 Open Access Article
Open Access ArticleA microfluidic chip-compatible bioassay based on single-molecule detection with high sensitivity and multiplexing†
Randall E.
Burton‡
*,
Eric J.
White‡
,
Ted R.
Foss
,
Kevin M.
Phillips
,
Robert H.
Meltzer
,
Nanor
Kojanian
,
Lisa W.
Kwok
,
Alex
Lim
,
Nancy L.
Pellerin
,
Natalia V.
Mamaeva
and
Rudolf
Gilmanshin§
U. S. Genomics, Inc., 12 Gill St., Suite 4700, Woburn, MA, USA 01801. E-mail: rburton@usgenomics.com
First published on 14th January 2010
Abstract
Many applications in pharmaceutical development, clinical diagnostics, and biological research demand rapid detection of multiple analytes (multiplexed detection) in a minimal volume. This need has led to the development of several novel array-based sensors. The most successful of these so far have been suspension arrays based on polystyrene beads. However, the 5 µm beads used for these assays are incompatible with most microfluidic chip technologies, mostly due to clogging problems. The challenge, then, is to design a detection particle that has high information content (for multiplexed detection), is compatible with miniaturization, and can be manufactured easily at low cost. DNA is a solid molecular wire that is easily produced and manipulated, which makes it a useful material for nanoparticles. DNA molecules are very information-rich, readily deformable, and easily propagated. We exploit these attributes in a suspension array sensor built from specialized recombinant DNA, Digital DNA, that carries both specific analyte-recognition units, and a geometrically encoded identification pattern. Here we show that this sensor combines high multiplexing with high sensitivity, is biocompatible, and has sufficiently small particle size to be used within microfluidic chips that are only 1 µm deep. We expect this technology will be the foundation of a broadly applicable technique to identify and quantitate proteins, nucleic acids, viruses, and toxins simultaneously in a minimal volume.
Introduction
Existing suspension arrays with high degrees of multiplexing (106–107) use electronic or graphical tag encoding,1 large particles (10–103 µm),1–9 and require large volumes (0.1–1 ml). Analysis of 10–100 µl samples is dominated by 1–10 µm bead arrays.10–13 Intensity-based encoding with such beads can theoretically achieve multiplexing up to 104, but practical applications have been limited to 102, using commercially available flow cytometers that combine high sensitivity of the measurements with high throughput.10,11 Most suggested suspension array schemes have not been implemented due to the difficulty of combining high multiplexing, small particle size, and availability of a high-throughput detection system.Here we introduce a novel suspension array technology using a specialized DNA molecule—Digital DNA—as a scaffold that carries both an identifier and receptors. Digital DNA molecules are identified by a graphical encoding scheme that permits a high degree of multiplexing and processed by a single-molecule reader previously used for DNA mapping (Fig. 1).14–16 Our current sample size is ∼20 µl, primarily due to the need for manual pipetting and cleanup on a macro-scale. The microfluidic analyzer processes a minute amount of liquid (∼5 nl for a 15 min run), potentially enabling assays on extraordinarily small samples, such as laser-capture tissue dissections or even single cells.
 | ||
| Fig. 1 Suspension array based on Digital DNA. (a) Digital DNA molecules are built of two types of blocks: 1 and 0, generating many distinct bar codes. Identification fluorophores (green stars) and antibodies are attached to block 1. (b) Analytes (circles) bind to the DNA-bound antibodies and serve as bridges for the red-labeled secondary antibodies (red stars). (c) DNA is then stretched within the microfluidic device and interrogated with light beams I–IV. (d) Representative signals of a single capture unit, based on Digital DNA 111111111111. Traces 1 and 4 are produced by the DNA backbone. Traces 2 and 3 are generated by fluorophores of the identification tags and secondary antibodies, respectively. | ||
Materials and methods
Digital DNA amplification and purification
Digital DNA was prepared using standard molecular biology techniques and propagated in a standard cloning strain of Escherichia coli (GeneHogs, Invitrogen). DNA was extracted in supercoiled form using a Qiagen Maxi-Prep kit, and then linearized by digestion with NheI and XhoI restriction enzymes (New England Biolabs), summarized in the Results and discussion section. Details of Digital DNA construction and preparation can be found in the ESI†.Conjugation of antibodies with cross-linker and fluorophores
Antibody stocks (4–10 mg ml−1) were transferred into buffer #1 (0.1 M NaHxPO4, 0.15 M NaCl, 10 mM EDTA, pH 7.2) using a Sephadex G50 spin column (GE Healthcare, Piscataway NJ). To produce the antibody derivative for cross-linking with bisPNA-hybridized DNA, we added a 4-fold molar excess of SATA (N-succinimidyl S-acetylthioacetate, Pierce) cross-linker from a 5 mM stock in DMSO. To produce fluorescently labeled secondary antibodies, a 10-fold molar excess of Alexa 647 SE was added from a 5 mg ml−1 stock in DMSO. In both cases, the reaction was allowed to proceed for 1 h at room temperature. Then the excess cross-linker or fluorescent dye was removed with Sephadex G50 spin column equilibrated with buffer #1 containing 1 mM NaN3.Conjugation control experiments required antibody modified with both SATA and Alexa 647. In this case, antibody was transferred into buffer #1 as described above, allowed to react for 30 min with a 4-fold molar excess of SATA, and then a 10-fold excess of Alexa 647 SE was added. The reaction was allowed to continue for another 30 min, and then the excess cross-linker and dye were removed from the antibody with a Sephadex G50 column as described above.
Assembly of capture units
Linear Digital DNA molecules were excised from circular plasmids by restriction endonuclease digestion. In a typical reaction, 40 µg of plasmid DNA were incubated with 50 units of NheI and 200 units of XbaI in 1× NEB buffer #2 containing 0.1 mg ml−1 BSA (New England Biolabs). The reaction continued for 1 h at 37 °C; the completion of the reaction was verified by agarose gel electrophoresis. The enzymes were heat-inactivated at 65 °C for 20 min. DNA was precipitated with 2.5 volumes of ethanol and 1/10 volume of 3 M sodium acetate (pH 5.5) and then resuspended in buffer H (10 mM NaHxPO4, 1 mM EDTA, pH 8.0).For hybridization, DNA was combined with bisPNA in buffer H at final concentrations of 32 µg ml−1 and 2 µM, respectively. This mixture was incubated for 1 h at 65 °C, placed on ice, and combined with an equal volume of 0.5 mM LC-SPDP (succinimidyl 6-[3′-(2-pyridyldithio)-propionamido] hexanoate, Pierce) in buffer #1. The reaction continued overnight at room temperature. Excess bisPNA and LC-SPDP were removed by 4 cycles of 10-fold concentration with a Microcon YM-30 centrifuge filter unit (Millipore, Billerica, MA) and addition of 10 mM NaHxPO4 (pH 7.2) buffer with 15 mM NaCl and 10 mM EDTA to the original volume.
Antibodies were conjugated to the DNA by combining 32 µg ml−1 SPDP-modified DNA with 2 µM SATA-modified antibody in buffer #1. This mixture was incubated at room temperature overnight, then excess antibody was removed from the DNA–antibody conjugate by dialysis on 0.1 micron pore size filter disks (Millipore, catalog #VCWP 025 00) against 10 mM NaHxPO4 (pH 7.2) buffer with 50 mM NaCl and 1 mM EDTA for 4–8 h, changing disk and buffer once.
Reagents for assay
Botulinum toxoid A (BT) antigen, along with antibodies against botulinum toxin A and bacteriophage MS2 were purchased from the DoD-CRP as maintained under the Joint Program Executive Offices for Chemical and Biological Defense, Chemical Biological Medical Systems, and Medical Identification and Treatment Systems—Critical Reagents Program (JPEOCBD–CBMS–MITS–CRP). Follicle-stimulating hormone (FSH) and the matched pair of monoclonal anti-FSH antibodies were purchased from Biospacific (Emeryville, CA). Bacteriophage MS2 was purchased from ATCC.Microfluidic chip
The fused silica chip used in this study is similar to the microfluidic device described earlier.17 It also employs a four-port design with two-dimensional hydrodynamic flow focusing. However, its geometry was modified to be compatible with different sizes of Digital DNA. It is 1 µm deep with a 5 µm wide interrogation channel. The taper shape provides W(x) ≈ 1/(ax + b)2 profile (W is the taper width, x is the coordinate along the flow direction, a = 0.010971, and b = 0.063246); the taper length is 35 µm. The width of the channel before the taper is 250 µm. The fluid resistance for the DNA injection path and the sheathing buffer channels were designed to obtain a focusing ratio of about 30 (the width of the sample flow is 1/30th of the width of the interrogation channel). The chips were manufactured by Micralyne Inc. (Edmonton, Canada).Sandwich immunoassay—simple procedure
Varying concentrations of target protein were combined with 5 µg ml−1 CU in 10 mM NaHxPO4 (pH 7.2) buffer with 150 mM NaCl and 10 mM EDTA at total volume of 20 µl. This reaction mixture was incubated for 2 h at room temperature, then signal unit (Alexa 647-labeled antibody) was added to a final concentration of 50 nM, and the reaction proceeded for another 2 h at room temperature. The products were combined with an equal volume of 10 mM NaHxPO4 (pH 7.2) buffer with 150 mM NaCl, 1 mM EDTA, and 0.25 mg ml−1 of BSA, then dialyzed for 4 h against 10 mM NaHxPO4 (pH 7.2), 50 mM NaCl, 1 mM EDTA on a regenerated cellulose membrane disk (0.1 micron pore cut-off, Millipore).18 After dialysis, the DNA concentration was determined by PicoGreen assay (Invitrogen), and the sample was intercalated with POPO-1 (cat #P-3580, Invitrogen) at a 5 : 1 base pair-to-intercalator ratio and diluted to a final DNA concentration of 1 µg ml−1.Sandwich immunoassay—mini-reactor cleanup
We have improved the cleanup of free secondary antibodies by replacing the drop-dialysis step with a membrane-based mini-reactor, which is described in more detail elsewhere.19 The reaction occurs on the surface of a regenerated cellulose membrane (YM-30, Millipore), with simultaneous flows applied through the membrane (normal flow) and across the surface (tangential flow). By applying a normal flow of 50 µl min−1 with a tangential flow of 450 µl min−1, we have been able to retain 80% of the DNA-based reagents with nearly complete cleanup of the secondary antibodies.Sandwich immunoassay—on-chip cleanup
We have adapted the “DNA prism” as previously described by Huang et al. as a method for the effective separation of DNA from non-bound signal units.20 A uniform electric field of 80 V cm−1 is generated within a field of micron-scale posts organized in a hexagonal close-packed configuration. This electric field oscillates in orientation, and thus drives DNA molecules along either angled (30°, 450 ms dwell time) or horizontal (−90°, 260 ms dwell time) orientations. Through these oscillations, long fragments of DNA remain trapped along the angled orientation due to “hooking” interactions with the posts, whereas detection antibodies, which are too small to interact with the posts, move along with the time-averaged field (−15°). For combined fractionation and direct linear analysis of long DNA samples, the DNA prism has been integrated in a lab-on-chip device that also includes sample concentration functions. The details of this device will be presented in a later publication. These chips were produced by multi-mask etching procedures using wet and reactive ion techniques in fused silica (Micralyne). Automated operation of the lab-on-chip device was accomplished using in-house developed hardware and control software.Single-molecule measurements
Intercalated CUs with bound antigens and secondary antibodies were directly introduced into a microfluidic chip, where they were stretched and conveyed to the detection zone for single-molecule mapping. Measurements were performed on a customized detection platform as described previously14,15 at a linear flow rate of ∼20 µm ms−1, and data were recorded at 30 kHz with avalanche photodiodes (SPCM-AQR-12-FPC, PerkinElmer). Differently from the published work, one more detection channel was added to detect third color fluorophores with single-molecule sensitivity. The color scheme used in this work has also been changed. DNA backbone was stained with POPO-1 intercalator (Invitrogen, Carlsbad CA), which was excited with blue laser light in two spots to determine the molecule velocity. Emissions of TMR (bisPNA tag) and Alexa 647 (secondary antibody) fluorophores were excited by green and red laser light, respectively. The new optical components required to implement this new arrangement and to optimize detection of the new red fluorphore are presented in Table 1.| Optical componenta | Detection channel | |||
|---|---|---|---|---|
| Blueb | Green | Red | ||
| a Dichroic mirrors are not included in the table. The first dichroic mirror in the system is Z442/532/638rpc, which directs all excitation laser beams into the system and lets the fluorescence out. The second dichroic mirror 515dcxr reflects the POPO-1 emission from the blue spots to the corresponding detectors. The third dichroic mirror 625dcxr reflects the TMR emission from the green spot to the corresponding detector. All dichroic mirrors are products of Chroma Technologies (Rockingham, VT). b Blue excitation beam of single laser is separated into two spots of focused light to excite the fluorescence of DNA backbone. | ||||
| Laser | Wavelength/nm | 445 | 532 | 635 |
| Model | 56RCS/S2770 | Compass 215M-20 | Cube 635-25 | |
| Manufacturer | Melles Griot | Coherent | Coherent | |
| Bandpass excitation filter | Part number | FF01-438/24-25 | FF01-632/22-25 | |
| Manufacturer | Semrock | Semrock | ||
| Excitation power per spot/mW | 0.2–0.5 | 2–5 | 1–3 | |
| Bandpass emission filter | Part number | HQ 480/40 | HQ 580/40 | HQ 680/40 |
| Manufacturer | Chroma | Chroma | Chroma | |
Data analysis
The single-molecule data were analyzed using an in-house software package, GeneEngineer. It located DNA events in the data stream by identifying correlated signals between the two blue laser spots, measured the velocity, average intensity and length of each molecule, and associated the appropriate green and red tag signals with each event, as described previously15,16 (Fig. 1d). Only those DNA molecules with lengths exceeding 80% of their expected contour lengths (0.34 nm per bp) were selected for further analysis. The extracted molecular traces were normalized to their lengths, aligned by their centers, and averaged to obtain unoriented maps. Therefore in all figures, the signal intensity is plotted as a function of the distance from the center of DNA molecule.Results and discussion
Construction of Digital DNA
Digital DNA molecules are built with standard cloning techniques and mass-produced in bacteria. The suspension array is created by combining several DNA species, each with a specific identification pattern and associated receptors. We have designed two 10 kb DNA blocks 0 and 1; block 1 has 6 closely spaced sites that each bind a fluorescently labeled bisPNA21,22 tag. As this spacing is well below the optical resolution of our DNA reader, the signals combine to generate a single, strong identification signal, and block 0 lacks these sites. Many different identification patterns can be produced by combining these blocks (Fig. 1a). Antibodies are then conjugated to the amino groups of the bisPNAs. Digital DNA molecules with attached fluorescent tags and antibodies are called capture units (CUs). Several CUs are combined with the sample, and then analytes bind to the antibodies of the corresponding CUs (Fig. 1b), followed by addition of red-labeled secondary antibodies. After excess secondary antibodies are removed, CUs are interrogated within the microfluidic device14,15 (Fig. 1c and d). We measure fluorescence (excited by the light beams I–IV) from identifying tags, secondary antibodies, and the intercalated DNA backbone in separate detection channels 1–4.Digital DNA encoding is information-rich; each block can have a 0 or 1 value. As described below, we use a rigorous analysis of both the green bar code and the blue intercalator signal to discriminate between even closely related bar codes. Our longest molecule includes 20 blocks; at this length, multiplexing up to ∼2N−1 = 5.2 × 105 is possible (the use of 2N−1 corrects for sequences that are mirror images of one another). Up to 2000 unique identifiers are possible with the dodecameric Digital DNA described here, which is sufficient for many applications. If the DNA is designed for independent traces in two colors, up to ∼22N−1 = 8.4 × 106 unique identifiers are possible. In practice, the number of CUs is limited by library construction. However, we have produced over 40 Digital DNAs differing in sizes and bar codes with our simple modular approach. Once a Digital DNA is cloned, the plasmid is easily propagated in bacterial culture. CU assembly involves similar steps to those used to attach antibodies in any suspension array. Therefore, production of Digital DNA suspension arrays is no more complicated than production of current bead-based arrays, while offering significant advantages for increased multiplexing, compatibility with lab-on-chip devices and significantly lower batch to batch variability compared to polystyrene beads.
Digital DNA plasmids are constructed via stepwise ligation of the 0 and 1 blocks. The construction of these blocks and the plasmid vector used to propagate the DNA is described in detail in the ESI†. To add a block to the growing construct, the block was excised from its carrier plasmid with BamHI and XbaI restriction enzymes, then ligated to complementary 5′ overhangs in the Digital DNA plasmid, which was previously cut with BamHI and NheI (Fig. 2a). After ligation, the BamHI-specific site remained, but the NheI and XbaI sites were not present. Plasmids of the proper length were identified by pulsed-field gel electrophoresis with the Chef Mapper system (Biorad) (Fig. 2b). EcoRI digestion was used to confirm the proper sequence of block order, which also was confirmed by single-molecule analysis of bisPNA tagged DNA.
 | ||
| Fig. 2 Assembly of Digital DNA. (a) Assembly schematic: a plasmid with several connected blocks 0 and 1 is cut with BamHI and NheI restriction endonucleases. The obtained overhangs hybridize with complementary overhangs of block 1 excised with BamHI and XbaI enzymes. The assembly is ligated to yield block 10 and ready for addition of the next block. The NheI and XbaI sites are lost after hybridization. (b) Control of sizes of assembled Digital DNAs. Every plasmid was cut either with XbaI restriction enzyme to linearize the circular plasmid (lanes 2, 4, 6, 8, 10, and 12) or with NheI and XbaI restriction enzymes to excise the vector (lanes 3, 5, 7, 9, 11, and 13). The analyzed samples are Digital DNAs 10011101 (lanes 2–5), 1001110110 (lanes 6–9), 100111010010 (lanes 10 and 11), and 100111011011 (lanes 12 and 13). Lanes 2, 3, 6, and 7 include 4-site series Digital DNA. Lanes 1 and 14 contain a size standard (low range PFG Marker, NEB). (c) Confirmation of sequence of the blocks in Digital DNA. A single EcoRI site is located in the carrier plasmid 31 bases away from the BamHI site. An additional EcoRI site is located in the identification segment of block 1 but is absent from block 0. Digestion of the Digital DNA constructs with EcoRI enables independent confirmation of their proper construction. Lane 1, size standard (1 kb extension ladder, Invitrogen); lane 2, Digital DNA 0111; lane 3, Digital DNA 00111; lane 4, Digital DNA 100111; lane 5, Digital DNA 100111010100; lane 6, Digital DNA 100111011011. Digested DNA was fractionated by pulsed-field gel electrophoresis and stained with SYBR Green I. | ||
Detection of protein targets
We use averaged unoriented maps for analysis.15 These averages exhibit Gaussian peaks with FWHH (full width at half-height) of 1.5 µm at the identifying tag positions (Fig. 3a), instead of the single-bin spikes observed for single-molecule traces (Fig. 1d). These averages are the superposition of direct and inverted patterns, as half of the molecules transit the read in a “head-first” orientation and the rest go through tail-first (Fig. 3a). | ||
| Fig. 3 Specificity of detection of botulinum toxoid A (BT) with (100111010100) Digital DNA. (a) Unoriented maps are a superposition of multiple head-first (HF) and tail-first (TF) single-molecule traces. (b–e) Unoriented maps of anti-BT CUs measured in green (identification) and red (secondary antibodies) channels either without targets (b), with 50 nM of BT (c and e) or 3 × 109 pfu of MS2 phage (d). The secondary antibodies (also referred to as signal units or SUs) were either anti-BT (b, d and e) or anti-MS2 (c). Every map is an average of 1600 to 4200 single traces. Signal intensity is plotted against the distance from the center of DNA molecule. | ||
We tested the sensor with a sandwich immunoassay for botulinum toxoid A (BT) against samples containing no antigen, the correct target (BT), or an incorrect antigen (bacteriophage MS2) (Fig. 3). In every case a correct (green) identification map was detected, as every CU carried these tags (Fig. 3b–e). However, the (red) assay map was only detected in the presence of the correct antigen and secondary antibody (Fig. 3e). In all other cases, the red signal was indistinguishable from background. Thus, a complete antibody sandwich can be formed only with correct antigen, attachment of the antibody to Digital DNA does not prevent binding, and the immuno-sandwich structure survives transport and stretching within the chip.
Classification of DNA mixtures in a multiplex format
The enormous information content in long DNA molecules opens up the potential for highly multiplexed immunoassays. A key requirement for exploiting this advantage is the ability to accurately classify single DNA molecules based on their bar codes. Digital DNA is designed to have very bright signals at the “1” positions, with little background fluorescence between peaks. In addition, the intercalator signal allows us to measure the length precisely and determine the ends of the molecule relative to the bar code, precluding misclassification due to under- or overstretching. We have tested various classification algorithms by setting up an experimental worst-case scenario. Two DNA species with distinct bar codes (#1: 100111010100 and #2: 111111111111) were combined in an equimolar ratio. The DNA with bar code #2 also carried Alexa 647-labeled antibodies conjugated to the bisPNA tag, generating a red bar code identical to the green identification bar code on these molecules (Fig. 4). The DNA bearing bar code #1 was left unmodified. This mimics the situation where one target for a multiplex assay is present in saturating amounts, and any misclassification of these heavily red-labeled DNAs could lead to a false-positive signal for another target. This misclassification would then be the limiting factor for the sensitivity of a multiplex assay. To ensure that only uniformly stretched DNA molecules were analyzed, we used the width of the intercalator signal to measure the length of each event, and selected only those events that are at or near the expected contour length for this DNA, as described previously.14–16 A total of 2165 DNA events were recorded for this mixture, and the individual bar codes for each molecule were compared to a calculated template for the expected bar code. The similarity between the observed bar code and the anticipated template was quantified using Pearson's linear correlation (r).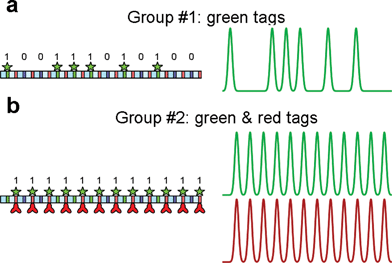 | ||
| Fig. 4 Discrimination of different DNA bar codes used to test single-molecule classification. Digital DNA (100111010100) was used to prepare both species. (a) To obtain group #1, bisPNA tag p2368 with TMR fluorophore was hybridized to the sites on the signal segments. (b) To obtain group #2, bisPNA tag p368 with TMR fluorophore was hybridized to the sites on the receptor segments. Since both block 0 and block 1 contain p368 sites, the resulting bar code is (111111111111). Alexa 647-labeled antibodies were conjugated to the bisPNA tag for group #2 molecules, generating a red bar code identical to the green identification bar code. | ||
In the simplest case, all molecules were included in the analysis; each molecule was assigned to either group #1 or #2 depending on which template was more correlated with the experimental bar code (Table 2). Because only group #2 molecules should have red signals, the classification performance could be independently verified. The molecule was considered incorrectly classified if either group #1 molecule carried at least one red peak >20 photons, or group #2 molecule carried no red peak above 20 photons. That way we determined that 148 molecules were misclassified, corresponding to 6.8% of the total.
| No filtering | Backbone filtera | r > 0.5 filterb | Backbone and r > 0.5 filters | |
|---|---|---|---|---|
| a Backbone filter excluded molecules if the profile of the fluorescence signal from the backbone-bound intercalator indicated stretching anomalies, usually molecules with a hairpin conformation. b Pearson's linear correlation (r) between the observed bar code and the anticipated template. c Incorrectly classified group #1 molecules exhibited fluorescence in red channel. Incorrectly classified group #2 molecules exhibited no fluorescence in red channel. d This is the fraction of the total molecules that passed the backbone and/or classification quality filters. | ||||
| Total events | 2165 | 2165 | 2165 | 2165 |
| Backbone OK | 2165 | 1562 | 2165 | 1562 |
| Classified as group #1 | 921 | 644 | 690 | 519 |
| Classified as group #2 | 1244 | 918 | 833 | 660 |
| Unclassified | 0 | 0 | 642 | 383 |
| Group #1 with redc | 99 | 51 | 6 | 3 |
| Group #2 without redc | 49 | 33 | 2 | 1 |
| % Misclassified | 6.8 | 5.4 | 0.5 | 0.3 |
| % Usable moleculesd | 100 | 72 | 70 | 54 |
To improve classification, we introduced extra criteria to eliminate poorly measured molecules. First, we applied a backbone filter, which excluded molecules if the fluorescence signal from the backbone-bound intercalator indicated stretching anomalies, caused by the inhomogeneous nature of DNA stretching in mixed elongational flow.16 The filter removed all molecules that contained any bins with fluorescence intensity exceeding the mean intercalator signal by at least 50%, eliminating molecules that have short folded-over regions on the DNA which are not big enough to affect the length measurement significantly, but could still affect classification of closely related bar codes. This filter removed 28% of the molecules. When the remaining traces were classified, the error dropped to 5.4%. In the second approach, we filtered out molecules with poor similarity to any of the expected bar codes; only traces with a correlation coefficient of at least 0.5 to one of the expected templates were kept for further analysis. This filter removed 30% of the observed molecules. When the remaining molecules were classified, the error rate dropped to 0.5%. Finally, if both filters were combined, 44% of the molecules were removed, and the classification error on the remainder was 0.3%. Ultimately, the fidelity of bar code classification depends on the efficiency and specificity of tag binding. BisPNA tags have proven to be extraordinarily versatile probes for detection of genomic DNA species, but they do bind reversibly and have significant affinity for single-mismatch sites. We have developed a new tagging technology based on photo-crosslinkable oligos that demonstrates excellent classification efficiency in a mixture of 12 different Digital DNA species.23 Work is ongoing to attach capture probes to these molecules to create a highly multiplexed assay.
To assess the assay performance in a mixture of antigens, we applied the mixture of CUs against BT and coat protein of the MS2 phage (anti-BT and anti-MS2, respectively) in various combinations (Fig. 5). We separated the signals by the number of the CU peaks (Fig. 5a). The sorted molecules exhibited correct green identification traces (Fig. 5b). Detection patterns for anti-BT and anti-MS2 CUs (Fig. 5c and d, respectively) were observed in the red channel only with correct antigens.
 | ||
| Fig. 5 Multiplexed detection of botulinum toxoid (BT) and MS2 virus. A mixture of (100111010100)-anti-MS2 and (111111111111)-anti-BT was used for the assay. (a) Detected CUs were grouped by the number of green peaks. (b) Each group had the expected ID barcode. Average detection channel traces were measured for (c) anti-BT and (d) anti-MS2, CUs with BT (red), MS2 (blue), BT + MS2 (black), or blank (green). Assays used 63 pM of each CU; 25 nM of each secondary antibody; 25 nM of each antigen, when included. 800 to 2400 single traces were averaged in each map. Signal intensity is plotted as in Fig. 3b–e. | ||
Assay sensitivity
We have detected follicle-stimulating hormone (FSH) at various concentrations to determine the assay sensitivity (Fig. 6a). The red detection threshold was determined as the average level of background in the red channel of five antigen-free samples plus two standard deviations. The detection limit was about 5 pM, on par with many bead-based immuno assays,24 although subpicomolar sensitivity was reported recently for bead arrays25 and single-molecule detection.26,27 Sensitivity can be improved by decreasing the concentration of CUs24 and with more efficient removal of secondary antibodies. When a specialized mini-reactor19 is used to rid the reaction mixture of uncomplexed secondary antibodies, the red signal from blank samples is dramatically reduced, allowing for confident detection of FSH at concentrations as low as 660 fM, corresponding to 6.6 × 10−17 mol of antigen (Fig. 6b). The signal increases linearly with analyte concentration over 3 log units, indicating that the working range of this technique will cover most biologically relevant assays. | ||
| Fig. 6 Dependence of the FSH assay signal on the antigen concentration. The normalized signal of the red channel was measured at different antigen concentrations. The concentrations of capture units and signal antibodies were 0.03 and 5 nM, respectively. The threshold level (dashed line) is set at the average red background level plus two standard deviations. The average pattern of red signal on Digital DNA at selected FSH concentrations is shown in the insets. Between 3800 and 8500 molecules were averaged in each case. Results are reported for an FSH sandwich immunoassay cleaned up with either drop-dialysis (a), or in a custom mini-reactor (b). | ||
Further improvements in assay sensitivity will involve integration of sample preparation onto the microfluidic chip itself, as this will allow for smaller volumes and the use of a minimal amount of the CUs. We have performed preliminary experiments with an on-chip device called the DNA prism.20 This device was originally designed for efficient size-separation of long DNA (100's of kb), but also works extremely well as a mechanism for cleanup of uncomplexed secondary antibodies from our Digital DNA-based immunoassay. In this experiment, a 20 µl reaction was processed using an integrated microfluidic chip that combines DNA stretching, cleanup with a DNA prism, and concentration on acrylamide gel surfaces.28 Details of this device will be published elsewhere. The prism consists of a hexagonal array of posts that interact with long DNA molecules as they are pulled through the device with an alternating pair of electric fields (Fig. 7a). In the first phase, voltage is applied at an angle across the postfield, pulling DNA through the channel between posts. The field then switches to a horizontal direction. Long DNA (>100 kb) hooks on the adjacent posts, preventing these molecules from following the horizontal field. In contrast, short DNA molecules and antibodies are small enough to avoid the posts, and are diverted from the long DNA stream in the horizontal direction. Over many such pulses, the Digital DNA is separated from the free antibodies, ultimately shunted into a different exit port. The port containing the Digital DNA molecules is fluidically connected with the DNA-stretching funnel, where analysis occurs. As shown in Fig. 5b, the red bar code on Digital DNA is barely seen over the background of red signal from free detection antibodies in the original mixture, preventing detection. After sample cleanup in the DNA prism, the background red signal from free secondary antibodies disappears, leaving only the optical background signal. Further work is necessary to assess the benefits of on-chip sample processing on assay sensitivity, but this proof-of-principle experiment demonstrates that all of the fundamental steps can be performed with an integrated microfluidic chip.
 | ||
Fig. 7 On-chip removal of secondary antibodies with DNA prism. A 20 µl reaction mix containing 3 × 109 pfu of MS2 phage was prepared as described in Materials and Methods, then run through a chip containing the DNA prism (a) and DNA-stretching fluidics. A control reaction was prepared at the same time, but not cleaned up in the DNA prism. The red signal from the control sample (average of 304 molecules) run directly through the stretching funnel (b, red line) is obscured by the intense noise from the uncomplexed secondary antibodies. In contrast, the sample run through the DNA prism (b, purple line) has a clear red bar code (average of 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 913 molecules) well above the background noise, indicating confident detection. 913 molecules) well above the background noise, indicating confident detection. | ||
Multiplexing, sensitivity and throughput
In addition to the advantages the DNA-based reagents provide for manipulation in microfluidic chips, the flexibility of the DNA coil allows for increased encoding capacity with minimal increase in assay volume. Ten extra blocks are needed to increase the encoding potential of a Digital DNA array 103-fold. If these are added to 12-meric Digital DNA, its contour length Lc will increase (12 + 10)/12 = 1.8 times and its volume, which scales as Lc3/2, will increase only 2.4 times. In contrast, bead assays with intensity encoding require a 103-fold larger bead to achieve the same increase in encodable signals, as the amount of impregnated fluorophore scales proportionally with particle volume. Both assay formats get an exponential encoding increase with the use of additional colors. Therefore, the Digital DNA-based assay can provide enormous encoding potential with a minimal increase of the “bead” volume.Particle volume is a fundamental limit to assay sensitivity, as it affects both sample size and required excitation volume. Macro-scale solution handling necessitates ≥10 µl samples, and includes a huge excess of targets over capture agents.24,25,29 Microchip assays reduce this waste, and allow for integration and automation of the assay. We can manipulate and measure the DNA “bead” in 1 µm deep chips, where the DNA is stretched into a linear conformation, then excited with a diffraction-limited laser spot (d ≈ 0.5 µm diameter). For a bead assay, the whole bead must be illuminated to excite the fluorescence of the secondary antibody. As noise is proportional to illuminated volume,30 the limiting signal-to-noise ratio for a Digital DNA-based assay is (D/d)3 = 103 times higher than that for the D = 5 µm beads typically used.24 Finally, using negatively charged DNA obviates the need for pre-blocking capture agents to reduce background signal, as CU's do not self-associate or adhere to the chip walls (a major problem for submicron polystyrene beads). DNA can also be manipulated within the chip by electric field, enabling extraction and purification protocols similar to those employed with magnetic beads, which facilitates automation and miniaturization of analysis.
A critical feature of any suspension array is the read rate for the encoded particles. Our reader scans DNA in constant hydrodynamic flow, which offers significant throughput advantages over other DNA-encoding strategies that require immobilization prior to measurement.31 For the experiments reported here, we were focused on proof-of-principle demonstration, not speed. As such, we drove the DNA through the chip at relatively low concentrations (0.5 ng µl−1) and speeds (15 µm ms−1). Reliable detection of targets in the duplex assay described in Fig. 4 required acquisition times of ∼10 min. The acquisition time will scale with the multiplexing of the assay, since ∼1000 molecules of each type need to be read to achieve reasonable confidence that a target is or is not present in the sample. Therefore, a 12-plex assay (typical for suspension array assays) using the same parameters would require an hour of read time, which may not be acceptable for some applications. Assay throughput in the chip can be improved by increasing the stretching velocity and/or decreasing DNA length. We currently drive DNA at 20 µm ms−1, but we have designed microchips for velocities up to 50 µm ms−1. As both identification sites and secondary antibodies carry multiple fluorophores, measurements can be performed at this velocity without considerable loss of sensitivity. In addition, our Digital DNA blocks can be shrunk from 10 kb to 4.5 kb (3 µm) given our current optical resolution. Shorter molecules can be read at higher concentrations, while still avoiding overlaps between adjacent molecules. The required size to achieve a given multiplexing can also be reduced with double-color coding; a trimer is sufficient to code more than a dozen targets with a 2-color encoding, which gives the overall length of 9 µm for this minimal DNA. With a DNA occupancy of 1/3 to prevent overlap in the detection channel (corresponding to a concentration of ∼6 ng µl−1) and 103 CUs per target, the minimum reading time for a 12-plex assay will be ∼6.5 s, insignificant compared to the tens of minutes to hours needed for sample preparation and incubation.24,25,32
Practical considerations
The ability to tolerate “dirty” samples is critical to the success of any assay. In our proof-of-principle experiments described here, we do not explicitly address this issue, but our preliminary experience suggests design constraints for sample cleanup prior to detection in the chip. First, any particles capable of clogging the microfluidics must be removed prior to introduction to the chip. Second, any background fluorescence in the sample must be mitigated before detection. Fluorescent species in the blue channel will interfere with our assessment of Digital DNA stretching. The presence of green-fluorescent particles would impact bar code classification, and red-fluorescing species would obscure target detection. For example, serum samples exhibit significant green fluorescence due to the presence of bilirubin protein.33 Both of the automated platforms for detection antibody cleanup, the mini-reactor and the DNA prism, have shown excellent performance to separate Digital DNA from small molecules and globular proteins, which encompass most of the autofluorescent background in typical samples. Demonstration of the robustness of these cleanup technologies to a variety of sample types is ongoing. Finally, the assay conditions must maintain the integrity of both our DNA “beads” and the capture agents, so components that may denature or degrade the nucleic acid and/or protein antibodies must be removed from the sample prior to analysis.Conclusion
We report a novel suspension array technology built around a specialized DNA molecule—Digital DNA. Since our assay components are based on proteins and nucleic acids, the particles are completely biocompatible. The technology offers a practically unlimited degree of multiplexing, small particle size, and a corresponding high-throughput reader. Preliminary experiments with automated cleanup of secondary antibodies show that detection of <1 pM target concentration is possible with this technology. Work is ongoing to build chips capable of performing the entire assay in a microfluidic environment, which we expect will further improve the sensitivity in terms of both target concentration and sample volume.Acknowledgements
We gratefully acknowledge D. Crothers, D. Whitney, and D. Hoey for critical comments on the manuscript. The FSH detection antibodies were suggested by E. Nalefski. This research was partially funded by NSF SBIR grant DMI-0320449 and US Department of Homeland Security, Science and Technology Directorate grant HSHQPA-05-9-0019.References
- K. Braeckmans, S. C. De Smedt, M. Leblans, R. Pauwels and J. Demeester, Nat. Rev. Drug Discovery, 2002, 1, 447–456 CrossRef CAS.
- E. J. Moran, S. Sarshar, J. F. Cargill, M. M. Shahbaz, A. Lio, A. M. M. Mjalli and R. W. Armstrong, J. Am. Chem. Soc., 1995, 117, 10787–10788 CrossRef CAS.
- K. C. Nicolaou, X.-Y. Xiao, Z. Parandoosh, A. Senyei and M. P. Nova, Angew. Chem., Int. Ed. Engl., 1995, 34, 2289–2291 CrossRef CAS.
- B. J. Battersby, D. Bryant, W. Meutermans, D. Matthews, M. L. Smythe and M. Trau, J. Am. Chem. Soc., 2000, 122, 2138–2139 CrossRef CAS.
- S. R. Nicewarner-Pena, R. G. Freeman, B. D. Reiss, L. He, D. J. Pena, I. D. Walton, R. Cromer, C. D. Keating and M. J. Natan, Science, 2001, 294, 137–141 CrossRef CAS.
- K. Braeckmans, S. C. De Smedt, C. Roelant, M. Leblans, R. Pauwels and J. Demeester, Nat. Mater., 2003, 2, 169–173 CrossRef CAS.
- H. Fenniri, S. Chun, L. Ding, Y. Zyrianov and K. Hallenga, J. Am. Chem. Soc., 2003, 125, 10546–10560 CrossRef CAS.
- Z.-l. Zhi, Y. Morita, Q. Hasan and E. Tamiya, Anal. Chem., 2003, 75, 4125–4131 CrossRef CAS.
- D. C. Pregibon, M. Toner and P. S. Doyle, Science, 2007, 315, 1393–1396 CrossRef CAS.
- J. P. Nolan and L. A. Sklar, Trends Biotechnol., 2002, 20, 9–12 CrossRef CAS.
- M. Han, X. Gao, J. Z. Su and S. Nie, Nat. Biotechnol., 2001, 19, 631–635 CrossRef CAS.
- H. Xu, M. Y. Sha, E. Y. Wong, J. Uphoff, Y. Xu, J. A. Treadway, A. Truong, E. O'Brien, S. Asquith, M. Stubbins, N. K. Spurr, E. H. Lai and W. Mahoney, Nucleic Acids Res., 2003, 31, e43 CrossRef.
- X. Su, J. Zhang, L. Sun, T.-W. Koo, S. Chan, N. Sundararajan, M. Yamakawa and A. A. Berlin, Nano Lett., 2005, 5, 49–54 CrossRef CAS.
- E. Y. Chan, N. M. Goncalves, R. A. Haeusler, A. J. Hatch, J. W. Larson, A. M. Maletta, G. R. Yantz, E. D. Carstea, M. Fuchs, G. W. Wong, S. R. Gullans and R. Gilmanshin, Genome Res., 2004, 14, 1137–1146 CrossRef CAS.
- K. M. Phillips, J. W. Larson, G. R. Yantz, C. M. D'Antoni, M. V. Gallo, K. A. Gillis, N. M. Goncalves, L. A. Neely, S. R. Gullans and R. Gilmanshin, Nucleic Acids Res., 2005, 33, 5829–5837 CrossRef CAS.
- J. W. Larson, G. R. Yantz, Q. Zhong, R. Charnas, C. M. D'Antoni, M. V. Gallo, K. A. Gillis, L. A. Neely, K. M. Phillips, G. G. Wong, S. R. Gullans and R. Gilmanshin, Lab Chip, 2006, 6, 1187–1199 RSC.
- J. R. Krogmeier, I. Schaefer, G. Seward, G. R. Yantz and J. W. Larson, Lab Chip, 2007, 7, 1767–1774 RSC.
- H. Riethman, B. Birren and A. Gnirke, in Analyzing DNA, ed. B. Birren, E. D. Green, S. Klapholz, R. M. Myers and J. Roskams, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1997, pp. 83–248 Search PubMed.
- E. T. Mollova, V. A. Patil, E. Protozanova, M. Zhang and R. Gilmanshin, Anal. Biochem., 2009, 391, 135–143 CrossRef CAS.
- L. R. Huang, J. O. Tegenfeldt, J. J. Kraeft, J. C. Sturm, R. H. Austin and E. C. Cox, Nat. Biotechnol., 2002, 20, 1048–1051 CrossRef CAS.
- E. Chan, U.S. Patent number 6355420, U.S. Patent and Trademark Office, U.S. Genomics, Inc., USA, 2002.
- P. E. Nielsen and M. Egholm, in Peptide Nucleic Acids. Protocols and Applications, ed. P. E. Nielsen, Horizon Bioscience, Norfolk, 2004, pp. 1–36 Search PubMed.
- V. Papkov, Y. Wu, T. Foss, A. L. Dow, E. J. White, N. L. Pellerin, N. Mamaeva, R. Gilmanshin and R. E. Burton, Biophys. J., 2009, 96(Suppl. 1), 25a CrossRef.
- D. A. A. Vignali, J. Immunol. Methods, 2000, 243, 243–255 CrossRef CAS.
- C. Heijmans-Antonissen, F. Wesseldijk, R. J. M. Munnikes, F. J. P. M. Huygen, P. van derMeijden, W. C. J. Hop, H. Hooijkaas and F. J. Zijlstra, Mediat. Inflamm., 2006, 2006, 1–8 Search PubMed.
- J. Todd, B. Freese, A. Lu, D. Held, J. Morey, R. Livingston and P. Goix, Clin. Chem. (Washington, D. C.), 2007, 53, 1990–1995 CrossRef CAS.
- H. Qiu, E. P. Ferrell, N. Nolan, B. H. Phelps, R. Tabibiazar, D. H. Whitney and E. A. Nalefski, Clin. Chem. (Washington, D. C.), 2007, 53, 2010–2012 CrossRef CAS.
- L. W. Kwok, Y. Zhou, B. Crane, L. Knaian, K. V. Vyavahare, R. H. Meltzer, J. Griffis, A. Edmonson, Q. Zhong, R. Allen, W. Mesadieu, J. W. Larson, J. R. Krogmeier and R. Gilmanshin, Biophys. J., 2009, 96(suppl. 1), 48a CrossRef.
- R. J. Fulton, R. L. McDade, P. L. Smith, L. J. Kienker and J. R. Kettman, Jr, Clin. Chem. (Washington, D. C.), 1997, 43, 1749–1756 CAS.
- M. Eigen and R. Rigler, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5740–5747 CAS.
- G. K. Geiss, R. E. Bumgarner, B. Birditt, T. Dahl, N. Dowidar, D. L. Dunaway, H. P. Fell, S. Ferree, R. D. George, T. Grogan, J. J. James, M. Maysuria, J. D. Mitton, P. Oliveri, J. L. Osborn, T. Peng, A. L. Ratcliffe, P. J. Webster, E. H. Davidson and L. Hood, Nat. Biotechnol., 2008, 26, 317–325 CrossRef CAS.
- M. T. McBride, S. Gammon, M. Pitesky, T. O'Brien, T. Smith, J. Aldrich, R. G. Langlois, B. Colston and K. S. Venkateswaran, Anal. Chem., 2003, 75, 1924–1930 CrossRef CAS.
- Y. Chen and J. Potter, Clin. Chem. (Washington, D. C.), 1992, 38, 2426–2430 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of Digital DNA construction and algorithms used to classify single molecule traces. See DOI: 10.1039/b922106a |
| ‡ These authors contributed equally to this work. |
| § All authors are currently or were employed by US Genomics, Inc. and involved in research on its behalf. R.E.B., E.J.W., T.R.F., R.H.M., N.V.M. and R.G. hold stock and/or stock options in U.S. Genomics, Inc. |
| This journal is © The Royal Society of Chemistry 2010 |