substrates by an inclined transfer method. 600–700 mL of toluene solution (0.25–0.3 mg mL−1) was spread onto the water in a Langmuir–Blodgett trough at room temperature (T = 24 °C). When the toluene evaporated, the film was compressed and then transferred onto the substrate typically at Π = 15 mN/m.19 The substrate was then dried for 12–18 h in a dark room at room temperature. The light source for UV irradiation was a high-pressure mercury lamp with color filters (peak wavelength: 330 nm, FWHM: 50 nm, 6.4 mW cm−2). The absorption spectrum was monitored by UV–vis spectrometry (Shimazu UV1800). The surface morphology was investigated by high-resolution SEM (Hitachi High-Technologies Corp.) and tapping-mode AFM (MFP-3D, Asylum Research, Santa Barbara, CA, USA). XPS (Theta Probe, Thermo Scientific Inc.) was used for surface chemical analysis using X-rays of Al Kα radiation (photon energy of 1486.6 eV and spot size of 400 μm). The Theta Probe XPS instrument collects photoelectrons emitted over an angular range of 23° < θ < 83°. For all XPS measurements, we employed exactly the same experimental conditions (scan duration time and energy step for data acquisition, X-ray spot size, and pass energy for the photoelectron analyzer). We set the relative positions between the sample and analyzer by using a fixed optical microscope, which enabled us to reproduce the distance between them with an accuracy of several tens of μm. Fluctuation of X-rays observed in the photoelectron intensity are usually less than 4% over 1 h. The binding energies of all XPS data are presented without normalization.
Results and discussion
Figs. 3 and 4 show the spectrum of Ag nanosheets deposited on TiO2 nanotube- and TiO2 particle (prepared from NDH-510C)-modified quartz substrates and their spectrum following periods of UV irradiation. All the spectra were taken using the corresponding TiO2 substrate as a reference, i.e., only the spectra of the Ag nanosheets are shown. In the case of TiO2 nanotubes, UV irradiation causes a rapid decrease of the plasmon absorption band at 470 nm and an increase in the absorption band in the long-wavelength region (550–1000 nm). This phenomenon is interpreted as the fusion of Ag nanoparticles resulting from the decomposition of the myristate cappings by photocatalytic reaction under UV irradiation. The following experiments with TiO2 particles were made for comparison and reveal how unique the photocatalytic properties of TiO2 nanotubes are. Here, only a decrease in the plasmon absorption band at 470 nm was detected but no increase in the absorption band in the long-wavelength region (550–1000 nm) was observed. On the other hand, UV irradiation caused no significant changes in the plasmon absorption bands for 100 min on substrates without TiO2. The amount of TiO2 nanotubes and TiO2 particles on the substrates are not the same even if we use the same adsorption time. Hence the direct comparison of reaction kinetics between the nanotubes and particles is not appropriate. However, the different shape of plasmon peaks after UV irradiation reveals fundamental differences in the photocatalytic reaction of TiO2 nanotubesversus particles. We performed measurements on substrates with higher surface density of TiO2 particles (prepared by 10 min adsorption) as well as with another particle sample (ST-01). The results were essentially the same as in Fig. 4, i.e., a decrease in the 470 nm peak without an increase in the long wavelength region (the data for ST-01 are available in the ESI† ).
 |
| | Fig. 3
Transmission spectrum change of Ag nanosheets deposited on TiO2 nanotube-modified quartz substrates by UV irradiation. Adsorption time allowed for TiO2 nanotubes on quartz substrates; (a) 1 min, (b) 3 min, (c) 6 min, and (d) 3 min + 3 min. A photograph of the sample substrates was taken after UV irradiation, (a)–(c) 100 min and (d) 60 min, in which only the center part of the surface (1 cm width) was exposed to UV light. | |
 |
| | Fig. 4
Transmission spectrum change of Ag nanosheets deposited on TiO2 particle-modified quartz substrates by UV irradiation. Adsorption time allowed for TiO2 nanotubes on quartz substrates; (a) 1 min, (b) 3 min, (c) 6 min, and (d) 10 min. A photograph of the sample substrates was taken after UV irradiation, (a)-(d) 100 min, in which only the center part on the surface (1 cm width) was exposed to UV light. | |
Fig. 5 shows SEM images of an Ag nanosheet on TiO2 nanotube-modified Si(100) wafer (thickness: 525 μm) before and after UV irradiation. In both images, the bright tubular structures (less than 1 μm in length) lying down on the surface are the TiO2 nanotubes at about 10% surface coverage.22 The length of TiO2 nanotubes, at around a micron, are shorter than expected. We offer two explanations, firstly the ultrasonication and annealing procedure ground down the TiO2 nanotubes which are brittle [see Fig. 2(b)], secondly the adsorption of short nanotubes may be favored during adsorption and spin-drying. In the magnified image before UV irradiation [the inset of Fig. 5(a)], the Ag nanosheet composed of densely packed Ag nanoparticles was clearly confirmed on the substrate between TiO2 nanotubes. The Ag nanosheet was totally decomposed after UV irradiation for 100 min, and only the enlarged particles formed by fusion of many nanoparticles [several tens of nm in diameter, the inset in Fig. 5(b)] were observed instead of the 5 nm particles on the surface. Tapping-mode AFM images of the same surface shown in Fig. 5 are shown in Fig. 6. In Fig. 6(a), TiO2 nanotubes are higher than the surrounding substrate and appear yellow in color. They are clearly distinguished from the flat Ag nanosheet (the red-colored background). Here the individual nanoparticles (d = 5 nm) could not be visualized at the resolution utilized, but the defects (dark-red, corresponding to Si(100) wafer) are seen. After UV irradiation, Fig. 6(b), the homogeneous Ag nanosheet has been converted into an inhomogeneous array of larger islands (seen as dots). This is perfectly consistent with the SEM image. Fig. 7 is a high-resolution SEM image of an Ag nanosheet on a TiO2 particle-modified Si(100) wafer after UV irradiation for 100 min. Here the absence of Ag nanoparticles was found only at the edge of the TiO2 particles (within ∼100 nm), while the other surface remains as it was, with no indication of decomposition, being covered with the densely packed Ag nanosheet. Taking the UV–vis absorption spectra in Fig. 4 into consideration, the absence of Ag nanoparticles at the edge of TiO2 particles seems to be the only reaction caused by the UV irradiation of TiO2 particles. We assume that Ag nanoparticles near TiO2 particles were fused and attached to TiO2 particles after decomposition. In the transmission UV–vis measurements, the area of attached TiO2 particles does not contribute to the absorption spectra since the light cannot penetrate through the TiO2 particles by light scattering. Thus the signals of fused Ag particles attached to TiO2 particles resulted in only a decrease of the original plasmon absorption band (470 nm) while no increase of the long-wavelength absorption appeared.
 |
| | Fig. 7
SEM images of an Ag nanosheet with TiO2 particles on TiO2 particle-modified naturally oxidized Si(100) wafer (1 min adsorption of TiO2 particles) after 100 min UV irradiation with a different magnification. | |
XPS surface analysis provides supplemental information concerning the decomposition of the myristate capping of the Ag nanoparticles under UV irradiation, where the decrease of the C1s signal and the increase of the O1s signal are clearly shown (Fig. 8). The fusion of Ag cores was also implied not only by the decrease of the Ag3d signal (less surface coverage) but also by the increase in the Ti2p and Si2p signals (TiO2/SiO2 are more exposed to the surface). We confirmed that the damage caused only by UV irradiation at 330 nm was negligibly small (see the ESI† ). Since our XPS data are taken under conditions at which quantitative discussion is possible, a notable change in relative signal intensities can be an evidence for photocatalytic chemical reaction on the substrate, since such response was not found on the surface without TiO2 nanotubes.
 |
| | Fig. 8
XPS signals of an Ag nanosheet deposited on TiO2 nanotube-modified naturally oxidized Si(100) wafer: (a) before UV irradiation, (b) after UV irradiation. | |
Although the photocatalytic reaction in this system is not perfectly clear yet, we presume the reaction to decompose myristate ligands as follows:
| R–COO–Ag + HO˙ → R˙ + CO2 + HO–Ag |
Here e−CB is an excited electron in the TiO2 conduction band, and h+VB is a hole in the TiO2 valence band. Ag is not a silver atom but a Ag nanoparticle as an adsorptive medium. R is an alkyl chain of myristate. Our previous FTIR study23 confirmed that myristates adsorb on Ag NPsvia ionic bonding. Probably, at first the excited electrons and holes react with O2 and H2O in air and produce superoxide radical anions, hydrogen peroxide and hydroxyl radicals. Next hydroxyl radicals react with myristate cappings and dissociate to CO2 and alkyl radicals . As for the mechanism of lateral remote oxidation, the following reactions are proposed:
(1) diffusion of H2O2 on the SiO2 surface and photodissociation to hydroxyl radicals.16c
(2) production of hydroxyl radicals from alkyl radicals
(3) radical chain reaction between radicals produced in (2) and R–COO–Ag.
Since the alkyl chains of myristate cappings are closely packed with an ‘interdigited structure’ in the Ag nanosheets, the chain reaction between neighboring myristates seems to occur without difficulty.
TiO2 nanotubes of only 10% surface coverage (average interparticle spacing is ≥1 μm) can decompose the Ag nanosheets across their entire surface and induce the complete shift of the plasmon absorption band to the near-IR region. On the other hand, lateral remoteoxidation by commercial TiO2 particles is effective only within a ∼100 nm zone. Based on these results, we can conclude that our TiO2 nanotubes have a quite high lateral remote photocatalytic activity compared with commercial TiO2 particles. The result is depicted in schematic form in Fig. 9. Macak et al. reported that TiO2 nanotubes fabricated by anodization exhibited significantly higher photocatalytic activity compared with a commercial TiO2 nanopowder (Degussa P25) against the decomposition of organic azodyes.11 They mentioned that the surface area of TiO2 tubes is rather less than the nanopowder, so that the higher efficiency of the nanotubes must originate for other reasons, although in their experiments using solution cells it was not clear whether the reaction proceeds by activation-control or diffusion-control. Majima et al. has investigated the photocatalytic reaction of TiO2 nanotubes and TiO2 powder precisely using time-resolved diffuse reflectance spectroscopy, and reported remarkably long-lived radical cations (originating from aromatic compounds) and trapped electrons produced by TiO2 nanotubes.13 They pointed out that the morphology of TiO2 (nanotube or powder) plays an important role in the charge recombination dynamics, with special attention to the porosity. The porosity is unimportant in our systems as our target molecules (myristates on Ag nanoparticles) are immobilized in the sheet and have no way to diffuse into pores. However, the radicals produced in our experiments using TiO2 nanotubes (hydroxyl or the other radicals originating from carboxylates), participating in lateral remoteoxidation seem to have a long life. We may need to consider the influence of Ag particles attaching to the tube after the initial UV exposure as well. Before the exposure, the TiO2 nanotubes and Ag nanoparticles are isolated by the myristate cappings. However, the situation as described by the term ‘Ag nanoparticle-loaded TiO2 nanotube’14 will be produced by decomposition of myristate cappings near the TiO2 nanotubes soon after the UV irradiation (and the enhancement effect is known to be significant with tubular TiO214a), although charge separation induced by plasmon excitation cannot be an explanation for the high photocatalytic activity observed in our experiments with only UV light irradiation (λ = 330 nm).24
Conclusions
Lateral remote photocatalytic reaction of TiO2 nanotubes fabricated by anodic oxidation was successfully characterized by use of Ag nanosheets as a marker. Here the fusion of Ag nanoparticles resulting from the decomposition of myristate cappings is clearly confirmed by UV–vis spectra and SEM/AFM images, even in regions of the surface a few microns away from any TiO2 nanotubes. This lateral remote reaction was not obvious for TiO2 particles. It may correlate to the different lifetime of radicals produced on each surface although a further investigation is necessary. Ag nanosheets, homogeneous on the nanoscale with a sharp plasmon absorption band and resistant to photobleaching, but highly sensitive to redox reactions, will be a powerful tool to develop new potential nanocatalytic materials, especially for substrate-based device applications.25
Acknowledgements
K.T thanks Dr V. Craig, Australian National University, for valuable discussions. This work is supported by a Grant-in-Aid for scientific Research No. 21310067 from the Ministry of Education, Science, Sport, and Culture.
Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CAS.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS.
-
(a) R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taha, Science, 2001, 293, 269 CrossRef CAS;
(b) H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483 CrossRef CAS;
(c) A. Ghicov, J. M. Macak, H. Tsuchiya, J. Kunze, V. Haeublein, L. Frey and P. Schmuki, Nano Lett., 2006, 6, 1080 CrossRef CAS.
- T. Ohno, M. Akiyoshi, T. Umebayashim, K. Asai, T. Mitsui and M. Matsumura, Appl. Catal., 2004, A265, 115.
-
(a) H. Irie, Y. Watanabe and K. Hashimoto, Chem. Lett., 2003, 32, 772 CrossRef CAS;
(b) T. Ohno, T. Tsubota, K. Kakiuchi and K. Sayama, Chem. Lett., 2004, 33, 1610 CrossRef CAS;
(c) S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908 CrossRef CAS.
- W. Y. Choi, A. Termin and M. R. J. Hoffmann, J. Phys. Chem., 1994, 98, 13669 CrossRef.
-
(a) M. Andersson, L. Osterlund and S. Ljungstrom, J. Phys. Chem. B, 2002, 106, 10674 CrossRef CAS;
(b) C. B. Almquist and P. Biswas, J. Catal., 2002, 212, 145 CrossRef CAS.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2005, 5, 191 CrossRef CAS.
- R. Beranek, H. Tsuchiya, T. Sugishima, J. M. Macak, L. Taveira, S. Fujimoto, H. Kisch and P. Schmuki, Appl. Phys. Lett., 2005, 87, 243114 CrossRef.
- J. M. Macak, M. Zlamal, J. Krysa and P. Schmuki, Small, 2007, 3, 300 CrossRef CAS.
- K. Ishibashi, R. Yamaguchi, Y. Kimura and M. Niwano, J. Electrochem. Soc., 2008, 155, K10 CrossRef CAS.
-
(a) T. Tachikawa, S. Tojo, M. Fujitsuka, T. Sekino and T. Majima, J. Phys. Chem. B, 2006, 110, 104055;
(b) K. Naito, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2009, 131, 934 CrossRef CAS.
-
(a) I. Paramasivalm, J. M. Macak and P. Schmuki, Electrochem. Commun., 2008, 10, 71 CrossRef CAS;
(b) D. Guin, S. V. Manorama, J. N. L. Latha and S. Singh, J. Phys. Chem. C, 2007, 111, 13393 CrossRef CAS;
(c) H. Li, X. Duan, G. Liu and X. Liu, J. Mater. Sci., 2008, 43, 1669 CrossRef CAS;
(d) K. Nishijima, T. Fukaahori, N. Murakami, T. Kamai, T. Tsubota and T. Ohno, Appl. Catal., A, 2008, 337, 105 CrossRef CAS;
(e) J. Yu, G. Dai and B. Huang, J. Phys. Chem. C, 2009, 113, 16394 CrossRef CAS;
(f) P. Wang, B. Huang, X. Qin, X. Zhang, Y. Dai, J. Wei and M.-H. Whangbo, Angew. Chem., Int. Ed., 2008, 47, 7931 CrossRef CAS.
- A. Mills, S. Hodgen and S. K. Lee, Res. Chem. Intermed., 2005, 31, 295 CrossRef CAS.
-
(a) T. Tatsuma, S. Tachibana, T. Miwa, D. A. Tryk and A. Fujishima, J. Phys. Chem. B, 1999, 103, 8033 CrossRef CAS;
(b) T. Tatsuma, S. Tachibana and A. Fujishima, J. Phys. Chem. B, 2001, 105, 6987 CrossRef CAS;
(c) W. Kubo and T. Tatsuma, J. Am. Chem. Soc., 2006, 128, 16034 CrossRef CAS;
(d) W. Kubo and T. Tatsuma, J. Mater. Chem., 2005, 15, 3104 RSC.
-
(a) H. Haick and Y. Paz, J. Phys. Chem. B, 2001, 105, 3045 CrossRef CAS;
(b) K. Kawahara, Y. Ohko, T. Tatsuma, A. Fujishima and Akira Fujishima, Phys. Chem. Chem. Phys., 2003, 5, 4764 RSC.
- C. D. Keum, N. Ishii, K. Michioka, P. Wulandari, K. Tamada, M. Furusawa and H. Fukushima, J. Nonlinear Opt. Phys. Mater., 2008, 17, 131 CrossRef CAS.
- K. Tamada, K. Michioka, X. Li, Y. Ikezoe, M. Saito and K. Otsuka, Proc. SPIE–Int. Soc. Opt. Eng., 2009, 72130E.
-
(a) H. Nakanishi, K. J. M. Bishop, B. Kowalczyk, A. Nitzan, E. A. Weiss, K. V. Tretiakov, M. M. Apodaca, R. Klajn, J. F. Stoddart and B. A. Grzybowski, Nature, 2009, 460, 371 CrossRef CAS;
(b) X. N. Xie, Y. Xie, X. Gao, C. H. Sow and A. T. S. Wee, Adv. Mater., 2009, 21, 3016 CrossRef CAS;
(c) X. N. Xie, X. Gao, D. Qi, Y. Xie, L. Shen, S.-W. Yang, C. H. Sow and A. T. S. Wee, ACS Nano, 2009, 3, 2722 CrossRef CAS.
- After 3 min adsorption, once dried then adsorption for a further 3 min.
- The thickness determined by surface plasmon technique also confirmed the surface coverage of TiO2 nanotubes is less than 10% after 1 min adsorption.
- K. Naoi, Y. Ohko and T. Tatsuma, J. Am. Chem. Soc., 2004, 126, 3664 CrossRef CAS.
- P. Wulandari, T. Nagahiro, K. Michioka, K. Tamada, K. Ishibashi, Y. Kimura and M. Niwano, Chem. Lett., 2008, 37, 888 CrossRef CAS.
-
(a) J. P. Lee and M. M. Sung, J. Am. Chem. Soc., 2004, 126, 28 CrossRef CAS;
(b) J. P. Bearinger, G. Stone, A. L. Hiddessen, L. C. Dugan, L. Wu, P. Hailey, J. W. Conway, T. Kuenzler, L. Feller, S. Cerritelli and J. A. Hubbell, Langmuir, 2009, 25, 1238 CrossRef CAS;
(c) N. Blondiaux, S. Zürcher, M. Liley and N. D. Spencer, Langmuir, 2007, 23, 3489 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.