The network formation assay: a spatially standardized neurite outgrowth analytical display for neurotoxicity screening†
Jean-Philippe
Frimat‡
a,
Julia
Sisnaiske‡
b,
Subanatarajan
Subbiah
c,
Heike
Menne
a,
Patricio
Godoy
b,
Peter
Lampen
a,
Marcel
Leist
d,
Joachim
Franzke
a,
Jan G.
Hengstler
b,
Christoph
van Thriel§
*b and
Jonathan
West§
*a
aISAS—Institute for Analytical Sciences, Otto-Hahn-Str. 6b, D-44227, Dortmund, Germany. E-mail: west@isas.de
bLeibniz Research Centre for Working Environment and Human Factors at the University of Dortmund (IfADo), Ardeystr. 67, D-44139, Dortmund, Germany. E-mail: thriel@ifado.de
cFakultät Bio- und Chemieingenieurwesen, Lehrstuhl für Systemdynamik und Prozessführung, Technische Universität Dortmund, D-44221, Dortmund, Germany
dDepartment of Biology, Universität Konstanz, Postfach M633, D-78457, Konstanz, Germany
First published on 4th January 2010
Abstract
We present a rapid, reproducible and sensitive neurotoxicity testing platform that combines the benefits of neurite outgrowth analysis with cell patterning. This approach involves patterning neuronal cells within a hexagonal array to standardize the distance between neighbouring cellular nodes, and thereby standardize the length of the neurite interconnections. This feature coupled with defined assay coordinates provides a streamlined display for rapid and sensitive analysis. We have termed this the network formation assay (NFA). To demonstrate the assay we have used a novel cell patterning technique involving thin film poly(dimethylsiloxane) (PDMS) microcontact printing. Differentiated human SH-SY5Y neuroblastoma cells colonized the array with high efficiency, reliably producing pattern occupancies above 70%. The neuronal array surface supported neurite outgrowth, resulting in the formation of an interconnected neuronal network. Exposure to acrylamide, a neurotoxic reference compound, inhibited network formation. A dose–response curve from the NFA was used to determine a 20% network inhibition (NI20) value of 260 µM. This concentration was approximately 10-fold lower than the value produced by a routine cell viability assay, and demonstrates that the NFA can distinguish network formation inhibitory effects from gross cytotoxic effects. Inhibition of the mitogen-activated protein kinase (MAPK) ERK1/2 and phosphoinositide-3-kinase (PI-3K) signaling pathways also produced a dose-dependent reduction in network formation at non-cytotoxic concentrations. To further refine the assay a simulation was developed to manage the impact of pattern occupancy variations on network formation probability. Together these developments and demonstrations highlight the potential of the NFA to meet the demands of high-throughput applications in neurotoxicology and neurodevelopmental biology.
Introduction
There is an urgent need for the widespread screening and identification of chemicals that pose a health risk to the human nervous system. Standard protocols for neurotoxic risk assessment involve the use of in vivo rodent models. This approach is lengthy, resource intensive and requires many animals, making it impractical for screening large numbers of chemicals. As a result there is currently a strong emphasis on substituting many animal experiments with simplified in vitro cell culture experiments for a high-throughput screening approach to chemical assessment.1–4In vitro nervous system models must accurately mimic the critical cellular events of neurodevelopment and plasticity. Axonal and dendritic outgrowths (collectively termed neurites) are defining morphological characteristics of the differentiated neuronal phenotypes that are essential for neuronal connectivity and network function. As such the outgrowths are hallmark neurodevelopmental end point indicators and are popularly used as the subject of measurement in the neurite outgrowth assay: the assay uses a sparsely seeded neuron culture to isolate individual neurons and their neurite outgrowths for ease of visualization and measurement. In its simplest form the assay measures the percentage of cells elaborating neurites or the number of neurites per cell.3 More quantitative methods involve neurite length measurements (e.g. longest, average, total) and the measurement of branching complexity.5 These methods are manually intensive, and instead high content screening platforms with automated image capture, processing and analysis have been developed for rapid data acquisition.6 Fixation and antibody staining are used to aid feature recognition, producing results with an equivalent accuracy to manual tracing.7,8 Despite this progress, the neurotoxicology endeavour would greatly benefit from a standardized in vitro analysis platform that provides a simple end point read-out for rapid and accurate high content screening.
Microtechnologies can be used to meet many of the demands of modern bioanalysis. In particular, microfabrication can be used for the parallel isolation and observation of cellular processes. In the field of neurobiology, microfluidic corridors and interconnected compartments have been applied to the study of axon guidance,9 and neuronal regeneration processes.10–12 For the spatiotemporal investigation of electrophysiological function, an ultimate neurodevelopmental end point, microelectrode arrays can be used.13–17 These have been coupled with microfluidic compartments for the aligned monitoring of neuronal network activity.18–20 Alternatively, cell patterning21–24 can be used to interface neurons with electrodes for probing potential propagation at defined locations through engineered neuronal architectures25,26 and even along single axons.27
The early work on patterning functional neurons involved photolithographic silane patterning,28–30 while more recently microcontact printing of cell adhesion materials has gained popularity.31–33 Between the complexities of cell–material interactions and microelectrode array techniques there is scope to apply cell patterning to address widespread and routine challenges in the life sciences. In this study we demonstrate the value of cell patterning for neurotoxicity testing. A neuronal microarray was produced by thin film PDMS printing34 and used as a simple neurite interconnection display for the rapid and sensitive dose-dependent measurement of acrylamide neurotoxicity. We have termed this novel approach as the network formation assay (NFA).
Concept
Neurite outgrowths serve as hallmark neurodevelopmental end point indicators which can be correlated, using biokinetic models, with in vivo observations35 and thereby provide predictive value for screening the neurotoxicity effects of test substances where the molecular targets and modes of action are unknown.1,3,36 Traditionally, neurite outgrowth assays involve the use of sparsely seeded cell cultures (for illustration see Fig. 1A), with the resource intensive task of identifying single neurons and measuring their outgrowths. The presented NFA combines the benefits of the neurite outgrowth assay with cell micropatterning to provide a spatially standardized analysis platform. A uniformly spaced hexagonal array of adhesion nodes is used to pattern neurons. Neurite outgrowths interconnect the cellular nodes and result in the development of a neuronal network (for illustration see Fig. 1B). Critically, the array system standardizes the neurite outgrowth length, thereby eliminating length measurements. The distance is also designed to satisfy standard neurite classification criteria which require a length equal to or greater than one or more cell body diameters.37–39 The use of analysis coordinates and a common outgrowth length both significantly streamline the identification and measurement effort for high-throughput neurotoxicology screening. | ||
| Fig. 1 Differentiated SH-SY5Y neurons cultivated on standard tissue culture substrates have an irregular distribution and elaborate complex and ‘chaotic’ neurite outgrowths (A). Cells were fixed and stained with Giemsa to aid visualization. Illustration of the network formation assay end point where hexagonally patterned neurons are interconnected by neurite outgrowths (B). | ||
Materials and methods
Thin film PDMS microcontact printing
In this study we have used a novel cell patterning technique based on thin film PDMS microcontact printing (µCP) on tissue culture substrates.34 PDMS (Sylgard® 184, Dow Corning) stamps were fabricated by moulding a microstructured SU-8 master with a degassed mixture of pre-polymer and curing agent (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, w/w) at 70 °C for 30 min. For inking the stamps a thin film deposit of liquid PDMS was prepared: an uncured PDMS pre-polymer and curing agent mixture (1:5, w/w) was dissolved in chloroform (1:10, w/w), and a 500 µL volume was applied to a glass substrate and spin-coated at 6000 rpm for 30 s. The chloroform evaporated to leave a uniform PDMS film. Inking the PDMS stamp with liquid PDMS was achieved by simple conformal contact of the stamp with the liquid PDMS film for ∼10 s. Excess liquid PDMS was removed from the stamp by printing on a sacrificial glass substrate followed by microcontact printing on the glass or tissue culture grade polystyrene substrates for ∼10 s. Thermal curing at 70 °C for 10 min was used to produce a stable thin film PDMS pattern with the perforations exposing areas of the underlying glass substrate. The PDMS micropatterning procedure is illustrated in Fig. 2.
:1, w/w) at 70 °C for 30 min. For inking the stamps a thin film deposit of liquid PDMS was prepared: an uncured PDMS pre-polymer and curing agent mixture (1:5, w/w) was dissolved in chloroform (1:10, w/w), and a 500 µL volume was applied to a glass substrate and spin-coated at 6000 rpm for 30 s. The chloroform evaporated to leave a uniform PDMS film. Inking the PDMS stamp with liquid PDMS was achieved by simple conformal contact of the stamp with the liquid PDMS film for ∼10 s. Excess liquid PDMS was removed from the stamp by printing on a sacrificial glass substrate followed by microcontact printing on the glass or tissue culture grade polystyrene substrates for ∼10 s. Thermal curing at 70 °C for 10 min was used to produce a stable thin film PDMS pattern with the perforations exposing areas of the underlying glass substrate. The PDMS micropatterning procedure is illustrated in Fig. 2.
 | ||
| Fig. 2 Thin film PDMS micropatterning. A PDMS stamp with a hexagonal array of recessed microstuctures is ‘inked’ with liquid PDMS from a spin-coated deposit by contact transfer (A). Microcontact printing is used to pattern the liquid PDMS on a culture substrate (B). Removal of the stamp followed by a thermal cure is used to produce a stable perforated PDMS micropattern (C). | ||
The surface free energy of the PDMS prints was evaluated by contact angle measurements of a 1 µL sessile water droplet. Angled SEM imaging (Quanta 200F, FEI) and white light interferometry (NewView 5000 Microscope, Zygo) were used to characterize the topology of the micropatterned thin film PDMS.
To demonstrate the widespread applicability of thin film PDMS printing various cell types were patterned on combinatorial arrays comprising adhesion islands with different dimensions and geometries. Neurotoxicity experiments used a hexagonal micropattern to provide a uniformly spaced array of 367 cell adhesion nodes. Each node had a diameter of 70 µm and was separated from neighbouring nodes by 100 µm. Narrow adhesion bridges between nodes were not included in the design. The absence of this feature allows unconstrained neurite outgrowth and also standardizes the neurite interconnection length by restricting the neurons to the adhesion nodes. The entire 20 × 20 mm pattern contained a 5 × 5 matrix of individual arrays.
Cell culture and imaging
Cell lines were purchased from DSMZ (Germany) and ECACC (UK), and all cell culture media and consumables were purchased from Sarstedt AG & Co. (Germany). The 7 human cell lines used in preliminary cell patterning studies were epithelial cells of colon origin (SW480, SW620, NCM460 and HT29), keratinocytes (HaCaT) and epithelial-like breast cancer cells (MCF-7 and BT474) as well as primary mouse hepatocytes and primary embryonic mouse neurons. All cells were cultured under standard conditions according to supplier recommendations. Typically, Dulbecco's modified Eagle medium (DMEM) supplemented with 10% (v/v) foetal bovine serum, 1% (v/v) Glutamax and 1% (v/v) penicillin and streptomycin was used. Cells were harvested using 0.25% (w/v) trypsin once 80% confluency was attained. Patterning involved seeding cells in a 1 mL suspension containing 2 × 105 cells and incubation for 12–24 h at 37 °C in a 5% CO2 atmosphere. The suspension was then removed, replaced with fresh media and incubated for up to a month, with 3–4 day periodic exchange of the media. In the neurotoxicity experiments, human SH-SY5Y neuroblastoma cells (DSMZ, Germany) were used. Cells were differentiated using 10 µM trans retinoic acid (Sigma) for 3 days and then harvested for seeding on the arrays. An inverted microscope (IX71, Olympus) or an environmental SEM (Quanta 200F, FEI) was used to image the cell patterns. The blue fluorescent nuclear dye 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI FluoroPure™ grade, Invitrogen) was used to ascertain the number of cells occupying individual nodes at 24 h after patterning. Cells were first fixed in 4% glutaraldehyde for 15 min, then washed twice for 5 min in 1× phosphate buffered saline (1× PBS), followed by stepwise alcohol dehydration. Samples were equilibrated in 1× PBS, immersed in deionized H2O with 30 nM DAPI for 1 min, followed by multiple 1× PBS rinses and finally dried in preparation for fluorescent microscopy.Neurotoxicity and cytotoxicity testing
To demonstrate the NFA the arrayed cells were exposed to acrylamide, a reference neurotoxic compound. Thin film PDMS patterns were prepared on glass substrates (1 × 26 × 26 mm) and transferred to Petri dishes (Ø 55 mm) for cell seeding. Following cell patterning a quality control procedure was undertaken. Only arrays with pattern occupancies in excess of 75% were used for subsequent neurotoxicity measurements. The differentiated SH-SY5Y neuron arrays were then cultured for 3 days in 7 mL of serum-containing media along with acrylamide at final concentrations ranging from 0–5 mM. Experiments were undertaken in triplicate, with array occupancy and neurite interconnection measurements taken daily. Array occupancy was defined as the percentage of the 367 cell adhesion nodes that contain one or more cells. The NFA involved a streamlined analysis approach, measuring the number of neurites connecting immediately neighbouring neuronal nodes and not connections to more distant nodes. Difficulties discerning multiple connections between the same neuronal node neighbours were also obviated by classifying these together as a single connection. To reduce the impact of sample–sample pattern occupancy variations values are converted to neurite interconnections per occupied neuronal node. To document the process cell arrays and neurite interconnections were imaged using an inverted microscope (IX71, Olympus).Cytotoxicity was measured using the CellTiter-Blue® Cell Viability Assay (Promega): a 100 µL volume of media containing 5 × 104 pre-differentiated SH-SY5Y cells was added per well of a 96 well plate. Cells were cultured in serum-containing media along with acrylamide at concentrations ranging from 0–500 mM for 24 h. Following exposure, a 20 µL volume of the CellTiter-Blue® resazurin reagent was added to the culture media and incubated for 4 h at 37 °C for conversion by viable cells into the fluorescent compound resorufin. Fluorimetry involved excitation at 540 nm and emission detection at 595 nm.
Pathway controls
Inhibitors of MAPK and PI-3K signaling pathways were used as positive controls for the NFA. Pathway control experiments involving the use of PD98059 (Sigma) to inhibit MEK1/2 signaling and LY29002 (Sigma) to inhibit PI-3K signaling were undertaken to evaluate the ability of the NFA to detect pathway specific mechanisms of neurite outgrowth inhibition. Pre-differentiated and patterned SH-SY5Y cells were exposed to the inhibitors at 5, 15.8 and 50 µM concentrations with network formation analysis at 24 h. To confirm pathway specific inhibition Western blot analysis was also undertaken: Each well of a 6 well plate was seeded with 5 × 105 pre-differentiated SH-SY5Y cells and exposed to the inhibitors at 5, 15.8 and 50 µM concentrations for 1 h, followed by harvesting and homogenization on ice in RIPA buffer (150 mM NaCl, 50 mM Tris–HCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 0.1% SDS, 1% NP-40 and 1% sodium deoxycholate, pH 7.4 with 0.5% (v/v) protease inhibitor cocktail (Sigma)). After centrifugation at 15000 g for 30 min at 4 °C the protein concentration was measured using the bicinchoninic acid (BCA) Protein Assay Kit (Pierce). Aliquots containing 50 µg of protein were separated on a 10% SDS polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Perkin-Elmer). The membrane was blocked in TBS-T (Tris-buffered saline with 0.1% (v/v) Tween-20) containing 5% (w/v) bovine serum albumin fraction V and incubated overnight with primary antibodies (rabbit anti-ERK1/2, rabbit anti-phospho-ERK1/2, rabbit anti-Akt, rabbit anti-phospho-Akt and rabbit anti-α-tubulin, all used at 1:1000). After washing in TBS-T, blots were incubated for 1 h with a secondary antibody (1:1000 anti-rabbit IgG) conjugated to horseradish peroxidase. All antibodies were obtained from Cell Signaling Technology, Inc. The antibody-labeled bands were detected by incubating the membrane with Western Lightning Chemiluminescence Reagent Plus (Perkin-Elmer), followed by imaging using a chemiluminescence documentation system (ChemiLux CSX-1400M, INTAS). The integrated density of the labeled bands was analyzed using ImageJ (NIH).
Network probability simulation
A simulation based on probability theory was developed to predict the impact of pattern occupancy on the probability of network formation. The simulation was undertaken using MATLAB®, and first involved defining the scale (in this case 367 adhesion nodes) and boundaries of the hexagonal array. The network comprised patterned neurons (i.e. occupied nodes) and interconnecting neurites which were encoded as numerical identities. In the initial condition, the array is completely occupied (i.e. all nodes contain neurons) and all possible connections between neighbouring nodes exist. A single node positioned within the array can share a maximum of 6 connections with neighbouring nodes (nodes at the array boundary can share a maximum of 3 or 4 connections). A uniform probability distribution function was then used to vacate a single node, and associated connections, followed by logging the number of remaining connections across the array. This process, with equal node selection probability, is iterated until the entire array is empty. To obtain results with statistical confidence the pattern depletion process (from 100% to 0%) was repetitively cycled 10000 times.
Results and discussion
Cell patterning
Digital hydrophilic–hydrophobic surfaces can be used for cell patterning.40 The novel thin film PDMS µCP technique used in this study was straightforward, reliably producing precision hydrophobic micropatterns on hydrophilic tissue culture substrates (Fig. 3A). The contact angle of the PDMS layer was 107.4° (SD ± 1.5°), consistent with the wetting properties of PDMS prepared by standard methods. The perforated PDMS film was ∼40 nm thick, only a small fraction of the height of an adherent cell such that the patterns are essentially planar in nature. Once the PDMS prints are prepared no further surface modifications were required. Cells were seeded in media containing serum and incubated overnight, followed by media exchange to reveal cellular patterns. Cells only grew on the exposed hydrophilic regions of the underlying glass or tissue culture grade polystyrene substrates. This provides identical physico- and biochemical surface adhesion conditions to standard tissue culture surfaces. Extracellular matrix proteins from the serum assemble on the exposed areas in a bioactive state to assist cell adhesion.41 In contrast, serum albumin becomes interfacially denaturated on hydrophobic surfaces,42,43 rendering them cell repellent. In this manner the hydrophobic PDMS-coated areas strongly resist cell adhesion, with pattern compliance maintained during long term (e.g. 2 weeks) cultivation. | ||
| Fig. 3 An array of square-shaped adhesion sites produced by thin film PDMS µCP (A). The method enables precision patterning for producing arrays of single SW480 epithelial cells (B), and even the geometric control of the adhesion footprint of a single SW480 epithelial cell (C). Reliable patterning over large areas was demonstrated with a reproduction of Da Vinci's Vitruvian Man using SW480 epithelial cells (D). | ||
To demonstrate the cell patterning capability we have prepared single cell arrays (Fig. 3B), sub-cellular resolution prints for controlling the geometry of the cellular adhesion footprint (Fig. 3C) and large area cellular displays (Fig. 3D). Thin film PDMS prints were successfully used for patterning a variety of cell types from diverse tissue origins. To date we have patterned the following human cell lines: MCF-7 and BT474 epithelial-like breast cancer cells, HaCaT keratinoctyes and SW480, SW620, NCM460 and HT29 colon epithelial cells (for illustration patterns of MCF-7 breast cancer and HT29 colon carcinoma cells are documented in Fig. S1†). Patterning primary cells is more challenging. Nevertheless we have used PDMS printing to pattern primary mouse hepatocytes on collagen-coated substrates and embryonic mouse neurons on poly-L-lysine-coated substrates. Thin film PDMS µCP is therefore widely applicable to many biological contexts and further lends itself to plasma-based PDMS–PDMS bonding for encapsulating cell patterns within microfluidic packages for the spatiotemporal manipulation of the (bio)chemical microenvironment. Importantly, thin film PDMS µCP is readily accessible to biologists and microengineers alike, and other imaginative uses of PDMS patterning for manipulating cell adhesion and behaviour can be anticipated.
SH-SY5Y cell patterning
For the neurotoxicity experiments the human SH-SY5Y neuroblastoma cell line was adopted to provide an authentic model with neurodevelopmental end points that recapitulate aspects of the human in vivo state. Differentiation with retinoic acid induces neurite outgrowth and the formation of functional synapses.44,45 In addition, retinoic acid differentiation leads to low levels of proliferation and promotes cell adhesion,46 both characteristics that favour cell patterning. Thin film PDMS printing was also successfully used for patterning the differentiated neuron-like SH-SY5Y cells. An angled SEM image of the hexagonally perforated PDMS film is shown in Fig. 4A along with an image of a patterned array of SH-SY5Y cells in Fig. 4B. The ability to pattern differentiated SH-SY5Y cells using thin film PDMS prints corroborates other reports of the cell repellent nature of untreated PDMS surfaces towards neural cells.40,47–50 | ||
| Fig. 4 Planar PDMS print imaged by angled SEM. The hexagonal array contains 70 µm diameter adhesion nodes separated from neighbouring nodes by 100 µm (A). Large area image of differentiated SH-SY5Y neurons patterned with an occupancy of 83% (B). Box and whiskers plot of the batch to batch (B1–4) pattern occupancy variation after 24 h of cultivation (C). Frequency distribution of the number of cells per adhesion node across an entire array (n = 3) (D). Cell number was inferred from DAPI-stained nuclei counts. | ||
The NFA relies on high pattern occupancy for neuronal interconnections to be formed between neighbouring adhesion nodes. The use of a 100 µm separation distance with the seeding of 2 × 105 differentiated SH-SY5Y cells was used to achieve high pattern occupancy and to enable adhesion nodes to be bridged by neurite interconnections, and not by cells. A single patterned chip contains 25 arrays, each with 367 adhesion nodes. The across chip, chip to chip and batch to batch pattern occupancy were reproducibly above 70% (see Fig. S2†). In comparison, the pattern occupancies obtained with other cell lines were even higher. For neurotoxicity experiments, only arrays with pattern occupancies higher than 75% were used. The mean pattern occupancy of arrays from four separate experiments ranged from 77.6% (SD ± 3.7%) to 84.4% (SD ± 2.1%). The pattern occupancy data are presented as a box and whiskers plot in Fig. 4C.
The adhesion nodes were 70 µm in diameter, sufficiently large to accommodate more than a single cell. In this way, all required cell signaling modalities can be retained. Following 24 h of patterned cultivation there were on average 7 cells per node. The combined frequency distribution data from three representative 367 node arrays are plotted in Fig. 4D. In summary, optimal conditions for reproducible SH-SY5Y cell patterning were established and used as a precondition for the NFA.
Neuronal network formation
The hexagonal array allows unconstrained neurite outgrowth and was ideally suited for observing the formation of interconnections between the patterned neurons. Following the initial day for cell patterning, the development of an SH-SY5Y neuronal network over a further 3 day period is shown in Fig. 5A–C. Larger images are available in Fig. S3–S5†. During the period of network formation the number of neurite connections across the array increased from 99.8 (SD ± 3.3) to 303.3 (SD ± 17.6). The data are plotted in Fig. 5D as the number of connections per node. During network formation the pattern occupancy also increased from 84.9 (SD ± 0.9%) to 94.1 (SD ± 1.4%). The ability of the differentiated SH-SY5Y cells to form a network of cellular nodes interconnected by neurite outgrowths demonstrates that the PDMS surface is permissive to neurite outgrowth, but not cell adhesion. This indicates that the processes of cell adhesion and neurite outgrowth involve different cell–material interactions. However, much like in vivo neuronal migration51 the neuronal array also has the potential for plasticity. Evidence from time-lapse microscopy demonstrates that neurite outgrowths can be used to translocate neurons to vacant nodes (a link to a time-lapse video can be found in the ESI†). This corroborates another report of array-based neuronal migration events.32 | ||
| Fig. 5 Microscopy time course panel of neuronal network development at 24 (A), 48 (B) and 72 (C) h. Connecting neurites are indicated with black arrows. To better discern the neurite connections larger images are available in Fig. S3–S5†. During the 3 day period, the mean number of connections per occupied node increased from 0.32 to 0.88 (D). | ||
Neurotoxicity testing
In the standard neurite outgrowth assay, cells are isolated to aid the identification of outgrowths by being sparsely seeded. In the NFA the neuronal arrays contain adhesion nodes separated by 100 µm. This condition standardizes the neurite interconnection length and satisfies the neurite classification criteria: a neurite outgrowth is typically defined as a process with a length equal to or greater than one or more times the cell body diameter.37–39 Moreover, neurite interconnection could serve as a higher indicator of functional potential than outgrowth alone. The principle advantages of registering neurons within a hexagonal array are the ease of locating neurons and their connections and eliminating the intensive task of measuring the neurite outgrowth length. Both features enable streamlined analysis for rapid and sensitive screening.To demonstrate the advantages of the neuronal array a dose–response experiment using the neurotoxin acrylamide was undertaken with differentiated SH-SY5Y neurons at passage 12. Dose dependency results following 24 h of exposure to acrylamide are compared with data from the CellTiter-Blue® Cell Viability Assay in Fig. 6. The NFA produced a sigmoidal dose–response curve over two orders of magnitude and was almost 10-fold more sensitive than the cytotoxicity test, revealing specific neurotoxicity effects whereas the viability assay only captures gross cytotoxic effects. The viability assay gave a 50% inhibition concentration (IC50) of 5.1 mM (95% CI 4.2–6.1). In contrast, the concentration that caused a 20% reduction in network formation (NI20) relative to controls was 0.26 mM (95% CI 0.12–0.46). These values are equivalent to the 20% SH-SY5Y neurite degeneration (ND20) acrylamide concentrations (0.25 and 0.21 mM) reported by Nordin-Andersson and colleagues.52,53 An additional NFA experiment involving SH-SY5Y cells at passage 10 gave an NI20 value of 0.28 mM (95% CI 0.17–0.38, Fig. S6†). This reproducibility and the narrow 95% confidence intervals demonstrate the robust character of the assay.
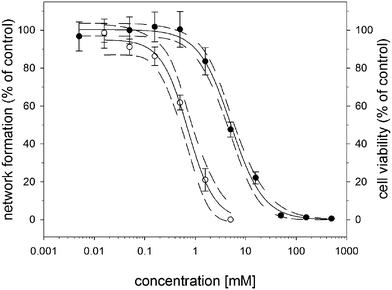 | ||
| Fig. 6 Sigmoidal acrylamide dose–response curves from the NFA (open circles) and the CellTiter-Blue® Cell Viability Assay (black circles). Data were obtained following 24 h of exposure to acrylamide using cells at passage 12. Data points with standard deviation are plotted along with non-linear regression curves for the mean (continuous line) and the 95% confidence intervals (dashed lines). | ||
The simplicity of the NFA analytical read-out enables rapid data acquisition, requiring only 3 h to manually undertake the dose–response analysis (367 nodes, with 7 concentrations in triplicate: 7707 nodes). In contrast, the manual assessment of neurite length is a labour intensive task requiring approximately 10 h for the analysis of just 200 cells.3 The positioning of neuronal cells and their neurite interconnections at pre-defined coordinates is also highly desirable for automated image capture and processing.
The NFA does not require fixation and staining, such that the analytical end points are not terminal. This feature allows kinetic information to be captured by periodically or continuously monitoring the neuronal network. The neuronal arrays were monitored daily for a 3 day period following exposure to acrylamide. Network formation continued on arrays exposed to ≤0.5 mM acrylamide, with neuronal networks recovering to control levels by day 3. These results demonstrate that sensitive neurotoxicity testing with the current protocol requires early stage analysis. Acrylamide concentrations of 1.58 and 5 mM were cytotoxic, with pattern occupancy depletion observed by day 2. Pattern occupancy can therefore be used as a surrogate cytotoxicity measure. In Fig. S7†, acrylamide concentration is plotted against pattern occupancy and was used to determine an IC50 of 3.1 mM, similar to the 5.1 mM value obtained by the CellTiter-Blue® Cell Viability Assay. In this way the NFA enables neurotoxic effects to be discerned from cytotoxic effects using the very same cells.
Pathway controls
Neurite outgrowth is known to depend on intact signaling via MAPK and Akt/PKB.54 Therefore, we used inhibitors of both pathways to assess whether they block neurite formation. Using a concentration range between 5 and 50 µM, PD98059 (an inhibitor of MAPK signaling) caused a concentration dependent decrease in ERK1/2 phosphorylation and LY29002 (an inhibitor of the PI-3K pathway) also caused a concentration dependent decrease in Akt phosphorylation (Fig. 7A–D). In the same concentration range neurite outgrowth was dose dependently inhibited by PD98059 and LY29002 (Fig. 7E and F). In contrast, cytotoxicity as evidenced by the pattern occupancy did not occur during the exposure period. The results demonstrate that the NFA measurement end point is pathway dependent, requiring intact MAPK as well as Akt signaling. In addition, the results demonstrate that the NFA faithfully reports MAPK and Akt pathway inhibition. Therefore, PD98059 (50 µM) and LY29002 (15.8 µM) represent adequate pathway controls for the NFA. | ||
| Fig. 7 Western blot image of the phospho-ERK1/2 dose–response to PD98059 along with total ERK1/2 and α-tubulin controls (A). Western blot image of the phospho-Akt dose–response to LY29002 along with total Akt and α-tubulin controls (B). Densitometry analysis of the phospho-ERK1/2 dose–response to PD98059 (C) and the phospho-Akt dose–response to LY29002 (D). NFA results (NF, white bars) of the dose–response to PD98059 (E) and LY29002 (F). Occupancy, a cytotoxicity indicator, is also presented (black bars). | ||
Network probability simulation
The presence of unoccupied adhesion nodes reduces the probability of neurite outgrowths to connect with neighbouring neuronal nodes. This has important implications when comparing data between arrays with disparate pattern occupancy values. To account for pattern occupancy variations in the experiments, neurite connection numbers were reported in terms of connections per node. This simple approach predicts a linear relationship between pattern occupancy and network formation probability. However, this approach neglects proximity effects which alter the probability of neurons forming neurite connections with directly neighbouring neurons. A simulation was developed to obtain a statistical appreciation of this phenomenon and to serve as a tool to aid data normalization. To evaluate all possible cell pattern distributions from 0 to 100% occupancy is impractical due to its combinatorial nature, and becomes an unfeasibly large calculation task for even small arrays. To solve this problem a simulation based on probability theory was developed to predict the impact of pattern occupancy on network formation probability.Results from a 10000 cycle simulation are presented in Fig. 8. The simulation was robust, producing error levels below 1% of the standard deviation in a repeat simulation. Plotting pattern occupancy against network formation probability produces an exponential profile. To validate the simulation, serial cell dilutions were patterned on the microarrays. Following a 24 h period for neurite outgrowth the number of connections was measured. The experimental data are also plotted in Fig. 8 as the number of connections per node (black circles) and support the prediction generated by the simulation. Like the simulation this indicates that the arrays are randomly occupied by neurons. Critically, network formation probability was especially sensitive to variations at high levels of pattern occupancy. For example, an array with 100% occupancy has more than twice as many connections than an array with 70% occupancy. It is therefore recommended that the simulation be used to aid data normalization in circumstances where there is a high variation in chip to chip pattern occupancy.
 | ||
| Fig. 8 Simulation of occupancy versus network formation probability with experimental results overlaid for verification. The 10000 cycle simulation mean (dotted line) is plotted with the 95% confidence intervals (continuous lines). The connection per occupied node prediction (dashed line) is plotted along with experimental data points (black circles) taken at 24 h. The array is not fully networked after 24 h, requiring data normalization based on the values obtained with 85% pattern occupancy. | ||
Assay development perspectives
Simple steps can be taken to develop the NFA analytical platform further. Growth supplements, differentiation cues and passage history55 should be evaluated as should other cell models. In particular, the assay should be applied to primary neurons which retain many in vivo morphological, biochemical and electrophysiological characteristics. Alternatively, assay standardization could benefit from using non-transformed and self-renewing human neural stem cells.3 For larger scale deployment it will be necessary to conform to assay standards defined by the neurotoxicology community and guidelines established by the regulatory authorities. To aid such wider uptake and provide further gains in throughout the cell patterning technique should be adapted to be compatible with the standard microtitre plate format.56 In addition to neurotoxic hazard screening, the NFA principle can be applied to drug development (e.g. screening for substances with neuroregenerative potency) and also for applications in neurodevelopmental biology.Conclusions
This paper introduces the network formation assay: a spatially standardized analytical display for high-throughput neurotoxicity screening. Thin film PDMS µCP, a simple, reliable and widely applicable cell patterning technique, was used to register human neurons within a hexagonal array. This approach standardizes the neurite outgrowth length and assigns assay coordinates to greatly streamline the analysis process. The simple assay enabled the reproducible, rapid and sensitive assessment of neurite outgrowth inhibition by the neurotoxic compound acrylamide and specific inhibitors of the MAPK and PI-3K signaling pathways. Straightforward developments are required for the widespread adoption of the assay to progress the neurotoxic hazard assessment effort as well as impact the field of neurodevelopmental biology.Acknowledgements
The authors are grateful to Uli Marggraf for SU-8 master fabrication, Norman Ahlman for white light interferometry, Maria Becker for SEM analysis, and Sasidhar Maddula and Cristina Cadenas for providing cell types. Financial support from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. HEALTH-F5-2008-201619 (ESNATS), the Deutsche Forschungsgemeinschaft (WE 3737/3-1), the Ministerium für Innovation, Wissenschaft, Forschung und Technologie des Landes Nordrhein-Westfalen and from the Bundesministerium für Bildung und Forschung is also gratefully acknowledged.References
- P. Lein, E. Silbergeld, P. Locks and A. M. Goldberg, Environ. Toxicol. Pharmacol., 2005, 19, 735–744 CrossRef CAS.
- S. Coecke, A. M. Goldberg, S. Allen, L. Bużańska, G. Calamandrei, K. Crofton, L. Hareng, T. Hartung, H. Knaut, P. Honegger, M. Jacobs, P. Lein, A. Li, W. Mundy, D. Owen, S. Schneider, E. Silbergeld, T. Reum, T. Trnovec, F. Monnet-Tschudi and A. Bal-Price, Environ. Health Perspect., 2007, 115(6), 924–931.
- N. M. Radio and W. R. Mundy, NeuroToxicology, 2008, 29, 361–375 CrossRef CAS.
- M. Leist, S. Kadereit and S. Schildknecht, ALTEX, 2008, 25(1), 17–24 Search PubMed.
- L. Vutskits, E. Gascon, E. Tassonyi and J. Z. Kiss, Toxicol. Sci., 2006, 91(2), 540–549 CrossRef CAS.
- D. Liu, H. B. McIlvain, M. Fennell, J. Dunlop, A. Wood, M. M. Zaleska, E. I. Graziani and K. Pong, J. Neurosci. Methods, 2007, 163, 310–320 CrossRef CAS.
- N. M. Radio, J. M. Breier, T. J. Shafer and W. R. Mundy, Toxicol. Sci., 2008, 105(1), 106–118 CrossRef CAS.
- P. Ramm, Y. Alexandrov, A. Cholewinski, Y. Cybuch, R. Nadon and B. J. Soltys, J. Biomol. Screening, 2003, 8, 7–18 CrossRef.
- H. Francisco, B. B. Yellen, D. S. Halverson, G. Friedman and G. Gallo, Biomaterials, 2007, 28(23), 3398–3407 CrossRef CAS.
- A. M. Taylor, S. W. Rhee, C. H. Tu, D. H. Cribbs, C. W. Cotman and N. L. Jeon, Langmuir, 2003, 19(5), 1551–1556 CrossRef CAS.
- A. M. Taylor, M. Blurton-Jones, S. W. Rhee, D. H. Cribbs, C. W. Cotman and N. L. Jeon, Nat. Methods, 2005, 2(8), 599–605 CrossRef CAS.
- Y.-T. Kim, K. Karthikeyan, S. Chirvi and D. P. Davé, Lab Chip, 2009, 9(17), 2576–2581 RSC.
- C. A. Thomas, P. A. Springer, G. E. Loeb, Y. Berwald-Netter and L. M. Okun, Exp. Cell Res., 1972, 74, 61–66 CrossRef.
- E. W. Keefer, A. Gramowski, D. A. Stenger, J. J. Pancrazio and G. W. Gross, Biosens. Bioelectron., 2001, 16, 513–525 CrossRef CAS.
- A. Tscherter, M. O. Heuschkel, P. Renaud and J. Streit, Eur. J. Neurosci., 2001, 14(2), 179–190 CrossRef CAS.
- E. van Vliet, L. Stoppini, M. Balestrino, C. Eskes, C. Griesinger, T. Sobanski, M. Whelan, T. Hartung and S. Coecke, NeuroToxicology, 2007, 28, 1136–1146 CrossRef CAS.
- J. Ban, P. Bonifazi, G. Pinato, F. D. Broccard, L. Studer, V. Torre and M. E. Ruaro, Stem Cells, 2007, 25(3), 738–749 Search PubMed.
- I. Suzuki, Y. Sugio, Y. Jimbo and K. Yasuda, Lab Chip, 2005, 5(3), 241–247 RSC.
- S. K. Ravula, M. A. McClain, M. S. Wang, J. D. Glass and A. B. Frazier, Lab Chip, 2006, 6(12), 1530–1536 RSC.
- F. Morin, N. Nishimura, L. Griscom, B. Le Pioufle, H. Fujita, Y. Takamura and E. Tamiya, Biosens. Bioelectron., 2006, 21(7), 1093–1100 CrossRef CAS.
- M. Mrksich, L. E. Dike, J. Tien, D. E. Ingber and G. M. Whitesides, Exp. Cell Res., 1997, 235, 305–313 CrossRef CAS.
- R. S. Kane, S. Takayama, E. Ostuni, D. E. Ingber and G. M. Whitesides, Biomaterials, 1999, 20, 2363–2376 CrossRef.
- A. Folch and M. Toner, Annu. Rev. Biomed. Eng., 2000, 2, 227–256 CrossRef CAS.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 CrossRef CAS.
- E. V. Romanova, K. A. Fosser, S. S. Rubakhin, R. G. Nuzzo and J. V. Sweedler, FASEB J., 2004, 18(11), 1267–1269 CAS.
- Y. Mourzina, D. Kaliaguine, P. Schulte and A. Offenhäusser, Anal. Chim. Acta, 2006, 575, 281–289 CrossRef CAS.
- F. Patolsky, B. P. Timko, G. Yu, Y. Fang, A. B. Greytak, G. Zheng and C. M. Lieber, Science, 2006, 313, 1100–1104 CrossRef CAS.
- D. Kleinfeld, K. H. Kahler and P. E. Hockberger, J. Neurosci., 1988, 8(11), 4098–4120 CAS.
- K. E. Healy, B. Lom and P. E. Hockberger, Biotechnol. Bioeng., 1994, 43, 792–800 CrossRef CAS.
- J. M. Corey, B. C. Wheeler and G. J. Brewer, IEEE Trans. Biomed. Eng., 1996, 43(9), 944–955 CrossRef CAS.
- Y. Nam, D. W. Branch and B. C. Wheeler, Biosens. Bioelectron., 2006, 22, 589–597 CrossRef CAS.
- A. Ruiz, L. Bużańska, D. Gilliland, H. Rauscher, L. Sirghi, T. Sobanski, M. Zychowicz, L. Ceriotti, F. Bretagnol, S. Coecke, P. Colpo and F. Rossi, Biomaterials, 2008, 29, 4766–4774 CrossRef CAS.
- P. Shi, K. Shen and L. C. Kam, Dev. Neurobiol., 2007, 67, 1765–1776 CrossRef CAS.
- J.-P. Frimat, M. M. Mariani, R. Kettler, P. Jacob, J. Franzke and J. West, Proceedings of Nanotech2008, Montreux, Switzerland, 2008 Search PubMed.
- A. Forsby and B. Blaauboer, Hum. Exp. Toxicol., 2007, 26, 333–338 CrossRef CAS.
- P. Lein, P. Locke and A. Goldberg, Environ. Health Perspect., 2007, 115(5), 764–768.
- T. Morooka and E. Nishida, J. Biol. Chem., 1998, 273(38), 24285–24288 CrossRef CAS.
- L. Adlerz, M. Beckman, S. Holback, R. Tehranian, V. C. Toro and K. Iverfeldt, Mol. Brain Res., 2003, 119, 62–72 CrossRef CAS.
- K. P. Das, T. M. Freudenrich and W. R. Mundy, Neurotoxicol. Teratol., 2004, 26, 397–406 CrossRef CAS.
- J.-P. Frimat, H. Menne, A. Michels, S. Kittel, R. Kettler, S. Borgmann, J. Franzke and J. West, Anal. Bioanal. Chem., 2009, 395(3), 601–609 CrossRef CAS.
- P. A. Underwood, J. G. Steele and B. A. Dalton, J. Cell Sci., 1993, 104(3), 793–803 CAS.
- J. Kim and G. A. Somorjai, J. Am. Chem. Soc., 2003, 125, 3150–3158 CrossRef CAS.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2005, 127, 8168–8173 CrossRef CAS.
- A. Adem, M. E. K. Mattsson, A. Nordberg and S. Påhlman, Dev. Brain Res., 1987, 33, 235–242 CrossRef CAS.
- S. Påhlman, S. Mamaeva, G. Meyerson, M. E. Mattsson, C. Bjelfman, E. Örtoft and U. Hammerling, Acta Physiol. Scand. Suppl., 1990, 592, 25–37 Search PubMed.
- S. Påhlman, A.-I. Ruusala, L. Abrahamsson, M. E. K. Mattsson and T. Esscher, Cell Differ., 1984, 14, 135–144 CrossRef CAS.
- M. N. De Silva, R. Desai and D. J. Odde, Biomed. Microdevices, 2004, 6, 219–222 CrossRef CAS.
- M. N. De Silva, J. Paulsen, M. J. Renn and D. J. Odde, Biotechnol. Bioeng., 2006, 93, 919–927 CrossRef.
- D. R. Reyes, E. M. Perruccio, S. P. Becerra, L. E. Locascio and M. Gaitan, Langmuir, 2004, 20(20), 8805–8811 CrossRef CAS.
- L. J. Millet, M. E. Stewart, J. V. Sweedler, R. G. Nuzzo and M. U. Gillette, Lab Chip, 2007, 7(8), 987–994 RSC.
- A. R. Kriegstein and S. C. Noctor, Trends Neurosci., 2004, 27(7), 392–399 CrossRef CAS.
- M. Nordin-Andersson, A. Forsby, N. Heldring, J. DeJongh, P. Kjellstrand and E. Walum, Toxicol. in Vitro, 1998, 12, 557–560 CrossRef CAS.
- M. Nordin-Andersson, E. Walum, P. Kjellstrand and A. Forsby, Cell Biol. Toxicol., 2003, 19(1), 43–51 CrossRef CAS.
- T. Nishimura, T. Ishima, M. Iyo and K. Hashimoto, PLoS One, 2008, 3(7), e2558 CrossRef.
- L. Bużańska, A. Ruiz, M. Zychowicz, H. Rauscher, L. Ceriotti, F. Rossi, P. Colpo, K. Domańska-Janik and S. Coecke, Acta Neurobiol. Exp., 2009, 69(1), 24–36 Search PubMed.
- A. Azioune, M. Storch, M. Bornens, M. Théry and M. Piel, Lab Chip, 2009, 9(11), 1640–1642 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: MCF-7 and HT29 cell patterns (Fig. S1), pattern occupancy variations (Fig. S2), neuronal network development (Fig. S3–5), array-based neuronal migration video, NFA reproducibility (Fig. S6), pattern occupancy can be used to measure cytotoxicity (Fig. S7). See DOI: 10.1039/b922193j |
| ‡ These authors contributed equally to the research. |
| § These senior authors contributed equally to the research. |
| This journal is © The Royal Society of Chemistry 2010 |