Microfluidic synthesis of pure chitosan microfibers for bio-artificial liver chip†
Kwang Ho
Lee‡
ae,
Su Jung
Shin
a,
Chang-Beom
Kim
a,
Jung Kyung
Kim
b,
Yong Woo
Cho
c,
Bong Geun
Chung
d and
Sang-Hoon
Lee
*a
aDepartment of Biomedical Engineering, College of Health Science, Korea University, Seoul, Korea. E-mail: dbiomed@korea.ac.kr; Fax: +82-2-921-6818; Tel: +82-2-940-2881
bSchool of Mechanical Engineering, Kookmin University, Seoul, Korea
cDepartment of Chemical Engineering, Hanyang University, Ansan, Korea
dDepartment of Bionano Engineering, Hanyang University, Ansan, Korea
eResearch Institute of Health Sciences, Korea University, Seoul, Korea
First published on 8th March 2010
Abstract
We developed microfluidic-based pure chitosan microfibers (∼1 meter long, 70–150 μm diameter) for liver tissue engineering applications. Despite the potential of the chitosan for creating bio-artificial liver chips, its major limitation is the inability to fabricate pure chitosan-based microstructures with controlled shapes because of the mechanical weakness of the pure chitosan. Previous studies have shown that chitosan micro/nanofibers can be fabricated by using chemicals and electrospinning techniques. However, there is no paper regarding pure chitosan-based microfibers in a microfluidic device. This paper suggests a unique method to fabricate pure chitosan microfibers without any chemical additive. We also analyzed the chemical, mechanical, and diffusion properties of pure chitosan microfibers. Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectrometry and electron spectroscopy for chemical analysis (ESCA) were used to analyze the chemical composition of the synthesized chitosan microfibers. We measured the mechanical axial-force and diffusion coefficient in pure chitosan-based microfibers using fluorescence recovery after photobleaching (FRAP) techniques. Furthermore, to evaluate the capability of the microfibers for liver tissue formation, hepatoma HepG2 cells were seeded onto the chitosan microfibers. The functionality of these hepatic cells cultured on chitosan microfibers was analyzed by measuring albumin secretion and urea synthesis. Therefore, this pure chitosan-based microfiber chip could be a potentially useful method for liver tissue engineering applications.
Introduction
The ability to create artificial bio-tissues or organs is of importance for various tissue engineering applications, such as regenerative liver tissue constructs.1–4 The liver, one of the most important organs in the human body, plays a significant role in controlling various metabolic functions, such as protein synthesis and detoxification.5,6 However, the engineering of 3D liver tissues which regulate specific functions still has many challenges,7–9 such as the inability to properly select scaffold materials that can control the desired shapes.10 Chitosan, a natural polymer, is one of the most suitable scaffold materials for liver tissue, because its structure is similar to glycosaminoglycans (GAGs) that are natural components of the liver extracellular matrix (ECM). Thus, chitosan has been widely used for liver tissue engineering applications.11–14 However, the major limitation of chitosan-based liver tissue constructs is the inability to fabricate pure chitosan-based microstructures with controlled shapes. It is probably due to their mechanical weakness. The control of the size, shape, and spatial distribution of the scaffold has been extremely important in designing and creating tissue constructs.15 An electrospun nano-scale chitosan fiber mesh has been previously fabricated for nerve regeneration.16 However, it was only used as a 2D scaffold and the mechanical strength was also weak. To improve the mechanical properties of chitosan, chitosan-cast film has been coated on electrospun chitosan nanofibers.17Although electrospinning techniques have been widely used to create micro/nanofibers of various materials, such as polymers, composites, and ceramics, for tissue engineering applications,18–21 they still have some limitations, such as the inability to control the direction of micro/nanofiber alignment. To address these limitations imposed by the electrospinning technique, we have previously developed microfluidic-based fiber fabrication methods for culturing the cells.22–26
In this study, we developed the microfluidic fabrication process for pure chitosan microfibers and characterized their mechanical, chemical, and diffusion properties. In general, the scaffold microfibers undergo diffusion in response to transport proteins or growth factors. The measurement of the diffusion coefficient is important to predict the accurate releasing profiles of protein or growth factors. However, the direct measurement of a microfiber's diffusion coefficient is still challenging. Here, we measured the diffusion coefficient of a single fiber using the fluorescence recovery after photobleaching (FRAP) method. In addition, the handling of fibers (e.g. continuous winding of fibers on the frame or culturing of HepG2 cells on fibers) is required for practical application. Although the mechanical strength (e.g. axial force) is important for the safe handling of chitosan microfibers, it is not easy to measure the axial force of single chitosan microfibers. Thus, we measured the axial force by using our lab-made apparatus. On the pure-chitosan microfibers, we cultured HepG2 cells and observed their hepatic function by measuring albumin secretion and urea synthesis. Furthermore, we fabricated a bio-artificial liver (BAL) chip by embedding wound fibers in a poly(dimethylsiloxane) (PDMS)-based fluidic channel and analyzed the albumin secretion and urea synthesis.
Materials and methods
Materials
Chitosan was purchased from Wako (Chitosan 1000, Osaka, Japan). Acetic acid (AA), sodium triphosphate pentabasic (STP), ethanol (EtOH), sodium hydroxide (NaOH), phosphate buffered saline (PBS) and fluorescein isothiocyanate (FITC)-dextran (70 kDa) were purchased from Sigma (St. Louis, Mo., USA). FITC-BSA was purchased from Invitrogen (Paisley, UK). Minimum essential medium (MEM) and fetal bovine serum (FBS) were purchased from Gibco (Carlsbad, CA, USA). Cell proliferation reagent WST-1 was purchased from Roche (Mannheim, Germany), and Live/Dead assay reagent was purchased from Molecular Probes (LIVE/DEAD®, Eugene, OR, USA). Formaldehyde was purchased from Electron Microscopy Sciences (Fort Washington, PA, USA). Albumin secretion and urea synthesis kits were purchased from Sicdia (Eiken, Japan). Six-well cell culture plates were purchased from Corning (Corning, NY, USA). Uncoated polystyrene and coverglass-bottom dishes were purchased from SPL (Pcheon, Korea).Equipment
Optical microscopic images were obtained by using an Axiovert 200M instrument (Carl Zeiss, Goettingen, Germany). Scanning electron microscope images were obtained by a JEOL instrument (S-4700, Hitachi Co., Japan). Axial force was measured by using a digital force gauge meter (Imada, Toyohashi, Japan). A Perkin-Elmer system (Series 2000, Beaconsfield, UK) was used for ATR-FTIR spectrometry. ESCA analysis was performed by PHI 5800 ESCA System (Perkin-Elmer, Chanhassen, MN, USA). FRAP tests were performed by using a LASER (35LAP43, CVI-Melles Griot, Carlsbad, CA, USA), inverted fluorescence microscope (Olympus, IX71, Tokyo, Japan), mechanical shutter (Uniblitz LS3, NY, USA), and intensified charge-coupled device (ICCD) camera (Dicam-Pro, Cooke Corp., MI, USA). The FITC-bovine serum albumin (BSA) laden fiber and Live/Dead assays were monitored by using an Axiovert 200M. WST-1 was measured by ELISA (Multiskan Ex., Thermo Labsystems, Helsingfors, Finland).Characterization of chitosan fibers
Diameter control of chip-based chitosan fibers was determined by core and sheath flow rates. As the core flow rate increased, at constant sheath flow rate, the diameter was increased. In contrast, as the sheath flow rate increased, at constant core flow rate, the diameter decreased. We utilized core flow rates of 4.2, 4.8 and 5.4 mL h−1 and sheath flow rates of 30, 40, and 50 mL h−1. The chitosan fibers were divided into 2 groups: those generated at a constant core flow rate of 4.2 mL h−1 (A group) and those generated at a constant sheath flow rate of 30 mL h−1 (B group).The axial force was measured by using a digital force gauge meter with a step motor controller. During this test, the chitosan fibers were immersed in PBS solution at room temperature. Both ends of the specimen were clamped with polymer grips, which allowed pull in the lateral direction (Fig. 1). An axial force was applied at a rate of 4.5 N s−1 until the fiber broke from its original shape.
 | ||
| Fig. 1 Schematic diagram of axial force measurement system. | ||
The diffusion coefficient of a chitosan fiber was quantitatively assessed by using the FRAP test. The wound fibers were washed twice with deionized water and loaded with FITC-dextran (70 kDa, 50 mg mL−1) for 1 day. The temperature surrounding the samples was maintained at 25 °C using a temperature controller and the samples were positioned on an inverted fluorescence microscope equipped with a 20 × 0.45 NA objective. The photobleaching beam consisted of an Ar-ion laser operated at 488 nm with 23 mW, and the sample was exposed to the beam for 257 ms using a shutter. The shutter was then closed to capture images of fluorescence recovery using an ICCD camera and AQM6 software. As soon as the shutter was closed, the fluorescence-labeled molecules were diffused into the center of the bleached area. To minimize the effect of photobleaching by the mercury lamp, we installed a neutral density filter. The fluorescence recovery process was observed in a real-time manner (Fig. 2). To analyze images taken every 257 ms, we used ImageJ software (http://rsb.info.nih.gov/ij/). The gray levels of the bleached and unbleached areas were measured from every image to compensate for photobleaching by the mercury lamp. To obtain the recovery curve, the fractional intensity (fk(t)) was calculated from eqn (1),
 | (1) |
 | (2) |
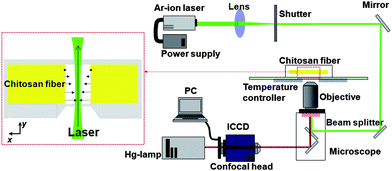 | ||
| Fig. 2 FRAP system for measurement of diffusion in chitosan fiber. | ||
The half-recovery time, τ1/2, and the measured initial spot radius, w, were used to calculate the diffusion coefficient (D). The half-recovery time was defined as the time at which the fluorescence intensity after photobleaching reached 50% of the fluorescence intensity before photobleaching. The radius of the beam spot was determined by the distance from the center of maximum intensity to the location at which the intensity was e−2 times the maximum intensity.
Fabrication of bio-artificial liver chip
We fabricated the PDMS-based BAL chip on which the three microfluidic channels (diameter: 1 mm) with inlet and outlet ports were engraved. We produced the BAL chip and frame using a soft lithographic method. We designed the BAL chip to embed the chitosan fiber wound PDMS frame. After embedding the fiber-wound frame, we seeded the HepG2 cells and perfused the culture medium at 0.1 mL h−1 using a syringe pump. The BAL chip was maintained in an incubator.Results and discussion
To generate chitosan microfibers with various diameters, we used the microfluidic fiber generation method (Fig. 3(a)) with a modification in the dimension of the glass pipette (tip size of pulled pipette ≈ 300 μm). We used 4% (w/v) chitosan solution in 2% (v/v) acetic acid (AA) as a core fluid and 10% (w/v) sodium triphosphate pentabasic (STP) as a sheath fluid to synthesize solidified chitosan fibers from the interface of the two liquids. By changing the flow rate of the core and sheath fluids, we changed the diameter of the chitosan microfibers. The extruded chitosan fibers were wound (fiber length: around 40 cm) onto the windowed polystyrene (PS) frame (12.35 × 10.25 mm, inset of Fig. 3(a)). Fig. 3(b) shows the fiber diameter relative to the flow rate of the core and sheath fluids and Fig. 3(c) shows scanning electron microscope (SEM) images of single and crossed fibers. By adjusting the flow rates of the core and sheath fluids, we generated fibers with 70 to 150 μm in diameter, and the fibers were continuously extruded without disconnection. The diameter of the fibers was measured from the microscopic image and commercial software (Adobe Photoshop), and the size was controlled by the applied flow rate (< ±10% error) as illustrated in Fig. 3(b). We measured the diameter for every 5 cm (we measured a total of 10 points for each sample) to investigate the size uniformity in length, and the diameter was almost uniform with ±3% error. For the measurement, we prepared two types (A and B group) of samples, and 5 samples were used per each type. Fig. 3(d) represents micrographs of chitosan fibers wound onto frames, showing the potential for stable handling of the generated fibers. Although we generated longer fibers by controlling the flow rate of the core and sheath fluid, the minimum diameter of chitosan fiber (∼70 μm) used in this study was larger than that of other fibers (alginate: 20 μm, PLGA: 20 μm) that we have previously generated in microfluidic devices.25,26 This discrepancy is probably due to the mechanical weakness of the chitosan. However, the size can be reduced if we optimize the material and flow rate. In general, it is difficult to measure the axial force of microscale chitosan fibers due to their mechanical weakness. To address this limitation, we developed the axial force measurement system as shown in Fig. 1. | ||
| Fig. 3 (a) Schematic of the synthesis of a chitosan microfiber-generating chip. The chip has two stages: generation of the core and sample flow, and winding of the fibers using an X–Y stage-based winder. For the synthesis of chitosan fibers, we used 4% w/w chitosan and 10% STP solutions as the core and sheath fluids, respectively. (b) Variation of controlled fiber diameter as a function of core and sheath flow rates (n = 3, * indicates P < 0.01). (c) SEM image of a chitosan fiber. (d) 1D and 2D wound chitosan fibers on cover glass-bottom dishes. (e) Fluorescent micrograph of FITC-BSA laden chitosan fibers. | ||
To analyze the mechanical properties of chitosan microfibers, we measured the axial force (Fig. 4(a)). As expected, the axial force of chitosan microfibers was low (0.02–0.04 N) and depended on the fiber diameter, indicating that the diameter of chitosan microfibers should be over a certain size for generating continuous production and fiber winding. This result shows that the minimum diameter of chitosan fibers is larger (∼70 μm) as compared to previous studies. To investigate the composition of the synthesized chitosan fibers, we performed attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectrometry analysis. Fig. 4(b) shows amide I and II stretches at 1640 cm−1 and 1575−1 cm, respectively, corresponding to peaks appearing in chitosan.27 From the ATR-FTIR analysis, we found the peaks of the functional groups of chitosan microfibers after the microfluidic fabrication.
 | ||
| Fig. 4 (a) Axial forces of synthesized chitosan fibers of three diameters (n = 3, * indicates P < 0.05). (b) ATR-FTIR surface analysis of a synthesized chitosan fiber. (c) FRAP data for diffusion of 70 kDa FITC-dextran in chitosan fibers; the fluorescence was fully recovered after photobleaching. (d) ESCA wide scan and C1s narrow scan with the curve fitting of chitosan. | ||
To analyze the diffusion properties within the microfibers, we measured the diffusion coefficient of microscale chitosan fibers by using a FRAP technique. Fig. 4(c) illustrates the fluorescence recovery curves of FITC-dextran (70 kDa) loaded chitosan fibers. The inset of Fig. 4(c) shows micrographs before and after laser radiation, and the black point represents the spot on which the laser was focused. We observed full recovery of fluorescence after photobleaching, showing that mobile fraction was 100%. The calculated diffusion coefficient (7.8 × 10−8 cm2 s−1) was higher than the diffusion coefficient of a chitosan sheet, indicating that diffusion into the fiber structure is more active.28 Electron spectroscopy for chemical analysis (ESCA) showed that most chitosan signals were obtained from carbon (52.82%), oxygen (38.03%), and nitrogen (38.03%) atoms (inset of Fig. 4(d)). The curve fitting with C1s high resolution showed three peaks, such as 286.6, 286.19, and 287.69 eV (Fig. 4(d)), confirming that the chemical structure of the chitosan fiber was corresponded to that of native chitosan. This result showed that the microfluidic fabrication process did not affect the chemical composition of the chitosan. It was also revealed that proteins and other molecules can be loaded into chitosan microfibers without changing any chemical compositions during the microfabrication process. It was confirmed by FITC-BSA laden chitosan microfibers (Fig. 3(e)).
The cell-laden chitosan microfiber was evaluated as a scaffold that could create liver tissue constructs. As a control, HepG2 cells were seeded at a density of 4.42 × 105 cells per well on uncoated polystyrene dishes (UPD) and gelatin coated polystyrene dishes (GCPD). The cells were cultured with 5 mL minimum essential medium (MEM). In a parallel study, cell-laden wound chitosan fibers were cultured in cover glass-bottom dishes. Fig. 5(a) shows a fluorescence micrograph of aggregated cells on the fiber. The cells were stained by a live/dead assay after culturing for 5 days in vitro, showing that cell viability was over 95%. Although cells cultured on Petri dishes did not self-aggregate (inset of Fig. 5(a)), cells cultured on chitosan microfibers were spontaneously self-aggregated, forming spheroid-like structures. The proliferation of HepG2 cells was measured by using the cell proliferation reagent (i.e. WST-1) on day 1, 3, and 5. Growth kinetics showed that cellular activity increased over time. On day 5, the cellular proliferation (23.5%) on the chitosan microfiber was higher as compared to UPD (p < 0.01). The hepatic function of HepG2 cells on the chitosan microfiber was evaluated by measuring albumin secretion and urea synthesis over 5 days.29 We found that HepG2 cells on chitosan fibers showed much higher albumin secretion as compared to the control. It was revealed that albumin secretion on chitosan microfibers was 27.7% and 16.9% higher than that on UPD and GCPD control, respectively (Fig. 5(c)). Similarly, the urea synthesis rate of cells on chitosan microfibers was 6% and 16% higher than on UPD and GCPD control (Fig. 5(d)). The liver function of hepatocytes could be improved by spheroid formation and glycosaminoglycans.11,30–32 Several approaches have been previously employed for the formation of spheroids.33,34 Thus, the self-formation of spheroids around chitosan fibers is a promising approach for enhancing hepatic function in vitro.
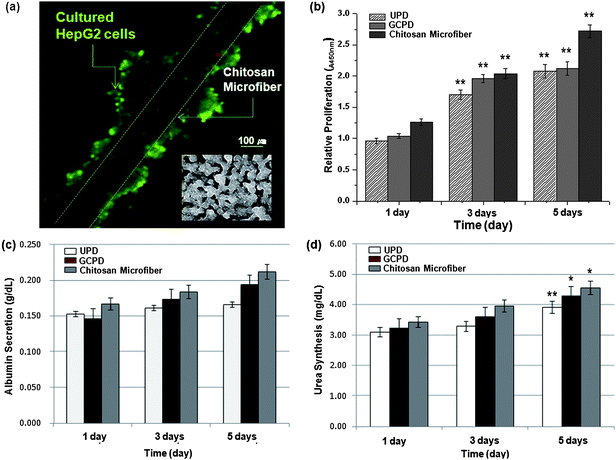 | ||
| Fig. 5 (a) Fluorescent micrograph of an aggregation of HepG2 cells on chitosan fibers and non-aggregation in Petri-dishes (inset). (b) Proliferation of HepG2 cells cultured on chitosan fibers for 5 days (n = 3, ** indicates P < 0.01). (c–d) Albumin secretion and urea synthesis levels synthesized in control HepG2 cells (UPD and GCPD) and cells cultured on chitosan fibers. Each point represents the mean of 5 samples (n = 5, * indicates P < 0.05, and ** indicates P < 0.01). | ||
As a proof-of-the concept, we applied chitosan fibers to a BAL chip. The PDMS microfluidic platform was fabricated (Fig. 6(a) and 6(b)). The chitosan fibers wound on a frame were embedded in a PDMS platform, and HepG2 cells were seeded and cultured forming clusters around the microfibers. The fresh medium was perfused at a flow rate of 0.1 mL h−1 and the albumin secretion and urea synthesis were measured at day 1, 3, and 5 after cell seeding. As shown in Fig. 6(c), albumin secretion was increased to 33%, while urea synthesis was increased to 27% after culturing for 5 days in vitro. Fig. 6(d) illustrates the HepG2 cells on the chitosan fiber in a BAL chip, showing that the cells were stably adhered to chitosan-based microfibers. Therefore, self-aggregated spheroids of HepG2 cells adhered on chitosan microfibers could be potentially useful for controlling hepatic function in a well-defined microenvironment.
 | ||
| Fig. 6 (a) Integration schematic of chitosan microfiber and PDMS microfluidic channel. (b) Picture of bio-artificial liver chip and inset shows the HepG2 cells clustered on the chitosan fibers. (c) Albumin secretion and urea synthesis at 1, 3, and 5 days after seeding (n = 5, * indicates P < 0.05). (d) Micrograph of clustered cells on the fibers after 5 days, showing a BAL chip. | ||
Conclusions
In this study, we prepared pure chitosan-based microfibers in a microfluidic device. ATR-FTIR spectrometry analysis and ESCA results showed that the chemical properties of the chitosan microfiber were not affected by the microfabrication process. We also measured the axial force and FRAP-based diffusion properties of pure chitosan microfibers. It was revealed that the mechanical axial force of pure chitosan was low (0.02–0.04 N) and that the diffusion within chitosan microfibers was more active as compared to the calculated diffusion coefficient. HepG2 cells cultured on chitosan-based microfibers were self-aggregated with a spheroid shape, showing a higher liver specific function that was confirmed by albumin secretion and urea synthesis. Therefore, our method to create pure chitosan-based microfibers in a microfluidic device could be a potentially useful tool for liver tissue engineering applications.Acknowledgements
This study was supported by a grant from the Korea Science and Engineer Foundation (KOSEF) (grant no. ROA-2007-000-20086-0) and this work was supported by the Korea Research Foundation Grant funded by the Korean Government (MEST) (KRF-2008-220-D00133). B. G. Chung is partially supported by Korea Industrial Technology Foundation (KOTEF) through the Human Resource Training Project for Strategic Technology.References
- L. G. Griffith and G. Naughton, Science, 2002, 295, 1009–1014 CrossRef CAS.
- R. C. Dutta and A. K. Dutta, Biotechnol. Adv., 2009, 27, 334–339 CrossRef CAS.
- W. L. Grayson, T. P. Martens, G. M. Eng, M. Radisic and G. Vunjak-Novakovic, Semin. Cell Dev. Biol., 2009, 20, 665–673 CrossRef CAS.
- R. F. Service, Science, 2008, 322, 1460–1461 CrossRef CAS.
- S. Diekmann, A. Bader and S. Schmitmeier, Cytotechnology, 2006, 50, 163–179 CrossRef CAS.
- A. Dash, W. Inman, K. Hoffmaster, S. Sevidal, J. Kelly, R. S. Obach, L. G. Griffith and S. R. Tannenbaum, Expert Opin. Drug Metab. Toxicol., 2009, 5, 1159–1174 Search PubMed.
- L. A. Furchtgott, C. C. Chow and V. Periwal, Biophys. J., 2009, 96, 3926–3935 CrossRef CAS.
- A. W. Duncan, C. Dorrell and M. Grompe, Gastroenterology, 2009, 137, 466–481 CrossRef.
- M. Ito, H. Nagata, S. Miyakawa and I. J. Fox, J. Hepato-Biliary-Pancreatic Surg., 2009, 16, 97–100 Search PubMed.
- J. M. Millis and J. E. Losanoff, Nat. Clin. Pract. Gastroenterol. Hepatol., 2005, 2, 398–405 Search PubMed quiz 434.
- P. Verma, V. Verma, P. Ray and A. R. Ray, In Vitro Cell. Dev. Biol.: Anim., 2007, 43, 328–337 Search PubMed.
- S. J. Seo, Y. J. Choi, T. Akaike, A. Higuchi and C. S. Cho, Tissue Eng., 2006, 12, 33–44 CrossRef CAS.
- Y. M. Elcin, V. Dixit and G. Gitnick, Artif. Organs, 1998, 22, 837–846 CrossRef CAS.
- S. S. Thorgeirsson, FASEB J., 1996, 10, 1249–1256 CAS.
- E. Lavik and R. Langer, Appl. Microbiol. Biotechnol., 2004, 65, 1–8 CAS.
- W. Wang, S. Itoh, A. Matsuda, S. Ichinose, K. Shinomiya, Y. Hata and J. Tanaka, J. Biomed. Mater. Res., Part A, 2008, 84A, 846–846 CrossRef CAS.
- A. Matsuda, G. Kagata, R. Kino and J. Tanaka, J. Nanosci. Nanotechnol., 2007, 7, 852–855 CrossRef CAS.
- J. D. Schiffman and C. L. Schauer, Polym. Rev., 2008, 48, 317–352 Search PubMed.
- W. E. Teo and S. Ramakrishna, Nanotechnology, 2006, 17, R89–R106 CrossRef CAS.
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- D. Liang, B. S. Hsiao and B. Chu, Adv. Drug Delivery Rev., 2007, 59, 1392–1412 CrossRef CAS.
- W. Jeong, J. Kim, S. Kim, S. Lee, G. Mensing and D. J. Beebe, Lab Chip, 2004, 4, 576–580 RSC.
- S. J. Shin, J. Y. Park, J. Y. Lee, H. Park, Y. D. Park, K. B. Lee, C. M. Whang and S. H. Lee, Langmuir, 2007, 23, 9104–9108 CrossRef CAS.
- C. Hwang, S. H. Lee and K. Sun, Proc. Micro TAS (Paris), 2007, 778–780 Search PubMed.
- C. M. Hwang, A. Khademhosseini, Y. Park, K. Sun and S. H. Lee, Langmuir, 2008, 24, 6845–6851 CrossRef CAS.
- K. H. Lee, S. J. Shin, Y. Park and S. H. Lee, Small, 2009, 5, 1264–1268 CrossRef CAS.
- G. Lawrie, I. Keen, B. Drew, A. Chandler-Temple, L. Rintoul, P. Fredericks and L. Grondahl, Biomacromolecules, 2007, 8, 2533–2541 CrossRef CAS.
- B. Falk, S. Garramone and S. Shivkumar, Mater. Lett., 2004, 58, 3261–3265 CrossRef CAS.
- K. Lee, B. G. Min, C. H. Mun, S. R. Lee and Y. S. Won, Blood Purif., 2008, 26, 491–497 CrossRef.
- W. B. Tsai and J. H. Lin, Acta Biomater., 2009, 5, 1442–1454 CrossRef CAS.
- E. Thedinga, A. Ullrich, S. Drechsler, R. Niendorf, A. Kob, D. Runge, A. Keuer, I. Freund, M. Lehmann and R. Ehret, ALTEX, 2007, 24, 22–34 Search PubMed.
- M. Khalil, A. Shariat-Panahi, R. Tootle, T. Ryder, P. McCloskey, E. Roberts, H. Hodgson and C. Selden, J. Hepatol., 2001, 34, 68–77 CrossRef CAS.
- Y. S. Torisawa, A. Takagi, Y. Nashimoto, T. Yasukawa, H. Shiku and T. Matsue, Biomaterials, 2007, 28, 559–566 CrossRef CAS.
- J. Fukuda, A. Khademhosseini, Y. Yeo, X. Yang, J. Yeh, G. Eng, J. Blumling, C. F. Wang, D. S. Kohane and R. Langer, Biomaterials, 2006, 27, 5259–5267 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Diagrams of measurement systems. See DOI: 10.1039/b924987g |
| ‡ Present address: Department of Mechanical Engineering and Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA |
| This journal is © The Royal Society of Chemistry 2010 |