The influence of EDTA and citrateanticoagulant addition to human plasma on information recovery from NMR-based metabolic profiling studies
Richard H.
Barton
ab,
Daniel
Waterman
c,
Frank W.
Bonner
c,
Elaine
Holmes
a,
Robert
Clarke
d,
the PROCARDIS Consortium†
e,
Jeremy K.
Nicholson
a and
John C.
Lindon
*a
aDepartment of Biomolecular Medicine, Faculty of Medicine, Imperial College London, Sir Alexander Fleming Building, South Kensington, London, UK SW7 2AZ. E-mail: j.lindon@imperial.ac.uk
bCentre for Integrated Systems Biology, Imperial College London, South Kensington, London, UK SW7 2AZ
cMetabometrix Ltd., Bioincubator, Bessemer Building, Prince Consort Road, London, UK SW7 2BP
dClinical Trial Service Unit, Richard Doll Building, University of Oxford, Old Road Campus, Headington, Oxford, UK OX3 7LF
eProcardis, Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford, UK OX3 7BN
First published on 28th September 2009
Abstract
The widely-used blood anticoagulantscitrate and EDTA give rise to prominent peaks in 1H NMR spectra of plasma samples collected in epidemiological and clinical studies, and these cause varying levels of interference in recovering biochemical information on endogenous metabolites. To investigate both the potential metabolic information loss caused by these substances and any possible inter-molecular interactions between the anticoagulants and endogenous components, the 1H NMR spectra of 40 split human plasma samples collected from 20 individuals into either citrate or EDTA have been analysed. Endogenous metabolite peaks were selectively obscured by large citrate peaks or those from free EDTA and its calcium and magnesium complexes. It is shown that the endogenous metabolites that give rise to peaks obscured by those from EDTA or citrate almost invariably also have other resonances that allow their identification and potential quantitation. Also, metabolic information recovery could be maximised by use of spectral editing techniques such as spin-echo, diffusion-editing and J-resolved experiments. The NMR spectral effects of any interactions between the added citrate or EDTA and endogenous components were found to be negligible. Finally, identification of split samples was feasible using simple multivariate statistical approaches such as principal components analysis. Thus even when legacy epidemiological plasma samples have been collected using the NMR-inappropriate citrate or EDTAanticoagulants, useful biochemical information can still be recovered effectively.
Introduction
The comprehensive and simultaneous measurement of multiple metabolites in biological fluids is frequently undertaken to characterise the responses to drugs, diet, lifestyle, environment, disease or genetic modulations.1 The role of metabonomics as a key component of systems biology has been expanding, in particular for understanding the complex interplay between humans and their gut microflora in diseases such as insulin resistance,2 and for population wide studies of metabolic phenotypes where metabolic connections have been made with important disease risk factors such as hypertension.3 Individual metabotype variation is also predictive of responses to drug exposure and toxicity as part of pharmacometabonomic studies.4Typically, multiparametric biofluid profiling utilises 1H NMR spectroscopy as a highly reproducible approach to measuring plasma metabolite profiles.5,6 There have been many previous studies of plasma using 1H NMR spectroscopy and most of the detectable NMR resonances in plasma have been assigned.5,6 The high degree of analytical reproducibility enables the technique to assess metabolic changes of interest over and above any natural biological variation. NMR based metabonomics has been used to study risk factors for coronary heart disease,7,8 to assess plasma lipoprotein classes9 and comprehensive protocols for NMR based metabonomics experiments have recently been published.10
Many blood samples collected for epidemiological or “bio-banking” purposes11 for subsequent use in phenotyping or disease risk studies have been collected into a variety of anticoagulants. These include lithium-heparin, citrate and the sodium salt of ethylenediamine-tetra-acetic acid (EDTA). Whilst heparin gives only weak and very broad NMR resonances from the polysaccharide moieties, citrate and EDTA have large sharp 1H NMR resonances at the concentrations typically used5,6 and it has been widely accepted that the presence of these peaks can severely inhibit information recovery in metabonomic studies.
Citrate gives rise to a characteristic AB doublet pattern in its 1H NMR spectrum, with the non-equivalent protons of each methylene group having typical chemical shifts of δ 2.54 and δ 2.68 with a 2JHH spin coupling constant of ca. 18 Hz. Since one of the carboxyl groups of citrate has a pKa close to physiological pH, the level of protonation is highly dependent on pH in this range. Hence since the protonated and deprotonated forms are in fast exchange on the NMR time scale, a population-weighted average spectrum of the two forms is seen, and this results in considerable pH dependence of the 1H NMR chemical shifts, this being particularly notable for the less shielded chemical shift.
The peaks generated by EDTA are more complex.12Free EDTA (H-EDTA3−) shows two singlets at ca. δ 3.2 from the four –NCH2CH2N– protons and a singlet of twice the intensity at ca. δ 3.6 from the eight –NCH2CO– hydrogens. However EDTA is strongly complexed in plasma with endogenous calcium and magnesium ions to form the Ca–EDTA2− and Mg–EDTA2− complex ions, which have distinct chemical shifts as they are in slow exchange on the NMR time scale. In some biological samples, it is also possible to observe the Zn–EDTA2− complex6 at ca. δ 2.86. In all of these complexes the –NCH2CH2N– protons remain equivalent and give rise to a singlet at δ 2.57 for the Ca–EDTA complex, and at δ 2.76 for the Mg–EDTA complex. However, due to the geometry of the metal complexes, the acetate protons are now no longer chemically or magnetically equivalent and give rise to AB patterns with characteristic 2JHH spin couplings, the appearance of which depends on the AB (inter-proton) chemical shift and the NMR observation frequency. These AB patterns are centered at δ 3.13 and 3.23 for the Ca–EDTA2− and Mg–EDTA2− complexes, respectively.
Although these NMR peaks can be used to quantitatively measure the Ca2+ and Mg2+ concentration in plasma,5,6 they cause interference to the observation of other 1H NMR signals from endogenous metabolites. Here we investigate whether use of these anticoagulants limits the value of 600 MHz 1H NMR spectroscopy for metabolic phenotyping. We have used a subset of blood plasma samples from the PROCARDIS study that were collected and then split for archive storage, and which used either citrate or EDTA as anticoagulants. The PROCARDIS study (www.procardis.org) was designed to assess genetic susceptibility to coronary heart disease (CHD) and also differences in levels of a wide range of intermediate phenotypes that might mediate any associations between genes and CHD risk.
Obviously, although any archived biobank samples could have been used, these were chosen because individual samples contained either citrate or EDTA. The purpose of the current study was not to quantitatively measure the levels of selected metabolites whose NMR peaks were partially obscured by those of citrate or EDTA because, in real biobank samples, the levels of both the endogenous metabolites and added substances could both vary widely, making such a task meaningless. Rather we have concentrated on determining how addition of such materials could affect a metabonomic classification analysis, as typically carried out.
Materials and methods
Sample preparation: samples were collected from 20 disease-free adults resident in Sweden or Germany, who were unaffected siblings in the PROCARDIS study. Some samples had a degree of cloudiness and two samples were very opaque. Some samples also presented some precipitate, which was avoided in the sample preparation procedure. Plasma samples had been stored at −80 °C prior to presentation for NMR analysis. Samples were thawed at room temperature and 350 μl aliquots were prepared in random sequence by the addition of 150 μl of isotonic saline (0.9% w/v NaCl) and 3 mM sodium azide in 80 : 20 H2O–D2O.NMR spectroscopy
A set of 1H NMR spectra were acquired on each sample, comprising a standard 1-D spectrum, a CPMG spin-echo spectrum, a diffusion-edited spectrum and high-resolution 2-dimensional J-resolved spectrum (JRES), all with water peak presaturation, and recorded on a Bruker Avance-600 spectrometer operating at 600.3 MHz, with the sample temperature held at 300 K.10Spectra were referenced to the chemical shift of the α-glucose anomeric proton doublet taken at δ 5.23, except for the diffusion-edited spectra, where the spectral reference (SR) offset value from a referenced CPMG co-acquisition on the same sample was used. For the standard spectrum, the pulse sequence was RD–90°–t1–90°–tm–90°–acquire FID, with water peak suppression achieved through irradiation of the water signal during tm and RD, using 64 scans, 8 dummy scans, spectrum width 20.02 ppm, 32k time and frequency domain data points, line broadening 1 Hz, relaxation delay (RD) 2 s, tm 150 ms, t1 3 μs. For the CPMG spectra, the pulse sequence was RD–90°–[τ–180°–τ]n–acquire FID, with water suppression achieved through irradiation of the water signal during RD, using 64 scans, 8 dummy scans, 32k time and frequency domain data points, line broadening 0.3 Hz, RD 2 s, 2nτ 80 ms (τ = 400 μs). For the diffusion-edited spectra, the pulse sequence was RD–90°–G1–180°–G1–90°–G2–Δ–90°–G1–180°–G1–90°–G2–τ–90°–acquire FID, with water suppression achieved through irradiation of the water signal during RD, using 128 scans, 8 dummy scans, spectrum width 20.02 ppm, 16k time and frequency domain data points, line broadening 1 Hz, RD 2 s, diffusion time (Δ) 80 ms, gradient strength (G1) 3.45 Gauss per cm, and eddy current delay (τ) 500 μs. For the 2-D JRES spectra, the pulse sequence was RD–90°–t1–180°–t1–acquire FID, with water suppression achieved through irradiation of the water signal during RD, using 4 scans per increment, 8 dummy scans, F2 spectrum width 10.01 ppm, F1 spectrum width 50 Hz, 8k and 50 time domain data points in F2 and F1, respectively, filled to 8k and 128 in the frequency domains, line broadening in F2 0.3 Hz, RD 2 s. The skyline projection along F2 was calculated after standard tilting and symmetrisation operations. All spectral phasing and baseline correction was carried out manually using the standard software from the instrument manufacturer (Bruker). The main data analyses were carried out on spectral data from the standard 1-D acquisition, using Excel 2003 (Microsoft Corporation, USA), Matlab R2007a (MathWorks Inc, Natick MA, USA), and SIMCA P+11.5 (Umetrics AB, Umeå, Sweden) software packages, with the other spectral acquisitions being used for enhanced data retrieval.Results
1H NMR spectra of plasma samples in the presence of anticoagulants
Typical 1H NMR spectra from plasma samples with added citrate or EDTA are shown in Fig. 1(a) and (b), respectively. For comparison, the corresponding spectrum of human serum from a different subject is also shown in Fig. 1(c) as this does not require the addition of an anticoagulant. The effects that are introduced by the use of citrate and EDTA comprise the obscuring of endogenous metabolite peaks due to overlap with high amplitude signals from the prominent EDTA or citrate peaks, and the potential for spectral perturbations caused by molecular interaction effects between the anticoagulants and endogenous components of the plasma.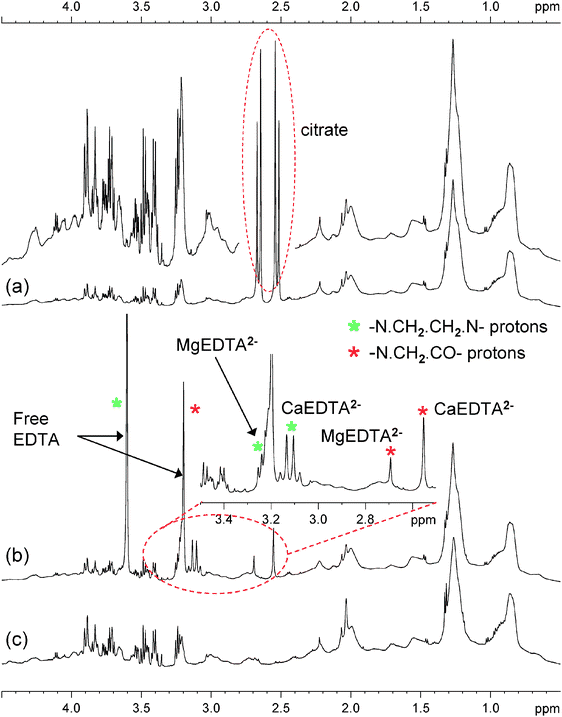 | ||
| Fig. 1 Partial 600 MHz 1H NMR spectrum (aliphatic region only) of (a) human plasma with added citrate, where a vertical expansion is also shown and the peaks from citrate are shown in the ellipse; (b) human plasma with added EDTA, where a horizontal expansion of the region shown in the ellipse is given and the various protons from EDTA and its complexes are also assigned; (c) human serum from a different subject without anticoagulants. The very broad baseline envelope arises from plasma proteins and the semi-broad peaks arise from lipoproteins. The sharp peaks are due to small molecule metabolites. Aromatic substances such as tyrosine and phenylalanine have peaks in the shift region δ 8.5–6.0, which is not shown here. | ||
Citrate is a normal component of plasma from the tricarboxylic acid cycle and gives a typical AB pattern of peaks in the 1H NMR spectra as shown in Fig. 1(a). Addition of extra citrate simply causes the existing citrate signals to have larger amplitudes. However, because of the Lorentzian line shape, markedly enhanced peak amplitudes give rise to an extended peak base, which may affect detection of small NMR signals lying close to the citrate peaks, e.g. those from methylamine and dimethylamine, and this might also subsequently affect any variance modelling studies. Citrate also complexes with Ca2+ and Mg2+, but these complexes are in fast exchange with free citrate on the NMR chemical shift time scale, and hence only one set of citrate peaks is observed at chemical shifts which are mole-fraction weighted averages of those from each of the species.5,6
In the case of EDTA, there is a more complex set of NMR peaks as described above, and this affects a greater number of regions of the spectrum (as seen in Fig. 1(b)), and thus there is greater peak overlap with more of the signals from endogenous compounds present in the plasma, and such overlaps could make information retrieval more difficult in these spectral regions. These signals are produced both by the protons of free EDTA itself, as well as the signals arising from chelated EDTA, where a range of bound endogenous metal ions found in the plasma are captured by the EDTA, a powerful metal sequestering agent.5 This binding affects both the chemical shift of the EDTA signals, and by molecular symmetry breaking, increases the number of signals superimposed onto the endogenous profile. Furthermore EDTA-related signals cover a wider range of shift values than citrate.
Potential peak overlap problems
The presence of significant peaks in the NMR spectra of plasma due to the presence of anticoagulants might cause difficulties relating to spectral regions where signal overlaps are not easily resolved. There is only a small positional variation in the NMR peak positions of citrate and particularly of EDTA for this set of samples (data not shown). However, the chemical shifts of the singlet peaks from free EDTA are in principle pH sensitive and thus could change position. Also the citrate NMR peak positions are themselves pH dependent since citrate has a pKa in the physiological range, and this is a common cause of variability in chemical shift (and also in principle, coupling constants).However, even with peaks having stable chemical shifts, the number of EDTA signals could induce problems in establishing the levels of some metabolites, particularly those which have characteristic signals which lie only in overlapped shift regions. It is a cumbersome procedure to attempt subtractive removal of such features in order to quantify other local metabolites. A partial candidate list of metabolites with signals falling in the regions where anticoagulant signals appear is provided in Table 1. Some endogenous metabolites with only single peaks will undoubtedly be obscured, and these include mainly methylamine and dimethylamine.
However, most endogenous metabolites have several NMR peaks and even though some might be obscured by the anticoagulant peaks, there are often other peaks observable in open regions of the spectra and which thus allow metabolite characterisation and quantitation, if required. Also included in Table 1 is information on how this can be achieved.
Interactions between anticoagulants and endogenous plasma components
The possible effects of anticoagulants on the structure of lipoproteins and on interactions with other endogenous substances were also investigated. For this purpose, the spectral dataset was divided into the two subsets, corresponding to the citrate- and EDTA-containing spectra, and the principal signals in each subset were probed using statistical total correlation spectroscopy (STOCSY).13 Selected signals are used as “driver” peaks to then determine any peak intensities across the sets of spectra that correlate with the variation in the “driver” peak intensity. By this means, any spectral variation of endogenous metabolite levels that correlate with the variation in the level of citrate or EDTA can be detected. This method provides a further insight into what effects if any, apart from the primary spectral features already discussed, are induced by the presence of anticoagulants. In each case, the diagram presented marks the driver signal peak used, and presents the results in a pseudo-spectral format. Hence the variation in endogenous profiles with the citrate (Fig. 2(a)) shows that added citrate does not have an effect on the observed metabolite or lipoprotein profile, since no high correlations are observed between the driver peak intensity and any other spectral feature. | ||
| Fig. 2 (a) STOCSY result based on the set of NMR spectra from citrate-containing plasma samples, with the correlation driven from the peak (arrowed) from citrate. The only significantly correlated signals are other peaks related to citrate itself (indicated as red regions). The calculated correlation coefficients are presented as a colour code according to the vertical scale for r2. The positional variability is greater for the low frequency component of the citrate AB system near δ 2.5 and results in a reduced correlation coefficient. (b) STOCSY result based on the set of NMR spectra from EDTA-containing plasma samples, with the correlation driven from the peak (arrowed) from Ca–EDTA2−. There is a positional variability of the less shielded component of the citrate AB system near δ 2.68, and this results in a reduced correlation coefficient with the δ 2.5 doublet. However, the same result is observed when either signal of the δ 2.5 doublet is used as the STOCSY driver, both with regard to the δ 2.68 doublet, and for the rest of the spectrum (data not shown). | ||
However, the various EDTA peaks showed correlated effects through the spectra. Whilst the free EDTA peak showed, as expected, only correlation with the other free EDTA signal, the Mg–EDTA2− singlet did not show any correlation to the other Mg–EDTA2− peak probably because of the lower signal-noise level combined with a greater degree of local spectral overlap. The result of STOCSY probing of the Ca–EDTA2− singlet at δ 2.55 shows not only the expected correlation with the well-resolved quartet from Ca–EDTA2− (Fig. 2(b)), but also a broader involvement throughout other spectral regions where protein signals arise, most visible in regions of the broad protein peak background, between δ 0.0–0.7 and δ 6.5–9.0, where there is little overlap with sharp peaks from low molecular weight metabolites, suggesting that the Ca–EDTA2− is also strongly associated with (non-lipoprotein) protein components of the plasma. Such proteins are known to be present in plasma at relatively high abundance and about 46% of plasma calcium is protein-bound in a variety of known species.14 Of the bound fraction, it has been shown that 32% of the calcium is bound to albumin , 45% to various globulin species and 23% to a variety of small molecule species. Whilst not implying that major structural effects result from this association, this observation is suggestive of wider plasma interactions due to EDTA mediated by calcium binding.
As an additional demonstration of the minor effects due to the presence of the anticoagulants, Fig. 3 shows the comparative positional change of citrate peak positions in the citrate-containing plasma, most probably due to pH changes effected by the high concentrations of citrate present in one of the two sample sets (the comparative low amplitude citrate peaks shown are from the EDTA spectral dataset). It is known that the less shielded pair of peaks is more affected by pH and metal complexation than the more shielded pair of peaks.5,6
 | ||
| Fig. 3 Overlaid 1H NMR spectra showing a small positional shift of the left hand pair of citrate peaks due to pH changes in the citrate-containing sample set. The much lower level peaks of citrate in the plasmas with no added citrate are also clearly seen. | ||
Projections of 2-dimensional J-resolved NMRspectra
A 2-D J-resolved spectrum is shown in Fig. 4(a). J-resolved spectra are edited to show mainly low molecular weight metabolites.6,15 The method can be used to resolve 1H–1H coupled spectral components where their multiplets are overlapped, by projecting each multiplet into a single peak at the chemical shift value, such that overlapped but shift-differentiated signals in standard 1-D spectra can be better resolved from one another. This editing technique is also particularly useful for spectral analysis in cases where there are many amino acids present, because their 1H NMR peaks show a number of complex multiplets and there is considerable peak overlap between species. The 2-D J-resolved spectrum can be projected onto the F2 axis, where every multiplet becomes a singlet, and this reduces spectral crowding and overlap of the various signals present. Examples of such 1-D spectra are shown in Fig. 4(b) and (c), for the corresponding citrate and EDTA pair-matched plasma J-resolved spectra, where in particular the quartet from Ca–EDTA2− can be resolved as a singlet with consequently reduced local overlap. | ||
| Fig. 4 (a) 600 MHz partial (aliphatic region only) J-resolved 2-D spectrum of a citrate-containing plasma sample. In this representation, all spin–spin coupled multiplets are rotated by 90° to the chemical shift axis reducing peak overlap. (b) 1-D skyline projection from a symmetrised 2-D J-resolved spectrum of a citrate-containing plasma sample. (c) 1-D skyline projection from a symmetrised 2-D J-resolved spectrum of an EDTA-containing plasma sample. Assignments are as marked. The profiles are generally close, apart from some minor shift variation and the significant peak at ca. δ 2.22 in the citrate-containing sample which is a contaminant present in all the citrate samples, with acetone as the suggested identity. Variations in ethanol are discussed in the text. Also very minor peaks of trace contaminants appear in the EDTA-associated samples. | ||
The rotation and projection of the Ca–EDTA2− signal allows access to signals close to δ 3.12 which would have been overlapped in other spectra such as the standard 1-D and CPMG spectra. There are time-related penalties in gaining this spectroscopic advantage, as well as a drop in the overall quantitative accuracy when compared with the standard 1-D method, which may be critical for some applications. However the profiles are very similar, with the exception of small shifts in some peaks, likely to be the effect of pH and/or ionic strength variation, as well as a peak (thought to be from acetone contamination introduced at the blood aliquoting stage) at ca. δ 2.22 which appears only in the citrate-containing samples. There are also some very small extra peaks associated with the EDTA-containing samples.
Protein binding of anticoagulants
The use of diffusion-edited spectroscopy is aimed at selective detection of macromolecular species, as opposed to J-resolved spectroscopy or CPMG spectra which show mainly peaks from low molecular weight species. The severity of the spectral editing can be adjusted by choosing different magnetic field gradient strengths, and in this study this was chosen to attenuate the peaks from the low molecular weight anticoagulants, and leave the lipoprotein and protein peaks unaffected. The resulting spectrum essentially presents a profile of the lipoprotein and proteinmacromolecular species present in the plasma sample and of small molecule metabolites that are bound to macromolecules and hence have a lowered diffusion coefficient. The diffusion-edited spectra for the same two samples given above are shown in Fig. 5 where some peaks from small molecules are observed. This is probably due to non-specific binding of the anticoagulants resulting in an averaged diffusion coefficient for free and bound forms weighted by their relative mole fractions. This interpretation is supported by observation of significant broadening in these peaks when compared with the standard 1-D NMR line widths for matching signals, where the freely mobile fraction of the substance dominates the signal amplitude. | ||
| Fig. 5 Diffusion-edited 1H NMR spectra for the same sample pair as in Fig. 4, showing small signals from EDTA and citrate at very high concentration, arrowed. The “choline” signal from the choline –N(CH3)3 moieties within phospholipids in lipoproteins is also marked, as it overlaps with a free EDTA signal. The general spectral profile reflects only macromolecular protein and lipoprotein signals from the plasmas. | ||
Pair-matching of split samples based on their NMR spectra
In an earlier study11 blind identification of split plasma samples based on their NMR spectra was carried out to examine the quality of aliquoting as well as effects of variation in the time from sample collection to low-temperature archiving. This procedure using iterative principal components analysis (PCA) was shown to be successful and specifically involved split samples of high-quality plasma. In view of the split samples collected into EDTA or citrate tubes, a similar method was used on the spectral data from this study.Since EDTA and citrate produce such strong spectral features, it was predictable that PCA would divide the sample group into 2 extremely well-separated classes, the model scores being dominated by the peaks from the respective anticoagulant employed (data not shown). Thus, the procedure was repeated using only partial spectral data from part of the aliphatic region, where the signal integrals are dominated by signals from plasma lipoproteins. This spectral cut-off effectively removes all the large EDTA- and citrate-related signals from the modelling.
In an unscaled PCA model, in the absence of anticoagulants, inter-sample variation in these lipoprotein signals would normally dominate the PCA loadings, and hence these peaks were expected to carry adequate information to implement the required pairing. The splits analysis was carried out with a reduced dataset employing only the shift region δ 0–2.4, and using a reduced data point resolution of 0.01 ppm and with total area normalisation over this spectral sub-region. By this means, it was possible to complete all 20 sample pairings from the single PCA model shown by simple visual inspection, Fig. 6(a). Comparison with the un-normalised data suggested that whilst paired lipoprotein profiles were also well matched, there were some variable concentration effects observable, and these were provisionally attributed to at-source aliquoting being started from the top of the centrifuge tube immediately post-centrifugation (data not shown).
 | ||
| Fig. 6 (a) PCA score plot (PC1 vs. PC2) for the entire spectra set, using data from the δ 0.0–2.4 region only and subjected to total peak area normalisation. The closeness of split sample pairings (one red and one black for each pair) and the ease of pair assignments are apparent. (b) 7-fold cross-validated predictive O-PLS-DA discriminant score plot showing how the samples can be separated based on country of origin (Swedes in red and Germans in blue), using only the aliphatic sub-region of the spectra dominated by lipoprotein signals. Both area-normalised data (R2X = 0.994, Q2Y = 0.73) and non-normalised data (R2X = 0.997, Q2Y = 0.49) results are given. An expansion of the central region is shown. One subject (2 samples) is misclassified in the non-normalised data modelling, probably due to an anomalous lipoprotein profile. | ||
A preliminary study to separate samples based on subject origin
Efforts were made to establish if chemometric techniques could be utilised to separate samples based on sample class. The most obvious class separation is based on sample origin and allows a comparison based on an equal number of samples in each class (n = 20 for both Swedes and Germans). A previous study has also investigated the metabolic profiles of biofluids from Swedish subjects.16An O-PLS discriminant chemometrics approach (O-PLS-DA) was employed,17 whereby the model is told which samples belong to which classes (i.e. Swedish or German) allowing the model to test how the spectra differ between the two classes. An O-PLS-DA loadings plot was then used to establish which areas of the spectra carried the most weight for establishing a difference between the specified classes. This analysis was applied to the same low-shift spectral region used for pair-matching, but was carried out at full spectral resolution.
It was observed that class differences were present in the LDLlipid regions of the spectra, being significantly higher in the 10 German individuals compared with the 10 Swedish individuals. The level of ethanol also varied between the samples, probably due to antiseptic ethanol swabbing at the phlebotomy point and differing levels of subcutaneous fat affecting ethanol absorption. Confirmatory STOCSY analyses (data not shown) were conducted to ensure that the across-group discriminant signals were a result of the difference in LDLlipid signals, as clearly the NMR spectral region is made up of contributions from multiple chemical species. STOCSY calculations conducted using the peaks at δ 0.81 and δ 1.19 (known to be from LDL) as the drivers confirmed that the discrimination is indeed due to LDL-related lipids and not the possible alternative candidate substances, 3-hydroxybutyrate or ethanol.
Finally cross-validated score plots for the O-PLS-DA discrimination using both area-normalised and non-normalised data (1 aligned, 5 orthogonal components) are shown in Fig. 6(b) and these illustrate the separation between the groups more visually (the central region is also shown expanded). Swedes are plotted in red and the Germans are in blue. One subject (2 samples) is misclassified in the non-normalised data modelling, probably due to an anomalous lipoprotein profile. It is likely that the discrimination can be improved using more complex chemometrics approaches, as well as the use of data from a wider spectral region, but the current result demonstrates the power of the technique even when using robust 7-fold cross-validated scores only. It should be noted that the use of cross-validated scores is also a useful indication that the model shown is not over-fitted, as otherwise there would be a major discrepancy in the group separation between using the direct scores and the cross-validated scores (data not shown). The result using cross-validated scores indicates that the model produced is therefore robust and not over-fitted.
Discussion
The present study has demonstrated that the anticoagulantscitrate and EDTA give rise to prominent NMR peaks in spectra and produce varying levels of difficulty in recovering information from NMR spectra of plasma. However, the degree of obscuration depends on the NMR chemical shift region being examined, as available current techniques, including spectral editing and mathematical data processing, go a considerable way towards redressing these difficulties. Incidentally, it has previously been shown that selected metabonomics analysis is possible in rabbit plasma samples collected with the addition of EDTA.18 Such examples are important to illustrate possibilities for the data mining of previously-established repositories of biosamples where the storage protocols used at the time have implications for both the instrumental analytical methods employed and for the quality of data extraction, factors which were not considered or anticipated at the time of archiving. It is noted that because analytical techniques continue to advance rapidly, this has to be factored into planning for the creation, and appropriate stable storage, of biosample archives.The identification of molecular species of interest, and the consequent determination of their concentrations in plasma, is thus generally feasible, apart from those with single NMR peaks close to the very largest amplitude anticoagulant signals. Most metabolites have several NMR peaks for quantitation purposes, and it is not usual for all to be obscured. In addition, identification of split samples was found to be feasible using simple multivariate statistical approaches, and it was also possible to identify the sample source, suggesting that more complex discriminations will be possible with larger sample sets, as well as more inclusive spectral data such as from the aromatic region of the NMR spectrum.
The main conclusion from this study is that analysis of previously-banked blood plasma samples that had been collected using either citrate or EDTA as an anticoagulant is feasible using NMR-based metabonomics. This ensures that future studies of disease risk, such as using established human biobanks for obesity and diabetes screening,3 or even wider metabolome association studies,4 become possible.
Acknowledgements
We are grateful to the participants and to the medical and nursing staff, who assisted in the PROCARDIS Consortium. The PROCARDIS project was funded by the British Heart Foundation, EC Sixth Framework Programme (LSHM-CT-2007-037273) and AstraZeneca AB. The PROCARDIS consortium involves the collaboration of the Department of Cardiovascular Medicine (Hugh Watkins, Fiona Green, Martin Farrall, John Peden), University of Oxford, with the Clinical Trial Service Unit (Rory Collins, Robert Clarke, Jemma Hopewell), University of Oxford and other university departments in Milan (Gianni Tognoni, Maria Grazia Franzosi), Münster (Udo Seedorf) and Stockholm (Anders Hamsten). The collaboration was extended in 2007 to include the Centre Nationale de Genotypage, Paris (Mark Lathrop), Ethox Centre, Oxford (Mike Parker), Metabometrix Ltd, London (Frank Bonner, John Lindon, Daniel Waterman), Barcelona (Jose Manuel Soria); Clinical Gene Networks, Stockholm, (Johan Björkegren); Milan (Elena Tremoli); and Digilab BioVisioN, Hannover (Petra Budde).References
- J. K. Nicholson, J. Connelly, J. C. Lindon and E. Holmes, Nat. Rev. Drug Discovery, 2002, 1, 153–161 CrossRef CAS.
- M.-E. Dumas, R. H. Barton, A. Toye, O. Cloarec, C. Blancher, A. Rothwell, J. Fearnside, R. Tatoud, V. Blanc, J. C. Lindon, S. C. Mitchell, E. Holmes, M. I. McCarthy, J. Scott, D. Gauguier and J. K. Nicholson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12511–12516 CrossRef CAS.
- E. Holmes, R. L. Loo, J. Stamler, M. Bictash, I. K. Yap, Q. Chan, T. Ebbels, M. De Iorio, I. J. Brown, K. A. Veselkov, M. L. Daviglus, H. Kesteloot, H. Ueshima, L. Zhao, J. K. Nicholson and P. Elliott, Nature, 2008, 453, 396–400 CrossRef CAS.
- T. A. Clayton, J. C. Lindon, H. Antti, C. Charuel, G. Hanton, J.-P. Provost, J.-L. Le Net, D. Baker, R. J. Walley, J. R. Everett and J. K. Nicholson, Nature, 2006, 440, 1073–1077 CrossRef CAS.
- J. K. Nicholson, M. J. Buckingham and P. J. Sadler, Biochem. J., 1983, 211, 605–615 CAS.
- J. K. Nicholson, P. J. D. Foxall, M. Spraul, R. D. Farrant and J. C. Lindon, Anal. Chem., 1995, 67, 793–811 CrossRef CAS.
- J. T. Brindle, H. Antti, E. Holmes, G. Tranter, J. K. Nicholson, H. W. L. Bethell, S. Clarke, P. M. Schofield, E. McKilligin, D. E. Mosedale and D. J. Grainger, Nat. Med. (N. Y.), 2003, 9, 477 CAS.
- H. L. Kirschenlohr, J. L. Griffin, S. C. Clarke, R. Rhydwen, A. A. Grace, P. M. Schofield, K. M. Brindle and J. C. Metcalfe, Nat. Med. (N. Y.), 2006, 12, 705–710 CrossRef CAS.
- E. J. Jeyarajah, W. C. Cromwell and J. D. Otvos, Clin. Lab. Med., 2006, 26, 847–870 CrossRef.
- O. Beckonert, H. C. Keun, T. M. D. Ebbels, J. Bundy, E. Holmes, J. C. Lindon and J. K. Nicholson, Nat. Protocols, 2007, 2, 1–12 CrossRef.
- R. H. Barton, J. K. Nicholson, P. Elliott and E. Holmes, Int. J. Epidemiol., 2008, 37, i31–i40 Search PubMed.
- D. P. Higham, J. K. Nicholson, J. Overnell and P. J. Sadler, Environ. Health Perspect., 1986, 65, 157–166 CrossRef CAS.
- O. Cloarec, M.-E. Dumas, A. Craig, R. H. Barton, J. Trygg, J. Hudson, C. Blancher, D. Gauguier, J. C. Lindon, E. Holmes and J. K. Nicholson, Anal. Chem., 2005, 77, 1282–1289 CrossRef CAS.
- B. Deng, P. Zhu, Y. Wang, J. Feng, X. Li, X. Xu, H. Lia and Q. Xu, Anal. Chem., 2008, 80, 5721–5726 CrossRef.
- S. Tiziani, A. Lodi, C. Ludwig, H. M. Parsons and M. R. Viant, Anal. Chim. Acta, 2008, 610, 80–88 CrossRef CAS.
- E. M. Lenz, J. Bright, I. D. Wilson, A. Hughes, J. Morrisson, H. Lindberg and A. Lockton, J. Pharm. Biomed. Anal., 2004, 36, 841–849 CrossRef CAS.
- J. Trygg, E. Holmes and T. Lundstedt, J. Proteome Res., 2007, 6, 469–479 CrossRef CAS.
- M. A. Constantinou, A. Tsantili-Kakoulidou, I. Andreadou, E. K. Iliodromitis, D. T. Kremastinos and E. Mikros, Eur. J. Pharm. Sci., 2007, 30, 303–314 CrossRef CAS.
Footnote |
| † Membership of the PROCARDIS Consortium is at www.PROCARDIS.org. |
| This journal is © The Royal Society of Chemistry 2010 |