Data-mining patent literature for novel chemical reagents for use in medicinal chemistry design
Jason G.
Kettle
*,
Richard A.
Ward
and
Ed
Griffen
AstraZeneca, Cancer and Infection Discovery, Mereside, Alderley Park, Macclesfield, SK10 4TG, United Kingdom. E-mail: jason.kettle@astrazeneca.com; Fax: +44(0)1625519749; Tel: +44(0)1625517920
First published on 13th October 2010
Abstract
The properties of any molecule are fixed at the point of design. Since reliance on commercially available reagents sets to probe SAR relationships may result in incomplete interrogation of property space, efforts to pursue novel proprietary reagent collections are of clear benefit. One such approach based on fragmentation and analysis of molecules described in patent and medicinal chemistry literature is described, highlighting an example of key secondary amines with potential for broad applicability across medicinal chemistry.
Introduction
High-throughput screening (HTS) has established itself as the principal hit-finding strategy for the majority of drug targets within major pharmaceutical companies. The increase in screening capacity afforded by the drive towards assay miniaturisation has meant most companies have embarked on programs to enhance both the size and quality of their screening collections.1 Experience with early HTS screening campaigns, together with a more detailed understanding of physicochemical space occupied by marketed oral drugs has led directly to concepts such as ‘lead-like’ chemical space2 and other measures of quality that are routinely applied across the discovery phases. Established parameters such as size and lipophilicity are now joined with a variety of calculated parameters designed to assess quality, and overall distance to an ideal candidate drug profile. Calculated models designed to predict cardiac liability3 and solubility4 for example are commonplace, with models generally built on internal corporate data. Additionally, sub-structural filters designed to flag or remove unwanted groups, such as those that may carry a genotoxicity5 or reactive metabolite6 risk are routinely applied.The properties and therefore quality of any molecule is fixed at the point of design. We were motivated therefore to look earlier, at the design process itself, and the feedstocks into this process, namely the chemical reagents used to synthesise molecules themselves. This was in part driven by frustration – for many of the commonly used reagent classes, utilised both in collection enhancement initiatives and traditional medicinal chemistry lead optimisation programmes, the diversity found from commercial sources can be limiting. Equally, in addition to limited structural diversity, the physicochemical property space afforded from commercial reagent sources can also be narrow. For certain reactive reagent classes such as sulfonyl halides for example, commercial sets focus on relatively stable and readily synthesisable lipophilic aliphatic or aromatic moieties, with more polar or basic groups, desirable from a medicinal chemistry perspective often poorly exemplified. We recognised that rapid access to non-commercial sets of medicinally relevant reagents may offer a significant competitive advantage. Commercial reagent sets are available to all competitors, are generally costly for truly innovative reagents and can be subject to limited availability and long lead times incompatible with project timelines. Consequently a reliance on such sets may lead to incomplete structure–activity (SAR) and structure–property (SPR) assessment unless significant time and effort is invested by project teams to synthesise reagents that address this.
Initiatives that aim to address the quality of reagent collections used in medicinal chemistry design would be expected to impact directly the quality of screening and project compounds. Access to a broad, diverse and novel reagent collection could deliver a competitive advantage through structural novelty, allowing for more thorough exemplification in intellectual property applications, and could give access to substructures known (either through literature or in-house experience) to help address key medicinal chemistry issues such as hERG activity, physical properties and DMPK liabilities. Such reagents could provide ready made isosteric replacements for activity-enhancing but often problematic groups such as phenols. Ultimately, a high quality reagent collection could result in shorter make-test cycles where time is not spent deriving novel reagents to probe project-specific SAR and SPR, allowing for interrogation of property space not readily available to competitors.
One of a number of approaches that AstraZeneca has taken to enhance it's global reagent collection has been to mine the information held within the structures of molecules that are disclosed in small-molecule patent applications and publications detailing biological activity. The aim was to fragment all bioactive molecules of interest and search for embedded reagents that it may be useful to procure. After property and novelty assessment and analysis of frequency of occurrence, this would lead to reagents that competitors are using to tune the properties of biologically relevant small molecules, have a synthetic route disclosed but cannot be purchased. This approach should yield reagents, which although exemplified as modulators of one particular target or targets, might have applicability and utility against others. Groups that have been found to impart beneficial non-efficacy properties, be it in terms of novelty, lower lipophilicity or metabolism burden for example, might reasonably be expected to do so independently of any given target. In particular, reagents that appear across multiple patent applications, and in particular, being exploited by several companies could be anticipated to be of even greater interest, the concept of ‘privileged fragments’7 extended to reagent sets. Herein we disclose one such example utilising a cyclic secondary amine query, since analysis of internal data indicates this to be the most used class of reagents in AstraZeneca drug discovery and collection enhancement initiatives, and also the query likely to generate the largest output.
The database that was utilised for this search was a composite of externally licensed and internally curated patent and target information,8 and whilst not comprehensive, covers a major part of the bioactive compound output and target landscape from 30 years of global drug research and development, encompassing over 2 million unique compounds and over 42,000 patents and 66,000 publications. A major component of this is a focus on target class coverage - kinases, phosphatases, proteases, GPCRs, nuclear hormone receptors, transporters, phosphodiesterases, ion-channels, lipases, and transferases are all exemplified. The approach should however be applicable to any medicinally relevant database of compounds.
Results and discussion
In this study we required an automated method to release the embedded secondary amines from the molecules in the database described above. For this process we chose the reaction transform language SMIRKS for speed of processing.9 The SMIRKS language uses a combination of SMARTS10 and SMILES11 notation to describe a specific transformation process. Three separate SMIRKS definitions (Fig. 1(a)) were encoded to identify the sub-structures of embedded cyclic secondary amines (on the left hand side of the SMIRKS) and converting these to the ‘free’ secondary amine on the right hand side. SMIRKS are written in two possible “dialects” where either the hydrogens are processed explicitly or edited and removed automatically. From experience we chose to use the explicit option as this reduces errors of incorrect protonation, and unlikely tautomers being proposed. Fig. 1(b) shows a schematic of how such a transformation may be visualised using traditional molecular reaction architecture. The atom mapping for the transformations are contained in the SMIRKS string and also shown on the schematic representation.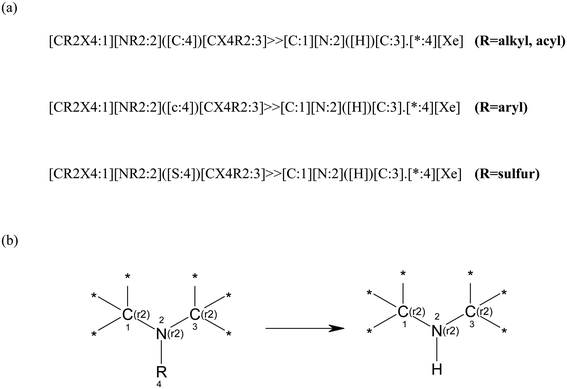 | ||
| Fig. 1 (a) SMIRKS definitions used to code the key transformations. (b) Schematic representation of the SMIRKS: (r2) designation indicates atom is contained within a ring attached to two other ring atoms. | ||
In the SMIRKS definitions, the embedded secondary amine is described by SMARTS and the transformation products are expressed by SMILES strings. We utilised three separate SMIRKS definitions to target molecules which were substituted on the nitrogen by carbon (non-aromatic), carbon (aromatic) and sulfur. This allows us to release the embedded secondary amine from N-alkyl, N-acyl, N-aryl and N-sulfur linked compounds. The SMIRKS definitions only process secondary amines where the nitrogen and two adjacent carbons (mapped as 1, 2 and 3 above) are located in a ring. No constraints were placed on ring size, although only two ring bonds were allowed for atoms 1, 2 and 3, and both carbons were fixed with sp3 centres.
A SMIRKS processor12 was compiled in-house to take the full database and process all compounds using the rules above. As shown in Scheme 1 this reads in a SMILES file from the database, removes the salts and then applies each of the SMIRKS in turn through an enumeration process. For compounds which contained more than one of the sub-structures in the SMIRKS, all possible permutations of transformation were generated from the initial input molecule. Where the SMIRKS were applicable, the resulting product contains the core and R-groups, with the ‘[*:4][Xe]’ on the right-hand side of the SMIRKS definition used as a tag. Any part of the product containing a Xe atom is then filtered out. This has the effect of removing the substituent R (mapped as atom 4) to leave only the reagent candidate released as a secondary amine, where the substituent is replaced by hydrogen in the procedure encoded by the SMIRKS. The SMIRKS processor was used only to identify the first generation products, meaning only the molecules from the database were processed with these SMIRKS, none of the generated products were further processed. The set of reagent candidate released secondary amines were then prioritised in the subsequent analyses.
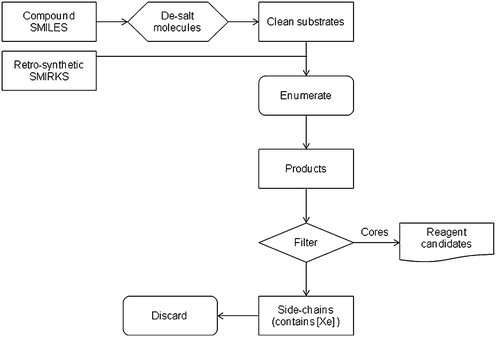 | ||
| Scheme 1 Schematic representing the fragmenting/enumerating process whereby the side-chains are removed from the amine cores. | ||
Scheme 2 illustrates the filtering procedures that were used to focus our search for secondary amines of interest. Application of the stripping/fragmentation steps on the complete list of 2,124,189 entries in the database led to 259,120 unique cyclic secondary amines which were grouped by frequency. In order to consider those that had significant exploitation in medicinal chemistry literature we kept only those with a frequency ≥ 5 (step 2). This reduced the number of amines dramatically to 16,232, in the process removing 199,200 amines with single occurrence. An exact match to the ACD database13 of commercially available reagents using in-house program FLUSH14 was then used to find and remove any amine that could be sourced commercially (step 3). Occurrence within ACD is a crude filter of commercial availability since not everything present can necessarily be sourced, and reagents without a presence may be obtained via non-ACD suppliers. Nevertheless, it is a quick and relatively simple filter to apply to large datasets such as this, and applying this matching process highlighted 2,734 reagents that were present in ACD.
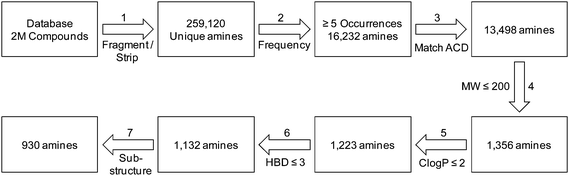 | ||
| Scheme 2 Schematic representing the filtering process to derive novel cyclic secondary amines disclosed in the medicinal and patent literature. | ||
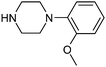
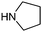
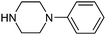
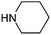
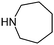
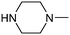
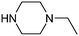
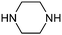
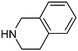
Table 1 shows the top 10 commercially available amines present in the search database along with frequency and as such this list presents few surprises. Morpholine (entry 1) is the most utilised amine in this data set with over 46,000 entries, presenting an often ideal balance of lipophilicity and basicity. As expected, the simplest of the cyclic secondary amine homologues such as pyrrolidine, piperidine and piperazine occur with high frequency. Indeed, piperazine as a substructure is represented in 5 of the top 10 most used commercially available amines (entries 4–7 and 9), ranging from unsubstituted to simple aryl substituted examples common to many GPCR ligands (entries 6 and 7). Sub-structural searching of the source data set indicates that piperazine as a motif occurs singly in 115,744 bioactive molecules, and twice in 2,212 molecules, making this, by some margin, the most commonly exploited amine. For this search no substituents were allowed on the piperazine ring carbons and as with the SMIRKS definitions only N-alkyl, N-acyl, N-aryl and N-sulfur substituents were included. Such frequency analysis may serve to focus efforts aimed at deriving novel proprietary amines for synthesis.
| Entry | Amine | Frequency | Entry | Amine | Frequency |
|---|---|---|---|---|---|
| 1 |
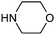
|
46,127 | 6 |
|
3,582 |
| 2 |
|
29,268 | 7 |
|
3,231 |
| 3 |
|
25,881 | 8 |
|
2,957 |
| 4 |
|
20,293 | 9 |
|
2,780 |
| 5 |
|
10,080 | 10 |
|
2,423 |
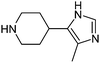
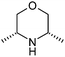
After removing the commercially available amines, and in order to narrow the search further, we then applied a variety of calculated property filters. Firstly we limited our search to only those amines with a molecular weight ≤ 200 Da (Scheme 1, step 4). This figure is clearly subjective, but was based on an analysis of in-house data that suggested the frequency of use of any reagent in library synthesis was negligible above this cut-off. In the event this filter removed the bulk of the remaining amines, reducing the list by 12,142 to 1,356. If the focus of this work had been analysis of fragments for fragment-based lead generation activities then a higher molecular weight cut-off may have been adopted. A ClogP ≤ 2 filter was also applied (step 5). The application of ClogP to this set of reagents is at first sight not especially relevant since the overall contribution to lipophilicity from a given reagent is entirely dependent on both the scaffold it is attached to, and, for amines, the nature of that attachment. Nevertheless this was a generous guide, removing only 133 of the most lipophilic amines. In medicinal chemistry design, control of the number of hydrogen-bond donors (HBD) is often of critical importance, particularly with regard to cell permeability, efflux and absorption processes.15 Retaining only those amines that had HBD ≤ 3 (step 6) removed a further 91 examples to give 1,132 secondary amines (since one donor would be lost on coupling to a given scaffold, allowing for the incorporation of a further 2 donors is also considered generous). In a final step 7, we removed certain substructures deemed to be less desirable when present in amines for organic synthesis, and these included acyl hydroxamates (51, derived from metalloproteinase inhibitors in the source data), esters/acids (117) and ketones (35). Three amines found to contain boron atoms were also removed to give a final count of 930 novel secondary amines. Table 2 shows the top 10 amines from this set, by frequency, together with information on number of WO patents that disclose their use, the number of unique applicants of these and the number of associated distinct drug targets.16
| Entry | Amine | Frequency | Patentsa | Applicantsb | Targetsc |
|---|---|---|---|---|---|
| a PCT International Applications only. b Unique PCT applicants. c Unique primary targets where disclosed. In this analysis, distinct kinases are considered as a single primary target for example, but kinase activators a different primary target. | |||||
| 11 |
|
203 | 22 | 13 | 9 |
| 12 |
|
195 | 3 | 2 | 2 |
| 13 |
|
161 | 18 | 11 | 10 |
| 14 |
|
146 | 4 | 3 | 1 |
| 15 |
|
146 | 2 | 1 | 1 |
| 16 |
|
127 | 11 | 6 | 6 |
| 17 |
|
121 | 48 | 27 | 14 |
| 18 |
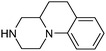
|
121 | 9 | 8 | 5 |
| 19 |
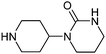
|
121 | 23 | 8 | 7 |
| 20 |
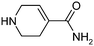
|
104 | 7 | 5 | 4 |
Frequency of occurrence of novel amines in this dataset may not necessarily reflect broad medicinal chemistry utility, especially where an entry is from a single organisation. Such examples may just reflect the patenting strategy in place for a given company (e.g. more comprehensive exemplification). Perhaps of more value are those reagents that appear not just across multiple patent applications, but from different companies, and crucially against a diversity of drug targets. While reagents arising from this analysis were assessed for synthesis against each of these parameters, ultimately it was an assessment of structural attractiveness by a team of medicinal chemists, in particular with a view to dis-similarity to existing internally available reagent sets, that was the principal selection criteria. Since most reagents derived in this manner should be target agnostic, even a reagent used a handful of times, by a single company against one target may ultimately prove of value in lead optimisation against unrelated targets, and could also potentially contribute to the resolution of unrelated medicinal chemistry issues.
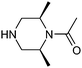
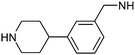
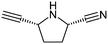
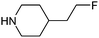
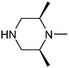
This list of most used non-commercial amines is dominated by 6-ring analogues based around piperidine and piperazine. Indeed the meso-2,6-dimethylpiperazine moiety occurs twice (entries 11 and 17) capped by acyl and methyl groups. The first of these for example is disclosed in 22 WO patent applications from 13 different organisations, covering a diverse array of 9 targets/target classes, figures that infer its ability to impart favourable properties within medicinal chemistry design sets. Entries in the table which feature a high number of examples or patents but few targets are indicative that the amine in question may be more closely linked to the pharmacophore required for modulation, and as such may have lower utility as a reagent in general design. The piperidine entry 12 is one such example, where the benzylamine moiety is known to occupy and form key hydrogen bonds in the S1 pocket of βII tryptase.17 Similarly pyrrolidine-nitrile entry 14 is the key warhead in an established series of DPP IV inhibitors,18 while novel fluoropiperidine entry 15 has been heavily exploited by Novartis as a P2 pocket ligand against thrombin.19 For reagents such as 12 to be of value as additions to novel reagent collections would of course require a suitable protecting group strategy be employed. In the work described below, we considered a tert-butoxycarbonyl mono-protection strategy for all unsymmetrical bis-amines of interest, and indeed against other reagent classes where the reactive functionality was incompatible with the presence of a base.
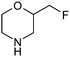
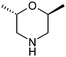
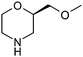
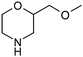
Table 1 highlighted morpholine as the key commercially available cyclic secondary amine exploited in bioactive molecules. In the field of kinase inhibition for example, morpholine is utilised both as basic solubilising side-chain such as seen in gefitinib,20 and also key pharmacophoric hinge-binding motif as seen in an array of recent lipid kinase inhibitors.21 Through sub-structural searching of the dataset it is possible to gain insight into additional novel analogues that are being exploited in drug design that might be applied against alternative targets. Table 3 highlights the top 10 by frequency of this useful sub-structure in the filtered list. The most used analogue (2S)-2-(methoxymethyl)morpholine, entry 21 also occurs in (R)- and racemic forms (entries 28 and 25 respectively) making this an ideal candidate in any collection of reagents. Simple chiral dimethyl substituted analogues also feature (entries 22, 23 and meso-form 24).
| Entry | Amine | Frequency | Entry | Amine | Frequency |
|---|---|---|---|---|---|
| 21 |
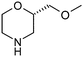
|
63 | 26 |
|
11 |
| 22 |
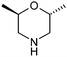
|
62 | 27 |
|
9 |
| 23 |
|
24 | 28 |
|
8 |
| 24 |
|
19 | 29 |
|
8 |
| 25 |
|
15 | 30 |
|
8 |
Analysis of this data by frequency of occurrence, and specific sub-structural queries on amines of interest as outlined above is of value when defining strategies to optimise collections of reagents for use in design. Perhaps of greater value is an understanding of those reagents that show value across a diversity of drug targets, or those that are under exploitation by a range of organisations, even where overall frequency may be low. Table 4 illustrates a selection of novel, non-commercial amines found through this analysis which are either utilised by several companies, or against a range of targets, or both. Entry 31, a piperidine with an unusual pendant 1,1-difluoromethylene group has been exploited by a range of companies against a wide diversity of drug targets. The novel fluorinated azetidine analogue entry 32, to date only disclosed by Vertex, has nevertheless been utilised by them in both kinase and GPCR programs. Entry 33 highlights a novel isostere of 4-phenylpiperidine/piperazine incorporating a polar phosphine oxide group utilised across several targets and research groups. Similarly, fluorinated pyrrolidine analogue entry 34 highlights a recurring theme in the extracted dataset, that of amines with pendant groups designed to modulate pKa, exploited here in ligands against kinases, integrases, proteases and ion channels.
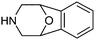
| Entry | Amine | Frequencya | Organisationb | Modulated Targets |
|---|---|---|---|---|
| a Frequency of substructure as extracted from SciFinder. This figure is greater than the frequency found by direct extraction from the database due to more comprehensive indexing and mirrors the data compiled in the remaining columns. b Excludes academic patent applications and literature. Patents from legacy (pre-merger) companies combined under current organisational structure where known. | ||||
| 31 |
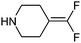
|
206 | Boehringer Ingleheim, Daichi-Sankyo, Epix, Helicon, Paratek, Sanofi-Aventis | Factor Xa, Fibrin, MAO-B, NCCa-ATP channel, Oxytocin receptor, Tetracycline antibiotics, Thrombin |
| 32 |
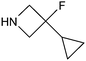
|
61 | Vertex | AUR kinase, CGRP receptor, PLK kinase |
| 33 |
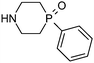
|
18 | Merck & Co, Panacea Biotec, Pharmacyclics | BTK kinase, HDAC, Oxazolidinone antibiotics |
| 34 |
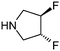
|
19 | Eisai, Merck & Co, Pfizer | 11 bHSD-1, Carbonic anhydrase, DPP IV, FAK kinase, HIV integrase, Kv1.5, Squalene synthase |
| 35 |
|
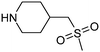
7 | BMS, Merck & Co, Pfizer, Roche, Tibotec | BACE, HIV protease, NK1/3, p53 |
It is perhaps informative to look at the context in which novel reagents such as these are incorporated into drug-like molecules. Fig. 2 highlights selected examples of the (3R,4R)-3,4-difluoropyrrolidine 34 as it occurs in the medicinal literature. This novel amine, whose synthesis is in fact disclosed22 has been heavily exploited by Pfizer23 against a range of diverse targets implicated in a variety of diseases such as cancer, infection and diabetes (entries 36 to 39 inclusive). Example 40, from Merck24 illustrates utility against potassium channel antagonists for cardiovascular therapy, an area also targeted with the squalene synthase inhibitor 41 from Eisai.25 Clearly in each case the primary pharmacology is driven by the scaffold to which the amine is appended, but its prevalence suggests an ability to contribute to the overall beneficial non-efficacy parameters required in any medicinal chemistry program.
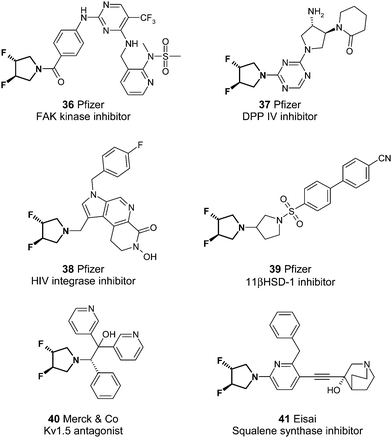 | ||
| Fig. 2 Selected exploitation of (3R,4R)-3,4-difluoropyrrolidine 34 against a diverse set of drug targets by a range of companies. | ||
Conclusion
In summary, we have outlined a simple and direct strategy for extraction of potentially high value reagents from patent and other medicinal literature, the application of which should allow for the creation of reagent inventories that are novel, probe chemical space not immediately accessible via commercial vendors and deliver more comprehensive scoping of project SAR and SPR relationships. Such a strategy might reasonably be applied to a range of similar or alternative datasets to derive not solely reagents, but also information on privileged structures or ideas to bias screening sets for fragment based lead generation activities for example. Indeed, an earlier study by Lewell et al. uses a related approach to generate fragments of the World Drug Index which may be considered either reagents or core templates and scaffolds.26 This approach splits query molecules into all possible fragments according to specific connectivity rules, whereas here, only the key fragment(s) relevant to the precise reagent query are extracted. Such considerations become important when larger datasets are queried (>2 million here compared to 50000). It is likely that the patent literature represents a more comprehensive distillation of the medicinal chemistry knowledge available than a focus only on marketed and developed drugs and drug candidates. Since most small molecule patent applications are concerned with one or a small number of scaffolds to which are appended common reagent sets, it may be argued this is a more relevant source when the query is, as here, specifically focused on reagents. In both these analyses, frequency of use is used as a surrogate marker of broad utility, although this assumption must be approached with caution. It is likely true that groups which carry particularly frequent liabilities can be propagated in medicinally chemistry design just as those with a desirable positive impact are. One preferred approach is to mine datasets where comprehensive effects on parameters of interest such as effect on hERG potency or metabolic liability can be analysed, and reagents prioritised based on an understanding of their probability of positively impacting each.27 Since such non-efficacy information is not disclosed in patent applications, frequency of usage is a reasonable compromise. In silico models for relevant parameters may be considered but largely this would have to be in the context of fully enumerated product structures rather than reagents themselves.In reagent exploitation, certain reagent classes lend themselves more suitable to this approach, for example amines, acids and sulfonyl halides where the bond retrosynthesis is mostly unambiguous, than for others. Fragmentation of datasets for aldehydes for example, which are generally incorporated into target molecules via reductive amination to give alkylamines, may lead to reagents which in reality were derived via alternative chemistries. Nevertheless, as a tool to generate ideas for reagents to synthesise, the approach remains valid. Over the past several years, in conjunction with efforts to enhance screening collections,28 AstraZeneca has engaged in a comprehensive effort to improve the quality and scope of reagent collections used in in-house project and library synthesis using a diversity of approaches including the technique outlined here. Knowledge of heavily exploited but non-commercial reagents has been used to secure both exact analogues and as inspiration for design and synthesis of entirely novel reagents to aid internal medicinal chemistry optimisation. These efforts have, to date, contributed in excess of one thousand novel reagents for exploitation across all AstraZeneca small-molecule synthesis sites, and begun to demonstrate broad impact across all early phases of drug discovery, including application in candidate drug molecules.
References
- (a) E. Jacoby, A. Schuffenhauer, M. Popov, K. Azzaoui, B. Havill, U. Schopfer, C. Engeloch, J. Stanek, P. Acklin, P. Rigollier, F. Stoll, G. Koch, P. Meier, D. Orain, R. Giger, J. Hinrichs, K. Malagu, J. Zimmermann and H.-J. Roth, Curr. Top. Med. Chem., 2005, 5, 397–411 CrossRef CAS; (b) A. Schuffenhauer, M. Popov, U. Schopfer, P. Acklin, J. Stanek and E. Jacoby, Comb. Chem. High Throughput Screening, 2004, 7, 771–781 CrossRef CAS; (c) D. H. Drewry and R. Macarron, Curr. Opin. Chem. Biol., 2010, 14(3), 289–298 CrossRef CAS.
- (a) P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS; (b) T. I. Oprea, A. M. Davis, S. J. Teague and P. D. Leeson, J. Chem. Inf. Comput. Sci., 2001, 41, 1308–1315 CrossRef CAS; (c) M. M. Hann and T. I. Oprea, Curr. Opin. Chem. Biol., 2004, 8, 255–263 CrossRef CAS.
- (a) M. J. Waring and C. Johnstone, Bioorg. Med. Chem. Lett., 2007, 17, 1759–1764 CrossRef CAS; (b) A. Stary, S. J. Wacker, L. Boukharta, U. Zachariae, Y. Karimi-Nejad, J. Aqvist, G. Vriend and B. L. de Groot, ChemMedChem, 2010, 5, 455–67 CrossRef CAS; (c) K.-M. Thai and G. F. Ecker, Bioorg. Med. Chem., 2008, 16, 4107–4119 CrossRef CAS.
- (a) D. Butina and J. M. R. Gola, J. Chem. Inf. Comput. Sci., 2003, 43, 837–841 CrossRef CAS; (b) O. Engkvist and P. Wrede, J. Chem. Inf. Comput. Sci., 2002, 42, 1247–1249 CrossRef CAS.
- R. D. Snyder, G. S. Pearl, G. Mandakas, W. N. Choy, F. Goodsaid and I. Y. Rosenblum, Environ. Mol. Mutagen., 2004, 43, 143–158 CrossRef CAS.
- (a) A. S. Kalgutkar and M. T. Didiuk, Chem. Biodiversity, 2009, 6, 2115–2137 CrossRef CAS; (b) A. S. Kalgutkar, I. Gardner, R. S. Obach, C. L. Shaffer, E. Callegari, K. R. Henne, A. E. Mutlib, D. K. Dalvie, J. S. Lee, Y. Nakai, J. P. O'Donnell, J. Boer and S. P. Harriman, Curr. Drug Metab., 2005, 6, 161–225 Search PubMed.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14(3), 347–361 CrossRef CAS.
- GVK Bio Databases. Target Inhibitor Databases. http://www.gvkbio.com/informatics.html.
- Daylight Chemical Information Systems, Inc. http://www.daylight.com/dayhtml_tutorials/languages/smirks/index.html .
- Daylight Chemical Information Systems, Inc. http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html .
- Daylight Chemical Information Systems, Inc. http://www.daylight.com/smiles .
- The SMIRKS processor was written by Ed Griffen using the Openeye toolkit: OEChem 1.6.1 ed.; http://www.eyesopen.com: OpenEye Scientific Software, Inc. Santa Fe, NM, USA, 2008 and scripted in Python: Python 2.5.2 ed.; http://www.python.org: Python Software Foundation, 2008. The libgen module was used to process the SMIRKS.
- Symyx®. Available Chemicals Directory. http://www.symyx.com/products/databases/sourcing/acd/index.jsp.
- N. Blomberg, D. A. Cosgrove, P. W. Kenny and K. Kolmodin, J. Comput.-Aided Mol. Des., 2009, 23, 513–525 CrossRef CAS.
- A. Seelig, Eur. J. Biochem., 1998, 251, 252–261 CrossRef CAS.
- Chemical Abstract Service. SciFinder. http://www.cas.org/products/scifindr/index.html.
- J. Levell, P. Astles, P. Eastwood, J. Cairns, O. Houille, S. Aldous, G. Merriman, B. Whiteley, J. Pribish, M. Czekaj, G. Liang, S. Maignan, J.-P. Guilloteau, A. Dupuy, J. Davidson, T. Harrison, A. Morley, S. Watson, G. Fenton, C. McCarthy, J. Romano, R. Mathew, D. Engers, M. Gardyan, K. Sides, J. Kwong, J. Tsay, S. Rebello, L. Shen, J. Wang, Y. Luo, O. Giardino, H.-K. Lim, K. Smith and H. Pauls, Bioorg. Med. Chem., 2005, 13, 2859–2872 CrossRef CAS.
- Z. Pei, X. Li, T. W. Von Geldern, K. Longenecker, D. Pireh, K. D. Stewart, B. J. Backes, C. Lai, T. H. Lubben, S. J. Ballaron, D. W. A. Beno, A. J. Kempf-Grote, H. L. Sham and J. M. Trevillyan, J. Med. Chem., 2007, 50, 1983–1987 CrossRef CAS.
- J. Ambler, L. Brown, X.-L. Cockcroft, M. Grutter, J. Hayler, D. Janus, D. Jones, P. Kane, K. Menear, J. Priestle, G. Smith, M. Talbot, C. V. Walker and B. Wathey, Bioorg. Med. Chem. Lett., 1999, 9, 1317–1322 CrossRef CAS.
- A. E. Wakeling, A. J. Barker, D. H. Davies, D. S. Brown, L. R. Green, S. A. Cartlidge and J. R. Woodburn, Breast Cancer Res. Treat., 1996, 38(1), 67–73 CrossRef CAS.
- (a) K. A. Menear, S. Gomez, K. Malagu, C. Bailey, K. Blackburn, X.-L. Cockcroft, S. Ewen, A. Fundo, A. Le Gall, G. Hermann, L. Sebastian, M. Sunose, T. Presnot, E. Torode, I. Hickson, N. M. B. Martin, G. C. M. Smith and K. G. Pike, Bioorg. Med. Chem. Lett., 2009, 19, 5898–5901 CrossRef CAS; (b) R. Frederick, C. Mawson, J. D. Kendall, C. Chaussade, G. W. Rewcastle, P. R. Shepherd and W. A. Denny, Bioorg. Med. Chem. Lett., 2009, 19, 5842–5847 CrossRef CAS.
- C. G. Caldwell, P. Chen, J. He, E. R. Parmee, B. Leiting, F. Marsilio, R. A. Patel, J. K. Wu, G. J. Eiermann, A. Petrov, H. He, K. A. Lyons, N. A. Thornberry and A. E. Weber, Bioorg. Med. Chem. Lett., 2004, 14, 1265–1268 CrossRef CAS.
- (a) M. J. Luzzio; C. L. Autry; S. K. Bhattacharya; K. D. Freeman-Cook; M. M. Hayward; C. A. Hulford; K. L. Nelson; J. Xiao; X. Zhao. PCT Int. Appl. WO2008129380 Search PubMed; (b) K. R. Dress; T. W. Johnson; M. B. Plewe; S. P. Tanis; H. Zhu. PCT Int. Appl. WO2007042883 Search PubMed; (c) H. Cheng; K. R. Dress; B. Huang; S. W. Kupchinsky; P. T. Q. Le; C. R. Smith; T. Wang; Y. Yang. PCT Int. Appl.. WO2006134481 Search PubMed; (d) J. W. Benbow; D. W. Piotrowski; Y. Hui. PCT Int. Appl. WO2007148185 Search PubMed.
- C. J. Dinsmore; J. M. Bergman; D. C. Beshore; B. W. Trotter; K. K. Nanda; R. Isaacs; L. S. Payne; L. A. Neilson; Z. Wu; M. T. Bilodeau; P. J. Manley; A. E. Balitza. PCT Int. Appl. WO2006015159 Search PubMed.
- T. Okada; N. Kurusu; K. Tanaka; K. Miyazaki; D. Shinmyo; H. Sugumi; H. Ikuta; H. Hiyoshi; T. Saeki; M. Yanagimachi; M. Ito. PCT Int. Appl. WO2001023383 Search PubMed.
- X. Q. Lewell, D. B. Judd, S. P. Watson and M. M. Hahn, J. Chem. Inf. Comput. Sci., 1998, 38, 511–522 CrossRef CAS.
- A. G. Dossetter, Bioorg. Med. Chem., 2010, 18, 4405–4414 CrossRef CAS.
- M. J. Stocks, G. R. H. Wilden, G. Pairaudeau, M. W. D. Perry, J. Steele and J. P. Stonehouse, ChemMedChem, 2009, 4, 800–808 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |