Synthesis of a sulfonato-salen-nickel(II) complex immobilized in LDH for tetralin oxidation
Samiran
Bhattacharjee
a,
Kwang-Eun
Jeong
b,
Soon-Yong
Jeong
b and
Wha-Seung
Ahn
*a
aDepartment of Chemical Engineering, Inha University, Incheon 402-751, Korea. E-mail: whasahn@inha.ac.kr; Fax: +82 32-872 0959; Tel: +82 32-866 0143
bApplied Chemistry and Engineering Division, KRICT, Yuseong 305-600, Korea
First published on 5th November 2009
Abstract
A novel sulfonato-salen-nickel(II) complex has been immobilized on a Zn(II)-Al(III) layered double hydroxide (LDH) host. XRD, FT-IR, TGA and UV-vis spectroscopy, as well as chemical analysis, confirmed the successful incorporation of the nickel-salen complex within the LDH structure. BET surface area measurements, SEM and TEM were also used to characterize the heterogenized catalyst. The sulfonato-salen-nickel(II) complex-immobilized material, LDH-[nickel-salen], was found to be effective in the oxidation of tetralin, where a combination of trimethylacetaldehyde and dioxygen at atmospheric pressure was employed as the oxidant. At 72.3% conversion, tetralin was converted to 1-tetralone with 72.2% selectivity at 70 °C after 7 h. Tetralin oxidation using tert-butyl hydroperoxide afforded a lower conversion and selectivity of 1-tetralone than with trimethylacetaldehyde and dioxygen as the oxidant. The effect of various reaction parameters on catalytic performance was also investigated. A hot filtration experiment coupled with a blank test revealed that oxidation proceeded mostly on nickel-salen sites in LDH-[nickel-salen]. A reaction mechanism is proposed based on the experimental results.
Introduction
Tetralin (1,2,3,4-tetrahydronaphthalene) oxidation produces 1-tetralone, an important source of synthetic precursors and a reactive intermediate for a wide range of products, including pharmaceuticals, dyes and agrochemicals.1,2 In addition, 1-tetralone and the by-product 1-tetralol can be used as additives to enhance the cetane number of diesel fuels for cleaner combustion.3 Selective oxidation of such benzylic hydrocarbons in the liquid phase has been investigated in conjunction with a variety of catalytic systems over recent decades, viz., using soluble metal (Ni, Mn and Cu) catalysts,4,5 2,4-dimethylpentane-2,4-diol cyclic chromate6 and Ni complexes with surface-active ligands.7 Transition metal-containing molecular sieves such as MAPO-5, MAPO-11 and MAPSO-34 (M = Cr, Co, Mn and V)8,9 can immobilize transition metals (viz., Cr, Ni, Cu, Fe, Co, Mn, La and Ru) on MCM-41 mesoporous silica;10–14 chromium-exchanged zeolite Y15 and ZSM-516 have also been studied. A recent catalyst test of tetralin oxidation over various transition metal-containing molecular sieve catalysts by us has shown that Cr-incorporated AlPO4-5 is more active than Fe- or Co-substituted catalysts.17 As a consequence, very recently, we reported that a mesoporous Cr(III) terephthalate metal–organic framework, MIL-101, affords excellent catalytic activity with high catalyst stability.18 However, despite the high activity and selectivity offered by liquid phase oxidation, leaching of the active metal species from the matrix during the catalytic reaction usually poses a problem in many heterogeneous catalysts.16,17,19 Further efforts to improve catalytic methods for benzylic oxidation with improved stability are highly desired.Schiff base ligands derived from salicylaldehyde and amines are known as salen ligands, and their transition metal complexes have been widely applied to the synthesis of fine chemicals by oxidation, showing high catalytic activity under both homogeneous and heterogeneous conditions.20–23 However, among these, nickel-salen-based complexes have not been extensively studied as catalysts compared to their Mn, Cr or Co analogues. While few studies of Ni complexes with salen ligands have been reported, in terms of their utility as catalysts, excellent catalytic activities in olefin epoxidation and phenol oxidation reactions have been reported.24–28 Our earlier work with surface-active Ni complex catalysts7 for tetralin oxidation in biphasic reactions also established the high potential of Ni-based catalytic systems and led us to test a nickel-salen complex immobilized on an inorganic solid support as a heterogeneous catalyst for tetralin oxidation in the liquid phase.
Layered double hydroxides (LDHs) are a family of anionic clays consisting of hydroxides of common metals, such as Mg and Al, and can be synthesized at ambient temperature and pressure from aqueous solutions. They have received increasing attention in recent years as catalysts and catalyst supports.29–35 More importantly, it has been claimed that LDH-hosted catalysts do not suffer from leaching of the active metal in epoxidation reactions, and that they can be reused several times without loss of activity.33,34 These findings prompted us to test the efficacy of an LDH-hosted nickel-salen complex as a catalyst for the oxidation of bicyclic arenes where dioxygen serves as an oxidant.
In this work, we describe the synthesis of a new catalyst, in which a nickel-salen complex, [NiL] [L = sulfonato-salen], is immobilized on a Zn-Al LDH by the anion exchange method. To gain insight into the structure of the nickel-salen complex intercalated into the LDH host, we performed detailed characterization, including X-ray diffraction (XRD), BET surface area measurements, scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The oxidation of tetralin was studied using in situ-generated acylperoxy radicals by dioxygen at atmospheric pressure in the presence of sacrificial aldehyde, as well as tert-butyl hydroperoxide. The influence of various reaction parameters, such as temperature and the amount of catalyst, on tetralin conversion was investigated; a plausible reaction mechanism is proposed based on the experimental results. To the best of our knowledge, the application of an intercalated system containing a salen-nickel(II) complex for the oxidation of tetralin has not been previously reported.
Experimental methods
Materials
Salicylaldehyde (98%, Aldrich), aniline (99.5%, Aldrich), 1,2-ethylenediamine (TCI), zinc(II) nitrate tetrahydrate (Aldrich), aluminium(III) nitrate nonahydrate (Aldrich), nickel(II) perchlorate hexahydrate (Aldrich), trimethylacetaldehyde (96%, Aldrich) and tert-butyl hydroperoxide (5–6 M solution in decane, Aldrich) were purchased and used without further purification.Catalyst preparation
Sodium salicylaldehyde-5-sulfonate (1) and [NiL] [L = sulfonato-salen ligand] (2) were prepared using methods previously reported in the literature.36,37 The immobilized benzoate ion on the LDH host, LDH-[C6H5COO] (3), was prepared according to a previously reported method.33 0.4 g of 2 was dissolved in de-carbonated water (300 cm3) and 1.0 g of 3 was added to the solution, which was stirred for 10 h at room temperature under a nitrogen atmosphere to produce the [NiL]2−-immobilized material, 4 (4). The orange solid was filtered off and washed with water and ethanol, and dried overnight at 60 °C.Catalyst characterization
X-Ray powder diffraction patterns were recorded on a Rigaku Miniflex diffractometer using Cu-Kα radiation (λ = 1.54 Å). FT-IR spectra of the catalyst samples were recorded using KBr discs on a Bruker VERTEX 80V FT-IR spectrophotometer. Thermogravimetric analyses in the range 25–850 °C were carried out on a TGA (Scinco S-1000) system under an air flow at a heating rate of 5.0 °C min−1. BET surface areas were determined on a Micromeritics ASAP 2000 surface analyzer at 77 K. UV-vis diffuse reflectance spectra were obtained on a Varian Cary 3E double-beam spectrophotometer using MgO as a reference at ambient temperature. Elemental analyses were performed using a FLASH EA 1112 elemental analyzer (Thermo Electron Corporation). Metal contents were determined on a Perkin-Elmer atomic absorption spectrometer (model: AAnalyst 400); samples for the measurements were dissolved in a mixture of HCl, HNO3 and hydrogen peroxide (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1). SEM micrographs were obtained on a Hitachi S-4200 instrument. TEM micrographs were obtained on a JEOL JEM-2100F instrument. Samples were dispersed in ethanol in an ultrasonic bath, and a drop of the supernatant suspension was placed onto a holey carbon coated grid and dried at 60 °C.
:2:1). SEM micrographs were obtained on a Hitachi S-4200 instrument. TEM micrographs were obtained on a JEOL JEM-2100F instrument. Samples were dispersed in ethanol in an ultrasonic bath, and a drop of the supernatant suspension was placed onto a holey carbon coated grid and dried at 60 °C.
Catalytic reaction
Tetralin oxidation reactions using t-BuOOH as the oxidant were carried out using a Chemistation PPS-2510 fitted with a condenser (Eyela). In a typical reaction, a mixture of 2 mmol tetralin, 5 cm3 chlorobenzene (solvent) and 0.02 g catalyst were placed in a Chemistation glass reactor and heated to the desired temperature. The oxidant (t-BuOOH, 4 mmol) was then added through a septum to the reactant mixture and stirred at 600 rpm. The oxidation of tetralin was carried out using dioxygen at atmospheric pressure in a twin-necked round flask equipped with a condenser. In a typical run, 2 mmol tetralin, 4 mmol trimethylacetaldehyde, 10 cm3 acetonitrile and 0.02 g catalyst were stirred while dioxygen was bubbled through at atmospheric pressure and kept at the desired reaction temperature. After completion of the reaction, the catalyst was filtered off, and the selectivity and conversion were measured using a GC (Acme 6000, Younglin, Korea) fitted with a high performance HP-5 capillary column (60 m, 0.32 mm, 0.25 μm) and an FID.Catalyst hot filtration experiments were carried out by separating the catalyst quickly from the reaction solution after 1 h of reaction while maintaining the reaction temperature (50 °C). The filtrate was further stirred at 50 °C for an additional duration of up to 7 h.
Results and discussion
Synthesis of 4
The synthesis route to the 4 catalyst is shown in Scheme 1. The host hydrotalcite-like material, 3, was prepared by the co-precipitation of a solution of zinc and aluminium nitrates with an aqueous solution of NaOH and benzoic acid.332 was intercalated into 3 at room temperature from an aqueous solution by exchange of the C6H5COO− ion to produce 4. Catalyst 4 is stable in air for several months, and remains stable in most organic solvents and water at elevated temperature.![Synthesis route to the LDH-[NiL] catalyst, 4.](/image/article/2010/NJ/b9nj00314b/b9nj00314b-s1.gif) | ||
| Scheme 1 Synthesis route to the LDH-[NiL] catalyst, 4. | ||
Characterization of [Ni(II)-(sulfonato-salen)]2− and the LDH-[Ni-(sulfonato-salen)] catalyst
The XRD patterns of 3 and 4 are shown in Fig. 1. The basal spacing of 3 was increased from 15.4 to 19.5 Å following the exchange process. The gallery height of catalyst 4 is 14.8 Å when the thickness of the brucite layers (4.7 Å) are subtracted. This increase in gallery height strongly suggests successful intercalation of the [NiL]2− ion into the LDH host. An elemental analysis (Table 1) of the catalyst was in good agreement with the formula unit [Zn1.87Al1.04(OH)5.81][NiL]0.41[C6H5COO]0.21·6H2O. TGA up to 250 °C revealed a sample weight loss of 18.0% (calculated loss of 6H2O: 18.2%). The [Al/(Zn + Al)] ratios of 3 and 4 were almost identical, indicating that leaching of neither Zn(II) nor Al(III) occurred during the exchange procedure. The FT-IR spectra of 2, 3 and 4 are shown in Fig. 2. These spectra confirm the intercalation of the nickel-salen complex into the LDH host via exchange of the C6H5COO− ion. The FT-IR spectrum of 4 shows strong bands at 1116 and 1035 cm−1 due to the antisymmetric and symmetric stretching modes of the SO32− moiety.38 This corresponds closely to the spectrum of the free sulfonato-salen-nickel(II) (2), which registers strong bands at 1111 and 1030 cm−1, thus indicating the presence of the [NiL]2− ion in the LDH host. Further support for this intercalation into the LDH host is provided by the νNi–N vibration of 4 at 465 cm−1.39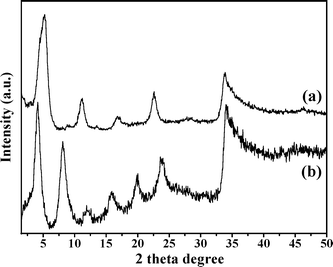 | ||
| Fig. 1 X-Ray powder patterns of (a) 3 and (b) 4. | ||
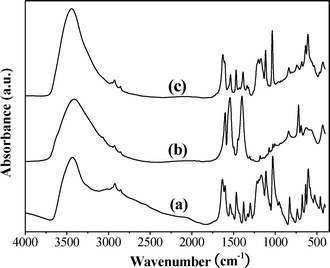 | ||
| Fig. 2 FT-IR spectra of (a) free 2, (b) 3 and (c) 4. | ||
The TGA curves of 2, 3 and 4 are shown in Fig. 3. The TGA diagram of free complex 2 shows three weight loss steps (Fig. 3(a)). The first step, up to 150 °C, is due to the removal of water molecules. The second step, which is observed in the temperature range 270–360 °C, may be due to the loss of a sulfonato group.40 The decomposition of the organic part of the chelate occurred in the temperature range 360–385 °C and resulted in the formation of nickel oxide at the final temperature.40 The weight loss behaviors of 3 and 4 revealed that the weight loss of both compounds occurred below 250 °C, which can be attributed to the loss of adsorbed and interlayer water molecules.41 The weight loss behavior of 3 (Fig. 3(b)) reflects a two-step weight loss in the temperature range 340–445 °C. In this case, the first loss may be due to the loss of the C6H5COO− ion and the second may result from dehydroxylation of the LDH layers.41 Collapse of the hydrotalcite structure in 3 occurred at 445 °C, and no further weight loss occurred up to 700 °C. On the other hand, 4 showed a significant weight loss between 330 and 415 °C, which may be due to decomposition of the organic part of the chelate in the [NiL] complex, as well as dehydroxylation of the layers. Gradual weight loss was observed up to 850 °C. Continuous weight loss behavior has also been found in other layered hydroxides, and was assigned to the continuous loss of volatile gas from the micropores of the oxide residue.41 Upon increasing the temperature in 4 from 415 °C, gradual weight loss was observed due to sintering of the oxide, as well as the collapse of the pores at elevated temperatures. Based on the decomposition temperatures, thermal stability in the solid state can be inferred in each case. Fig. 3 shows that [NiL]2− intercalated compound 4 is thermally less stable than benzoate intercalated LDH material 3, whereas 4 showed a higher thermal stability than free complex 2.

N2 adsorption–desorption isotherms at 77 K for 3 and 4 are shown in Fig. 4. The isotherms of 3 and 4 show a type II pattern according to the IUPAC classification,42 with a hysteresis loop beginning at 0.40 and 0.15 P/P0 for 3 and 4, respectively. The BET surface area of benzoate anion-pillared hydrotalcite 3 (32.3 m2 g−1) is marginally lower than that of 4 (38.4 m2 g−1), indicating that intracrystalline gallery surfaces have been created in the latter, and that 4 is slightly more accessible due to pillaring of the Ni(II) complex.
 | ||
| Fig. 4 N2 adsorption–desorption isotherms at 77 K of (a) 3 and (b) 4. | ||
SEM images of 3 and 4 are shown in Fig. 5. Similar agglomerates with a granular morphology and individual particle sizes as small as 100 nm or less are observed in the SEM images in both cases (Fig. 5(a) and (b)). These results indicate that the morphology of the host hydrotalcite-like material, 3, is not significantly influenced by the intercalation of the nickel-salen complex into the LDH host by partial substitution of benzoate ions.
 | ||
| Fig. 5 Scanning electron micrographs of (a) 3 and (b) 4. | ||
Fig. 6(a) and (b) present TEM images of 3 and 4. Both images show agglomerates of small, smooth, rounded particles of variable shape and size. Particle sizes typically fall in the 10–100 nm range for both materials. The parallel, linear, dark features seen in both images are evidence of a layered structure in each, indicating that the LDH structure remains intact during the ion-exchange process. Interestingly, one of the TEM images of 4 (Fig. 6(c)) appears to show single-layer scroll- or nanotube-like structures (lower right) and multi-layer nanotubes (top right), both viewed end-on and in a large crystal (centre), where the layer structure is clear (parallel dark lines).
 | ||
| Fig. 6 Transmission electron micrographs of (a) 3, (b) and (c) 4. | ||
The UV-vis diffuse reflectance spectra of 2, 3 and 4 are shown in Fig. 7. The free Ni(II) complex, 2, shows two absorption maxima above 400 nm (Table 1), where that at 535 nm may represent the lowest energy d–d transition of Ni(II). Earlier studies37 reported similar band spectra in this region for Ni(II)-salen complexes in solution. The UV-vis spectrum of 4 showed identical features to the free complex, indicating that no change at the Ni(II) coordination centre took place during intercalation. The electronic spectrum of 3 did not show any band maxima in the 300–800 nm region (Fig. 7(c)).
 | ||
| Fig. 7 Diffuse reflectance UV-vis spectra of (a) free 2, (b) 4 and (c) 3. | ||
Tetralin oxidation over the LDH-[Ni-(sulfonato-salen)] catalyst
Benzylic oxidation reactions are commonly carried out using t-BuOOH as the oxidant.43 The liquid phase oxidation of tetralin over heterogeneous catalysts affords 1-tetralone and 1-tetralol the as major products, and 1-naphthol and naphthalene as by-products.17 Initially, tetralin oxidation using t-BuOOH as the oxidant was carried out under varying reaction temperatures from 70 to 100 °C in a chlorobenzene medium. As shown in Table 2, tetralin conversion gradually increased from 36 to 45% as the temperature was increased from 70 to 100 °C. The selectivity for 1-tetralone reached 66% at 70 °C and then slightly dropped to 64% at 100 °C. The reaction was then carried out with a varying amount of catalyst from 5.0 to 40.0 mg with a tetralin : t-BuOOH molar ratio of 1:2 at 70 °C. Tetralin conversion gradually increased with increasing amounts of catalyst, while the selectivity for 1-tetralone and 1-tetralol remained virtually unaltered (Table 2). It was also found that non-catalytic thermal oxidation takes place in the presence of t-BuOOH, leading to 12% tetralin conversion at 70 °C after 8 h.
| Temp./°C | Catalyst amount/mg | Conversion (%) | Selectivity (%)c | |||
|---|---|---|---|---|---|---|
| 1-Tlone | 1-Tlol | Nthol | Nlene | |||
| a Reaction conditions: tetralin (2 mmol), t-BuOOH (4 mmol), chlorobenzene (5 cm3), 8 h. b Without catalyst (in the presence of t-BuOOH) conversion = 12%. c 1-Tetralone, 1-tetralol, 1-naphthol and naphthalene are denoted as Tlone, Tlol, Nthol and Nlene, respectively. | ||||||
| 70b | 5.0 | 24.4 | 66.8 | 25.7 | 6.1 | 1.4 |
| 20.0 | 36.2 | 66.2 | 25.9 | 6.0 | 1.9 | |
| 40.0 | 39.1 | 66.0 | 26.2 | 5.8 | 2.0 | |
| 80 | 20.0 | 39.9 | 65.5 | 26.8 | 5.4 | 2.3 |
| 100 | 20.0 | 45.2 | 64.2 | 24.9 | 6.8 | 4.1 |
In situ-generated acylperoxy radicals from an aldehyde and dioxygen were also considered as an alternative oxidant. The LDH-[NiL] catalyst, 4, was thus tested in the oxidation of tetralin using dioxygen (1 atm) in the presence of trimethylacetaldehyde. The effect of the reaction temperature on the oxidation of tetralin was investigated between 25–70 °C. As presented in Table 3, the conversion of tetralin gradually increased with increasing temperature. With an increase in temperature from 25 to 70 °C, the total reaction time was shortened and the selectivity for 1-tetralone decreased from 79 to 72% at 70 °C, and where tetralin conversion levelled off at 72%. The reaction was then carried out with a varying amount of catalyst from 5.0 to 40.0 mg at 50 °C. According to Table 3, the conversion gradually increased from 53 to 68% with increases in the catalyst amount, while the selectivity for 1-tetralone and 1-tetralol remained virtually unaltered. Therefore, in situ-generated acylperoxy radicals proved to be a more effective oxidant than t-BuOOH for tetralin oxidation. A group of researchers have reported that the epoxidation of styrene using in situ-generated acylperoxy radicals from a dioxygen/isobutylraldehyde oxidant over an immobilized Cu(II)-perchlorophthalocyamine complex on MCM-41 as the catalyst gave a higher yield of epoxide than when t-BuOOH was employed as the oxidant.44 Thermal oxidation was found to be promoted by changing the oxidant from t-BuOOH to dioxygen/trimethylacetaldehyde (see Table 3).
| Temp./°C | Time/h | Catalyst amount/mg | Conversion (%) | Selectivity (%)f | |
|---|---|---|---|---|---|
| Tlone | Tlol | ||||
| a Reaction conditions: tetralin (2 mmol), trimethylacetaldehyde (4 mmol), acetonitrile (10 cm3) and dioxygen (1 atm). b Without catalyst (in the presence of dioxygen and trimethylacetaldehyde) conversion = 15%. c Second run. d Third run. e Fourth run. f 1-Tetralone and 1-tetralol are denoted as Tlone and Tlol, respectively. | |||||
| 25 | 18 | 20.0 | 37.2 | 77.0 | 22.1 |
| 50b | 8 | 5.0 | 52.5 | 78.6 | 21.0 |
| 20.0 | 65.6 | 78.8 | 20.8 | ||
| 20.0c | 64.8 | 78.6 | 21.0 | ||
| 20.0d | 64.5 | 78.3 | 20.3 | ||
| 20.0e | 64.5 | 78.4 | 20.6 | ||
| 40.0 | 67.7 | 79.1 | 20.5 | ||
| 70 | 7 | 20.0 | 72.3 | 72.2 | 21.0 |
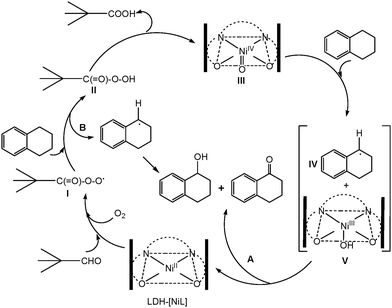
Based on the experimental results and evidence from the reported data, a plausible reaction mechanism is suggested in Scheme 2. The mechanism involves the initial activation of dioxygen in the presence of the aldehyde by the catalyst to produce the corresponding peroxy acid; a similar pathway was reported by Mukaiyama et al.45 using Mn(III)-salen complexes under homogeneous conditions. The oxidation of alkenes and alkanes using the Ni(II) complex as a catalyst under homogeneous conditions suggests the formation of either oxo-Ni(IV) or Ni(III)–O˙ as intermediate species, the formation of these species being dependent on the nature of the ligand field and the ability of the oxygen atom donor.46,47 In case of the LDH hosted nickel-salen system, we propose that trimethylacetaldehyde is initially converted to highly active acylperoxy radical I, leading to peroxy acid II, and then forms a higher valent nickel-oxo speices III. This species reacts with tetralin to form radical IV and nickel(III)-hydroxy complex V.46 In addition to hydrogen atom abstraction from C–H bonds by the high valence nickel(III)-oxo species (path A), the reaction may simultaneously produce the C12H11˙ radical by donating hydrogen atoms to acylperoxy radicals (path B, Scheme 2) to form the corresponding products.
It is important to verify the stability of the heterogeneous catalyst under the given reaction conditions, since metal leached from the catalyst could be responsible for the observed catalytic behavior.16,19 The stability of the catalyst was tested by performing repeat reaction cycles under the same reaction conditions as described above. At the end of each reaction cycle, the catalyst was recovered by filtration, washed with acetonitrile, dried and reused. The results are shown in Table 3. Tetralin conversion dropped from 65.6 to 64.8% after the first run and remained almost identical after the second run. Further experiments on the stability of the catalyst during tetralin oxidation were performed by the hot filtration technique. The catalyst was filtered off at 50 °C after 1 h in order to avoid re-adsorption of leached nickel onto the catalyst surface. The reaction still proceeded, but with a substantially lower conversion than in the presence of the catalyst (Fig. 8). This higher conversion (3.3%) after hot filtration relative to the blank run conversion after an 8 h reaction may be contributed-to by the initially formed active radical intermediates remaining in the filtrate,48 as well as by a very minor homogeneous contribution. The FT-IR spectrum of the solid catalyst after reuse was indistinguishable from that of the fresh catalyst (Fig. 9). This observation indicates that the catalyst remains unchanged during the oxidation reaction. Therefore, the catalytic activity seen in the present catalyst must be mostly attributable to the nickel-salen center in 4.
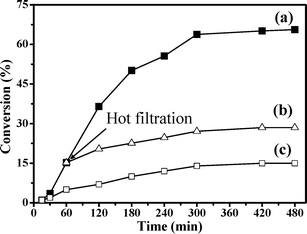 | ||
| Fig. 8 Tetralin oxidation at 50 °C: (a) fresh catalyst, (b) filtrate (filtered 4 after a 1 h reaction time) and (c) without catalyst (in the presence of trimethylacetaldehyde and dioxygen). | ||
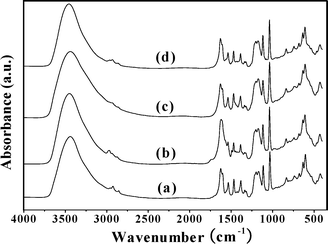 | ||
| Fig. 9 FT-IR spectra of (a) fresh 4 and the reused catalyst after the (b) first run, (c) second run and (d) fourth run. | ||
Conclusions
A sulfonato-salen-nickel(II) compound was successfully intercalated into a Zn(II)-Al(III) LDH host at room temperature from an aqueous medium and used as a catalyst for the oxidation of tetralin. The in situ-generated acylperoxy radical (via a dioxygen/trimethyl-acetaldehyde oxidant system) was found to be a more efficient method than t-BuOOH for transforming tetralin into 1-tetralone using LDH-[nickel-salen] as the catalyst. Recycling and hot filtration experiments indicated that the oxidation occurred over LDH-[nickel-salen] through mostly heterogeneous nickel-salen centers and thermal autooxidation, but a homogeneous contribution could not be entirely excluded. The formation of acylperoxy radicals and a high-valent nickel-oxygen species was proposed as the mechanism responsible for the oxidation of tetralin over the LDH hosted Ni-complex catalyst.This work was supported by the Korea Energy Management Corporation (KEMCO) through the Energy and Resources Development Program (2008).
References
- G. A. Wächter, R. W. Hartman, T. Sergejew, G. L. Grun and D. Ledergerber, J. Med. Chem., 1996, 39, 834 CrossRef CAS.
- H. G. Franck and J. W. Stadelhofer, in Industrial Aromatic Chemistry, Springer-Verlag, Berlin, Heidelberg, 1988, pp. 313 Search PubMed.
- R. G. Tailleur, G. A. Salva and G. Garcia, Fuel, 2009, 88, 744 CrossRef CAS.
- A. E. Woodward and R. B. Mesrobian, J. Am. Chem. Soc., 1953, 75, 6189 CrossRef CAS.
- Y. Kamiya and K. U. Ingold, Can. J. Chem., 1964, 42, 2424 CrossRef CAS.
- J. Muzart, Chem. Rev., 1992, 92, 113 CrossRef CAS.
- Y. M. Chung, W. S. Ahn and P. K. Lim, J. Catal., 1998, 173, 210 CrossRef CAS.
- P. Tian, Z. Liu, Z. Wu, L. Xu and Y. He, Catal. Today, 2004, 93–95, 735 CrossRef CAS.
- H. E. B. Lempers and R. A. Sheldon, Appl. Catal., A, 1996, 143, 137 CrossRef CAS.
- W. A. Carvalho, M. Wallan and U. Schuchardt, J. Mol. Catal. A: Chem., 1999, 144, 91 CrossRef CAS.
- V. Pârvulescu and B. L. Su, Catal. Today, 2001, 69, 315 CrossRef CAS.
- A. Sakthivel, S. K. Badamali and P. Selvam, Catal. Lett., 2002, 80, 73 CrossRef CAS.
- V. Pârvulescu, C. Anastasescu and B. L. Su, J. Mol. Catal. A: Chem., 2004, 211, 143 CrossRef CAS.
- S. K. Mohapatra and P. Selvam, J. Catal., 2007, 249, 394 CrossRef CAS.
- O. B. Ryan, D. E. Akporiaye, K. H. Holm and M. Stöcker, Stud. Surf. Sci. Catal., 1997, 108, 369 CAS.
- Z. Lounis, A. Riahi, F. Djafri and J. Muzart, Appl. Catal., A, 2006, 309, 270 CrossRef CAS.
- R. A. Shaikh, G. Chandrasekar, K. Biswas, J. S. Choi, W. J. Son, S. Y. Jeong and W. S. Ahn, Catal. Today, 2008, 132, 52 CrossRef CAS.
- J. Kim, S. Bhattacharjee, K. E. Jeong, S. Y. Jeong and W. S. Ahn, Chem. Commun., 2009, 3904 RSC.
- H. E. B. Lempers and R. A. Sheldon, J. Catal., 1998, 175, 62 CrossRef CAS.
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801 CrossRef CAS.
- M. J. Sabater, A. Corma, A. Domenech, V. Fornes and H. Garcia, Chem. Commun., 1997, 1285 RSC.
- W. F. Holderich and F. Kollmer, in Catalysis, ed. J. J. Spivey, Royal Society of Chemistry, Cambridge, 2002, vol. 16, pp. 43 and references therein Search PubMed.
- H. Zhang, S. Xiang and C. Li, Chem. Commun., 2005, 1209 RSC.
- H. Yoon and C. T. Burrows, J. Am. Chem. Soc., 1988, 110, 4087 CrossRef CAS.
- D. Chatterjee and A. Mitra, J. Mol. Catal. A: Chem., 1999, 144, 363 CrossRef CAS.
- D. Chatterjee, S. Mukherjee and A. Mitra, J. Mol. Catal. A: Chem., 2000, 154, 5 CrossRef CAS.
- M. R. Maurya, S. J. J. Titinchi and S. Chand, J. Mol. Catal. A: Chem., 2003, 201, 119 CrossRef CAS.
- R. Ferreira, H. Garcia, B. De Castro and C. Freire, Eur. J. Inorg. Chem., 2005, 4272 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- A. Cervilla, A. Corma, V. Fornes, E. Llopis, P. Palanca, F. Rey and A. Ribera, J. Am. Chem. Soc., 1994, 116, 1595 CrossRef CAS; A. Cervilla, A. Corma, V. Fornes, E. Llopis, F. Perez, F. Rey and A. Ribera, J. Am. Chem. Soc., 1995, 117, 6781 CrossRef CAS.
- F. Trifiro and A. Vaccario, in Comprehensive Supramolecular Chemistry, Pergamon, Elsevier Science, Oxford, 1996, vol. 7, pp. 251 Search PubMed.
- S. Xiang, Y. Zhang, Q. Xin and C. Li, Chem. Commun., 2002, 2696 RSC.
- S. Bhattacharjee, T. J. Dines and J. A. Anderson, J. Catal., 2004, 225, 398 CrossRef CAS.
- B. M. Choudary, T. Ramani, H. Maheswaran, L. Prashant, K. V. S. Ranganath and K. Vijay Kumar, Adv. Synth. Catal., 2006, 348, 493 CrossRef CAS.
- R. J. Chimentao, S. Abello, F. Medina, J. Llorca, J. E. Sueiras, Y. Cesteros and P. Salagre, J. Catal., 2007, 252, 249 CrossRef CAS.
- K. J. Berry, F. Moya, K. S. Murray, A. B. van der Bergen and B. O. West, J. Chem. Soc., Dalton Trans., 1982, 109 RSC.
- M. Botsivali, D. F. Evans, P. H. Missen and M. W. Upton, J. Chem. Soc., Dalton Trans., 1985, 1147 RSC.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, in The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, San Diego, 1991, pp. 245 Search PubMed.
- B. S. Garg and D. N. Kumar, Spectrochim. Acta, Part A, 2003, 59, 229 CrossRef CAS.
- A. A. El-Bindary and A. Z. El-Sonbati, Pol. J. Chem., 2000, 74, 615 CAS.
- S. V. Prasanna, P. V. Kamath and C. Shivakumara, Mater. Res. Bull., 2007, 42, 1028 CrossRef CAS.
- B. C. Lippens and J. H. De Boer, J. Catal., 1965, 4, 319 CrossRef.
- R. A. Sheldon, J. Mol. Catal. A: Chem., 1996, 107, 75 CrossRef CAS and references therein.
- P. Karandikar, M. Agashe, K. Vijayamohanan and A. J. Chandwadkar, Appl. Catal., A, 2004, 257, 133 CrossRef CAS.
- T. Yamada, K. Imagawa, T. Nagata and T. Mukaiyama, Chem. Lett., 1992, 2231 CAS.
- J. D. Koola and J. K. Kochi, Inorg. Chem., 1987, 26, 908 CrossRef CAS.
- H. Yoon, T. R. Wagler, K. J. O’Conner and C. J. Burrows, J. Am. Chem. Soc., 1990, 112, 4568 CrossRef CAS.
- F. X. Llabrés i Xamena, O. Casanova, R. G. Tailleur, H. Garcia and A. Corma, J. Catal., 2008, 255, 220 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |