Recent advances in the chemistry and biology of stable synthetic Lipoxin analogues
Colm D.
Duffy
and
Patrick J.
Guiry
*
Centre for Synthesis and Chemical Biology, UCD School of Chemistry and Chemical Biology, Conway Institute of Biomolecular and Biomedical Research, Belfield, Dublin 4, Ireland. E-mail: patrick.guiry@ucd.ie; Fax: (+353)-1-7162501
First published on 29th September 2010
Abstract
Lipoxins are naturally occurring signalling molecules which play an integral role in the resolution of inflammation. In this review, we highlight the synthetic and biological developments of novel stable Lipoxin analogues, which show resistance to enzymatic metabolism. This article aims to illustrate the major and noteworthy synthetic obstacles and achievements which have dominated this active area of research over the past twenty five years. We examine the synthetic routes to stable Lipoxin analogues and the evaluation of their biological potency in an ongoing effort to provide novel therapeutic agents to combat an array of inflammatory diseases.
 Colm Duffy | Colm Duffy was born in Dublin, Ireland, and graduated from UCD in 2006 with a 1st class Honours B.Sc. in Chemistry. He was awarded an Ad Astra Scholarship to carry out his PhD with Professor Pat Guiry where his research focused on the synthesis of biologically active natural product analogues. Having recently completed his doctoral studies, he now carries out postdoctoral research at The University of Manchester with Professor John Sutherland on prebiotic chemistry and the origins of life. In January 2011, he will continue his research at the MRC Laboratory of Molecular Biology (LMB) in Cambridge. |
 Pat Guiry | Pat Guiry was awarded his B.Sc. and PhD degrees from UCD, with Professor Dervilla Donnelly (UCD), Sir Derek Barton (Texas A&M) and Dr Jean-Pierre Finet (Marseille) as his PhD supervisors. He carried out postdoctoral research in asymmetric catalysis with Dr John Brown FRS (Oxford University). Returning to UCD in 1993, he was the recipient of President's Research/Teaching Awards in 1996/2000 and was a Visiting Professor at the University of Toronto (2004). He was the Chief Executive of the Conway Institute at UCD 2004-5 and is currently the Director of the CSCB and Full Professor of Synthetic Organic Chemistry since 2006. |
Intoduction
Lipoxin A4 (LXA4) and Lipoxin B4 (LXB4), Fig. 1, are biologically important oxygenated derivatives of arachidonic acid which were discovered and identified from human leukocytes by Serhan and Samuelsson in 1984.1,2
Lipoxins (LX) are trihydroxytetraene-containing eicosanoids, which are produced by the sequential actions of lipoxygenases (LO) during a series of complex cellular interactions.1 LO are a family of iron-containing enzymes, which are known to catalyse the oxygenation of unsaturated fatty acids and lipids. The combined oxygenase activity of 5-LO, 12-LO and 15-LO leads to the biosynthesis of LXA4 and LXB4.3 The 15-epi-LX, or aspirin-triggered Lipoxins (ATL), differ only in the stereochemistry at C15 and are produced by aspirin-acetylated cyclooxygenase-2 (COX-2).4
Once biosynthesised, enzymatically derived LXA4 and LXB4 are known to possess potent and selective anti-inflammatory activity.5 They act as so-called “stop signals” by activating the receptor FPR2/ALX to prevent the migration of neutrophils to sites of inflammation.6,7 During injury, an inflammatory response is triggered and a cascade of cellular events occurs at the site of inflammation which includes the migration of neutrophils. These neutrophils accumulate usually within one hour of the injury and this process is regarded as the most important process that leads to inflammation.8 Monocytes also accumulate at the site and develop into larger macrophages which cause the phagocytosis of apoptotic polymorphonuclear leukocytes (PMNs). Lipoxins have been previously shown to have the ability to regulate PMNs, chemotaxis, adhesion and transmigration.9 It has been demonstrated that Lipoxins resolve inflammation by promoting nonphlogistic phagocytosis of apoptotic PMN by macrophages in vitro and in vivo.10
Their isolation in 1984 prompted the search for new pharmacological drug candidates based on these potential therapeutic agents. As with many natural products, minimal quantities result from isolating these compounds from natural sources. This inspired the development of efficient synthetic routes for their preparation. Extensive spectroscopic and chromatographical evidence, combined with comparisons of biological activities, proved LXA4 to be (5S,6R,15S)-trihydroxy-(7E,9E,11Z,13E)-icosatetraenoic acid,11 and LXB4 to be (5S,14R,15S)-trihydroxy-(6E,8Z,10E,12E)-icosatetraenoic acid.12
The accumulation of LXA4 and LXB4 at the site of inflammation is short lived. As with all autocoids, LX are rapidly metabolised in vivo into inactive metabolites, Scheme 1.13
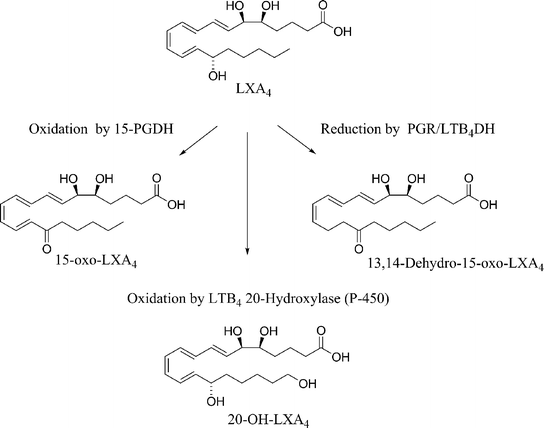 | ||
| Scheme 1 Rapid metabolism of LXA4. | ||
Lipoxin A4 is converted by specific leukocytes into 15-oxo-LXA4, 13,14-dihydro-15-oxo-LXA4 and 13,14-dihydro-LXA4 and oxidation can also occur at C20. This instability issue is a major obstacle to the application of these compounds as important pharmacological agents.
Structure–activity relationships of natural Lipoxins have been extensively reported which show that certain functionalities and stereocentres are extremely important in order to retain biological activity, Fig. 2.13,14
 | ||
| Fig. 2 Structure–activity relationships. | ||
The Lipoxin metabolites, discussed above, dramatically reduce the bioactivity of this class of compounds and render them poor potential pharmacological agents. In light of the findings associated with the stabilisation and market value of the synthetic prostaglandin and prostacyclin analogues,15 it was thought that a similar approach could be beneficial with respect to the native Lipoxins.
Design, synthesis and biological evaluation of stable Lipoxin analogues
The rationale behind synthetic efforts mimicking the core structure of the native LXA41 by replacing certain functionalities with chemically stable motifs was to (a) retain the potent biological activity, (b) aid in the study of LX receptors, (c) elucidate LX agonist properties and (d) identify the actual biological roles of LX. These stable analogues will be sub-divided into three distinct categories (A, B and C), based on the target area being modified, Fig. 3. The strategies include (A) structural modifications of the C15–20 chain;3,16,17 (B) replacement of the triene with chemically stable aromatic/heteroaromatic systems;18–20 and (C) modifications of the C1–8 unit.21 While excellent reviews have extensively covered the synthesis and biological relevance of the native LX and their stereoisomers,16,22 this article will focus on the synthesis and biological evaluation of stable LX analogues.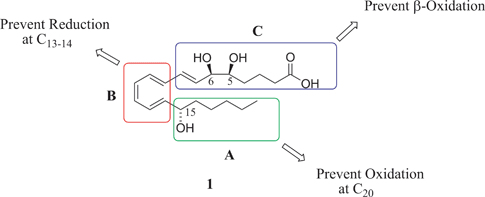 | ||
| Fig. 3 Targeted domains for modifications of the native Lipoxin A41. | ||
(A) Structural modifications of the C15–20 chain
The desire to prevent oxidation at C15–20 led to the design of the first LXA4 analogues which showed resistance to oxidation.23 Replacement of this alkyl chain with several different groups furnished a number of analogues with increased pharmacokinetic profiles. Structural adaptations incorporated 15-deoxy-LXA42, 15-(R/S)-methyl 3, 16-phenoxy 4 and 15-cyclohexyl 5 into the C15–20 chain, Fig. 4.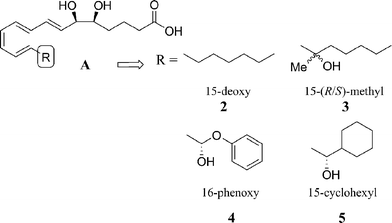 | ||
| Fig. 4 Design of C15–20 stable analogues.23 | ||
The synthetic routes used for these analogues were not reported in the literature, although they were clearly constructed by using previously reported syntheses for the related native LX.24 The authors observed that these structural modifications dramatically increased biostability compared to the native LX by preventing dehydrogenation by differential HL-cells and recombinant 15-hydroxyprostaglandin dehydrogenase. The bioactivity was also secured in the 15-(R/S)-methyl 3, 16-phenoxy 4 and 15-cyclohexyl 5 analogues due to their ability to prevent PMN transmigration and adhesion in leukocyte migration. The 15-deoxy-LXA42 showed the least activity, suggesting that the hydroxyl group at C15 is essential for the preservation of bioactivity.
Alternative analogues have been developed which resulted in enhanced bioactivity compared to the native LX. These designs, initially developed by Petasis and Serhan,3,16 include the addition of a fluoro 6 and trifluoromethyl 7 group onto the 16-phenoxy analogue 4, Fig. 5.25
 | ||
| Fig. 5 Fluoro and trifluoromethyl stable analogues.16,25 | ||
The para-fluorophenoxy analogue 6 (termed ATLa) has proven itself to be an extremely potent derivative as it inhibited tumor necrosis factor (TNF)-α-induced leukocyte recruitment into the dorsal air pouch.25 It was also found to suppress both LTB4- and PMA-induced recruitment when applied to mouse ear skin. Furthermore, this analogue has shown potential as an anticancer agent, as it inhibits endothelial cell proliferation leading to suppressed angiogenesis at the 1 to 10 nM range.26 Realising the potential of these fluorinated analogues, a number of research groups began to develop efficient synthetic routes to these biologically important derivatives. The key synthetic transformations combine a cis-reduction of an alkyne, a palladium-catalysed Sonogashira reaction and a Wadsworth–Emmons alkene transformation, Scheme 2.
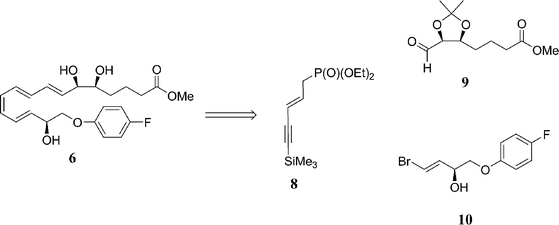 | ||
| Scheme 2 Retrosynthetic analysis of para-fluorophenoxy analogue 6. | ||
Phillips and co-workers reported the synthesis of the para-fluorophenoxy analogue 6 by adopting a chiral pool strategy,17 starting from 2-deoxy-D-ribose 11.27 This approach has the advantage of using a readily available starting material which incorporates the two stereocentres which will ultimately appear at C5 and C6. Protection of 2-deoxy-D-ribose 11 was achieved through its propylidine acetal 12 using 2-methoxypropene and pyridinium p-toluenesulfonate (PPTS) in ethyl acetate at room temperature, giving a 43% yield, Scheme 3. A Wittig reaction of methyl(triphenylphosphoranylidine)acetate and the aldehyde form of 12, followed by a catalytic hydrogenation using 10% Pd/C furnished alcohol 13 in high yields of 81% and 87%, respectively. Oxidation of 13 using Swern conditions afforded aldehyde 9 in 86% yield. This was subjected to a Wadsworth–Emmons transformation with phosphonate 8 and deprotected using KF and 18-crown-6 to form the key intermediate 14 in 99% yield.
 | ||
| Scheme 3 Formation of key intermediate 14.17 | ||
Phosphonate 8 was itself assembled by the treatment of alkyne 15 with ethylmagnesium chloride and quenching with chlorotrimethylsilane followed by an Appel-type reaction, giving the corresponding bromide in 90% and 74% yields, respectively, Scheme 4. This bromide was subjected to Arbusov reaction conditions to afford 8 in 90% yield.
 | ||
| Scheme 4 Formation of phosphonate 8.17 | ||
The synthesis of the Sonogashira coupling partner 10 was accomplished in five steps, beginning with the alkylation of p-fluorophenol with 3-chloropropane-1,2-diol 16 in 56% yield, Scheme 5. Cleavage of the diol with silica-supported sodium periodate in dichloromethane afforded aldehyde 17 in 98% yield. Addition of lithium 2-trimethylsilylacetylide to 17, followed by treatment with NaOH to remove the TMS group, gave alkyne 18 in 76% yield. Vinylstannane 19 was constructed by treating 18 with tri-n-butyltin hydride. Addition of NBS in dichloromethane to 19 gave the vinylbromide 20 in 95% yield. An attempted kinetic resolution of vinylstannane 19 using Sharpless epoxidation, followed by treatment of the unreacted alcohol with NBS to give 10, proceeded with poor ee. Subsequently, racemic 20 was resolved with chiral supercritical fluid chromatography to give vinylbromide 10 in 42% yield and 99% ee.
 | ||
| Scheme 5 Synthesis of Sonogashira coupling partner 10.17 | ||
The Sonogashira reaction, employing Pd(PPh3)4 and CuI in the presence of n-propylamine at room temperature, was used to cross-couple vinylbromide 10 and the terminal alkyne 14, resulting in the formation of 21 in 75% yield, Scheme 6. The catalyst loading was not given for this Sonogashira coupling. The acid sensitive acetal group was cleaved by the addition of methanolic HCl to give the corresponding diol. At this stage Lindlar's catalyst can be employed to access the C11–12cis-double bond. However, problems have arisen with this method including over-reduction and isomerisation of the C11–12trans-double bond isomer during the synthesis of other Lipoxin analogues.28 Selective cis-reduction with an activated zinc alloy has previously been described by Boland,29 and this protocol afforded the para-fluorophenoxy Lipoxin analogue 6 in 80% yield. Activation of the zinc requires the addition of 2 N HCl for 1–2 min for a clean reaction to take place.
 | ||
| Scheme 6 Synthesis of para-fluorophenoxy Lipoxin analogue 6.17 | ||
In a similar synthetic approach, starting from 2-deoxy-D-ribose 11, Petasis and co-workers synthesised stable Lipoxin analogues varying at the C15–20 chain, via the introduction of aliphatic, aromatic and fluoroaromatic groups, Scheme 7.16 The synthetic strategy incorporates a Wittig reaction for the construction of the C7–8 double bond, a Sonogashira reaction followed by a cis-reduction of the alkyne to establish the C11–12 double bond. Simple structural variations of the Sonogashira coupling partners gave rise to many synthetic analogues.
 | ||
| Scheme 7 Synthesis of aliphatic, aromatic and fluoroaromatic LXA4 analogues.16 | ||
The precise details of the synthesis, including % yields and mol% of catalysts, were not reported as this was part of a review article. The tert-butyldimethylsilyl-protected aldehyde 22 was accessed through the chiral pool strategy using 2-deoxy-D-ribose 11. Compound 23, previously prepared,28 was reacted with 22 in a Wittig reaction. Double bond isomerisation with I2 in dichloromethane followed by removal of the trimethylsilyl group by AgNO3 and KCN in EtOH, THF and H2O gave the alkyne coupling partner 24. Reaction conditions employed for the Sonogashira reaction included Pd(PPh3)4, CuI in n-propylamine followed by the addition of the corresponding vinyl bromide or iodide. The tert-butyldimethylsilyl protecting groups were cleaved using TBAF in THF, followed by reduction of the alkyne, by either H2 in the presence of Lindlar's catalyst, or by selective cis-reduction with an activated zinc alloy, to afford the series of analogues 26. The 15-cyclohexyl, 15-cyclooctyl and the 16-phenoxy analogues were all found to retain the native Lipoxin bioactions. The inactivation by 15-PDGH and P-450-mediated ω-oxidation were hindered due to the absence of the free ω-alkyl chain. These analogues, of type 26, were also found to be extremely useful in studying the exact binding site in vivo.30 The fluorinated analogues were found to be the most stable and active in vivo.25
(B) Structural modifications of the triene
In recent years, researchers have focused their attention on modifying the triene structure of the Lipoxin A4 and B4 framework. Derivitisation of this part of the molecule has major advantages in terms of (i) considerably increasing the stability of the molecule towards enzymatic decomposition (ii) development of a short and economical synthesis in an effort to access and screen numerous analogues to further tune the pharmacological profile, and (iii) prevention of the double bond isomerisation as described above. Significant advances in the area include the substitution of the triene with aromatic (27–32)18–20 and heteroaromatic rings (32),31Fig. 6. | ||
| Fig. 6 Analogues designed and synthesised by the groups of Petasis (27–31) and Guiry (27, 32). | ||
Petasis and colleagues were the first researchers who successfully managed to stabilise the native LXA41 by the design and synthesis of LX analogues in which the triene was replaced by a more durable benzene ring.20,32 Their synthetic route allowed for the synthesis of an array of analogues (27–31) Fig. 6. Compounds 28–31 were designed from a strategy combining domain modifications (A) and (B), Fig. 3.
The synthesis of 28 and 29 relied on two sequential Suzuki–Miyaura coupling reactions, Scheme 8. The first combines 2-bromophenylboronic acid 34 and vinyl iodide 33, which was constructed by a Takai olefination of 22.28 Suzuki–Miyaura reaction conditions incorporated Pd(PPh3)4 and K2CO3 using dioxane as the solvent at 60 °C furnished 35 in 70% yield. The catalyst loading of palladium was not reported in this coupling reaction.
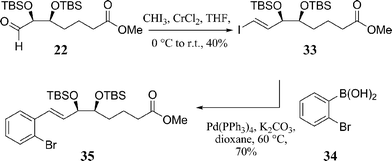 | ||
| Scheme 8 Synthesis of key intermediate 35. | ||
Boronic esters 38 and 39 were both synthesised from the corresponding alkynes 36 and 37, respectively, Scheme 9. Compound 37 was synthesised by the protection of the corresponding alcohol.33,34
 | ||
| Scheme 9 Synthesis of key intermediates 38 and 39.20 | ||
The second Suzuki–Miyaura coupling combined aryl bromide 35 and boronic esters 38 and 39 in the presence of Pd(PPh3)4 and K2CO3 using a mixture of dioxane and water as the solvent at 80 °C, giving 40 and 41 in moderate yields, Scheme 10. Deprotection followed with the use of TBAF in THF affording triol 29 and diol 28 in excellent yields.
 | ||
| Scheme 10 Synthesis of key intermediates 28 and 29.20 | ||
Petasis also described an interesting and alternative generation of 29 involving a novel and time-conserving one-pot boronic acid Heck-type coupling, Scheme 11. Both alkenes 42 and 43 were prepared from their corresponding aldehyde precursors by way of an extremely useful titanium-mediated methylenation developed by Petasis and Bzowej.35 Firstly, boronic acid 34 reacts with olefin 42 and reactivity is observed solely at the boronic acid position. In the same reaction vessel, a second Heck reaction occurs under reaction conditions reported by Jeffery,36 using Pd(OAc)2, NaHCO3, Bu4NCl, PPh3 in acetonitrile at 60 °C, giving 40 in 47% yield.
 | ||
| Scheme 11 Alternative one-pot synthesis of 29.20 | ||
Petasis then described the first reported synthesis of a novel meta-LXA4 analogue 30 using a related synthetic pathway starting from 3-bromophenylboronic acid 44, Scheme 12. The vinyl iodide derivative 33 was coupled to 44 by way of a palladium-catalysed Suzuki–Miyaura reaction affording 45 in 70% yield. This aryl bromide 45 was further reacted in a consecutive Suzuki–Miyaura reaction with boronic ester 39, followed by deprotection with TBAF to give the meta-LXA4 analogue 30 in 42% yield over the final two steps.
 | ||
| Scheme 12 Synthesis of a meta-LXA4 analogue 30.20 | ||
The LXA4 analogue 31 was prepared in order to determine the impact of increasing the chain length of the analogues on its ability to act as an agonist in the known receptor site of FPR2/ALX. The vinylboronic acid 47 was synthesised by hydroboration of the available 2-bromophenyl alkyne 46 using the reaction conditions reported by Matteson and co-workers, Scheme 13.37 The Suzuki–Miyaura reaction of 47 with vinyl bromide 48, prepared previously,38 gave the aryl bromide 49 in 65% yield. Conversion of this aryl bromide 49 to its pinacol boranate 50 using bis-pinacolato diboron, Pd(dppf)Cl2 and AcOK in dimethysulfoxide at 80 °C proceeded in 40% yield. Boronate 50 was coupled with vinyl iodide 33 by a Suzuki–Miyaura reaction to give the silyl-protected intermediate which was then deprotected to furnish the novel analogue 31 in 43% yield over the final two steps.
 | ||
| Scheme 13 Synthesis of LXA4 analogue 31.20 | ||
In addition, Petasis also outlined a non-stereoselective (at the benzylic position) synthesis of the benzene-containing LXA4 analogue 27, Scheme 14.20 The Grignard derivative of bromopentane was prepared and reacted with the Weinreb amide derived from acid chloride 51 to give the aryl ketone in 70% yield. This ketone was then reduced using NaBH4 in MeOH, followed by silyl protection to furnish 52 in high yield. Aryl bromide 52 was converted to its corresponding boronate 53 in a modest 40% yield. The trans-olefin was constructed by the Suzuki–Miyaura coupling of boronate 53 and vinyl iodide 33 and the epimeric triol 27 was produced in 95% yield after removal of the silyl ethers.
 | ||
| Scheme 14 Non-stereoselective synthesis of LXA4 analogue 27.20 | ||
Each new stable LXA4 analogue compiled by Petasis and co-workers (27–31) was subjected to enzymatic stability examinations in order to accurately demonstrate their resistance to rapid metabolism by recombinant eicosanoid oxido-reductase (EOR). These compounds were compared to the native LXA41 to determine which was metabolised the fastest, Fig. 7.
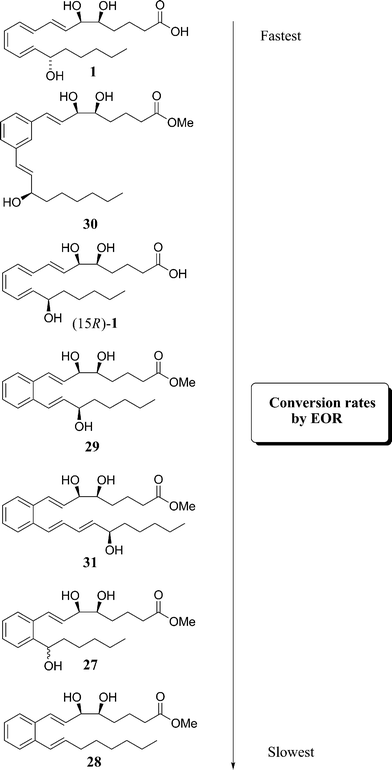 | ||
| Fig. 7 Enzymatic metabolism by eicosanoid oxido-reductase.20 | ||
The deactivation was monitored by the production of the co-factor NADH. As expected, analogue 28 was the slowest to be metabolised due to the absence of a hydroxyl group on the lower chain.
These new compounds were also tested for their ability to inhibit PMN infiltration by comparison of zymosan A induced-peritonitis in mice, Fig. 8.
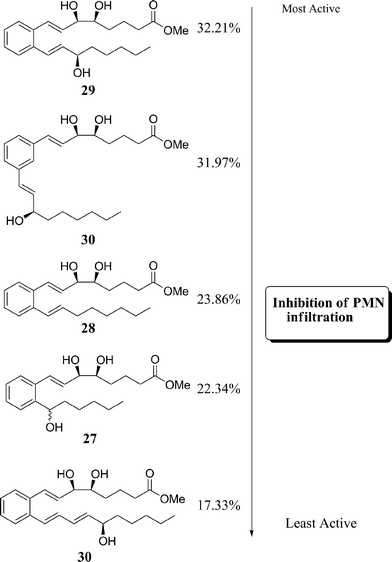 | ||
| Fig. 8 Activity of stable analogues to inhibit PMN infiltration in vivo.20 | ||
All of the above new stable analogues from Petasis were found to be potentially effective anti-inflammatory agents as they increased the inhibition of PMN by up to 32% in the case of 29. This level of activity is significant as LX and their analogues possess comparable potency to current non-steroidal anti-inflammatory drugs on the market. For example, the anti-inflammatory drug indomethacin, Fig. 9, reduces PMN infiltration by 35–40% in the same model of peritonitis.39
 | ||
| Fig. 9 Current non-steroidal anti-inflammatory drug indomethacin. | ||
Further to this, the aromatic analogue 29 displayed therapeutic ability to reduce PMN infiltration in murine hind-limb ischemia-induced lung injury comparable to synthetic analogues that lack the additional benzene ring moiety.32,40 Compound 29 was also shown to regulate the production of important cytokines and chemokines known to be fundamental in the inflammatory process.41,42 A decrease in MIP-2, TNF-α, and IFN-γ was observed and no effect was observed on the levels of RANTES or SDF-1.
Subsequently, Guiry and co-workers described a stereoselective synthesis of the LXA4 and LXB4 analogues 27 and 54 respectively, Fig. 10, using Sharpless asymmetric epoxidation, palladium-mediated Heck coupling and diastereoselective reduction reactions as the key synthetic transformations.19 These reactions provided enantio- and diastereoselective generation of each stereocentre and complete control for the formation of the trans olefin. In a similar synthetic route Guiry and co-authors synthesised a novel pyridine-containing LXA432 that was also found to possess important biological properties.31

The first stereoselective route to the novel aromatic analogue 27 described by Guiry and co-workers employed the commercially available divinylcarbinol 55 as the starting material, Scheme 15.19
 | ||
| Scheme 15 Synthesis of key intermediate 42.19,31 | ||
This allylic alcohol 55 was subjected to Sharpless asymmetric epoxidation reaction conditions to give the chiral epoxide 56 in 85% yield and with an enantiomeric excess of greater than 99%. Ring opening of 56 with the Grignard derivative of 57 in the presence of a catalytic amount of CuI afforded the desired diol 58 in 82% isolated yield. This diol required an acid stable protecting group as the acidic Jones' reagent was applied to cleave the dioxane in the following transformation. The diol protection was successfully achieved by the addition of acetyl chloride and pyridine in THF at 0 °C to give the bisacetate in 97% yield. The addition of Jones' reagent in acetone for 2 h yielded the corresponding acid 59, which was esterified using diazomethane in diethyl ether. A change of protecting group strategy was employed at this stage as the bis-acetate methyl ester was an unsuitable coupling partner for the Heck reaction. For this reason, deprotection with NaOMe in MeOH followed by reprotection with a tert-butyldimethylsilyl group was necessary in order to afford the bis-silyl ether 42 in high yield. This protected olefin was then successfully applied in a palladium-mediated Heck reaction in both the benzene- and pyridine-containing LXA4 analogues, 27 and 32, respectively. The authors also found that zirconium tetrachloride was an efficient catalyst for a one-pot protection/deprotection synthetic methodology and used this for the synthesis of 42.43 This protocol also led to the synthesis of 6-acetoxy-5-hexadecanolide, a component of mosquito oviposition attractant pheromones,44 and also a microwave-assisted asymmetric synthesis of exo- and endo-brevicomin.45
The preparation of aryl bromide 61 required as the other Heck coupling partner was achieved through the addition of the Grignard derivative of 1-bromopentane 60 to acid chloride 51, Scheme 16. The reaction was performed at −78 °C to prevent any of the double addition product forming. An initial screening of Heck reaction conditions revealed that tributylamine, with its high boiling point, afforded the coupled product 62 in a very high yield (88%). Reduction of this ketone was achieved using sodium borohydride giving rise to a mixture of epimeric alcohols which were easily separated by column chromatography. The authors also employed (−)-β-chlorodiisopinocampheylborane to give alcohol 63 in 67% yield and with a 92% diastereomeric excess. Finally, this alcohol was deprotected using p-toluenesulfonic acid in MeOH giving the triol (1S)-27 in 84% yield. This triol and the (1R)-27 analogue were both converted to their corresponding acids by LiOH in a mixture of methanol and water and were also investigated for their ability to aid in the resolution of inflammation.
 | ||
| Scheme 16 Synthesis of aromatic LXA4 (1S)-27. | ||
The stereoselective synthesis of the aromatic LXB4 analogue (5S)-54 exploited a similar synthetic route, assembling the trans double bond via a palladium-catalysed Heck reaction with aryl bromide 66, Scheme 17. The aryl bromide 66, required for the Heck reaction, was formed through a Sonogashira coupling of 1-bromo-2-iodobenzene 64 and the commercially available terminal alkyne 65, followed by oxidation with sulfonic acid and esterification.
 | ||
| Scheme 17 Synthesis of Heck coupling partner 66.19 | ||
Another epoxide ring opening reaction via Grignard chemistry produced the olefin Heck coupling partner 67, Scheme 18.
 | ||
| Scheme 18 Stereoselective synthesis of aromatic LXB4 analogue (5S)-54.19 | ||
The Heck reaction proceeded under similar reaction conditions to those employed for the synthesis of aromatic LXA4 (1S)-27, furnishing 68 in 41% yield. Asymmetric reduction of ketone 68 was again accomplished by way of Brown's (−)-β-chlorodiisopinocamphylborane to give the alcohol in 67% yield with a de value of 97%. The final step was acetal cleavage using 2 N HCl in THF at room temperature to furnish triol (5S)-54 in 59% yield. These new aromatic analogues possess great potential as therapeutic agents as the modular synthetic approach to these compounds renders them extremely accessible and their pharmacodynamics can be further tuned by the addition of known classical bioisosteres.
The novel aromatic LXA4 analogues (1S)-27 and (1R)-27 promoted increased clearance of apoptotic PMNs when compared to the effect of the native LXA4. The aromatic LXB4 (5S)-54 analogue also stimulated phagocytosis of apoptotic PMNs with a maximum effect observed at 10−11 M. In addition to this, both analogues (27 and 54) caused F-actin rearrangement which has also been observed with the native compounds.46 Phagocytosis of PMNs was inhibited by pre-treatment with the pan-FPR inhibitor Boc2. This strongly suggests that the effect of these analogues is mediated by the activation of the LX receptor. These analogues were also screened for their ability to stimulate adherence of monocytes to a matrix such as laminin, which is a previously known property of the native LX and also some of the synthetically stable analogues.47,48 In the experiments, the acids did not exhibit an increase in phagocytosis over the same concentration range as the methyl esters.19 This lack of activity was attributed to the fact that the esters act as prodrugs, converting in vivo to the free acid and evoking LX-mediated biological actions.7
Bannenberg and co-workers also showed that oral administration of LXA4 has the ability to inhibit leukocyte infiltration in zymosan A-induced peritonitis.40 Guiry and co-workers found that their (1R)-27 analogue caused a significant decrease in neutrophil accumulation at 50 μg kg−1 while the (1S)-27 analogue also showed a decrease at the highest dose tested.19
A similar synthetic strategy as shown above for the synthesis of the benzene-containing LXA4 analogues was applied by Guiry and co-authors for the preparation of the novel pyridine-containing LXA4 analogue 32. This pyridine-containing LXA4 analogue was designed to allow for a Structure–Activity Relationship study, whereby the effect of the decreased electron density of the heteroaromatic ring and how the extra heteroatom may alter its ability to accept hydrogen-bonds from the known receptor, FPR2/ALX, could be determined.
The construction of the Heck coupling partner ketone 71, Scheme 19, was assembled achieved via a regiospecific pyridine lithiation of commercially available 3-bromopyridine 69.49 The oxidation method of choice for the preparation of ketone 71 was pyridinium chlorochromate (PCC) in the presence of glacial acetic acid which afforded a 70% yield.
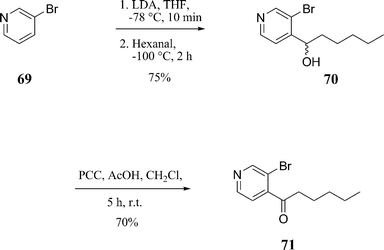
Ketone 71 was coupled to olefin 42via a palladium-catalysed Heck reaction using allylpalladium chloride dimer, tri-o-tolylphosphine, sodium acetate as the base in toluene and dimethylacetamide (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 115 °C for 12 h giving 72 in a 82% yield, Scheme 20.50 Compound 72 was reduced to give the alcohols (1S)-73 and (1R)-73 in high de. These alcohols were finally deprotected using para-toluene sulfonic acid in MeOH to give the required triols (1S)-32 and (1R)-32.
:1) at 115 °C for 12 h giving 72 in a 82% yield, Scheme 20.50 Compound 72 was reduced to give the alcohols (1S)-73 and (1R)-73 in high de. These alcohols were finally deprotected using para-toluene sulfonic acid in MeOH to give the required triols (1S)-32 and (1R)-32.
 | ||
| Scheme 20 Synthesis of pyridine-containing LXA4 analogues. | ||
These compounds showed anti-inflammatory characteristics by displaying increased potency in the clearance of apoptotic leukocytes. Furthermore, these analogues were analysed by their effect on cytokine production by macrophages, a property of the native LX.42,51–53
Compound (1S)-32 had a more potent effect on IL-12p40 production, with significant suppression of this cytokine at 10 μM, 1 μM and 1 nM. Both (1R)-32 and (1S)-32 suppressed LPS-induced production of IL-1β at both 1 μM and 1 nM concentrations.
 | ||
| Fig. 11 Inversion of stereocentre at C6.32 | ||
The C5 and C6 hydroxyl groups have displayed resistance to enzymatic metabolism by EOR, therefore rendering this an undesirable part of the Lipoxin structure to alter. However, Guilford and co-workers discovered β-oxidation can occur at C3 in the para-fluorophenoxy analogue 6, Scheme 21.21
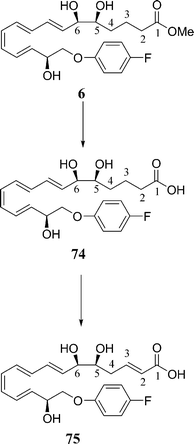 | ||
| Scheme 21 In vivo metabolism of para-fluorophenoxy analogue 6.21 | ||
Their stability experiments carried out on plasma samples revealed an unexpected result—the para-fluorophenoxy analogue 6 was converted into the corresponding acid 74 followed immediately by β-oxidation to furnish the 2,3-dehydro analogue 75. The assignment of this structure was aided with direct comparisons to the lipid metabolisms of the prostaglandin and the leukotriene pathways previously reported in the literature.55,56 With these findings in hand, Guilford designed and synthesised two new LXA4 analogues (76 and 77) by directly replacing the methylene group at C3 with an oxygen to prevent this β-oxidation and hence to enhance the metabolic and chemical stability, Fig. 12.21 The design of these analogues combines the useful strategy of domain modifications (C) and (A), Fig. 3, modifications of the upper and lower chain.
 | ||
| Fig. 12 Design of stable analogues 76 and 77 by preventing β-oxidation.21 | ||
The stereoselective synthesis of 76 and 77 relies upon a Wittig reaction of a known enyne reagent,57 a palladium-catalysed Sonogashira coupling reaction and an activated zinc reduction of an alkyne. A successful chiral pool strategy was utilised in order to achieve the correct stereochemistry at C5 and C6, as key intermediates for the Sonogashira coupling reaction were obtained from L-Rhamnose 78, Scheme 22.
 | ||
| Scheme 22 Synthesis of key intermediate 81.21 | ||
L-Rhamnose 78 was reacted with sulfuric acid, copper sulfate and cyclohexanone at room temperature for 16 h to afford the corresponding protected cyclohexlidene ketal 79 in 57% yield. This was reduced using NaBH4 in methanol to give the triol 80 in 88% yield. Phase transfer conditions were employed to prepare the required ester which was converted into the corresponding aldehyde 81 in 92% yield using sodium metaperiodate in a mixture of water and acetone. A Wittig coupling of aldehyde 81 and the protected alkyne 82 yielded a 2:1 of mixture of E,E and E,Z isomers as determined by 1H NMR spectroscopic analysis. This mixture was dissolved in dichloromethane and treated with iodine to give the required protected E,E-dienyne in 49% yield, Scheme 23. This was further deprotected using TBAF in THF, giving the required terminal alkyne 83 in 99% yield.
 | ||
| Scheme 23 Synthesis of key intermediate 83.21 | ||
The synthesis of the Sonogashira coupling partner 87, Scheme 24, proceeded with the conversion of carboxylic acid 84 into its acid chloride by treatment with oxalyl chloride and a catalytic amount of DMF, followed by direct preparation of the Weinreb amide.
 | ||
| Scheme 24 Synthesis of vinyl bromide 87 for Sonogashira coupling. | ||
This amide was treated with a solution of ethynylmagnesium bromide to furnish the target ketone 85 in 59% yield over three steps. Ketone 85 was reduced using R-Alpine-Borane, although with a modest ee value of between 60 and 70%. This problem was overcome by the conversion of the alcohol to its dinitrobenzoyl derivative followed by a recrystallisation to give ee values greater than 98%. This ester was deprotected using K2CO3 in MeOH, followed by bromination using NBS and silver nitrate to form the chiral alcohol 86 in 79% yield over the final two steps. Reduction of 86 using lithium aluminium hydride and aluminium chloride gave the vinyl bromide 87, the substrate for a subsequent Sonogashira coupling reaction, Scheme 25.
 | ||
| Scheme 25 Synthesis of stable analogues 76 and 77.21 | ||
The Sonogashira coupling of 87 and 83 gave the required alkyne in 50% yield. Cleavage of the acetal protecting group with AcOH gave diol 88 in 58% yield. Diol 88 was hydrolysed under basic conditions affording 76 in 58% yield. Reduction using activated zinc, followed by hydrolysis furnished 77 in a low 30% yield.
The natural LX along with stable analogues provide anti-inflammatory benefits in several models of induced skin inflammation.58 With this information in hand, β-oxidation-resistant analogues 76 and 77 were analysed in a calcium ionophore model topically applied to the mouse ear skin. This study revealed comparable potency to the native analogues, by inhibiting edema formation along with a decrease in neutrophil and granulocyte infiltration. Moreover, compounds 76 and 77 have demonstrated the ability to promote the resolution of colitis induced by the hapten trinitrobenzene sulfonic acid which is a model of Crohn's disease.59,60
Conclusion
Modifications of three key target areas on the LX structure have resulted in the development of Lipoxin analogues displaying increased bioactivity and bioavailability compared to the native LX. The potential biological applications of these stable LX analogues have resulted in a number of efficient synthetic routes being developed for their preparation. Replacement of the C15–20 chain by cyclohexyl- and phenoxy-groups and later the further derivitisation of these analogues with fluoro-groups, gave rise to compounds which showed increased biostability and displayed potential anticancer properties. Petasis pioneered the research involving stabilisation of this key C15–20 chain. Modification of the triene structure, which is present in the native LX, has been an active area of research, also in the Petasis group who were the first to report such analogues. Incorporation of benzene or a heteroaromatic ring in place of this triene structure has given a number of enhanced properties, including stability towards enzymatic decomposition. Guiry and co-workers reported the first stereocontrolled synthesis of a benzene-containing analogue and found that it enhanced the phagocytosis of PMN by macrophages. They also later published the synthesis of a novel analogue, where the triene had been replaced by a pyridine ring. They found that both epimers displayed potent anti-inflammatory characteristics. There have been fewer reports of structural modifications of the upper chain of the LX, mainly due to the importance of retaining the hydroxyl groups in order to maintain bioactivity. Guilford incorporated oxygen into the upper chain, replacing the β-methylene group. This resulted in an analogue that displayed resistance to β-oxidation, leading to heightened metabolic and chemical stability. This derivative also showed potential in the treatment of Crohn's disease.This article reports a concise review of the synthetic and biological developments of novel stable Lipoxin analogues. The major and noteworthy synthetic obstacles and achievements were outlined and discussed. There is an ongoing effort to provide novel therapeutic agents to combat an array of inflammatory diseases and it is hoped that this timely review will help to stimulate the design and biological evaluation of novel Lipoxin analogues.
References
- C. Serhan, M. Hamberg and B. Samuelsson, Biochem. Biophys. Res. Commun., 1984, 118, 943–949 CrossRef CAS.
- C. Serhan, M. Hamberg and B. Samuelsson, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 5335–5339 CAS.
- C. N. Serhan, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2005, 73, 141–162 CrossRef CAS.
- J. Claria and C. N. Serhan, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9475–9479 CrossRef CAS.
- B. McMahon and C. Godson, Am. J. Physiol.: Renal Physiol., 2004, 286, 189F–F201 CrossRef.
- (a) C. Brink, S.-E. Dahlén, J. Drazen, J. F. Evans, D. W. P. Hay, S. Nicosia, C. N. Serhan, T. Shimizu and T. Yokomiao, Pharmacol. Rev., 2003, 55, 195–227 CrossRef CAS; (b) R. D. Ye, F. Boulay, J. M. Wang, C. Dahlgren, C. Gerard, M. Parmentier, C. N. Serhan and P. M. Murphy, Pharmacol. Rev., 2009, 61, 119–161 CrossRef.
- N. Chiang, C. Serhan, S.-E. Dahlén, J. M. Drazen, D. W. P. Hay, G. N. Rovati, T. Shimizu, T. Yokomiao and C. Brink, Pharmacol. Rev., 2006, 58, 463–487 CrossRef CAS.
- N. A. Campbell and J. B. Reece, in Biology, 6th edn, Benjamin Cummings, San Francisco, 2001, 901–903 Search PubMed.
- C. Godson, S. Mitchell, K. Harvey, N. A. Petasis, N. Hogg and H. R. Brady, J. Immunol., 2000, 164, 1663–1667 CAS.
- S. Mitchell, G. Thomas, K. Harvey, D. C. Cottell, K. Reville, G. P. Berlasconi, N. A. Petasis, L. Erwig, A. J. Rees, J. Savill, H. R. Brady and C. Godson, J. Am. Soc. Nephrol., 2002, 13, 2497–2507 CrossRef CAS.
- C. N. Serhan, K. C. Nicolaou, S. E. Webber, C. A. Veale, S.-E. Dahlén, T. J. Puustinen and B. Samuelsson, J. Biol. Chem., 1986, 261, 16340–16345 CAS.
- C. N. Serhan, M. Hamberg, B. Samuelsson, J. Morris and D. G. Wishka, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 1983–1987.
- B. Clish, D. Levy and N. Chiang, J. Biol. Chem., 2000, 275, 25372–25380 CrossRef.
- N. Chiang, I. Fierro, K. Gronert and C. Serhan, J. Exp. Med., 2000, 191, 1197–1207 CrossRef CAS.
- P. W. Collins and S. W. Djuric, Chem. Rev., 1993, 93, 1533–1564 CrossRef CAS.
- N. A. Petasis, I. Akritopoulou-Zanze, V. V. Fokin, G. Bernasconi, R. Keledjian, R. Yang, J. Uddin, K. C. Nagulapalli and C. N. Serhan, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2005, 73, 301–321 CrossRef CAS.
- E. D. Phillips, H.-F. Chang, C. R. Holmquist and J. P. McCauley, Bioorg. Med. Chem. Lett., 2003, 13, 3223–3226 CrossRef CAS.
- N. A. Petasis Preparation of lipoxin benzoanalogs for use in pharmaceutical compositions for the treatment of various diseases, including inflammation, autoimmune disease and abnormal cell proliferation, USP2005203184, 2005 Search PubMed.
- T. P. O'Sullivan, K. S. A. Vallin, S. T. A. Shah, J. Fakhry, P. Maderna, M. Scannell, A. L. F. Sampaio, M. Perretti, C. Godson and P. J. Guiry, J. Med. Chem., 2007, 50, 5894–5902 CrossRef CAS.
- N. A. Petasis, R. Keledjian, Y.-P. Sun, K. C. Nagulapalli, E. Tjonahen, R. Yang and C. N. Serhan, Bioorg. Med. Chem. Lett., 2008, 18, 1382–1387 CrossRef CAS.
- W. J. Guilford, J. G. Bauman, W. Skuballa, S. Bauer, G. P. Wei, D. Davey, C. Schaefer, C. Mallari, J. Terkelsen, J.-L. Tseng, J. Shen, B. Subramanyam, A. J. Schottelius and J. F. Parkinson, J. Med. Chem., 2004, 47, 2157–2165 CrossRef CAS.
- K. C. Nicolaou, J. Y. Ramphal, N. A. Petasis and C. N. Serhan, Angew. Chem., Int. Ed. Engl., 1991, 30, 1100–1116 CrossRef.
- C. N. Serhan, J. F. Maddox, N. A. Petasis, I. Akritopoulou-Zanze, A. Papayianni, H. R. Brady, S. P. Colgan and J. L. Madara, Biochemistry, 1995, 34, 14609–14615 CrossRef CAS.
- S. E. Webber, C. A. Veale and K. C. Nicolaou, Adv. Exp. Med. Biol., 1988, 229, 61–77 CAS.
- C. B. Clish, J. A. O'Brien, K. Gronert, G. L. Stahl, N. A. Petasis and C. N. Serhan, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8247–8252 CrossRef CAS.
- I. M. Fierro, J. L. Kutok and C. N. Serhan, J. Pharmacol. Exp. Ther., 2002, 300, 385–392 CrossRef CAS.
- A. R. Rodríguez and B. W. Spur, Tetrahedron Lett., 2001, 42, 6057–6060 CrossRef CAS.
- K. C. Nicolaou, C. A. Veale, S. E. Webber and H. Katerinopoulos, J. Am. Chem. Soc., 1985, 107, 7515–7518 CrossRef CAS.
- W. Boland, N. Schruer, C. Sieler and M. Feigel, Helv. Chim. Acta, 1987, 70, 1025–1040 CrossRef CAS.
- T. Takano, S. Fiore, J. F. Maddox, H. R. Brady, N. A. Petasis and C. N. Serhan, J. Exp. Med., 1997, 185, 1693–1704 CrossRef CAS.
- C. D. Duffy, P. Maderna, C. McCarthy, C. E. Loscher, C. Godson and P. J. Guiry, ChemMedChem, 2010, 5, 517–522 CrossRef CAS.
- Y.-P. Sun, E. Tjonahen, R. Keledjian, M. Zhu, R. Yang, A. Recchiuti, P. S. Pillai, N. A. Petasis and C. N. Serhan, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2009, 81, 357–366 CrossRef CAS.
- K. C. Nicolaou and S. E. Webber, J. Am. Chem. Soc., 1984, 106, 5734–5736 CrossRef CAS.
- M. M. Midland, D. C. McDowell, R. L. Hatch and A. Tramontano, J. Am. Chem. Soc., 1980, 102, 867–869 CrossRef CAS.
- N. A. Petasis and E. I. Bzowej, J. Am. Chem. Soc., 1990, 112, 6392–6394 CrossRef CAS.
- T. Jeffery, Tetrahedron, 1996, 52, 10113–10130 CrossRef.
- R. Soundararajan and D. S. Matteson, J. Org. Chem., 1990, 55, 2274–2275 CrossRef.
- N. A. Petasis and I. A. Zavialov, Tetrahedron Lett., 1996, 37, 567–570 CrossRef CAS.
- S. Hong, K. Gronert, P. R. Devchand, R.-L. Moussignac and C. N. Serhan, J. Biol. Chem., 2003, 278, 14677–14687 CrossRef CAS.
- G. Bannenberg, R.-L. Moussignac, K. Gronert, P. R. Devchand, B. A. Schmidt, W. J. Guilford, J. G. Bauman, B. Subramanyam, H. D. Perez, J. F. Parkinson and C. N. Serhan, Br. J. Pharmacol., 2004, 143, 43–52 CrossRef CAS.
- C. N. Serhan, S. Hong, K. Gronert, S. P. Colgan, P. R. Devchand, G. Mirick and R.-L. Moussignac, J. Exp. Med., 2002, 196, 1025–1037 CrossRef CAS.
- S. Sodin-Semrl, B. Taddeo, D. Tseng, J. Varga and S. Fiore, J. Immunol., 2000, 164, 2660–2666 CAS.
- S. Singh, C. D. Duffy, S. T. A. Shah and P. J. Guiry, J. Org. Chem., 2008, 73, 6429–6432 CrossRef CAS.
- S. Singh and P. J. Guiry, Eur. J. Org. Chem., 2009, 1896–1901 CrossRef CAS.
- S. Singh and P. J. Guiry, J. Org. Chem., 2009, 74, 5758–5761 CrossRef CAS.
- P. Maderna, D. C. Cottell, G. Berlasconi, N. A. Petasis, H. R. Brady and C. Godson, Am. J. Pathol., 2002, 160, 2275–2283 Search PubMed.
- J. F. Maddox and C. N. Serhan, J. Exp. Med., 1996, 183, 137–146 CrossRef CAS.
- J. F. Maddox, M. Hachicha, T. Takano, N. A. Petasis, V. V. Fokin and C. N. Serhan, J. Biol. Chem., 1997, 272, 6972–6978 CrossRef CAS.
- G. W. Gribble and M. G. Saulnier, Heterocycles, 1993, 35, 151–169 CrossRef CAS.
- N. Robert, C. Hoarau, S. Celanire, P. Ribereau, A. Godard, G. Queguiner and F. Marsais, Tetrahedron, 2005, 61, 4569–4576 CrossRef CAS.
- S.-H. Wu, P. Y. Liao, L. Dong and Z. Q. Chen, Inflammation Res., 2008, 57, 430–437 CrossRef CAS.
- Y. Decker, G. McBean and C. Godson, Am. J. Physiol.: Cell Physiol., 2009, 296, C1420–1427 CrossRef CAS.
- J. Aliberti, C. Serhan and A. Sher, J. Exp. Med., 2002, 196, 1253–1262 CrossRef CAS.
- B. Samuelsson, S. E. Dahlen, J. A. Lindgren, C. A. Rouzer and C. N. Serhan, Science, 1987, 237, 1171–1176 CrossRef.
- C. K. Ellis, M. D. Smigel, J. A. Oates, O. Oelz and B. J. Sweetman, J. Biol. Chem., 1979, 254, 4152–4163 CAS.
- Y. Guindon, D. Delorme, C. K. Lau and R. Zamboni, J. Org. Chem., 1988, 53, 267–275 CrossRef CAS.
- R. A. Robinson, J. S. Clark and A. B. Holmes, J. Am. Chem. Soc., 1993, 115, 10400–10401 CrossRef CAS.
- A. J. Schottelius, C. Giesen, K. Asadullah, I. M. Fierro, S. P. Colgan, J. Bauman, W. Guilford, H. D. Perez and J. F. Parkinson, J. Immunol., 2002, 169, 7063–7070 CAS.
- S. Fiorucci, J. L. Wallace, A. Mencarelli, E. Distrutti, G. Rizzo, S. Farneti, A. Morelli, J.-L. Tseng, B. Suramanyam, W. J. Guilford and J. F. Parkinson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15736–15741 CrossRef CAS.
- W. J. Guilford and J. F. Parkinson, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2005, 73, 245–250 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |