The missing Zinc: p53 misfolding and cancer
Stewart N.
Loh
Department of Biochemistry & Molecular Biology, State University of New York Upstate Medical University, 750 East Adams Street, Syracuse, NY 13210, USA
First published on 18th May 2010
Abstract
The p53 tumor suppressor is a transcription factor that contains a single zinc ion near its DNA binding interface. Zn2+ is required for site-specific DNA binding and proper transcriptional activation. In addition to its functional significance, zinc plays a dominant role in determining whether p53 folds productively or misfolds. Insufficient zinc and excess zinc cause p53 to misfold by distinct mechanisms which both result in functional loss. The zinc-binding status of p53 in the cell is impacted significantly by the presence of tumorigenic mutations and by metal ion homeostasis. This review discusses mechanisms by which zinc modulates folding and misfolding of p53, how improper metal binding and release leads to loss of function and cancer, and how misfolding can be rescued by metallochaperones.
 Stewart N. Loh | Stewart Loh received a BS in Chemistry at the University of Utah in 1987 and a PhD in Biochemistry at the University of Wisconsin-Madison in 1993, under the direction of John Markley. He was a Damon Runyon-Walter Winchell Postdoctoral Fellow at Stanford University in the laboratory of Robert Baldwin. He joined the faculty at the State University of New York Upstate Medical University in 1996, where he has held the rank of Professor since 2007. His research interests are protein folding and the design of protein-based molecular switches. |
Introduction
Zinc is the second most abundant transition metal in the human body. Between 3% and 10% of human genes likely encode for zinc-binding proteins, making this family the most abundant type of metalloprotein in the metazoan proteome.1–3 Zinc binding can serve both structural and catalytic functions. Zn2+ prefers to coordinate to O, N, and S atoms and as such it is well-suited to bind proteins and stabilize their native folds. In this structural role, the four tetrahedral coordination sites are occupied by amino acid groups, which are frequently the side chains of cysteine and histidine. All six major classes of enzymes contain examples of zinc-binding proteins.4 As a catalytic metal it coordinates a water molecule and activates it for ionization, polarization, or displacement.Transcription factors are especially reliant on zinc for function. Nearly half of eukaryotic transcription factors bind zinc1,4 and in most of these instances the metal is used to maintain structure. The p53 tumor suppressor is one such example that plays a particularly dominant role in human health. The p53 tetramer coordinates a single Zn2+ in each of its four identical subunits. In response to DNA damage, telomere degradation, or other oncogenic stress signals, p53 activates transcription of genes involved in cell cycle arrest, DNA repair, apoptosis, and senescence. It can also enter mitochondria and promote apoptosis by a transcription-independent mechanism.5 Given the fact that p53 directs these numerous response pathways, it is not surprising that loss of p53 function leads to uncontrolled cell growth. p53 is the single most frequently mutated protein in human cancer. One in two tumors contains mutant p53 (most often of the missense variety) and mutation-induced loss of function is associated with all major forms of the disease.6–8 Restoring function to mutant p53 offers the potential for eliminating a wide variety of tumor types bearing different genetic lesions, as was demonstrated recently in mice.9,10
p53 (393 amino acids) is comprised of three functionally distinct domains. The N-terminal domain (residues 1–67) is responsible for transactivation and is largely disordered. The central DNA binding domain (DBD; residues 94–312) binds the single zinc ion. Oligomerization is encoded by the tetramerization domain (residues 325–355). p53 activates transcription most efficiently when it is tetrameric,11,12 although it can exist in the cell in monomic and dimeric forms as well.13,14 This review focuses primarily on the isolated DBD fragment, which is monomeric, because the majority of structural and biophysical studies of p53 have been carried out on this domain. Nevertheless, available data indicate that many of the basic properties of DBD are preserved in dimeric and tetrameric p53. X-ray structures of the DBD fragment,15 DBD dimer,16 and DBD tetramer17–19 indicate that monomeric and oligomeric DBD share essentially identical zinc- and DNA-binding features. Thermodynamic stability of the DBD fragment is comparable to that of DBD within the p53 tetramer.20 DBD and full-length p53 exhibit similar folding (and misfolding) mechanisms with one notable difference. As described below, isolated DBD exhibits significant misfolding under moderately destabilizing conditions (e.g. physiological temperature, elevated pressure, or in the presence of tumorigenic mutations). Misfolding is even more pronounced in full-length p53;21 it is apparently enhanced by intramolecular association of DBDs within the same tetramer.20 Since improper zinc binding is known to promote misfolding of isolated DBD (vide infra), it is likely that the effects of zinc loss and misligation discussed here are equally deleterious to the full-length p53 tetramer, if not more so.
Over 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tumorigenic germline mutations in p53 have been identified to date (International Agency for Research on Cancer p53 database; www-p53.iarc.fr). More than 90% of these mutations are found in DBD. They act by compromising DNA binding activity via one of two general mechanisms.22,23 The first is global destabilization of DBD. Strongly destabilizing mutations can cause p53 to unfold while less severe mutations can lead to aggregation or increased proteosomal degradation. The second mechanism is direct reduction of DNA binding affinity. These mutations are found at the DNA binding interface or near the zinc binding site. Biochemical experiments establish that loss of Zn2+ compromises DNA binding activity in vitro.24–26 Patient studies suggest a clinical parallel: point mutations in the zinc-binding loops (vide infra) are associated with inferior prognosis for cancers of the breast,27–30 prostate,31,32 non-small cell lung,33 and esophagus.34
000 tumorigenic germline mutations in p53 have been identified to date (International Agency for Research on Cancer p53 database; www-p53.iarc.fr). More than 90% of these mutations are found in DBD. They act by compromising DNA binding activity via one of two general mechanisms.22,23 The first is global destabilization of DBD. Strongly destabilizing mutations can cause p53 to unfold while less severe mutations can lead to aggregation or increased proteosomal degradation. The second mechanism is direct reduction of DNA binding affinity. These mutations are found at the DNA binding interface or near the zinc binding site. Biochemical experiments establish that loss of Zn2+ compromises DNA binding activity in vitro.24–26 Patient studies suggest a clinical parallel: point mutations in the zinc-binding loops (vide infra) are associated with inferior prognosis for cancers of the breast,27–30 prostate,31,32 non-small cell lung,33 and esophagus.34
Structures of zinc-bound and zinc-free DBD
Chelation experiments carried out in 1993 first demonstrated that p53 is a zinc-dependent metalloprotein.24 Solved the following year, the landmark X-ray structure of the DBD/DNA complex revealed why the metal is necessary for site-specific DNA binding.15 The DNA contact interface consists of two loops (L1 and L3) and a short helix (H2) (Fig. 1A). L1 and H2 bind in the major groove while L3 interacts with the minor groove. The tetrahedrally-coordinated zinc ion is coordinated to the side chains of Cys176 and His179 in L2 and Cys238 and Cys242 in L3. Zn2+ bridges the two loops and appears to hold L3 in the proper orientation for minor groove binding. p53 is distinct from the larger class of zinc-finger transcription factors, although it is sometimes mistakenly classified as such. Zinc finger proteins use the metal to stabilize a short recognition helix which docks into the major groove. Helical stabilization is achieved by the use of two Cys/His residues spaced one helical turn apart to coordinate Zn2+. The remaining two ligands are Cys/His residues spaced closely together in a β-strand; the two secondary structures are connected by a short linker. By contrast, the pairs of zinc-binding residues in p53 are separated by a large portion of the DBD structure, and the recognition motif stabilized by zinc is a loop that interacts with the minor groove. | ||
| Fig. 1 X-ray crystal structures of human DBD (A), the zinc-binding region of mouse DBD (B), and the zinc-binding region of mouse apoDBD (C). The zinc ion is shown in green and Cys and His residues are colored red and blue, respectively. In panel A, the zinc-coordinating residues in human DBD are Cys176, His179, Cys238, and Cys242; the loops and helices that interact with DNA or zinc are indicated. | ||
Wild-type (WT) DBD binds zinc tightly at low temperatures. Recombinant protein expressed in E. coli (20 °C) purifies with one equivalent of bound Zn2+. Zinc remains bound to WT DBD after several days of incubation with excess EDTA.35 The dissociation rate was reported to be ∼10−6 s−1 at 10 °C.36 Zinc binding affinity decreases markedly with temperature as well as in the presence of mutations. At physiological temperature, WT DBD loses zinc to chelators on the timescale of minutes.26 The R175H variant, which is among the most oncogenic of the hot-spot mutants,37 loses Zn2+ within seconds at 20 °C. These observations, together with measurements of thermodynamic stability and unfolding rates of DBD,38,39 suggest that the rate of metal ion abstraction is at least partially limited by DBD unfolding. If so, then most tumorigenic mutations will exacerbate zinc loss because the majority of these alterations destabilize DBD22,23 as well as increase its unfolding rate.39
The effect of zinc removal on DBD structure has been characterized by both experimental and computational methods. Zinc remains bound to DBD (at micromolar protein concentration) even when the protein is unfolded in 6 M urea.22 However, affinity is weak compared to that of the native protein and the metal can be extracted by briefly exposing DBD to moderately destabilizing conditions (e.g. pH 4.3) in the presence of EDTA.26 Zinc-free DBD (apoDBD) is folded but it is significantly less stable than DBD (ΔG° = 10 kcal mol−1 and 6 kcal mol−1 for DBD and apoDBD, respectively).26,40,41 Surprisingly, hot-spot mutations that lower stability of DBD have little or no effect on apoDBD.26 This finding argues that destabilizing mutations may have the secondary effect of increasing the percentage of apoDBD in the cell. Two-dimensional NMR spectra reveal that many resonances do not shift upon zinc abstraction.26 These residues map to the major groove-binding L1 loop and H2 helix as well as to numerous positions in the central β-sandwich. Extensive chemical shift differences are observed in the L2 and L3 loops as well as portions of the β-sandwich proximal to the zinc binding site. A similar result was obtained by molecular dynamics simulations of DBD and apoDBD.42 The greatest difference in structure between the two proteins was localized to L2. L2 appears to be flexible even when zinc is bound, as the H1 helix (which is embedded in L2) was observed to transiently unfold and dislocate His179 from the metal binding site. This unfolding event was suggested to represent the pathway for spontaneous zinc release. Significantly, the simulations of the apoDBD/DNA complex found that Arg248 was displaced from the minor groove. The functional consequence of this dislocation is discussed below.
The 1.5 Å X-ray structure of mouse apoDBD was solved in 2008.43 DBD crystals were grown in the absence of reducing agent, and this allowed a disulfide bond to form between the Zn2+-coordinating side chains of Cys173 and Cys239 (equivalent to Cys176 and Cys242 in human DBD). Consequently, the protein lacked zinc but bore a crosslink between the L2 and L3 loops. Fig. 1B and Fig. 1C compare the zinc binding regions of DBD44,45 and apoDBD, respectively, for the mouse protein. Consistent with the two previous studies, the influence of zinc is mainly localized to L3. The structures of apoDBD and DBD superimpose with a Cα root mean-square deviation of 0.536 Å if the L3 loop is excluded from comparison. L3 is displaced significantly in the apoDBD structure with Arg245 (equivalent to Arg248 in human DBD) once again out of position for DNA binding.
What is the functional consequence of zinc loss? It does not eliminate DNA binding but it does significantly diminish DNA binding specificity. DBD binds the gadd45 sequence (Kd = 0.89 μM) 11-fold more tightly than a nonconsensus oligonucleotide (Kd = 9.6 μM).46 By contrast, apoDBD exhibits the same Kd (6.0 μM) towards the gadd45 and nonconsensus oligonucleotides.26 Available data suggest that displacement of Arg248 from the minor groove, which is observed in all apoDBD structures, is chiefly responsible for the lack of discrimination. The hot-spot R248Q mutation is known to inhibit DNA binding.23 Arg248 is located in an especially narrow region of the minor groove which corresponds to a minimum of negative electrostatic potential.19 A recent analysis of protein-DNA complexes in the PDB suggests that this type of interaction plays a critical role in minor groove recognition and overall DNA binding specificity.47
Zinc and folding
Tumorigenic p53 mutations fall into two broad categories: those that destabilize the molecule, and those that directly impair DNA binding. There is substantial evidence, however, that misfolding is a significant cause of functional loss, particularly for the first class of mutations. It is therefore of interest to understand the kinetic folding and unfolding mechanisms of p53 and how Zn2+ influences these processes.DBD folds by an unusually complex mechanism. At minimum, refolding from urea is modeled by two parallel pathways and no less than six intermediates.38 The unique feature of DBD folding is that native molecules form in four distinct tracks. One-fourth of molecules fold with a half-time of 8 s. This rate is relatively representative for a protein the size and complexity of DBD.48 The remaining population folds considerably more slowly; approximately equal fractions fold with half-times of 30 s, 4.3 m, and several hours. It is clear that p53 falls victim to more kinetic traps than do most proteins. The destabilizing mutations G245S, R249S, and R282Q have no effect on DBD folding rates or amplitudes. Instead, they accelerate unfolding. This finding suggests that these and other tumorigenic mutations contribute to loss of p53 function by causing DBD to cycle unusually rapidly between native and unfolded states. A sizeable fraction of DBD becomes trapped with each unfolding-folding cycle. Accumulation of these trapped species is manifested by loss of DNA binding activity and aggregation in vitro. In the cell, these misfolded proteins are likely to aggregate and/or be degraded before they can be resolved.
Is metal ligation responsible for the preponderance of kinetic traps? Does Zn2+ help or hinder DBD folding? In some cases, metals or other cofactors accelerate folding by possibly stabilizing the transition state ensemble.49–51 Metal binding can also benefit folding by reducing the populations of aggregation-prone intermediate states, as has been demonstrated for superoxide dismutase.49,52–54 On the other hand, metals can frustrate productive folding by binding to and stabilizing non-native conformations. In the case of p53, zinc does not simplify the DBD folding reaction. ApoDBD and DBD exhibit similar folding rates and amplitudes.36 The main difference is that native apoDBD forms slightly faster than native DBD. For example, 80% of apoDBD molecules reach the native state within one minute while that figure is only 48% for DBD.36 Zn2+ ligation (or misligation) may thus exacerbate one or more of the existing kinetic traps but it does not cause them. Rather, heterogeneity appears to be an inherent feature of the p53 folding landscape.
The folding mechanisms of apoDBD and DBD are shown in simplified form in Fig. 2A and Fig. 2B, respectively. Although zinc stays associated to DBD when the protein is unfolded in urea, it is not known whether the metal remains bound by the native residues or is coordinated to non-native ligands. The former scenario is assumed in Fig. 2B. The top set of grey arrows represents folding from unfolded to native states (all four folding tracks are condensed into a single arrow for simplicity). The bottom set of grey arrows indicate tetramerization and DNA binding. In summary, the data rule out the scenario that Zn2+ accelerates or is required for folding.
 | ||
| Fig. 2 Schematic of zinc-dependent folding and misfolding of DBD. The major folding (top set of grey arrows) and DNA binding (bottom set of grey arrows) reactions of DBD are shown in conditions of insufficient zinc (A), normal zinc (B), and excessive zinc (C). Zinc ions are depicted as grey spheres. MC indicates a metallochaperone such as metallothionein or nitriloacetate. In both panel A and in panel B, a significant percentage of DBD misfolds with each folding-unfolding cycle; for clarity this process is not shown. | ||
Zinc and misfolding
DBD is a relatively unstable protein at physiological temperature. Although it is quite stable at low temperatures (ΔG° = 10 kcal mol−1 at 10 °C),22 its stability decreases markedly with temperature and it exhibits an apparent melting temperature of ∼45 °C.39,55 p53 misfolding is consequently exacerbated by conditions which destabilize DBD by even a moderate amount. For example, WT DBD remains soluble and functional for many days at 4 °C, but it aggregates visibly within hours at 37 °C20,26,56 and loses DNA binding activity on the same time scale.21 Destabilizing mutations hasten the aggregation rate by 3 to 4-fold.56 Similarly, elevated pressure destabilizes DBD and causes it to form fibrillar aggregates.57,58 Destabilizing conditions likely promote misfolding by two related mechanisms: by speeding up the rate of cycling between native and unfolded states (Fig. 2), and by increasing the equilibrium population of partially folded forms, which tend to be aggregation-prone. Recent evidence suggests that misfolding is even more pronounced in the full-length p53 tetramer, because the high local concentration of DBDs causes partially unfolded DBDs to form nonfunctional complexes within a single tetramer.20 Consistent with that hypothesis, full-length p53 loses DNA binding activity significantly faster than DBD at 37 °C.21The mechanisms by which zinc binding and loss affect p53 function extend beyond direct modulation of the DNA binding interface. Insufficient as well as excess zinc cause DBD to misfold by distinct pathways. Zinc loss mimics tumorigenic mutations, elevated temperature, and elevated pressure in that it destabilizes DBD and destabilization is manifested by faster unfolding rates.36,38 The resulting acceleration of the unfolding-folding cycle leads to accumulation of misfolded protein. For example, like mutant DBD, WT apoDBD aggregates within minutes at physiological temperature.26 In fact, a small amount of apoDBD can nucleate aggregation of DBD.26 This finding implies that if one subunit of the p53 tetramer loses zinc it can rapidly lead to aggregation of functional DBDs in the same tetramer and between tetramers.
Excess zinc causes pronounced misfolding by an altogether different means. When ZnCl2 is added to unfolded DBD at greater than a 2:1 metal:protein ratio, folding is completely arrested (Fig. 3). If the excess metal is not removed within a short time then the protein precipitates irreversibly (Fig. 2C). Similarly, native DBD aggregates visibly in the presence of a two-fold or greater excess of zinc.36 In both cases, DBD-DBD association is presumably triggered by additional zinc ions coordinating to other Cys and/or His residues, of which there are 10 and 9, respectively (Fig. 1A). This problem is especially pronounced during folding, most likely because the plasticity of the nascent structures allows for a multitude of opportunistic binding sites to form. In any event, these misligation events lead to precipitation by either inducing DBD to adopt an aggregation-prone conformation, or by bridging multiple DBDs with shared zinc ions.
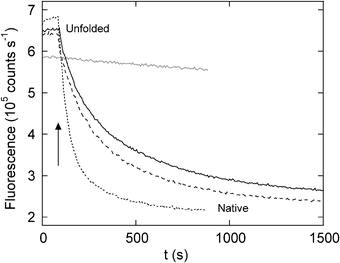 | ||
| Fig. 3 Rescue of DBD misfolding by metal chelators monitored by Trp fluorescence. DBD (0.75 μM) was refolded in the presence of 16 μM ZnCl2. After 80 s of stalled folding (indicated by the arrow), NTA (black line), EGTA (long dashes), EDTA (short dashes), or nothing (grey line) was added to the solution. Chelator concentrations are 0.5 mM. Figure reprinted with permission from the publisher.36 | ||
Metallochaperone-assisted folding
Fig. 2 implies that p53 can exist in functional form only if exactly one equivalent of free Zn2+ (per DBD monomer) is present (Fig. 2B). A deficit of zinc results in loss of DNA binding specificity (Fig. 2A); a surplus causes misfolding and aggregation (Fig. 2C). Clearly this criterion is not met in vivo. Protein metallochaperones (MC), chiefly the metallothioneins (MT), serve to buffer zinc ions in the cell.59,60 Small molecules can possess MC activity as well and rescue DBD misfolding in vitro. The strategy is to choose a chelator whose affinity for zinc is less than that of the native site but greater than that of nonphysiological sites. In this way the MC serves as both sink and source of zinc ions. The MC protects against misfolding by sequestering zinc during the critical early stages of folding, when the protein is flexible and particularly vulnerable to misligation. The MC transfers Zn2+ to the protein only after it has safely attained the native state and the high-affinity pocket is formed.Both EDTA and EGTA rescue misfolding of DBD. When folding is stalled by excess ZnCl2, addition of these chelators allows folding to proceed to completion (Fig. 3). However, EDTA (Kd = 10−15 M) and EGTA (Kd = 10−12 M) bind Zn2+ too tightly to be useful: apoDBD is the end product of folding in this case. Nitriloacetate (NTA; Kd = 10−8 M), however, restores correct folding and regenerates functional DBD with high efficiency (Fig. 3). Importantly, once DBD is regenerated, NTA has the requisite affinity for zinc to prevent metal-induced aggregation. Fig. 3 suggests that the non-physiological sites bind Zn2+ with Kd values in the range of 10−6 to 10−9 M. A small amount of Zn2+/NTA present in the solution (e.g. 30 μM ZnCl2 and 75 μM NTA) exerts a generally protective effect; it guards against misfolding during unfolding/folding cycles, discourages zinc-induced aggregation of native DBD, and supplies metal ions to DBD for proper DNA binding.36
Zinc homeostasis, p53, and cancer
Zinc binding status of p53 in vivo is governed by several factors including the presence of DBD mutations, redox state of Cys residues in DBD, and zinc bioavailability in the cell. As discussed above, destabilizing mutations are generally expected to promote zinc loss. Oxidative stress is another potential factor in metal dissociation. Reducing agents are required for site-specific DNA binding, and oxidation or alkylation of Cys residues abrogates this binding.61–63 Not surprisingly, substituting any of the zinc-coordinating cysteines (Cys176, Cys238, and Cys242) or Cys275 with serine impairs DNA binding in vivo.64 Individual mutation of the remaining six Cys residues does not inhibit DNA binding. The C176S mutant accumulates in nuclei of neuroblastoma cells and is able to bind DNA but not activate transcription.65 p53 is believed to be maintained in the reduced state by the thioredoxin/thioredoxin reductase system. Thioredoxin or the redox-active protein Ref-1 promotes p53 binding to DNA in vitro66,67 and overexpressing these proteins in mammalian cells stimulates p53-dependent transcription.66 Conversely, deleting the thioredoxin reductase gene in yeast inhibits p53 activity and recent evidence suggests that this effect is due either to oxidation of the zinc-bound Cys residues or to oxidation of an upstream activator.64 It is perhaps not coincidental that two of the metal-coordinating cysteines had spontaneously formed a disulfide bond in the X-ray structure of apoDBD (Fig. 1C).Zinc homeostasis likely figures prominently in the outcome of p53 folding in vivo. The concentration of available zinc in the cell is largely dependent on two factors: dietary zinc intake, and the presence of MCs and other metal-binding proteins. With respect to the first point, epidemiological studies have yielded conflicting correlations between intracellular zinc concentration and cancer. Low circulating zinc concentrations are associated with increased cancer risk.68,69 Reduced cellular zinc levels have been implicated in prostate and other cancers.70,71 Other studies, however, conclude that high zinc diets promote prostate cancer.72 Nevertheless, zinc depletion experiments reveal some consistent trends when p53 is examined. Rats fed a zinc-deficient diet had elevated p53 concentrations in their livers, and zinc repletion reduced p53 to control levels.73 Total DNA binding activity of p53 in liver extracts was unchanged, however, suggesting that the protein may be misfolded in the zinc-deficient rats. Depleting zinc from the growth medium causes a similar increase in p53 levels in a variety of cultured cell types74–80 and in at least some cases, much of the accumulated p53 appears to be nonfunctional.74,75 Treating cells with the membrane-permeant chelator TPEN causes p53 to amass in misfolded form;25 p53 activity can then be restored by removal of TPEN or supplementation with zinc.81 Most available data are consistent with the view that zinc loss leads to p53 misfolding and aggregation in the cell.
Regardless of dietary intake, the zinc-bound status of p53 is strongly influenced by MCs such as MT. MT binds up to seven zinc ions with Kd values from 10−8 M–10−12 M.60,82 Despite the high abundance of Zn2+, MCs limit its free concentration in the cell to the low picomolar range.83,84 ApoMT binds to p53, suggesting that it can abstract zinc from DBD.85 Indeed, expression of MT : p53 at a 10:1 ratio inhibits p53 activity in cultured cells.81 Decreasing MT expression to match that of p53 enhances transcriptional activity by a factor of 2–3. This same effect was demonstrated recently by using interfering RNA to deplete homeodomain-interacting protein kinase-2 (HIPK2) in prostate cancer cells.86 HIPK2 knockdown induces p53 misfolding in the cell. Zinc supplementation reactivates p53 and suppresses tumor growth. Further analysis determined that p53 misfolding is caused by increased MT expression, which results from HIPK2 depletion.87 These results gain significance in light of the fact that MTs are upregulated in a variety of malignancies.60 MT appears to modulate p53 activity by a zinc exchange mechanism much like that of NTA.
Conclusions and perspective
Proper p53 folding and function requires precise control of zinc availability and binding. Loss of zinc is doubly deleterious. ApoDBD cannot distinguish between consensus and nonconsensus DNA sequences, and it is less stable than DBD. The latter property, in combination with the kinetic traps inherent to the p53 folding mechanism, leads to misfolding by increasing the cycling rate between folded and unfolded states. Excess free zinc induces aggregation by coordinating to non-physiological binding sites in either an intra- or inter-molecular fashion. With regard to cancer, the two factors most likely to promote improper zinc binding and associated loss of p53 function are the presence of destabilizing DBD mutations and alteration of zinc bioavailability. Loss of protein stability is the cause of 75% of monogenic diseases88 and many tumorigenic mutations likewise destabilize DBD. Accordingly, a longstanding therapeutic goal has been to develop small molecules that bind to p53 and stabilize the native structure.89–95 Such molecules will guard against zinc loss as well, and this strategy remains at the forefront for treating p53-related malignancies. Chemical chelators such as NTA are effective at preventing misfolding in vitro. The cell-permeant chelator 1,10-phenanthroline has been shown to activate p53 in mouse cells.96 The challenge for metallochaperone-based rescue of p53 function is to strike the appropriate balance between metal abstraction and donation. Robust buffering will likely require agents that bind zinc with a range of affinities. This condition may be met by metallochaperones such as MT or by a combination of small-molecule chelators.References
- C. Andreini, L. Banci, I. Bertini and A. Rosato, Counting the zinc-proteins encoded in the human genome, J. Proteome Res., 2006, 5, 196–201 CrossRef CAS.
- A. I. Anzellotti and N. P. Farrell, Zinc metalloproteins as medicinal targets, Chem. Soc. Rev., 2008, 37, 1629–1651 RSC.
- W. Maret, Metallothionein redox biology in the cytoprotective and cytotoxic functions of zinc, Exp. Gerontol., 2008, 43, 363–369 CrossRef CAS.
- K. A. Jackson, R. A. Valentine, L. J. Coneyworth, J. C. Mathers and D. Ford, Mechanisms of mammalian zinc-regulated gene expression, Biochem. Soc. Trans., 2008, 36, 1262–1266 CrossRef CAS.
- D. R. Green and G. Kroemer, Cytoplasmic functions of the tumour suppressor p53, Nature, 2009, 458, 1127–1130 CrossRef CAS.
- J. M. Nigro, S. J. Baker, A. C. Presinger, J. M. Jessup, R. Hostetter, K. Cleary, S. H. Bigner, N. Davidson, S. Baylin, P. Devilee and B. Vogelstein, Mutations in the p53 gene occur in diverse human tumour types, Nature, 1989, 342, 705–708 CrossRef CAS.
- S. Jones, X. Zhang, D. W. Parsons, J. C. Lin, R. J. Leary, P. Angenendt, P. Mankoo, H. Carter, H. Kamiyama, A. Jimeno, S. M. Hong, B. Fu, M. T. Lin, E. S. Calhoun, M. Kamiyama, K. Walter, T. Nikolskaya, Y. Nikolsky, J. Hartigan, D. R. Smith, M. Hidalgo, S. D. Leach, A. P. Klein, E. M. Jaffee, M. Goggins, A. Maitra, C. Iacobuzio-Donahue, J. R. Eshleman, S. E. Kern, R. H. Hruban, R. Karchin, N. Papadopoulos, G. Parmigiani, B. Vogelstein, V. E. Velculescu and K. W. Kinzler, Core signaling pathways in human pancreatic cancers revealed by global genomic analyses, Science, 2008, 321, 1801–1806 CrossRef CAS.
- D. W. Parsons, S. Jones, X. Zhang, J. C. Lin, R. J. Leary, P. Angenendt, P. Mankoo, H. Carter, I. M. Siu, G. L. Gallia, A. Olivi, R. McLendon, B. A. Rasheed, S. Keir, T. Nikolskaya, Y. Nikolsky, D. A. Busam, H. Tekleab, L. A. Diaz, Jr., J. Hartigan, D. R. Smith, R. L. Strausberg, S. K. Marie, S. M. Shinjo, H. Yan, G. J. Riggins, D. D. Bigner, R. Karchin, N. Papadopoulos, G. Parmigiani, B. Vogelstein, V. E. Velculescu and K. W. Kinzler, An integrated genomic analysis of human glioblastoma multiforme, Science, 2008, 321, 1807–1812 CrossRef CAS.
- W. Xue, L. Zender, C. Miething, R. A. Dickins, E. Hernando, V. Krizhanovsky, C. Cordon-Cardo and S. W. Lowe, Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas, Nature, 2007, 445, 656–660 CrossRef CAS.
- A. Ventura, D. G. Kirsch, M. E. McLaughlin, D. A. Tuveson, J. Grimm, L. Lintault, J. Newman, E. E. Reczek, R. Weissleder and T. Jacks, Restoration of p53 function leads to tumour regression in vivo, Nature, 2007, 445, 661–665 CrossRef CAS.
- T. Kawaguchi, S. Kato, K. Otsuka, G. Watanabe, T. Kumabe, T. Tominaga, T. Yoshimoto and C. Ishioka, The relationship among p53 oligomer formation, structure and transcriptional activity using a comprehensive missense mutation library, Oncogene, 2005, 24, 6976–6981 CrossRef CAS.
- J. A. Pietenpol, T. Tokino, S. Thiagalingam, W. S. El-Deiry, K. W. Kinzler and B. Vogelstein, Sequence-specific transcriptional activation is essential for growth suppression by p53, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1998–2002 CrossRef CAS.
- E. Natan, D. Hirschberg, N. Morgner, C. V. Robinson and A. R. Fersht, Ultraslow oligomerization equilibria of p53 and its implications, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14327–14332 CrossRef CAS.
- K. Sakaguchi, H. Sakamoto, M. S. Lewis, C. W. Anderson, J. W. Erickson, E. Appella and D. Xie, Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53, Biochemistry, 1997, 36, 10117–10124 CrossRef CAS.
- Y. Cho, S. Gorina, P. D. Jeffrey and N. P. Pavletich, Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations, Science, 1994, 265, 346–355 CrossRef CAS.
- W. C. Ho, M. X. Fitzgerald and R. Marmorstein, Structure of the p53 core domain dimer bound to DNA, J. Biol. Chem., 2006, 281, 20494–20502 CrossRef CAS.
- M. Kitayner, H. Rozenberg, N. Kessler, D. Rabinovich, L. Shaulov, T. E. Haran and Z. Shakked, Structural basis of DNA recognition by p53 tetramers, Mol. Cell, 2006, 22, 741–753 CrossRef CAS.
- K. A. Malecka, W. C. Ho and R. Marmorstein, Crystal structure of a p53 core tetramer bound to DNA, Oncogene, 2009, 28, 325–333 CrossRef CAS.
- M. Kitayner, H. Rozenberg, R. Rohs, O. Suad, D. Rabinovich, B. Honig and Z. Shakked, Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs, Nat. Struct. Mol. Biol., 2010, 17, 423–429 CrossRef CAS.
- D. J. Lubin, J. S. Butler and S. N. Loh, Folding of tetrameric p53: oligomerization and tumorigenic mutations induce misfolding and loss of function, J. Mol. Biol., 2010, 395, 705–716 CrossRef CAS.
- S. Bell, C. Klein, L. Müller, S. Hansen and J. Buchner, p53 contains large unstructured regions in its native state, J. Mol. Biol., 2002, 322, 917–927 CrossRef CAS.
- A. N. Bullock, J. Henckel, B. S. DeDecker, C. M. Johnson, P. V. Nikolova, M. R. Proctor, D. P. Lane and A. R. Fersht, Thermodynamic stability of wild-type and mutant p53 core domain, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14338–14342 CrossRef CAS.
- A. N. Bullock, J. Henckel and A. R. Fersht, Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy, Oncogene, 2000, 19, 1245–1256 CrossRef CAS.
- N. P. Pavletich, K. A. Chambers and C. O. Pabo, The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots, Genes Dev., 1993, 7, 2556–2564 CrossRef CAS.
- C. Méplan, M.-J. Richard and P. Hainaut, Redox signalling and transition metals in the control of the p53 pathway, Biochem. Pharmacol., 2000, 59, 25–33 CrossRef CAS.
- J. S. Butler and S. N. Loh, Structure, function, and aggregation of the zinc-free form of the p53 DNA binding domain, Biochemistry, 2003, 42, 2396–2403 CrossRef CAS.
- J. Alsner, V. Jensen, M. Kyndi, B. V. Offersen, P. Vu, A. L. Borresen-Dale and J. Overgaard, A comparison between p53 accumulation determined by immunohistochemistry and TP53 mutations as prognostic variables in tumours from breast cancer patients, Acta Oncologica, 2008, 47, 600–607 CrossRef CAS.
- M. Olivier, A. Langerod, P. Carrieri, J. Bergh, S. Klaar, J. Eyfjord, C. Theillet, C. Rodriguez, R. Lidereau, I. Bieche, J. Varley, Y. Bignon, N. Uhrhammer, R. Winqvist, A. Jukkola-Vuorinen, D. Niederacher, S. Kato, C. Ishioka, P. Hainaut and A. L. Borresen-Dale, The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer, Clin. Cancer Res., 2006, 12, 1157–1167 CrossRef CAS.
- H. J. van Slooten, M. J. van De Vijver, A. L. Borresen, J. E. Eyfjord, R. Valgardsdottir, S. Scherneck, J. M. Nesland, P. Devilee, C. J. Cornelisse and J. H. van Dierendonck, Mutations in exons 5-8 of the p53 gene, independent of their type and location, are associated with increased apoptosis and mitosis in invasive breast carcinoma, J. Pathol., 1999, 189, 504–513 CrossRef CAS.
- M. Gentile, M. Bergman Jungestrom, K. E. Olsen, P. Soderkvist and S. Wingren, p53 and survival in early onset breast cancer: analysis of gene mutations, loss of heterozygosity and protein accumulation, Eur. J. Cancer, 1999, 35, 1202–1207 CrossRef CAS.
- A. Russo, V. Bazan, B. Iacopetta, D. Kerr, T. Soussi and N. Gebbia, The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment, J. Clin. Oncol., 2005, 23, 7518–7528 CrossRef CAS.
- A. Russo, M. Migliavacca, I. Zanna, M. R. Valerio, M. A. Latteri, N. Grassi, G. Pantuso, S. Salerno, G. Dardanoni, I. Albanese, M. La Farina, R. M. Tomasino, N. Gebbia and V. Bazan, p53 mutations in L3-loop zinc-binding domain, DNA-ploidy, and S phase fraction are independent prognostic indicators in colorectal cancer: a prospective study with a five-year follow-up, Cancer Epidemiol. Biomarkers Prev., 2002, 11, 1322–1331 CAS.
- V. Skaug, D. Ryberg, E. H. Kure, M. O. Arab, L. Stangeland, A. O. Myking and A. Haugen, p53 mutations in defined structural and functional domains are related to poor clinical outcome in non-small cell lung cancer patients, Clin. Cancer Res., 2000, 6, 1031–1037 CAS.
- C. Kihara, T. Seki, Y. Furukawa, H. Yamana, Y. Kimura, P. van Schaardenburgh, K. Hirata and Y. Nakamura, Mutations in zinc-binding domains of p53 as a prognostic marker of esophageal-cancer patients, Jpn J. Cancer Res., 2000, 91, 190–198 CAS.
- M. Lokshin, Y. Li, C. Gaiddon and C. Prives, p53 and p73 display common and distinct requirements for sequence specific binding to DNA, Nucleic Acids Res., 2007, 35, 340–352 CAS.
- J. S. Butler and S. N. Loh, Zn2+-dependent misfolding of the p53 DNA binding domain, Biochemistry, 2007, 46, 2630–2639 CrossRef CAS.
- D. P. Liu, H. Song and Y. Xu, A common gain of function of p53 cancer mutants in inducing genetic instability, Oncogene, 2010, 29, 949–956 CrossRef CAS.
- J. S. Butler and S. N. Loh, Kinetic partitioning during folding of the p53 DNA binding domain, J. Mol. Biol., 2005, 350, 906–918 CrossRef CAS.
- J. S. Butler and S. N. Loh, Folding and misfolding mechanisms of the p53 DNA binding domain at physiological temperature, Protein Sci., 2006, 15, 2457–2465 CrossRef CAS.
- Y. Xue, S. Wang and X. Feng, Effect of metal ion on the structural stability of tumour suppressor protein p53 DNA-binding domain, J. Biochem., 2009, 146, 193–200 CrossRef CAS.
- Y. Xue, S. Wang and X. Feng, Influence of magnesium ion on the binding of p53 DNA-binding domain to DNA-response elements, J. Biochem., 2009, 146, 77–85 CrossRef CAS.
- J. Duan and L. Nilsson, Effects of Zn2+ on DNA recognition and stability of the p53 DNA binding domain, Biochemistry, 2006, 45, 7483–7492 CrossRef CAS.
- E. Kwon, D. Y. Kim, S. W. Suh and K. K. Kim, Crystal structure of the mouse p53 core domain in zinc-free state, Proteins, 2008, 70, 280–283 Search PubMed.
- W. C. Ho, C. Luo, K. Zhao, X. Chai, M. X. Fitzgerald and R. Marmorstein, High-resolution structure of the p53 core domain: implications for binding small-molecule stabilizing compounds, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 1484–1493 CrossRef.
- K. Zhao, X. Chai, K. Johnston, A. Clements and R. Marmorstein, Crystal structure of the mouse p53 core DNA-binding domain at 2.7 Å resolution, J. Biol. Chem., 2001, 276, 12120–12127 CrossRef CAS.
- A. Friedler, L. O. Hansson, D. B. Veprintsev, S. M. Freund, T. M. Rippin, P. V. Nikolova, M. R. Proctor, S. Rüdiger and A. R. Fersht, A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 937–942 CrossRef CAS.
- R. Rohs, S. M. West, A. Sosinsky, P. Liu, R. S. Mann and B. Honig, The role of DNA shape in protein-DNA recognition, Nature, 2009, 461, 1248–1253 CrossRef CAS.
- D. N. Ivankov, S. O. Garbuzynskiy, E. Alm, K. W. Plaxco, D. Baker and A. V. Finkelstein, Contact order revisited: influence of protein size on the folding rate, Protein Sci., 2003, 12, 2057–2062 CrossRef CAS.
- C. Kayatekin, J. A. Zitzewitz and C. R. Matthews, Zinc binding modulates the entire folding free energy surface of human Cu,Zn superoxide dismutase, J. Mol. Biol., 2008, 384, 540–555 CrossRef CAS.
- A. D. Pandit, B. A. Krantz, R. S. Dothager and T. R. Sosnick, Characterizing protein folding transition States using Psi-analysis, Methods Mol. Biol., 2007, 350, 83–104 CAS.
- N. A. Bushmarina, C. E. Blanchet, G. Vernier and V. Forge, Cofactor effects on the protein folding reaction: acceleration of α-lactalbumin refolding by metal ions, Protein Sci., 2006, 15, 659–671 CrossRef CAS.
- J. A. Rumfeldt, J. R. Lepock and E. M. Meiering, Unfolding and folding kinetics of amyotrophic lateral sclerosis-associated mutant Cu,Zn superoxide dismutases, J. Mol. Biol., 2009, 385, 278–298 CrossRef CAS.
- K. Teilum, M. H. Smith, E. Schulz, L. C. Christensen, G. Solomentsev, M. Oliveberg and M. Akke, Transient structural distortion of metal-free Cu/Zn superoxide dismutase triggers aberrant oligomerization, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18273–18278 CrossRef CAS.
- V. K. Mulligan, A. Kerman, S. Ho and A. Chakrabartty, Denaturational stress induces formation of zinc-deficient monomers of Cu,Zn superoxide dismutase: implications for pathogenesis in amyotrophic lateral sclerosis, J. Mol. Biol., 2008, 383, 424–436 CrossRef CAS.
- K. H. Khoo, A. C. Joerger, S. M. Freund and A. R. Fersht, Stabilising the DNA-binding domain of p53 by rational design of its hydrophobic core, Protein Eng., Des. Sel., 2009, 22, 421–430 CrossRef CAS.
- A. Friedler, D. B. Veprintsev, L. O. Hansson and A. R. Fersht, Kinetic instability of p53 core domain mutants, J. Biol. Chem., 2003, 278, 24108–24112 CrossRef.
- D. Ishimaru, L. R. Andrade, L. S. P. Teixeira, P. A. Quesado, L. M. Maiolino, P. M. Lopez, Y. Cordeiro, L. T. Costa, W. M. Heckl, G. Weissmüller, D. Foguel and J. L. Silva, Fibrillar aggregates of the tumor suppressor p53 core domain, Biochemistry, 2003, 42, 9022–9027 CrossRef CAS.
- D. Ishimaru, A. P. Ano Bom, L. M. Lima, P. A. Quesado, M. F. Oyama, C. V. de Moura Gallo, Y. Cordeiro and J. L. Silva, Cognate DNA stabilizes the tumor suppressor p53 and prevents misfolding and aggregation, Biochemistry, 2009, 48, 6126–6135 CrossRef CAS.
- M. O. Pedersen, R. Jensen, D. S. Pedersen, A. D. Skjolding, C. Hempel, L. Maretty and M. Penkowa, Metallothionein-I+II in neuroprotection, BioFactors, 2009, 35, 315–325 Search PubMed.
- T. Eckschlager, V. Adam, J. Hrabeta, K. Figova and R. Kizek, Metallothioneins and cancer, Curr. Protein Pept. Sci., 2009, 10, 360–375 CrossRef CAS.
- P. Hainaut and J. Milner, Redox modulation of p53 conformation and sequence-specific DNA binding in vitro, Cancer Res., 1993, 53, 4469–4473 CAS.
- T. R. Hupp, D. W. Meek, C. A. Midgley and D. P. Lane, Activation of the cryptic DNA binding function of mutant forms of p53, Nucleic Acids Res., 1993, 21, 3167–3174 CrossRef CAS.
- R. Rainwater, D. Parks, M. E. Anderson and P. Tegtmeyer, Role of cysteine residues in regulation of p53 function, Mol. Cell Biol., 1995, 15, 3892–3903 CAS.
- C. S. Stoner, G. D. Pearson, A. Koc, J. R. Merwin, N. I. Lopez and G. F. Merrill, Effect of thioredoxin deletion and p53 cysteine replacement on human p53 activity in wild-type and thioredoxin reductase null yeast, Biochemistry, 2009, 48, 9156–9169 CrossRef CAS.
- A. Wolff, A. Technau, C. Ihling, K. Technau-Ihling, R. Erber, F. X. Bosch and G. Brandner, Evidence that wild-type p53 in neuroblastoma cells is in a conformation refractory to integration into the transcriptional complex, Oncogene, 2001, 20, 1307–1317 CrossRef CAS.
- M. Ueno, H. Masutani, R. J. Arai, A. Yamauchi, K. Hirota, T. Sakai, T. Inamoto, Y. Yamaoka, J. Yodoi and T. Nikaido, Thioredoxin-dependent redox regulation of p53-mediated p21 activation, J. Biol. Chem., 1999, 274, 35809–35815 CrossRef CAS.
- L. Jayaraman, K. G. Murthy, C. Zhu, T. Curran, S. Xanthoudakis and C. Prives, Identification of redox/repair protein Ref-1 as a potent activator of p53, Genes Dev., 1997, 11, 558–570 CrossRef CAS.
- N. Leone, D. Courbon, P. Ducimetiere and M. Zureik, Zinc, copper, and magnesium and risks for all-cause, cancer, and cardiovascular mortality, Epidemiology, 2006, 17, 308–314 CrossRef.
- T. Wu, C. T. Sempos, J. L. Freudenheim, P. Muti and E. Smit, Serum iron, copper and zinc concentrations and risk of cancer mortality in US adults, Ann. Epidemiol., 2004, 14, 195–201 CrossRef.
- R. B. Franklin and L. C. Costello, Zinc as an antitumor agent in prostate cancer and in other cancers, Arch. Biochem. Biophys., 2007, 463, 211–217 CrossRef CAS.
- M. Murakami and T. Hirano, Intracellular zinc homeostasis and zinc signaling, Cancer Sci., 2008, 99, 1515–1522 CrossRef CAS.
- S. Gallus, R. Foschi, E. Negri, R. Talamini, S. Franceschi, M. Montella, V. Ramazzotti, A. Tavani, L. Dal Maso and C. La Vecchia, Dietary zinc and prostate cancer risk: a case-control study from Italy, Eur. Urol., 2007, 52, 1052–1056 CrossRef CAS.
- Y. Song, S. W. Leonard, M. G. Traber and E. Ho, Zinc deficiency affects DNA damage, oxidative stress, antioxidant defenses, and DNA repair in rats, J. Nutr., 2009, 139, 1626–1631 CrossRef CAS.
- M. S. Clegg, L. A. Hanna, B. J. Niles, T. Y. Momma and C. L. Keen, Zinc deficiency-induced cell death, IUBMB Life, 2005, 57, 661–669 Search PubMed.
- M. Yan, Y. Song, C. P. Wong, K. Hardin and E. Ho, Zinc deficiency alters DNA damage response genes in normal human prostate epithelial cells, J. Nutr., 2008, 138, 667–673 CAS.
- A. A. Alshatwi, C. T. Han, N. W. Schoene and K. Y. Lei, Nuclear accumulations of p53 and Mdm2 are accompanied by reductions in c-Abl and p300 in zinc-depleted human hepatoblastoma cells, Exp. Biol. Med. (Maywood), 2006, 231, 611–618 Search PubMed.
- E. Ho, C. Courtemanche and B. N. Ames, Zinc deficiency induces oxidative DNA damage and increases p53 expression in human lung fibroblasts, J. Nutr., 2003, 133, 2543–2548 CAS.
- E. Ho and B. N. Ames, Low intracellular zinc induces oxidative DNA damage, disrupts p53, NFkappa B, and AP1 DNA binding, and affects DNA repair in a rat glioma cell line, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16770–16775 CrossRef CAS.
- J. C. Fanzo, S. K. Reaves, L. Cui, L. Zhu and K. Y. Lei, p53 protein and p21 mRNA levels and caspase-3 activity are altered by zinc status in aortic endothelial cells, Am. J. Physiol. Cell Physiol., 2002, 283, C631–638 CAS.
- J. C. Fanzo, S. K. Reaves, L. Cui, L. Zhu, J. Y. Wu, Y. R. Wang and K. Y. Lei, Zinc status affects p53, gadd45, and c-fos expression and caspase-3 activity in human bronchial epithelial cells, Am. J. Physiol. Cell Physiol., 2001, 281, C751–757 CAS.
- C. Méplan, M.-J. Richard and P. Hainaut, Metallogregulation of the tumor supressor protein p53: zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells, Oncogene, 2000, 19, 5227–5236 CrossRef CAS.
- A. Krezel and W. Maret, Dual nanomolar and picomolar Zn(II) binding properties of metallothionein, J. Am. Chem. Soc., 2007, 129, 10911–10921 CrossRef CAS.
- R. Bozym, T. K. Hurst, N. Westerberg, A. Stoddard, C. A. Fierke, C. J. Frederickson and R. B. Thompson, Determination of zinc using carbonic anhydrase-based fluorescence biosensors, Methods Enzymol., 2008, 450, 287–309 CAS.
- R. A. Bozym, R. B. Thompson, A. K. Stoddard and C. A. Fierke, Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor, ACS Chem. Biol., 2006, 1, 103–111 CrossRef CAS.
- E. A. Ostrakhovitch, P. E. Olsson, S. Jiang and M. G. Cherian, Interaction of metallothionein with tumor suppressor p53 protein, FEBS Lett., 2006, 580, 1235–1238 CrossRef CAS.
- R. Puca, L. Nardinocchi, H. Gal, G. Rechavi, N. Amariglio, E. Domany, D. A. Notterman, M. Scarsella, C. Leonetti, A. Sacchi, G. Blandino, D. Givol and G. D’Orazi, Reversible dysfunction of wild-type p53 following homeodomain-interacting protein kinase-2 knockdown, Cancer Res., 2008, 68, 3707–3714 CrossRef CAS.
- R. Puca, L. Nardinocchi, G. Bossi, A. Sacchi, G. Rechavi, D. Givol and G. D’Orazi, Restoring wtp53 activity in HIPK2 depleted MCF7 cells by modulating metallothionein and zinc, Exp. Cell Res., 2009, 315, 67–75 CrossRef CAS.
- P. Yue, Z. Li and J. Moult, Loss of protein structure stability as a major causative factor in monogenic disease, J. Mol. Biol., 2005, 353, 459–473 CrossRef CAS.
- F. M. Boeckler, A. C. Joerger, G. Jaggi, T. J. Rutherford, D. B. Veprintsev and A. R. Fersht, Targeted rescue of a destabilized mutant of p53 by an in silico screened drug, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10360–10365 CrossRef CAS.
- V. J. Bykov, J. M. Lambert, P. Hainaut and K. G. Wiman, Mutant p53 rescue and modulation of p53 redox state, Cell Cycle, 2009, 8, 2509–2517 Search PubMed.
- J. M. Lambert, P. Gorzov, D. B. Veprintsev, M. Soderqvist, D. Segerback, J. Bergman, A. R. Fersht, P. Hainaut, K. G. Wiman and V. J. Bykov, PRIMA-1 reactivates mutant p53 by covalent binding to the core domain, Cancer Cell, 2009, 15, 376–388 CrossRef CAS.
- J. M. Lambert, A. Moshfegh, P. Hainaut, K. G. Wiman and V. J. Bykov, Mutant p53 reactivation by PRIMA-1(MET) induces multiple signaling pathways converging on apoptosis, Oncogene, 2009, 29, 1329–1338.
- A. C. Joerger, H. C. Ang and A. R. Fersht, Structural basis for understanding oncogenic p53 mutations and designing rescue drugs, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15056–15061 CrossRef CAS.
- A. C. Joerger, H. C. Ang, D. B. Veprintsev, C. M. Blair and A. R. Fersht, Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations, J. Biol. Chem., 2005, 280, 16030–16037 CrossRef CAS.
- A. C. Joerger and A. R. Fersht, Structure-function-rescue: the diverse nature of common p53 cancer mutants, Oncogene, 2007, 26, 2226–2242 CrossRef CAS.
- Y. Sun, J. Bian, Y. Wang and C. Jacobs, Activation of p53 transcriptional activity by 1,10-phenanthroline, a metal chelator and redox sensitive compound, Oncogene, 1997, 14, 385–393 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |